

Abstract
The tumor suppressor, microRNA-34 (miR-34), a transcriptional target of TP53, functions in a positive feedback loop to activate TP53. Although miR-34 can inhibit cancer cells carrying TP53 mutations, this feedback to TP53 may be a prerequisite for full miR-34 function and may restrict its therapeutic application to patients with intact TP53. To investigate the functional relationships between TP53 and miR-34, and that of other TP53-regulated miRNAs including miR-215/192, we have used a panel of isogenic cancer cell lines that differ only with respect to their endogenous TP53 status. miR-34–induced inhibition of cancer cell growth is the same in TP53-positive and TP53-negative cells. In contrast, miR-215/192 functions through TP53. In the absence of TP53, miR-34, but not miR-215/192, is sufficient to induce an upregulation of the cell cycle-dependent kinase inhibitor p21CIP1/WAF1. We identify histone deacetylase 1 (HDAC1) as a direct target of miR-34 and demonstrate that repression of HDAC1 leads to an induction of p21CIP1/WAF1 and mimics the miR-34 cellular phenotype. Depletion of p21CIP1/WAF1 specifically interferes with the ability of miR-34 to inhibit cancer cell proliferation. The data suggest that miR-34 controls a tumor suppressor pathway previously reserved for TP53 and provides an attractive therapeutic strategy for cancer patients irrespective of TP53 status.
Introduction
MicroRNA-34 (miR-34) is a potent tumor suppressor that shows a loss of function in many solid and hematological cancer types.1,2,3,4 It inhibits a broad range of cancer cells, presumably by repressing a plethora of oncogenes that control proliferation, senescence, apoptosis, and metastasis.5,6 miR-34 can also interfere with the growth of cancer stem cells,7,8 providing a strong rationale for the development of a miR-34 therapy. Evidence for the therapeutic application of miR-34 has been generated in murine tumor models of lung, liver, prostate, and lymphoma that showed robust tumor inhibition in response to the systemic delivery of nanoparticles loaded with synthetic miR-34 mimics.6,8,9,10,11
Much insight into the role of miR-34 has been added by recent reports demonstrating that the tumor suppressor TP53 (p53) transcriptionally induces the expression of all three miR-34 family members – miR-34a/b/c.12,13,14,15,16 TP53 also elevates the endogenous levels of miR-215, miR-192, and miR-194, all of which have the ability to inhibit cancer cell growth in culture.17,18,19 Although miR-215 and miR-192 are encoded on separate genomic loci, they share identical seed sequences (90.5% overall sequence homology) and are collectively referred to as miR-215/192. For some miRNAs, the positive regulation between TP53 and miRNA is reciprocal – miR-215/192 stimulates TP53 activity by repressing MDM2 (also referred to as HDM2), a ubiquitin ligase that negatively regulates TP53 stability via proteasomal degradation.19,20,21 Similarly, miR-34a activates TP53 in a positive feedback loop by repressing SIRT1 (silent information regulator 1), a nicotinamide adenine dinucleotide-dependent deacetylase that deactivates TP53, MDM4, a MDM2-like protein that negatively regulates TP53 transactivation, and YY1, a transcription factor that binds to a subset of TP53 DNA binding sites.22,23,24
While available data support the view that TP53 enhances the inhibitory activity of miR-215/192,19 a requirement for TP53 in miR-34–induced tumor suppression is controversial and the actual contribution of TP53 is unknown. Although previous studies suggest that miR-34 is also effective in cancer cells expressing mutated TP53,8,10,19 it is nevertheless possible that TP53 is a functional requirement for the full antiproliferative phenotype of miR-34. Given the high mutation rate of TP53 in cancer, this prerequisite may substantially limit the application of a miR-34–based therapy to patients with intact TP53. Here, we investigated the role of TP53 under physiological conditions and directly addressed TP53-dependent and -independent effects by using a panel of isogenic cancer cell lines in which the two TP53 alleles have been sequentially inactivated via targeted homologous recombination.25,26 We show that the miR-34–induced inhibition of cancer cell proliferation is the same in TP53-deficient and TP53 wild-type cells. These effects depend on the cell cycle-dependent kinase inhibitor p21CIP1/WAF1 (p21, CDKN1A) that is upregulated by a TP53-independent mechanism and involves the miR-34–mediated repression of histone deacetylase 1 (HDAC1). Therefore, p21 appears to be a critical effector molecule downstream of miR-34 and illustrates how miR-34 bypasses TP53 to function as a TP53-independent tumor suppressor.
Results
Inhibition of cancer cell proliferation by miR-34 is independent of TP53
Isogenic cells used in this study were derived from the MCF10A breast cancer and the colorectal carcinoma cell lines SW48, HCT116, RKO, and DLD-1 (Supplementary Table S1). In these cells, TP53 is either wild-type (+/+), heterozygous (+/−), or homozygously inactivated (−/−).25,26 Parental DLD-1 cells (DLD-1S241/SIL) do not express a functional TP53 protein due to the S241F/SIL TP53 genotype in which one allele is mutated and the other is epitopically silenced. Therefore, DLD-1+/SIL cells, in which the point mutation has been corrected by site-directed mutagenesis, serves as the DLD-1 reference line with intact TP53.25 Each nonisogenic cell line displays mutations in other tumor suppressor genes and oncogenes which may influence the expression and function of miRNAs (Supplementary Table S1).
To confirm the serial inactivation of TP53 in isogenic cell lines, we induced a TP53 response by exposing the cells to the DNA-damaging agent etoposide for 28 hours and collected total RNA. A quantitative reverse transcription-PCR (qRT-PCR) analysis showed an allele-dependent increase in TP53 mRNA and TP53-regulated target genes according to their genotype (Supplementary Figure S1). TP53 mRNA was not detectable in TP53−/− cells. Increased mRNA levels of TP53-regulated genes are similar to published data27 and varied between cell lines, presumably due to cell-type–specific regulation of these genes. Likewise, the induction of TP53-regulated miRNAs, miR-34a/b/c, miR-192, miR-194, and miR-215 was dependent on the cell line – all cell lines but DLD-1+/SIL lacked miR-34b/c expression, and miR-215 was solely detectable in SW48 and DLD-1 cells.
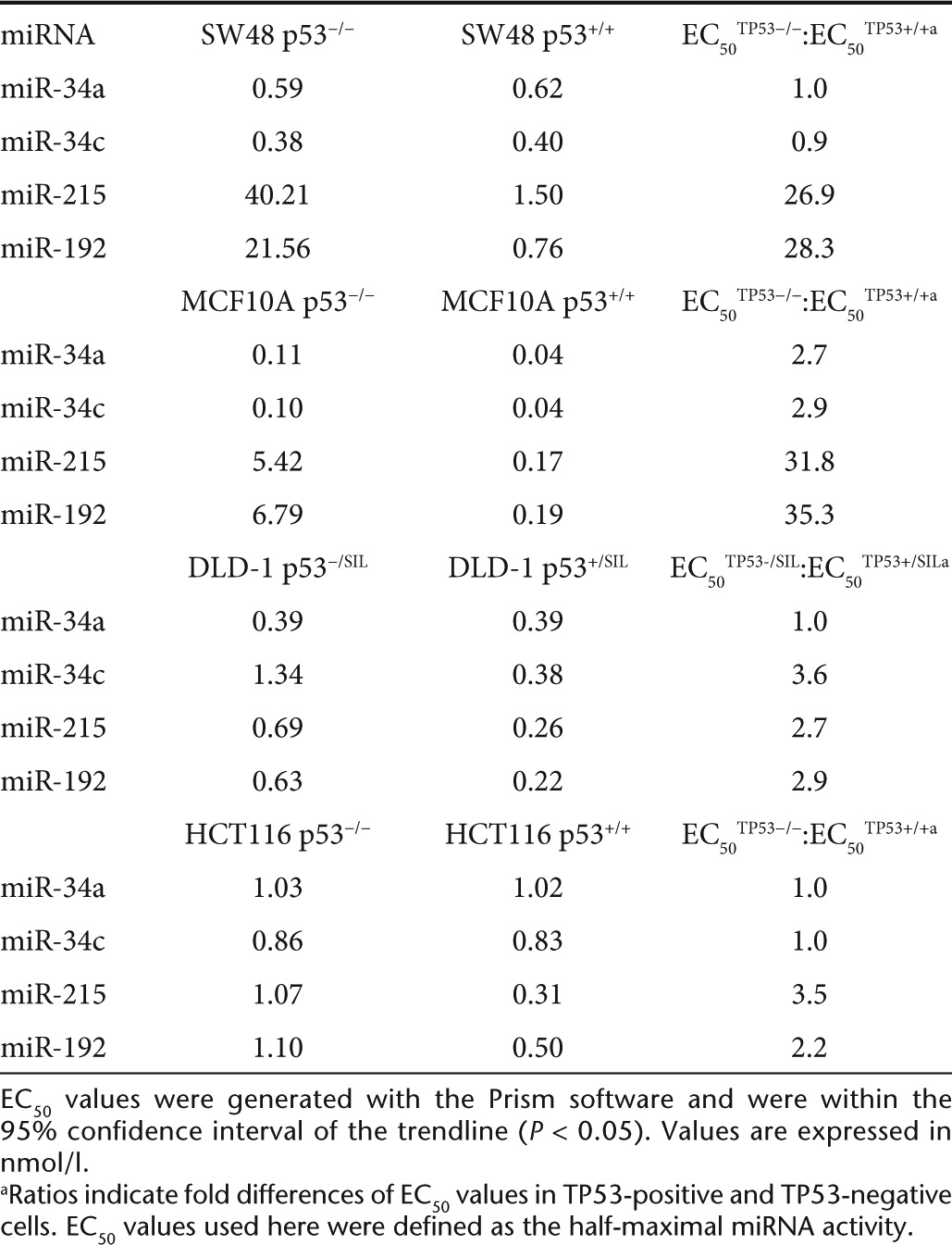
Next, we transfected isogenic cells with mimics of miR-34a, miR-34c, miR-192, miR-194, and miR-215. The miRNAs were used in a serial dilution to generate dose–response curves and to calculate EC50 values. As negative controls, mock-transfected cells and cells transfected with a miRNA carrying a scrambled sequence were used (miR-NC). After 3–4 days of incubation, cellular proliferation was assessed using AlamarBlue (Invitrogen, Carlsbad, CA). As shown in Figure 1, miRNAs mimics inhibited cellular proliferation by ~40–80% compared with controls. TP53 enhanced the ability of miR-215 and miR-192 to inhibit cancer cells which was greatest in MCF10A and SW48 cells with EC50 values ~28- 35-fold lower compared with TP53−/− cells (Table 1). In contrast, the inhibitory activity of miR-34a and miR-34c was the same in TP53-positive and TP53-negative cells (Figure 1 and Table 1; Supplementary Figure S2). RKO cells showed greater inhibition in the absence of TP53, further demonstrating that TP53 is not a prerequisite for the miR-34–induced phenotype (Supplementary Figure S3). Interestingly, in the presence of intact TP53, the maximal inhibitory activity of miR-215/192 was greater than the maximal activity of miR-34a/c, suggesting that miR-215/192 functions in the TP53 positive feedback loop and takes advantage of ancillary pathways exclusively regulated by TP53.19
Figure 1.

Inhibition of cancer cell proliferation by miRNAs in the presence or absence of TP53. TP53 genotype-dependent effects of miRNAs in isogenic cancer cell lines. Cells expressing (+/+; +/SIL) or lacking functional (−/− −/SIL) TP53 were transfected with increasing concentrations of miRNAs ranging from 0.01 to 30 nmol/l. After 3 days, cellular proliferation was determined. Data are normalized to mock-transfected cells. Averages, standard deviations, and nonlinear regression trendlines are shown.
Table 1. EC50 values of miRNAs in isogenic cancer cells.
miR-34a induces p21CIP1/WAF1 in the absence of TP53
To understand the miR-34–induced phenotype in TP53-positive and TP53-deficient cells, we determined the expression levels of genes involved in the TP53/miR-34 axis. An explanation for the TP53-independent effects is the possibility that these cells do not express endogenous SIRT1 or MDM4. However, as confirmed by qRT-PCR, both TP53+/+ and TP53−/− cells carry detectable SIRT1 and MDM4 mRNA levels, suggesting that the TP53-independent phenotype is not due to an absence of these gene products (Figure 2a). Rather, both mRNAs were reduced in cells transfected with miR-34, in accordance with experimental data showing that SIRT1 and MDM4 are directly targeted by this miRNA.22,23 Similarly, MET, a miR-34a target, and BCL2, a miR-215 target, were specifically downregulated in cells transfected by the respective miRNAs (Figure 2a). TP53 mRNA levels were not detectable in SW48−/− cells in accord with its defined genotype. In SW48+/+ cells, TP53 mRNA levels were constant and is in agreement with the hypothesis that the positive feedback loop to TP53 by these miRNAs does not require TP53 de novo synthesis but occurs post-transcriptionally. This is further corroborated by the observation showing that miR-215 induces the expression of p21CIP1/WAF1 (p21, CDKN1A) in TP53-positive cells, but fails to do so in TP53-deficient cells. Unexpectedly, miR-34a was able to induce p21 not only in TP53+/+ cells, but also in TP53-deficient cells (Figure 2a,b).
Figure 2.

Induction of p21CIP1/WAF1 by miR-34a in TP53-deficient cells. (a) Endogenous expression levels of target genes functioning in the miR-34a/TP53 axis were determined by quantitative reverse transcription-PCR (qRT-PCR) using RNA from SW48−/− and SW48+/+ cells transfected with either miR-34a or miR-215. Statistical significance of differential gene expression was determined by Student's t test (*P < 0.5; **P < 0.1; miRNA versus mock). (b) qRT-PCR results showing p21 mRNA levels in isogenic cell lines transfected with miR-34a. (c) Nonlinear regression analysis of p21 expression levels and proliferation rates in RKO−/− cells transfected with increasing concentrations of miR-34a ranging from 0.01 to 30 nmol/l. All values are normalized to those in mock-transfected cells (=1). Averages are shown. Standard deviations are included but are too small to be visible in the graph. *Statistical significance of p21 mRNA expression in miR-34a– versus miR-NC–transfected cells (Student's t test; P < 0.05). n, not detected.
HDAC1 is a direct target of miR-34a
A plausible explanation for the TP53-independent upregulation of p21 is a potential involvement of other TP53 family members, TP63 and TP73. Both proteins play roles distinct from TP53; however, they also control a set of genes that overlaps with that of TP53.28 To test this hypothesis, we measured endogenous mRNA levels of TP63 and TP73 in TP53 wild-type and TP53-deficient cells that had been transfected with miR-34a. However, none of the cells showed detectable mRNA levels of TP63 or TP73, suggesting that an involvement of these gene products is unlikely (data not shown).
Next, we focused on regulatory mechanisms that are independent of TP53 and searched for nuclear regulators that can control p21 expression. One candidate of interest was HDAC1 because it has a putative miR-34a binding site in its 3′ untranslated region (UTR) that is conserved across mammals and also present in other vertebrates (Figure 3a, Supplementary Figure S4). HDAC1 is downregulated in cells transfected with miR-34a (Figure 3b), and has previously been implicated in the transcriptional regulation of p21 in the absence of TP53.29 To establish whether HDAC1 is directly repressed by miR-34a, we examined whether miR-34a can repress a luciferase reporter that is fused to the entire HDAC1 3′ UTR. We transiently expressed this reporter in SW48−/− and H1299 cells that lack TP53 and express low levels of endogenous miR-34a (Supplementary Figure S1; ref. 10). Then, cells were transfected with miR-34a or miR-215, the latter of which is not predicted to repress HDAC1 and was used as a negative control. As shown in Figure 3c, transfection of miR-34a diminished luminescence by ~50% in both cell lines relative to controls. This repression was completely abolished upon mutation of the miR-34a binding site (Figure 3c), suggesting that the HDAC1 3′ UTR is directly targeted by miR-34a at this site. To further evaluate if the miR-34a–dependent repression of HDAC1 is reflected in human tumor specimens, we examined a cohort of 14 non–small-cell lung cancer samples previously used to document reduced miR-34a expression levels.10 Tumor HDAC1 mRNA and miR-34a levels were determined by qRT-PCR and normalized to the levels in their respective normal adjacent tissues. An analysis by the Pearson's method showed a statistically significant inverse correlation between HDAC1 mRNA and miR-34a levels (Figure 3d), supporting a role for miR-34a in the regulation of HDAC1 in human tumors.
Figure 3.

HDAC1 is a direct target of miR-34a. (a) miR-34a binding site in the 3′ UTR of the HDAC1 transcript. Base pairing of miR-34a with wt and mut HDAC1 3′ UTR sequences is shown. Lower case, miR-34a residues; upper case, mRNA residues; yellow, bases presumably involved in base pairing; bold, miRNA seed sequence; underlined, mut residues. (b) Quantitative reverse transcription-PCR (qRT-PCR) analysis showing HDAC1 mRNA levels in isogenic SW48 cells transfected with miR-34a. Values are normalized to those in mock-transfected cells. (c) miR-34a represses a luciferase transcript fused to the HDAC1 3′ UTR in SW48−/− colon cancer and H1299 lung cancer cells. Relative light units were normalized to those in miR-215–transfected cells (100%). P values were derived from two-tailed Student's t tests. (d) Inverse correlation of HDAC1 mRNA and miR-34 levels in a set of 14 tumors from non–small-cell lung carcinoma patients. Endogenous expression levels were determined by qRT-PCR. Correlation coefficient was generated by the Pearson's method; the P value was calculated by F test (GraphPad). CDS, coding sequence; HDAC1, histone deacetylase 1; mut, mutated; NS, nonsignificant; UTR, untranslated region; wt, wild-type.
Inhibition of HDAC1 mimics the miR-34a phenotype
Previous results implicated HDAC1 in the regulation of the p21 gene. For instance, HDAC1-deficient embryonic stem cells show elevated levels of p21, and inhibition of HDAC1 using the HDAC inhibitor trichostatin A can induce p21 expression in the absence of TP53.30,31 To confirm the TP53-independent induction of p21 upon depletion of HDAC1, we transfected TP53-negative cells with an small interfering RNA (siRNA) directed against HDAC1 and evaluated cell lysates by Western blotting. The results were compared to cells transfected with miR-34a or miR-215. Two cell lines were tested and included mock- and miR-NC–treated cells as negative controls. As expected, MET was solely downregulated in cells transfected with miR-34a, and HDAC1 protein was reduced by both miR-34a and the HDAC1 siRNA (Figure 4a; Supplementary Figure S5). Of note, both oligonucleotides induced a marked increase of p21 protein expression in these cells. This observation was in stark contrast to miR-215 that failed to induce p21 in TP53-deficient cells. However, transfection of miR-215 into TP53-positive cells led to an increase of p21 protein in TP53-positive cells (Supplementary Figure S6) in accord with the hypothesis that the miR-215–dependent induction of p21 is mediated by TP53 as a result of the positive feedback loop from miR-215 to TP53.19
Figure 4.

Knock-down of HDAC1 mimics the miR-34a phenotype. (a) Knock-down of HDAC1 by small interfering RNA (siRNA) and miR-34a induces p21CIP1/WAF1 expression in TP53-negative cells. Protein lysates from cells transfected with miR-34a, miR-215, or si-HDAC1 were probed by Western blotting. Met was used as a positive control for miR-34a transfection; actin was used as a loading control. (b) Knock-down of HDAC1 by siRNA similarly inhibits cellular proliferation of SW48−/− and SW48+/+ cells. Cells were transfected with siRNAs and miRNAs, and cellular proliferation was measured 3 days thereafter. Values are normalized to mock (=100%). Averages and standard deviations are shown. P values are derived from two-tailed Student's t tests. The dotted line denotes the level cellular proliferation in cells transfected with miR-34a. HDAC1, histone deacetylase 1.
To explore whether inhibition of HDAC1 can mimic the miR-34a phenotype, we measured the proliferation effects of an siRNA against HDAC1 in both TP53-positive and TP53-deficient SW48 cells. Cells were also transfected with a series of other siRNAs directed against gene products that can antagonize TP53 function. These genes include YY1, MDM4, and SIRT1, as well as a few others that are either validated or predicted miR-34a targets and were repressed in miR-34a–transfected cells (data not shown). Transient transfection of siRNAs led to >80% knock-down of target mRNAs as confirmed by qRT-PCR (Supplementary Figure S7). As controls, cells were also transfected with miR-34a and miR-215. We sought to identify siRNAs that yield a level of cancer cell inhibition that is similar in both cell lines. As expected, miR-34a equally inhibited SW48+/+ and SW48−/− cells, and the activity of miR-215 was dependent on TP53 (Figure 4b). Most siRNAs failed to reduce cellular proliferation in either cell type, including the siRNA against SIRT1 and YY1. Knock-down of MDM4 was able to inhibit proliferation of SW48+/+ cells but had no effect in SW48−/− cells. This is reminiscent of the miR-215 phenotype and confirms the role of MDM4 in modulating TP53 transactivation rather than DNA regulation.32 In contrast, knock-down of HDAC1 inhibited cancer cell growth that – similarly to miR-34a – was the same in both isogenic cell lines. Similar results were obtained from cells treated with trichostatin A (Supplementary Figure S8) further corroborating a role for HDAC1 in mediating a miR-34a response through p21.
Depletion of p21CIP1/WAF1 interferes with miR-34a–induced inhibition of cancer cell proliferation
The dose–response data generated in various cell lines suggest that p21 expression is a key event during miR-34a–induced inhibition of cancer cell proliferation. Expression levels of p21 markedly correlated with the ability of miR-34a to inhibit TP53-positive and TP53-negative cells. For instance, the inhibitory activity of miR-34a was the same in MCF10A and SW48 cells and correlated with similar p21 expression levels in both TP53−/− and TP53+/+ cells (Figures 1 and 2). HCT116 cells displayed greater p21 mRNA levels in TP53+/+ compared with TP53−/− cells, in accord with the slightly increased inhibitory activity of miR-34a in TP53+/+ cells at higher miR-34a concentrations (30 nmol/l, Figure 1). In RKO cells, p21 levels were higher in TP53−/− cells and mirrored the greater inhibition of proliferation in RKO−/− versus RKO+/+ cells (Supplementary Figure S3). The induction of p21 was also evident at low miR-34a concentrations and inversely correlated with inhibition of cell proliferation (Figure 2c).
To address whether p21 expression is required for the miR-34a–induced phenotype, we performed interference assays by cotransfecting cells with miR-34a and an siRNA directed against p21. As controls, cells were transfected with miR-34a, miR-215, or miR-NC. Each miRNA was supplemented with negative control oligo such that the total amount of transfected RNA equals the one of the miR-34a/si-p21 combination. The downregulation of targeted genes was verified by Western analysis (Figure 5a; Supplementary Figure S9). miR-34a alone reduced proliferation of RKO cells by ~20–30% and is in agreement with previous results (Figure 5b). In contrast, miR-34a in combination with the p21 siRNA had no effect on cancer cell proliferation despite the fact that miR-34a actively led to repression of HDAC1. This result suggests that p21 expression is indeed a necessary factor in mediating a miR-34 tumor suppressor response. The p21-dependent phenotype was reproducible in isogenic SW48 cancer cells (Supplementary Figure S10).
Figure 5.

Depletion of p21CIP1/WAF1 interferes with miR-34–induced inhibition of cancer cell proliferation. miRNA mimics and small interfering RNAs were transiently transfected into isogenic RKO cells as shown in the graph. Protein expression was verified by (a) Western analysis, and proliferation was assessed by (b) AlamarBlue. Proliferation data are normalized to cells transfected with negative control (100%). Averages and standard deviations are shown. P values were derived from two-tailed Student's t tests. HDAC1, histone deacetylase 1.
Discussion
The current paradigm views miR-34 as a cellular effector molecule that functions downstream of TP53 by repressing genes involved in cell cycle progression and apoptosis. Our data, however, suggest that miR-34 assumes a more central role that is independent of and in parallel to TP53 (Figure 6). Support for this hypothesis is provided here, showing that the inhibitory activity of miR-34 is the same in TP53-positive and TP53-negative cancer cells. These results pertain to a miR-34 mimic but may also shed light on the role of endogenous miR-34. Overexpression of miR-34 is sufficient to induce p21, a potent tumor suppressor otherwise known to be transcriptionally regulated by TP53 and necessary for the TP53 response. The primary functions of p21 involve cell cycle arrest by inhibiting cyclin-dependent kinases (CDKs) and blockage of DNA synthesis by binding to proliferating cell nuclear antigen.33 However, p21 can also inhibit other oncogenic pathways, including those regulated by WNT4, STAT3, MYC, and TERT.33 Thus, a key function of TP53 is evidently shared with miR-34. The existence of a separate miR-34 pathway is further illustrated by the TP53-independent transcriptional regulation of the miR-34a gene,34 as well as observations made in miR-34 knock-out mice that show an intact TP53 response in the absence of miR-34.35 miR-34 and TP53 may create an interface of two pathways with overlapping functions and activate each other reciprocally – TP53 via transcription and miR-34 via post-transcriptional repression of SIRT1, YY1, and MDM4 (Figure 6).
Figure 6.

TP53 and miR-34 function in parallel to activate p21 and to induce cell cycle arrest. TP53 transcription increases miR-34 expression, and miR-34 activates TP53 via repression of SIRT1, MDM4, and YY1. p21 expression can be directly induced by TP53, or indirectly by miR-34 through repression of HDAC1. Both TP53 and miR-34 may exert tumor suppression functions via p21 and other separate pathways. HDAC1, histone deacetylase 1.
In TP53-deficient cells, the miR-34–induced expression of p21 is an indirect effect of HDAC1 repression. HDAC1 has previously been implicated in the regulation of the p21 gene (CDKN1A). Supporting evidence comes from (i) HDAC1-deficient embryonic stem cells, that show elevated levels of p21, and (ii) p53-mutated human osteosarcoma cells in which p21 expression was induced after treatment with the HDAC inhibitor trichostatin A.30,31 In these studies, two Sp1 binding sites in the CDKN1A promoter were identified as trichostatin A-responsive elements, suggesting that – in the absence of TP53 – Sp1 is the transcription factor that controls the activation of the CDKN1A gene. Our data indicate that p21 is a functional prerequisite for miR-34a function. However, the effects of p21 depletion varied between cell lines. p21 depletion completely abolished the antiproliferative activity of miR-34a in RKO cells and merely weakened it in SW48 cells. Likewise, the inhibitory activity of miR-34a was not fully reduced but significantly lessened in TP53-negative Hep3B hepatocarcinoma cells that lack p21 (data not shown). In contrast, work by He et al. did not reveal a p21-dependent miR-34a phenotype in HCT116p21−/− cells.14 Therefore, the effects of p21 depletion appear to vary across cell lines. It is possible that the miR-34 phenotype is additionally controlled by other molecular events that are subject to change in cancer. Although deletion of CDKN1A can lead to spontaneous tumor formation in mice, somatic loss-of-function mutations in human cancer are rare.36 However, reduced expression has been noted in colorectal, cervical, esophageal, and lung cancers, and in some of these, this is due to the hypermethylation of the CDKN1A promoter.33,37 Thus, a miR-34a mimic may be less active in cancers with silenced CDKN1A which should be considered as a predictive biomarker for a miR-34 therapy.
The miR-34–specific induction of p21 offers an explanation for its invariable ability to inhibit TP53 wild-type and TP53-deficient cells. This is in stark contrast to miR-215/192 that is unable to induce p21 in the absence of TP53 and, consequently, has reduced inhibitory activity in TP53−/− cells. The data generated with miR-215/192 fit a model described previously in which miR-215/192 functions in a positive feedback loop to TP53 via repression of MDM2.19 Interestingly, the reported positive feedback from miR-34 to TP53 via SIRT1, YY1, or MDM4 does not seem to contribute to the antiproliferative miR-34 phenotype despite the fact that these gene products were downregulated by miR-34a. Given the modest effects on cellular proliferation of siRNAs against these gene products, it is possible that they do not participate in an immediate antiproliferative miR-34 response but may reveal added effects after prolonged repression.
Taken together, our data demonstrate that miR-34a–induced inhibition of cancer cell proliferation is independent of TP53 and suggests that a miR-34 therapy is effective in cancer patients irrespective of TP53 status. The ability of miR-34 to repress HDAC1 and to induce p21 significantly strengthens its position as a central tumor suppressor and complements its function in other important oncogenic pathways. Clinical studies may provide further insight whether TP53, HDAC1, or CDKN1A can predict therapeutic responses to miR-34.
Materials and Methods
Cell culture, oligos, and proliferation assays. Isogenic cancer cells derived from the MCF10A breast cancer and the SW48, HCT116, DLD-1, and RKO colorectal cancer cell lines were obtained from Horizon Discovery (Cambridge, UK) and are described in refs. 25,26. Synthetic miRNA mimics and siRNAs were purchased from Life Technologies (Ambion, Austin, TX). For stimulation of the TP53 pathway, cells were pretreated with 10 µmol/l etoposide for 28 hours, and RNA was harvested for qRT-PCR analysis. Optimal transfection conditions using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or RNAiMAX (Invitrogen) were determined for each cell line using an siRNA against EG5, a spindle protein required for proliferation.38 Reverse transfections were done in duplicates or triplicates and carried out as previously described.39 Briefly, 5 µl of oligo solution in RNAse-free water was added to 20 µl of OptiMEM (Invitrogen) per well containing a constant amount of Lipofectamine 2000 (SW48, MCF10A, DLD-1, HCT116) or RNAiMAX (RKO). The mixture was incubated for 20 minutes at room temperature to form lipid–RNA complexes. Then, 75 µl of cells suspended in medium were added to reach a final concentration of 6,000–10,000 cells per well, depending on the growth rate of each cell line. After ~18 hours, the supernatant was removed and replaced with fresh media. Cellular proliferation was determined using AlamarBlue (Invitrogen) 3–4 days post-transfection.40 The AlamarBlue substrate is metabolically converted into a fluorescent product in viable cells that is proportional to the number of living cells. Nonlinear regression and EC50 values were calculated using the GraphPad (Prism) software (version 6.01; Graphpad Software). All EC50 values were within the 95% confidence interval (P < 0.05) of the regression trendline.
Quantitative reverse transcription-PCR. Total RNA from cultured isogenic cancer cell lines was isolated using the mirVANA PARIS RNA isolation kit (Ambion) following the manufacturer's instructions. For qRT-PCR detection of miRNAs, 10 ng of total RNA and miRNA-specific RT-primers (assay IDs: hsa-miR-34a, 000426; hsa-miR-215, 000518; hsa-miR-192, 000491; hsa-miR-194, 000492; hsa-miR-34b, 002102; hsa-miR-34c, 000428; TaqMan miRNA Assay; Applied Biosystems, Foster City, CA) were heat-denatured at 70 °C for 2 minutes and reverse-transcribed using MMLV reverse transcriptase (cat. no. 28025-021, Invitrogen). miRNA expression levels were determined by PCR using Platinum Taq Polymerase reagents (Invitrogen) and the ABI Prism 7900 SDS instrument (Applied Biosystems). PCR reactions were performed by heating samples to 95 °C for 1 minutes, followed by incubating the samples at 95 °C for 5 seconds, and 60 °C for 30 seconds during multiple cycles. The house-keeping miRNAs miR-191 and miR-103 (assay IDs: 002299 and 000439) were amplified as internal references to adjust for well-to-well RNA input variances.41 Raw Ct (cycle threshold) values were normalized to the geometric mean of house-keeping miRNAs Cts and expressed as fold differences relative to those in untreated, miR-NC, or mock-transfected cells.
For detection of human mRNAs, cDNA was generated using 10 ng total RNA with random decamers (AM5722G; Ambion) as previously described.42 Gene-specific amplification was carried out using the following TaqMan Gene Expression Assays (Invitrogen): MDM4, Hs00159092_m1; XBP1, Hs00231936_m1; HDAC1, Hs02621185_m1; SNAI2, Hs00950344_m1; p21, Hs00385782_m1; PUMA (BBC3), Hs00248075_m1; MDM2, Hs00234753_m1; MYCT1, Hs00228305_m1; ATF6, Hs0023586_m1; SIRT1, Hs01009005_m1; MYCN, Hs00232074_m1; and TP53, Hs00153340_m1. mRNA levels of house-keeping GAPDH and cyclophilin A (TaqMan IDs: GAPDH, Hs99999905_m1; CYCLO A, Hs99999904_m1; Invitrogen) were used as loading controls. Raw Cts were normalized to those of house-keeping mRNAs and analyzed as described above.
Site-directed mutagenesis. Human HDAC1 3′ UTR Lenti-reporter-Luciferase vector (pLenti-UTR-Luc HDAC1, HDAC1 wt) encoding the luciferase reporter fused to the entire 3′ UTR of human HDAC1 was purchased from Applied Biological Materials (Richmond, BC). Two rounds of mutagenesis were performed to introduce 6 point mutations in the miR-34 binding site of HDAC1 3′ UTR (HDAC1 mutated). For site-directed mutagenesis, the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) was used following the manufacturer's instructions. In the first round, the following primers were used: 5′-CCTCAAGTGAGCCAAGAAACAATAACTGCCCTCTGTCTGTC-3′ and 5′-GACAGACAGAGGGCAGTTATTGTTTCTTGGCTCACTTGAGG-3. A positive clone was verified by sequencing (University of Texas, Austin, TX) and used as a template for the second round of mutagenesis using the following primers: 5′-GGCCTCAAGTGAGCCAAAAAtAAATAACTGCCCTCTGTCTGTC-3′ and 5′-GACAGACAGAGGGCAGTTATTTATTTTTGGCTCACTTGAGGCC-3′. All vectors used in transfections were verified by sequencing.
Luciferase reporter assays. SW48−/− and H1299 cells were reverse transfected with 1 nmol/l or 10 nmol/l miR-34a, respectively, in 96-well plate using Lipofectamine 2000 (Life Technologies). As controls, cells were transfected with miR-NC at the same concentrations. The next day, cells were forward transfected with each 100 ng of HDAC1 wt or HDAC1-mutated luciferase plasmids. After 48 hours, cell lysates were prepared and quantified using the BCA system from Pierce (Thermo Scientific, Rockford, IL). Luminescence was determined using the PolarStar OptiMA plate reader (BMG Labtech, Ortenberg, Germany) and the Luciferase Assay System (Promega, Madison, WI). Luminescence was normalized to total protein input.
Western analysis. 200,000 SW48 and RKO cells were seeded in six-well plates and reverse-transfected with miRNA mimics and siRNAs in six-well plate using 2.5-µl Lipofectamine 2000 or RNAiMAX. After 3 days, cell lysate were collected in RIPA buffer (Cell Signaling, Danvers, MA), and protein concentrations were measured using the BCA assay kit from Thermo Scientific. Each 2.5 µg of total cell lysate was loaded on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and then transferred to a polyvinylidene difluoride membrane. The membrane was blotted with primary antibody specific for p21, HDAC1, c-MET, and actin (Cell Signaling) overnight at 4 °C. The membrane was washed in 1× phosphate-buffered saline containing 0.2% Tween-20 and incubated with a horseradish peroxidase-conjugated secondary antibody at room temperature for 1 hour. After washing with 1× phosphate-buffered saline containing 0.2% Tween-20, the membrane was incubated with ECL detection reagent (EMD Millipore, Darmstadt, Germany) and protein bands were visualized using the AFP X-ray film developer (AFP Image, Elmsford, NY). Western data were quantified using the AlphaImager EC instrument from Cell Biosciences (Santa Clara, CA).
Human tissue samples. Non–small-cell lung carcinoma tumor samples and the corresponding normal adjacent tissues were purchased from ProteoGenex (Culver City, CA) and the National Disease Research Interchange (Philadelphia, PA). Staging information and miR-34a levels are reported in ref. 10. HDAC1 mRNA and miR-34a levels were determined by qRT-PCR and expressed as relative expression between each tumor and normal adjacent tissue pair. Linear regression was calculated using GraphPad.
Statistical analysis. Statistical analyses were done using the Excel and GraphPad (Prism) software. Averages and standard deviations were calculated from duplicate or triplicate experiments. P values were generated by two-tailed Student's t test or F test as indicated in the figure legends.
SUPPLEMENTARY MATERIAL Figure S1. Induction of TP53 genes by etoposide. Figure S2. Dose-dependent effects of miR-34c and miR-192 in cancer cells with or without functional TP53. Figure S3. Dose-dependent effects of miR-34a and miR-34c in isogenic RKO cells. Figure S4. Conservation of miR-34a binding sites in the HDAC1 3′ UTR across vertebrates. Figure S5. Quantification of Western data shown in Figure 4A using the AlphaImager EC instrument from Cell Biosciences. Figure S6. miR-34a-induced repression of HDAC1 and induction of p21CIP1/WAF1 in TP53-positive cells. Figure S7. Endogenous mRNA expression levels in isogenic SW48 cells transiently transfected with siRNAs. Figure S8. Effects of Trichostatin A on isogenic cancer cells. Figure S9. Quantification of Western data shown in Figure 5A using the AlphaImager EC from Cell Biosciences. Figure S10. Depletion of p21 interferes with miR-34a–induced inhibition of cell proliferation in SW48 cells. Table S1. TP53 genotypes of isogenic cancer cell lines.
Acknowledgments
The authors thank Peter K Vogt and Jay Stoudemire for critical reading of the manuscript, and Michael Omotola and Kevin Kelnar for technical assistance. This work was supported by grants from the National Institutes of Health (1R43CA134071 and 1R43CA137939 to A.G.B.) and a commercialization grant from the Cancer Prevention and Research Institute of Texas (CPRIT). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. J.Z., P.L., and A.G.B. are employees of Mirna Therapeutics which develops miRNA-based therapies. The other author declares no conflicts.
Supplementary Material
References
- Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo E, Navarro A, Viñolas N, Marrades RM, Diaz T, Gel B, et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–1909. doi: 10.1093/carcin/bgp219. [DOI] [PubMed] [Google Scholar]
- Chim CS, Wong KY, Qi Y, Loong F, Lam WL, Wong LG, et al. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis. 2010;31:745–750. doi: 10.1093/carcin/bgq033. [DOI] [PubMed] [Google Scholar]
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet. 2012;3:120. doi: 10.3389/fgene.2012.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, et al. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. 2011;19:1116–1122. doi: 10.1038/mt.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig VJ, Tzankov A, Flori M, Schmid CA, Bader AG, Müller A. Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia. 2012;26:2421–2424. doi: 10.1038/leu.2012.110. [DOI] [PubMed] [Google Scholar]
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- Braun CJ, Zhang X, Savelyeva I, Wolff S, Moll UM, Schepeler T, et al. p53-Responsive microRNAs 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68:10094–10104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges SA, Biery MC, Kim SY, Schelter JM, Guo J, Chang AN, et al. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008;68:10105–10112. doi: 10.1158/0008-5472.CAN-08-1846. [DOI] [PubMed] [Google Scholar]
- Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–381. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandke P, Wyatt N, Fraser J, Bates B, Berberich SJ, Markey MP. MicroRNA-34a modulates MDM4 expression via a target site in the open reading frame. PLoS ONE. 2012;7:e42034. doi: 10.1371/journal.pone.0042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QR, Yu LR, Tsang P, Wei JS, Song YK, Cheuk A, et al. Systematic proteome analysis identifies transcription factor YY1 as a direct target of miR-34a. J Proteome Res. 2011;10:479–487. doi: 10.1021/pr1006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA. 2009;106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MB, Vitolo MI, Mohseni M, Rosen DM, Denmeade SR, Park BH, et al. Deletion of p53 in human mammary epithelial cells causes chromosomal instability and altered therapeutic response. Oncogene. 2010;29:4715–4724. doi: 10.1038/onc.2010.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol. 2010;2:a004887. doi: 10.1101/cshperspect.a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:2669–2679. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, et al. Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites. Biochem Biophys Res Commun. 1997;241:142–150. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Krummel KA, Lee CJ, Liu CW, Rodewald LW, Tang M, et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9:273–285. doi: 10.1016/j.ccr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- Concepcion CP, Han YC, Mu P, Bonetti C, Yao E, D'Andrea A, et al. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 2012;8:e1002797. doi: 10.1371/journal.pgen.1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- Teramen H, Tsukuda K, Tanaka N, Ueno T, Kubo T, Ando M, et al. Aberrant methylation of p21 gene in lung cancer and malignant pleural mesothelioma. Acta Med Okayama. 2011;65:179–184. doi: 10.18926/AMO/46629. [DOI] [PubMed] [Google Scholar]
- Weil D, Garçon L, Harper M, Duménil D, Dautry F, Kress M. Targeting the kinesin Eg5 to monitor siRNA transfection in mammalian cells. BioTechniques. 2002;33:1244–1248. doi: 10.2144/02336st01. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- Nakayama GR, Caton MC, Nova MP, Parandoosh Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J Immunol Methods. 1997;204:205–208. doi: 10.1016/s0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
- Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–852. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.