

Abstract
Klotho is a recently discovered anti-aging gene and is primarily expressed in kidneys. In humans, the klotho level decreases with age whereas the prevalence of chronic kidney disease (CKD) increases with age. Diabetic nephropathy is the most common form of CKD, which leads to end-stage renal disease. A decrease in klotho has been found in kidneys of patients with diabetic nephropathy. The purpose of this study is to assess whether klotho gene deficiency affects early diabetic nephropathy in a mouse of model of type 1 diabetes induced by streptozotocin (STZ). Male KL+/− mutant and wild-type mice (6–8 weeks) were injected with multiple low doses of STZ. Renal functions and renal blood flow were assessed. Kidneys were collected for histological examination and molecular assays of TGFβ1 and mammalian targets of rapamycin (mTOR) signaling. Klotho deficiency in KL+/− mutant mice exacerbated STZ-induced increases in urine albumin, blood urea nitrogen, expansion of mesangial matrix in renal glomeruli, and kidney hypertrophy, suggesting a protective role of klotho in kidney function and structure. Klotho deficiency did not affect renal blood flow. Notably, klotho deficiency significantly increased phosphorylation of Smad2, indicating enhanced TGFβ1 signaling in kidneys. Klotho deficiency also increased phosphorylation of mTOR and S6 (a downstream effector of mTOR), indicating enhanced mTOR signaling in kidneys of early diabetic mice. Thus, klotho gene deficiency may make kidneys more susceptible to diabetic injury. Klotho gene deficiency exacerbated early diabetic nephropathy via enhancing both TGFβ1 and mTOR signaling in kidneys.
Overexpression of klotho extended life span in mice, whereas mutation of the klotho gene caused multiple premature-aging phenotypes and shortened lifespan (1, 2). Thus, klotho is an anti-aging gene. The mouse klotho (also called α-klotho) gene contains 5 exons and 4 introns and encodes a single-pass transmembrane protein with 1014 amino acids (3). Klotho is predominately expressed in the kidney and brain choroid plexus (1). Most amino acids in the klotho peptide reside in the amino-terminal extracellular domain, which is followed by a 21-amino-acid transmembrane domain, and an 11-amino-acid short intracellular carboxy terminus (1). There are 2 forms of klotho, the full-length klotho (135 kDa) and the short-form klotho (65 kDa) that is generated by alternative RNA splicing or proteolytic cleavage (1, 4, 5).
In humans, the klotho level decreases with age (3, 6), whereas the prevalence of chronic kidney diseases (CKDs) increases with age (7–13). At age 70 years, the klotho level is about half of what it is at age 40 years (6). The kidney function declines in the aged population (7–11, 14). The prevalence of CKD including diabetic nephropathy is higher in the aged than in the young population (12–14). Therefore, CKD is an aging-related kidney disorder. Arterial stiffening, hypertension, and angiogenesis are associated with CKD (15–17). CKD eventually results in multiple organ dysfunction, leading to heart failure and stroke. Diabetic nephropathy is the most common cause of end-stage renal disease (ESRD) (18). It is projected that 30% to 40% of patients with type 1 diabetes and 5% to 10% of patients with type 2 diabetes eventually develop ESRD (18). Conventional therapies such as strict glycemic control and other antimetabolic treatments do not completely stop the progression of diabetic nephropathy in diabetic patients (18). Early alterations in diabetic nephropathy include glomerular hyperfiltration, glomerular and tubular epithelial hypertrophy, and the development of microalbuminuria, followed by the development of glomerular basement membrane thickening, the accumulation of mesangial matrix, and overt proteinuria, eventually leading to glomerulosclerosis and ESRD (18).
Diabetic nephropathy is the most common form of CKD. Klotho expression levels were decreased in kidneys of patients with early diabetic nephropathy (19, 20). Klotho deficiency may serve as a biomarker as well as a pathogenic factor for the progression of renal disease and further complications (21). However, little is known about whether klotho deficiency affects the development of early diabetic nephropathy. Both TGFβ1 signaling and mammalian targets of rapamycin (mTOR) signaling have been considered to be crucial for the development of diabetic nephropathy (22, 23). Whether klotho affects TGFβ1 and mTOR signaling in diabetic nephropathy has never been investigated. Multiple low doses of streptozotocin (STZ) have been shown to cause selective β-cell destruction and subsequently modest hyperglycemia leading to functional damage (eg, albuminuria) and histological lesions in kidneys (ie, diabetic nephropathy) (24, 25). The purpose of this study was to investigate a hypothesis that klotho gene deficiency enhances TGFβ1 and mTOR signaling and promotes the development of early diabetic nephropathy in a mouse model of type 1 diabetes induced by multiple low doses of STZ.
Materials and Methods
Animal studies
Male heterozygous klotho (KL+/−) mutant mice with more than 9 generations on a 129/Sv background were used (1, 26), whereas wild-type (WT) littermate 129/Sv mice were used as control for KL+/− mutant mice. Heterozygous mutant mice were used because they have half the klotho expression levels seen in WT mice, which mimics the decline in klotho levels associated with human aging. Klotho homozygous mutants (KL−/−) were not used because they demonstrate early and extensive aging phenotypes and die before the age of 8 weeks. As a result, klotho homozygous mice do not survive STZ challenge. Briefly, all mice were housed in cages at room temperatures (25°C ± 1°C) and were provided with Purina laboratory chow (No. 5001) and tap water ad libitum throughout the experiment. This study was carried out according to the guidelines of the National Institutes of Health on the care and use of laboratory animals. This project was approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center.
Briefly, 8- to 10-week-old body weight-matched KL+/− mutant and WT male mice were injected with multiple low doses of STZ (40 μg/g/d ip for 5 days) or citrate buffer, respectively. Body weight was monitored weekly. Blood glucose was measured from the tail vein blood using a ReliOn Ultima glucose reader (Solartek Products, Inc). The mice were fasted for 3 hours before glucose measurement. Blood pressure was monitored in some animals during weeks 2 and 4 after treatments using the tail-cuff method as we described previously (27, 28).
Measurements of renal arterial blood flow
Three and 5 weeks after the initial injections of STZ, right renal arterial blood flow was measured using a Doppler signal processing workstation (DSPW; Indus Instruments) (29). Briefly, all mice were anesthetized with ketamine (90 mg/kg body weight) and xylazine (10 mg/kg body weight) via ip injection. Each mouse was then removed, weighed, and taped prone to electrocardiogram electrodes on a heated procedure board (Indus Instruments). A 2-mm diameter, 20-MHz Doppler probe was used. Data were analyzed using the manufactorer's software DSPW.
Tissue collection
At the end of week 5 after STZ injections (when diabetic nephropathy could be measured), animals were euthanized with an overdose of sodium pentobarbital (100 mg/kg, ip) and blood was collected. Plasma samples were stored at −80°C. After blood collection, animals were perfused transcardially using heparinized saline. The left kidney of each animal was placed in 4% PBS-buffered paraformaldehyde for 24 hours and then embedded in paraffin. The other kidney was stored at −80°C until use.
Measurements of albumin
Spot urine for each animal was collected during the fifth week after the initial injections of STZ. Urinary albumin excretion was measured with a mouse-specific microalbuminuria ELISA kit (Albuwell M; Exocell) according to the manufacturer's instructions. Urine creatinine was measured using a QuantiChrom creatinine assay kit (DICT-500; BioAssay Systems).
Morphological investigations
A series of cross- sections of the left kidney (3 to 5 μm) were cut. Tissue sections (4 to 8 specimens per group) were stained with periodic acid Schiff (PAS) and Masson's trichrome, respectively.
Briefly, kidney sections (5 μm) were stained with Masson's trichrome. Images of cortex from 3 to 5 consecutive sections for each animal were collected at equal exposure conditions under Nikon Eclipse Ti microscopy (magnification ×100). The fraction area for collagenous components in cortex was obtained using NIS-Elements BR version 3.0 (Nikon). Images of kidney sections were also collected at equal exposure conditions under a higher magnification (×400).
For sections (3 μm) stained with PAS, images of 20 glomeruli of each animal were collected at equal exposure conditions and at magnification ×400 under a microscope (Nikon Eclipse Ti). Mesangial matrix area was defined by PAS-positive and nuclei-free area in the mesangium. The glomerular area was defined by tracing along the borders of the capillary loop. Relative mesangial area (defined as fraction area of mesangial matrix area over glomerular area) was obtained using Image J (National Institutes of Health freeware).
Western blotting
Western blot was performed as described in our previous studies (30–32). Briefly, kidneys were lysed in RIPA buffer containing protease inhibitor cocktail, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 2mM sodium vanadate, 1mM EDTA, and 1mM EGTA. The protein concentration was measured using the Pierce bicinchoninic acid assay (Thermo Scientific). The lysates (30 μg protein per well) under reduced conditions were directly subjected to SDS-PAGE (4%–20% Tris-HCl precast gel; Bio-Rad) followed by Western blotting with antibodies against klotho (R&D Systems), phospho- (p-)Akt (Ser 473) (Cell Signaling), p-mTOR (Cell Signaling), p-S6 (Ser 235/236) (Cell Signaling), p-Smad2 (Ser 465/467) (Cell Signaling), LC3 (U.S. Biological), or TGFβ1 (Santa Cruz Biotechnology; TGFβ1 and its precursor). The blot was then rinsed and reprobed with antibodies against Akt (Cell Signaling), mTOR (Cell Signaling), Smads 2 (Cell Signaling), S6 (Cell Signaling), or β-actin for the loading control.
Statistical analysis
Data were analyzed using a one-way ANOVA. The Newman-Keuls procedure was used to assess differences between means. Data are expressed as mean ± SEM. P < .05 was considered significant.
Results
Klotho deficiency exacerbated kidney functional damage in an STZ-induced mouse model of diabetes
To study the roles of endogenous klotho in the development of early diabetic nephropathy, we used a model of mild diabetes induced by multiple low doses of STZ in KL+/− mutant and their littermate WT mice with a 129/Sv background (33). Both WT and KL+/− mutant mice developed hyperglycemia as early as 3 weeks after the initial injection of STZ (Supplemental Figure 1B, published on The Endocrine Society's Journals Online website at http://endo.endojournals.org). The STZ challenge caused more severe hyperglycemia in KL+/− mutant mice vs the WT mice (Figure 1A and Supplemental Figure 1B).
Figure 1.
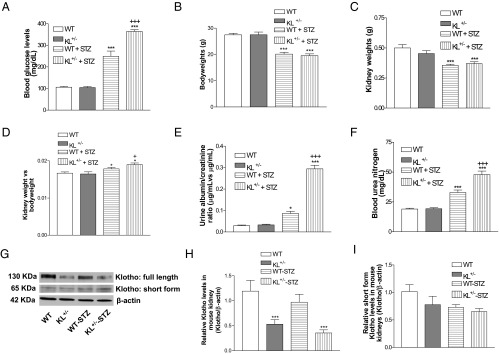
Effects of klotho deficiency on blood glucose levels, kidney weights, urine albumin, and BUN levels in STZ-induced diabetic mice. KL+/− mutant and WT male mice were injected with STZ or citrate buffer. A, Blood glucose levels 5 weeks after the initial injections of STZ. B–D, Body weights (B), kidney weights (C), and normalized kidney weights (D) 5 weeks after the initial injection of STZ. E, Urine albumin during the fifth week after the initial injection of STZ. F, Blood urea nitrogen levels 5 weeks after the initial STZ injections. G, Renal klotho expression levels were measured 5 weeks after the initial injections of STZ. H, Quantification of full-length klotho. I, Quantification of short-form klotho. Klotho was normalized with β-actin, which was then calculated as fold changes vs the control WT group. Data are shown as mean ± SEM; n = 8 animals per group. *, P < .05; **, P < .01; ***, P < .001 vs the WT group; +, P < .05; +++, P < .001 vs the WT-STZ group.
There was a significant decrease in body weights in STZ-injected mice vs animals without STZ injections (Figure 1B). The STZ injections also decreased the kidney weights in both KL+/− and WT mice (Figure 1C). Interestingly, there was a significant increase in normalized kidney weights in STZ-injected mice compared with buffer-injected mice (Figure 1D). In addition, KL+/− mice had greater normalized kidney weights compared with the WT mice after injection with STZ (Figure 1D), suggesting that klotho gene deficiency exacerbated renal hypertrophy in diabetic mice.
Injections of STZ caused an increase in urine albumin levels in both WT and KL+/− mutant mice, and STZ injections generated higher urine albumin levels in KL+/− mutant mice compared with WT mice (Figure 1E). STZ injections also increased blood urea nitrogen (BUN), levels whereas STZ injections produced greater BUN levels in KL+/− mutant mice compared with WT mice (Figure 1F). These data strongly suggested that KL +/− mutant mice are susceptible to STZ-induced damage of kidney functions.
Furthermore, KL+/− mutant mice had short-form klotho (full-length, 130 kDa) in kidneys (Figure 1H). However, renal expression of short-form klotho (65 kDa) was not different between KL+/− mice and WT mice (Figure 1I).
Klotho deficiency exacerbated glomerular mesangial matrix expansion in diabetic kidneys
STZ injections led to glomerular mesangial matrix expansion as indicated by PAS staining in both WT and KL+/− mutant mice (Figure 2, A and B). Klotho deficiency in KL+/− mutant mice further enhanced glomerular mesangial expansion compared with WT mice in response to STZ injections (Figure 2, A and B), suggesting that klotho deficiency aggravated STZ-induced glomerular damage in diabetic kidney.
Figure 2.
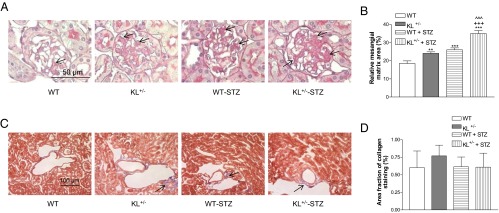
Effects of klotho deficiency on mesangial matrix expansion and collagen deposition in diabetic kidneys. KL+/− mutant and WT male mice were injected with STZ or citrate buffer. Animals were killed 5 weeks after the initial injections. A, Representative photomicrographs of PAS-stained kidney sections. Arrows indicate PAS-positive mesangial material (pink). B, Quantitative measurements of mesangial matrix expansion. Relative mesangial matrix area is expressed as PAS-positive mesangial matrix per total glomerular tuft cross-sectional area. An average value was obtained from analyses of 20 glomeruli per mouse. C, Representative photomicrographs of Masson's trichrome-stained kidney sections. Arrows indicate trichrome-positive collagenous components in cortical interstitium (light blue). D, Quantitative analysis of collagenous components in renal cortex. Images of cortex from 3 consecutive sections for each animal, collected at equal exposure conditions under Nikon Eclipse Ti microscopy, were used in the analysis. Data are shown as mean ± SEM; n = 8. **, P < .01; ***, P < .001 vs the WT group; +++, P < .001 vs the WT-STZ group; ^^^, P < .001 vs the KL+/− group.
No obvious change in trichrome staining as an index of renal global collagen accumulation at this stage has been observed (Figure 2, C and D), suggesting that these animals were still in the early stage of development of diabetic nephropathy.
Klotho deficiency enhanced TGFβ1 signaling in diabetic kidneys
To further explore the mechanisms of klotho deficiency-induced exacerbation of diabetic kidney damage, we assessed TGFβ1 expression in diabetic kidneys. The TGFβ1 signaling may be involved in the induction of glomerular fibrosis in diabetic nephropathy (22). Phosphorylation of Smad2 in kidneys was increased in diabetic mice vs the control (Figure 3A). KL+/− mutant mice displayed greater Smad2 phosphorylation levels in kidneys compared with those of WT mice after induction of diabetes with STZ (Figure 3A), indicating that klotho deficiency further enhanced TGFβ1 signaling. Neither injections of STZ nor the genotype difference affected protein levels of Smad2 in kidneys (Figure 3A). STZ injections increased TGFβ1 levels in kidneys compared with WT mice injected with buffer alone (Figure 3B).
Figure 3.
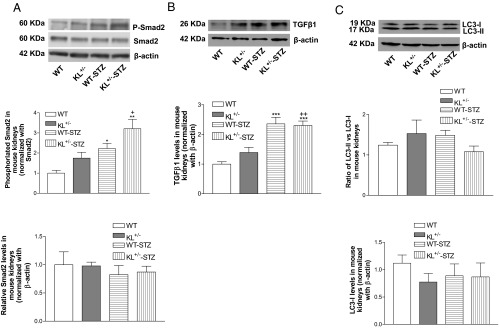
Effects of klotho deficiency on p-Smad2, Smad2, TGFβ1, and LC3 in diabetic kidneys. KL+/− mutant and WT male mice were injected with STZ or citrate buffer. Animals were killed 5 weeks after the initial injections. A, Representative blots of p-Smad2 and Smad2 and quantification of p-Smad2 and Smad2. B, Representative blots of TGFβ1 and quantification of TGFβ1. C, Representative blots of LC3 and quantification of LC3 (ratio of LC3-II to LC3-I; LC3-I). p-Smad2 was normalized with Smad2, which was then calculated as fold change of the control WT group. Smad2, TGFβ1, and LC3-I was normalized to β-actin, respectively, which was as then calculated as fold change of the control WT group. Data are shown as mean ± SEM; n = 4 to 5. *, P < .05; **, P < .01; ***, P < .001 vs the WT group; +, P < .05; ++, P < .01 vs the WT-STZ group.
The autophagy signaling is lower in diabetic nephropathy (34). It was reported that upregulation of autophagy is involved in the amelioration of diabetic nephropathy by food restriction (35). LC3 has been considered as a marker of autophagy (36). Unexpectedly, STZ injections did not affect either the levels of LC3-II or the ratio of LC3-II to LC3-I in kidneys (Figure 3C). Klotho deficiency in kidneys did not alter the LC3 levels (Figure 3C).
Klotho deficiency enhanced mTOR signaling in diabetic kidneys
Diabetic mice had significantly higher levels of phosphorylation of mTOR in kidneys compared with nondiabetic controls (Figure 4A). Klotho deficiency further enhanced kidney mTOR phosphorylation in diabetic mice (Figure 4A). In addition, diabetic mice displayed increased phosphorylation of S6, a downstream effector of mTOR, compared with nondiabetic controls (Figure 4B). Klotho deficiency in KL+/− mutant mice further enhanced renal phosphorylation of S6 in diabetic kidneys (Figure 4B).
Figure 4.
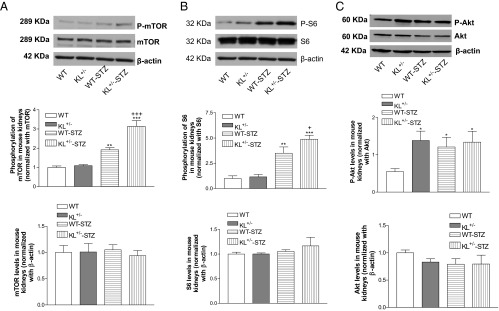
Effects of klotho deficiency on p-mTOR, mTOR, p-S6, S6, p-Akt, and Akt levels in diabetic kidney. A, Representative blots of p-mTOR and mTOR and quantification of p-mTOR and mTOR. B, Representative blots of p-S6 and S6 and quantification of p-S6 and S6. C, Representative blots of p-Akt and Akt and quantification of p-Akt and Akt. Phosphorylated protein was normalized with its total protein, which was then calculated as fold change of the control WT group. mTOR, S6, or Akt was normalized with β-actin, respectively, which was then calculated as fold change of the control WT group. Data are shown as mean ± SEM; n = 4 to 5. *, P < .05; **, P < .01; ***, P < .001 vs the WT group; +, P < .05; +++, P < .001 vs the WT-STZ group.
Deficiency of klotho also caused an increase in phosphorylation of Akt compared with WT mice (Figure 4C). Phosphorylation of Akt was increased in kidneys of diabetic mice vs the WT mice without treatments (Figure 4C). However, deficiency of klotho did not further increase Akt phosphorylation in mice treated with STZ (Figure 4C).
Neither STZ injections nor klotho deficiency affected renal arterial blood flow
It was reported that the early signs of glomerular hyperfiltration result from decreased resistance in both the afferent and efferent arterioles of the glomerulus (37). We used the Doppler signal processing workstation to study the renal arterial blood flow. Surprisingly, STZ did not affect renal arterial blood flow at week 3 or 5 after treatment (Tables 1 and 2), suggesting that short-term injections of STZ did not cause renal hemodynamic changes in this animal model. Thus, early diabetic nephropathy may not be due to renal ischemia. Klotho deficiency did not alter renal arterial blood flow in either group (Tables 1 and 2).
Table 1.
Renal Arterial Blood Flow in STZ-Treated Mice in Week 3
| WT (n = 8) | KL+/− (n = 7) | WT-STZ (n = 8) | KL+/−-STZ (n = 9) | |
|---|---|---|---|---|
| Heart rate (beats/min) | 279.96 ± 18.08 | 291.54 ± 22.20 | 260.95 ± 12.32 | 239.70 ± 12.57 |
| R-R interval (ms) | 221.16 ± 14.38 | 213.44 ± 16.46 | 234.23 ± 12.24 | 257.65 ± 14.36 |
| Peak velocity (cm/s) | 25.48 ± 2.32 | 21.61 ± 3.09 | 24.39 ± 1.81 | 24.92 ± 1.17 |
| Minimum flow velocity (cm/s) | 10.66 ± 0.54 | 10.33 ± 1.11 | 8.84 ± 0.69 | 9.03 ± 0.61 |
| Mean flow velocity (cm/s) | 18.37 ± 1.47 | 15.32 ± 1.65 | 16.27 ± 1.46 | 15.64 ± 1.24 |
| Pulsatility index | 0.90 ± 0.14 | 0.92 ± 0.19 | 1.08 ± 0.14 | 1.15 ± 0.16 |
| Resistivity index | 0.51 ± 0.04 | 0.53 ± 0.05 | 0.57 ± 0.04 | 0.57 ± 0.03 |
Table 2.
Renal Arterial Blood Flow in STZ-Treated Animals in Week 5
| WT (n = 5) | KL+/− (n = 5) | WT-STZ (n = 5) | KL+/−-STZ (n = 5) | |
|---|---|---|---|---|
| Heart rate (beats/min) | 286.05 ± 11.09 | 234.59 ± 6.76 | 260.46 ± 10.69 | 267.46 ± 21.43 |
| R-R interval (ms) | 211.12 ± 7.93 | 256.93 ± 7.40 | 240.14 ± 12.50 | 231.23 ± 17.62 |
| Peak velocity (cm/s) | 24.39 ± 3.12 | 26.32 ± 0.84 | 23.23 ± 2.67 | 30.12 ± 3.39 |
| Minimum flow velocity (cm/s) | 13.31 ± 2.42 | 8.78 ± 0.46 | 8.99 ± 0.85 | 8.27 ± 0.85 |
| Mean flow velocity (cm/s) | 14.18 ± 2.05 | 16.42 ± 0.78 | 17.93 ± 2.04 | 20.25 ± 1.16 |
| Pulsatility index | 0.96 ± 0.06 | 0.86 ± 0.02 | 0.91 ± 0.07 | 1.08 ± 0.10 |
| Resistivity index | 0.53 ± 0.01 | 0.51 ± 0.01 | 0.51 ± 0.01 | 0.54 ± 0.01 |
Discussion
Klotho is a recently discovered anti-aging gene (1, 2). In humans, the klotho level decreases with age (3, 6) whereas the prevalence of CKD, including diabetic nephropathy, increases with age (12–14). However, whether klotho affects CKD is not clear. Diabetic nephropathy is the most common form of CKD. The klotho expression levels were decreased in kidneys of patients with early diabetic nephropathy (19, 20). One of the novel findings of this study is that renal klotho deficiency in KL+/− mutant mice exacerbated early diabetic nephropathy (impaired renal function and glomerular mesangial matrix expansion) in type 1 diabetes induced by STZ (Figures 1 and 2). Thus, klotho deficiency may make the kidneys more susceptible to diabetic injury. To mitigate nonspecific cytotoxicity, we used multiple low doses of STZ, which induce moderate hyperglycemia in 129/Sv mice (25). It is noted that KL+/− mutant mice displayed greater hyperglycemia in response to injection of STZ. Hyperglycemia induced by STZ injections is closely related to diabetic nephropathy (38). However, strict glycemic control does not completely stop the progression of diabetic nephropathy in diabetic patients (18), suggesting that other factors other than hyperglycemia may be involved in diabetic nephropathy. Klotho deficiency-induced exacerbation of diabetic nephropathy may not be due to renal ischemia because renal blood flow was not altered in KL+/− mice (Tables 1 and 2). The adverse effects of klotho deficiency may be independent of blood pressure because blood pressure was not affected by klotho deficiency (Supplemental Figure 1A). On the other hand, klotho deficiency itself causes oxidative damage (3, 39) that may contribute to the exacerbation of diabetic nephropathy. A further study is needed to address the relative importance of klotho deficiency alone in the development of diabetic nephropathy by controlling blood glucose levels (euglycemia) in all groups.
Conventional therapies such as strict glycemic control, antihypertensive treatment, and other antimetabolic treatments do not completely stop the progression of diabetic nephropathy in diabetic patients (18). Our results demonstrated that klotho may have a protective role in diabetic nephropathy. We reported that in vivo klotho gene delivery attenuated oxidative stress (39) and kidney damage (30) in spontaneous hypertensive rats. Therefore, promoting klotho expression in kidneys may have preventive and therapeutic potential for early diabetic nephropathy. This hypothesis warrants further investigations.
Recent studies indicated that renal klotho expression was decreased in early diabetic nephropathy in humans (19, 20). It was reported that the serum level of soluble klotho also declined in early CKD in diabetic patients, which paralleled with microalbuminuria (40). Klotho deficiency was proposed to be a biomarker as well as a pathogenic factor for the progression of renal disease and the further complications (21). Although these studies demonstrated an association between klotho deficiency and early diabetic nephropathy, the cause-effect relationship remains unknown. This study demonstrated that klotho deficiency exacerbated early diabetic nephropathy (Figures 2 and 3). Another interesting finding is that the adverse effects of klotho gene deficiency on early diabetic nephropathy may be mediated by TGFβ1 and mTOR signaling in kidneys.
This study reveals for the first time that deficiency of renal klotho in KL+/− mutant mice increased phosphorylation of Smad2, a key downstream signaling of TGFβ1, in diabetic kidney. This result supports a notion that endogenous klotho in kidney may be an important negative regulator of the TGFβ1 signaling in diabetic mice. TGFβ1 has been shown to be linked to renal fibrosis in diabetic nephropathy in animals and humans (41–43). Suppression of TGFβ1 inhibited hyperglycemia-induced collagen synthesis and prevented glomerular fibrosis and renal insufficiency in db/db mice (42, 44). Klotho was reported to suppress renal fibrosis induced by unilateral ureteral obstruction, likely by inhibiting TGFβ1 signaling (45).
Interestingly, this study demonstrated that renal klotho deficiency augmented mTOR signaling including increased phosphorylation of mTOR and phosphorylation of S6, a downstream effector of mTOR complex 1 in diabetic kidneys. Several lines of evidences have shown that activation of mTOR increases the synthesis of matrix proteins that contributes to basement membrane thickening and glomerular mesangial matrix expansion (46). Rapamycin not only reduces renal mTOR activity in a rat model of STZ-induced diabetes, but also ameliorates the glomerular changes, including hypertrophy, basement membrane thickening, and mesangial matrix accumulation (23). It was reported that activity of mTOR complex 1, a kinase that senses nutrient availability, was enhanced in the podocytes of diabetic animals (47). A number of studies have shown that activation of mTOR plays a pivotal role in renal hypertrophy and podocyte injury, which may contribute to the progressive loss of renal function in diabetic nephropathy (23). KL+/− mutant mice showed exacerbated kidney damage (Figures 1 and 2) that is likely attributed to klotho deficiency-induced enhancement of mTOR signaling. Another study is needed to address how klotho regulates mTOR signaling.
In summary, anti-aging gene klotho deficiency exacerbated early diabetic nephropathy including higher albuminuria levels, higher BUN levels, greater glomerular mesangial expansion, and enhanced kidney hypertrophy. Klotho gene deficiency increased TGFβ1 and mTOR signaling in kidneys, which may contribute to the exacerbation of early diabetic nephropathy in KL+/− mice.
Acknowledgments
This work was supported by National Institutes of Health Grants DK 093403, HL105302, and HL102074.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BUN
- blood urea nitrogen
- CKD
- chronic kidney disease
- ESRD
- end-stage renal disease
- mTOR
- mammalian targets of rapamycin
- p-
- phospho-
- PAS
- periodic acid Schiff
- STZ
- streptozotocin
- WT
- wild-type.
References
- 1. Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51 [DOI] [PubMed] [Google Scholar]
- 2. Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone klotho. Science. 2005;309:1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Sun Z. Current understanding of klotho. Ageing Res Rev. 2009;8:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104:19796–19801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shiraki-Iida T, Aizawa H, Matsumura Y, et al. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998;424:6–10 [DOI] [PubMed] [Google Scholar]
- 6. Xiao NM, Zhang YM, Zheng Q, Gu J. Klotho is a serum factor related to human aging. Chin Med J (Engl). 2004;117:742–747 [PubMed] [Google Scholar]
- 7. Anderson S, Brenner BM. Effects of aging on the renal glomerulus. Am J Med. 1986;80:435–442 [DOI] [PubMed] [Google Scholar]
- 8. Anderson S, Brenner BM. The aging kidney: structure, function, mechanisms, and therapeutic implications. J Am Geriatr Soc. 1987;35:590–593 [DOI] [PubMed] [Google Scholar]
- 9. Anderson S, Halter JB, Hazzard WR, et al. Prediction, progression, and outcomes of chronic kidney disease in older adults. J Am Soc Nephrol. 2009;20:1199–1209 [DOI] [PubMed] [Google Scholar]
- 10. Anderson S, Rennke HG, Zatz R. Glomerular adaptations with normal aging and with long-term converting enzyme inhibition in rats. Am J Physiol. 1994;267:F35–F43 [DOI] [PubMed] [Google Scholar]
- 11. Weinstein JR, Anderson S. The aging kidney: physiological changes. Adv Chronic Kidney Dis. 2010;17:302–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goetz FC, Jacobs DR, Jr, Chavers B, Roel J, Yelle M, Sprafka JM. Risk factors for kidney damage in the adult population of Wadena, Minnesota. A prospective study. Am J Epidemiol. 1997;145:91–102 [DOI] [PubMed] [Google Scholar]
- 13. Heras Benito M, Fernández-Reyes Luis MJ, Sánchez Hernández R, et al. Elderly patients with chronic kidney disease: what is their course at one year [in Spanish]? Nefrologia. 2008;28:325–328 [PubMed] [Google Scholar]
- 14. Abdelhafiz AH, Brown SH, Bello A, El Nahas M. Chronic kidney disease in older people: physiology, pathology or both? Nephron Clin Pract. 2010;116:c19–c24 [DOI] [PubMed] [Google Scholar]
- 15. Chung AW, Yang HH, Kim JM, et al. Upregulation of matrix metalloproteinase-2 in the arterial vasculature contributes to stiffening and vasomotor dysfunction in patients with chronic kidney disease. Circulation. 2009;120:792–801 [DOI] [PubMed] [Google Scholar]
- 16. Chung AW, Yang HH, Sigrist MK, et al. Matrix metalloproteinase-2 and -9 exacerbate arterial stiffening and angiogenesis in diabetes and chronic kidney disease. Cardiovasc Res. 2009;84:494–504 [DOI] [PubMed] [Google Scholar]
- 17. Porazko T, Kuniar J, Kusztal M, et al. Increased aortic wall stiffness associated with low circulating fetuin A and high C-reactive protein in predialysis patients. Nephron Clin Pract. 2009;113:c81–c87 [DOI] [PubMed] [Google Scholar]
- 18. Ruggenenti P, Remuzzi G. Nephropathy of type-2 diabetes mellitus. J Am Soc Nephrol. 1998;9:2157–2169 [DOI] [PubMed] [Google Scholar]
- 19. Asai O, Nakatani K, Tanaka T, et al. Decreased renal α-Klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. Kidney Int. 2012;81:539–547 [DOI] [PubMed] [Google Scholar]
- 20. Kim MK, Chung SW, Kim DH, et al. Modulation of age-related NF-κB activation by dietary zingerone via MAPK pathway. Exp Gerontol. 2010;45:419–426 [DOI] [PubMed] [Google Scholar]
- 21. Hu MC, Kuro-o M, Moe OW. The emerging role of klotho in clinical nephrology. Nephrol Dial Transplant. 2012;27:2650–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney. II. Pathophysiology and therapeutic implications. Am J Physiol Renal Physiol. 2012;303:F180–F191 [DOI] [PubMed] [Google Scholar]
- 24. Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417 [DOI] [PubMed] [Google Scholar]
- 25. Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology. 2007;12:261–266 [DOI] [PubMed] [Google Scholar]
- 26. Hsieh CC, Kuro-o M, Rosenblatt KP, Brobey R, Papaconstantinou J. The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging. 2010;2:597–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin Y, Sun Z. Thyroid hormone potentiates insulin signaling and attenuates hyperglycemia and insulin resistance in a mouse model of type 2 diabetes. Br J Pharmacol. 2011;162:597–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun Z, Bello-Roufai M, Wang X. RNAi inhibition of mineralocorticoid receptors prevents the development of cold-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1880–H1887 [DOI] [PubMed] [Google Scholar]
- 29. Reddy AK, Taffet GE, Li YH, et al. Pulsed Doppler signal processing for use in mice: applications. IEEE Trans Biomed Eng. 2005;52:1771–1783 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X, Skelley L, Wang B, Mejia A, Sapozhnikov V, Sun Z. AAV-based RNAi silencing of NADPH oxidase gp91(phox) attenuates cold-induced cardiovascular dysfunction. Hum Gene Ther. 2012;23:1016–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin Y, Sun Z. Antiaging gene klotho enhances glucose-induced insulin secretion by up-regulating plasma membrane levels of TRPV2 in MIN6 β-cells. Endocrinology. 2012;153:3029–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214–F222 [DOI] [PubMed] [Google Scholar]
- 34. Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes. 2012;61:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabet Res. 2011;908185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Las G, Shirihai OS. The role of autophagy in beta-cell lipotoxicity and type 2 diabetes. Diabetes Obes Metab. 2010;12(Suppl 2):15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab. 2008;4:444–452 [DOI] [PubMed] [Google Scholar]
- 38. Brosius FC., 3rd New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev Endocr Metab Disord. 2008;9:245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y, Kuro-o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell. 2012;11:410–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kacso IM, Bondor CI, Kacso G. Soluble serum klotho in diabetic nephropathy: relationship to VEGF-A. Clin Biochem. 2012;45:1415–1420 [DOI] [PubMed] [Google Scholar]
- 41. Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res. 2009;41:585–593 [DOI] [PubMed] [Google Scholar]
- 42. Lehmann R, Schleicher ED. Molecular mechanism of diabetic nephropathy. Clin Chim Acta. 2000;297:135–144 [DOI] [PubMed] [Google Scholar]
- 43. Zhu Y, Usui HK, Sharma K. Regulation of transforming growth factor beta in diabetic nephropathy: implications for treatment. Seminars in nephrology. 2007;27:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor-β1 (TGF-β1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mariappan MM. Signaling mechanisms in the regulation of renal matrix metabolism in diabetes. Exp Diabet Res. 2012:749812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]