

Abstract
Both anatomically modern humans and the gastric pathogen Helicobacter pylori originated in Africa, and both species have been associated for at least 100,000 years. Seven geographically distinct H. pylori populations exist, three of which are indigenous to Africa: hpAfrica1, hpAfrica2, and hpNEAfrica. The oldest and most divergent population, hpAfrica2, evolved within San hunter-gatherers, who represent one of the deepest branches of the human population tree. Anticipating the presence of ancient H. pylori lineages within all hunter-gatherer populations, we investigated the prevalence and population structure of H. pylori within Baka Pygmies in Cameroon. Gastric biopsies were obtained by esophagogastroduodenoscopy from 77 Baka from two geographically separated populations, and from 101 non-Baka individuals from neighboring agriculturalist populations, and subsequently cultured for H. pylori. Unexpectedly, Baka Pygmies showed a significantly lower H. pylori infection rate (20.8%) than non-Baka (80.2%). We generated multilocus haplotypes for each H. pylori isolate by DNA sequencing, but were not able to identify Baka-specific lineages, and most isolates in our sample were assigned to hpNEAfrica or hpAfrica1. The population hpNEAfrica, a marker for the expansion of the Nilo-Saharan language family, was divided into East African and Central West African subpopulations. Similarly, a new hpAfrica1 subpopulation, identified mainly among Cameroonians, supports eastern and western expansions of Bantu languages. An age-structured transmission model shows that the low H. pylori prevalence among Baka Pygmies is achievable within the timeframe of a few hundred years and suggests that demographic factors such as small population size and unusually low life expectancy can lead to the eradication of H. pylori from individual human populations. The Baka were thus either H. pylori-free or lost their ancient lineages during past demographic fluctuations. Using coalescent simulations and phylogenetic inference, we show that Baka almost certainly acquired their extant H. pylori through secondary contact with their agriculturalist neighbors.
Author Summary
Genetic analyses of Helicobacter pylori have illuminated human migrations and the history of human infection by these bacteria. Both humans and H. pylori originated in Africa, and have been intimately associated for at least 100,000 years. We hypothesized that communities who still live in relative isolation might provide further details about the evolutionary history of H. pylori in Africa. We therefore investigated H. pylori within Baka Pygmies of southeast Cameroon, who live as hunter-gatherers in the tropical rainforest, and compared those bacteria to H. pylori from neighboring farming populations of non-Baka ethnicities. Unexpectedly, Baka Pygmies were much less commonly infected (20.8%) than the non-Baka (80.2%). H. pylori from hunter-gatherers and agriculturalists were genetically very similar and ancient H. pylori lineages were not identified in Baka. We used an epidemiological model to show that demographic factors including small population size and low life expectancy can account for the low infection rate among Baka Pygmies, and that this low rate could have been attained within a few hundred years of secondary contact with their neighbors. We also suggest that the ancestors of the Baka Pygmies were initially H. pylori-free or that their ancestral bacteria have been lost through past demographic fluctuations.
Introduction
Helicobacter pylori is a major gastric pathogen that infects more than half the world's human population. Infection is almost always life-long, generally acquired during early childhood and results in gastric inflammation, which remains asymptomatic in most individuals. 10–15% of infected individuals develop gastric or duodenal ulcers, and infection with H. pylori is also a cause of gastric cancer and mucosa-associated lymphoid tissue (MALT) lymphoma [1]. Transmission within families plays a dominant role in areas with better sanitary conditions, but extra-familial horizontal transmission is typical in rural areas of the developing world [2], [3]. As a result, developing countries have infection rates that are much higher than the global average, reaching >90% in many areas [4]–[7].
The association of H. pylori with humans is ancient, dating back at least 100,000 years (100 kyr) [4]. Africa is the ancestral home of humans and H. pylori alike, and three distinct African populations have been identified within this bacterial species [8]. Infected anatomically modern humans carried H. pylori with them when they left Africa about 60,000 years ago (60 kya) [8], and diverse demographic and selective pressures subsequently resulted in four additional, geographically-associated populations in Eurasia, Sahul and the Americas [8]–[10]. The genetic diversity of H. pylori reflects historical human demographic events, including the peopling of the Pacific [10], the colonization of the Americas [9] and migrations in Iran [11], northern India [12], Malaysia [13] and Southeast Asia [14], [15]. Until recently, however, little attention was paid to demographic movements during early human history in Africa. The three H. pylori populations that are indigenous to Africa have been designated hpNEAfrica, hpAfrica1, and hpAfrica2 [8], [9] (Figure 1A). hpAfrica1 has been subdivided into two subpopulations: hspSAfrica, found mainly in South Africa, and hspWAfrica, which is found in both West and South Africa. hpNEAfrica was isolated from Nilo-Saharan speakers in Ethiopia, Somalia, Sudan, Nigeria and Algeria. hpAfrica2, the most genetically distinct of all H. pylori populations, was isolated only from individuals in South Africa, Namibia and southern Angola [4], [9], [16].
Figure 1. The sources of African H. pylori isolates.

(A) The proportions of haplotypes from the different African H. pylori populations at each sampling location are displayed as pie charts, whose size is proportional to the total number of haplotypes. Colors indicate STRUCTURE population assignments. All hpAfrica1 and hpNEAfrica isolates indicated were used in the corresponding STRUCTURE analyses. Country code: DZ, Algeria; AO, Angola; BF, Burkina Faso; CM, Cameroon; ET, Ethiopia; GM, Gambia; MA, Morocco; NA, Namibia; NG, Nigeria; SN, Senegal; SO, Somalia; ZA, South Africa; SD, Sudan. (B) The distributions of the Cameroonian haplotypes at the two sampling locations are displayed as pie charts. The size corresponds to the total number of haplotypes. The Baka villages are located at distances of 18–19 km from Abong Mbang and 6–14 km around Yokadouma, respectively. The non-Baka participants lived either in Abong Mbang or Yokadouma or in villages located within a distance of less than 50 km from Abong Mbang, or 36 km from Yokadouma.
The existence of these multiple populations of H. pylori reflects diversification in Africa after its initial acquisition by humans at least 100 kya. Thereafter, H. pylori diverged into two super-lineages, one which became hpAfrica2, and a second which eventually diversified into all other extant populations. hpAfrica2 evolved within the click-speaking San [4], a group of hunter-gatherers indigenous to southern Africa who represent one of the deepest branches of the human population tree [17]–[22]. Genome-wide analyses of numerous human populations have revealed that other hunter-gatherer populations such as Pygmies, Sandawe and Hadza also branched early in African prehistory, suggesting a common and ancient origin for hunter-gatherers [20], [23], [24]. Pygmy hunter-gatherers diverged from the ancestors of contemporary agriculturalist Africans about 56–70 kya [25], [26]. Population splits within each hunter-gatherer group, however, are more recent. Molecular data suggest that the San split into two groups only 32–47 kya [4], [27] whereas Pygmies separated into ancestral Western and Eastern populations ∼20 kya [25] (see Figure S1).
From a bacterial perspective, only the hpAfrica2 super-lineage has been found to be associated with hunter-gatherers thus far. The populations hpNEAfrica and hpAfrica1 comprise the African component of the second H. pylori super-lineage, sharing a common ancestor 36–52 kya [4]. Therefore they may reflect more recent human demographic events in Africa. We find hpNEAfrica to be endemic among eastern Nilo-Saharan speakers in northeast Africa [8]. This language group is thought to have originated in northeast Africa, expanding westward during the humid Holocene period (6–9 kya) when the central Sahara and beyond was inundated with rivers and wetlands [28] (Figure S1). Since only few hpNEAfrica strains have been isolated from elsewhere in Africa, it has remained unclear whether hpNEAfrica strains from Nigeria and Algeria represent an ancient westward expansion of Nilo-Saharans or modern migrations. hpAfrica1 appears to be associated with the more recent sub-Saharan migrations of a highly successful group of agriculturalists whose languages belong to a closely related language family commonly referred to as Bantu - officially the Benue-Congo subfamily of the Niger-Congo language family. Population expansions of Bantu speakers from their homeland in modern Cameroon/Nigeria spread their languages and agriculture through most of sub-Saharan Africa in the last 3–6 kyr [29]. Bantu speakers are thought to have migrated in two waves that skirted the dense tropical rainforests, down the west coast towards southern Africa and to the eastern side of Africa followed by further southward migrations [30], [31] (Figure S1), although the details remain controversial [31], [32]. The west and south African subpopulations within hpAfrica1 might parallel these human migrations [9].
Considering the ancient origins of Pygmy mitochondrial and Y-chromosome lineages, and by analogy with the association between hpAfrica2 and San hunter-gatherers, we hypothesized that Pygmies might be infected with a currently unknown, Pygmy-specific population of H. pylori derived from one of the two super-lineages. Similar to the San, we also anticipated that Pygmies may have acquired populations of H. pylori from their agriculturalist neighbors, with whom they have been in contact for 3–6 kyr [33], [34], and from whom they acquired the Bantu language. We therefore tested these hypotheses in a population genetic study of the H. pylori infecting the Baka, a subgroup of the Western Pygmies from two populations in Cameroon. Baka inhabit the rainforests of Cameroon, Congo and Gabon and constitute one of the largest of all Pygmy groups, with a population size of 30–40,000 [35]. The prevalence and population structure of the H. pylori infecting the Baka and other Pygmies was previously unknown. Their Bantu-speaking, agriculturalist neighbors all have high H. pylori infection rates [36], but again the population structure of those bacteria was unknown. We therefore also screened H. pylori from neighboring non-Baka of multiple ethnic groups in order to quantify the levels of bacterial gene flow between hunter-gatherers and agriculturalists.
We obtained several interesting results: 1) the prevalence of H. pylori infection among Baka Pygmies is much lower than among neighboring non-Baka agriculturalists, 2) H. pylori from the Baka did not define a new population, 3) Baka acquired their H. pylori infections from non-Baka neighbors, 4) acquisition of H. pylori by the Baka was recent, and post-dated the 3–6 kyr of secondary contact between Baka and non-Baka, 5) the frequency of H. pylori infection of the Baka is probably limited by population size and other demographic factors.
Results
Infection rates
We analyzed H. pylori prevalence in 178 Cameroonians, including 77 Baka Pygmies and 101 non-Baka agriculturalists, by bacterial culture from gastric mucosal biopsies obtained through esophagogastroduodenoscopy (Table 1). Among the Baka, 46.8% were male; the mean age of the Baka participants was 38.9 years. Among non-Baka, 21.8% were male, with a mean age of 46.0 years. Similar inter-sample age differences were present for males and females. We found a high prevalence of H. pylori infection (81/101; 80.2%) within the non-Baka (Table 1), which is concordant with infection rates in Cameroon among patients [5], [36] and asymptomatically infected individuals [37]. However, the infection rate among Baka Pygmies was significantly lower (16/77; 20.8%; Chi-square test: p<0.001) than observed among non-Baka, and this low rate was consistent between the two sampling locations, Abong Mbang (10/42; 23.8%) and Yokadouma (6/35; 17.1%), although they are separated by a distance of over 200 km (Figure 1B). Baka women were more frequently infected than men (24.4% vs. 16.7%), and non-Baka men were more frequently infected than non-Baka women (90.9% vs. 77.2%). However, these differences were not significant (Fisher exact test: p>0.05). A multiple logistic regression (controlling for sex and age) suggests that in this sample, non-Baka individuals were about 16 times more likely to be infected than Baka (OR = 16.1, 95% CI 7.3–35.6, p<0.001).
Table 1. Assignment of Cameroonian H. pylori isolates to populations and subpopulations.
| Location | Ethnicity | Language subgroup | No. of individuals | No. of unique haplotypes assigned to H. pylori population/subpopulation1 | |||||
| total | H. pylori | hpNEAfrica | hpAfrica1 | hpEurope | |||||
| neg. | pos. | hspWAfrica | hspSAfrica | ||||||
| Hunter-Gatherers | 77 | 61 | 16 | 8 | 6 | 0 | 0 | ||
| Abong Mbang | Baka | Ubangi | 42 | 32 | 10 | 5 | 4 | 0 | 0 |
| Yokadouma | Baka | Ubangi | 35 | 29 | 6 | 3 | 2 | 0 | 0 |
| Agriculturalists | 101 | 20 | 81 | 65 | 39 | 1 | 2 | ||
| Abong Mbang | 41 | 8 | 33 | 22 | 21 | 1 | 0 | ||
| Badjoue | Benue-Congo | 5 | 0 | 5 | 5 | 1 | 0 | 0 | |
| Bamileke | Benue-Congo | 7 | 1 | 6 | 2 | 10 | 0 | 0 | |
| Bassa | Benue-Congo | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| Betsi | Benue-Congo | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| Bikele | Benue-Congo | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| Boulou | Benue-Congo | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Kako | Benue-Congo | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Maka | Benue-Congo | 17 | 2 | 15 | 10 | 6 | 1 | 0 | |
| Menemo | Benue-Congo | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Moghamo | Benue-Congo | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Pompom | Benue-Congo | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| Wimboum | Benue-Congo | 1 | 0 | 1 | 0 | 2 | 0 | 0 | |
| Yebekolo | Benue-Congo | 2 | 1 | 1 | 1 | 0 | 0 | 0 | |
| Zime | Benue-Congo | 1 | 0 | 1 | 2 | 0 | 0 | 0 | |
| Yokadouma | 60 | 12 | 48 | 43 | 18 | 0 | 2 | ||
| Bamileke | Benue-Congo | 7 | 1 | 6 | 0 | 8 | 0 | 0 | |
| Bapele | Benue-Congo | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| Baya | Ubangi | 2 | 0 | 2 | 4 | 0 | 0 | 0 | |
| Bimou | Benue-Congo | 9 | 1 | 8 | 9 | 1 | 0 | 2 | |
| Bogandou | Benue-Congo | 3 | 1 | 2 | 1 | 2 | 0 | 0 | |
| Boman | Benue-Congo | 2 | 2 | 0 | 0 | 0 | 0 | 0 | |
| Boulou | Benue-Congo | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| Foulbe | Atlantic | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| Kako | Benue-Congo | 3 | 0 | 3 | 3 | 0 | 0 | 0 | |
| Konabembe | Benue-Congo | 7 | 2 | 5 | 5 | 1 | 0 | 0 | |
| Maka | Benue-Congo | 1 | 0 | 1 | 2 | 0 | 0 | 0 | |
| Mvonvon | Benue-Congo | 21 | 4 | 17 | 17 | 4 | 0 | 0 | |
| Ndjem | Benue-Congo | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Toupouri | Adamawa | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| Total | 178 | 81 | 97 | 73 | 45 | 1 | 2 | ||
Seven of the ten identical haplotypes were isolated from individuals of different ethnicity and count more than once, thus, the total number of isolates in this table is 121 instead of 113.
Endoscopic evaluations revealed that 80% of all study participants showed evidence of gastroduodenal pathology (Table S1). None of the observed pathologies showed a significant correlation with H. pylori infection status, in agreement with prior results from developing countries [6]. Moreover, the prevalence of pathologies was not significantly different between Baka and non-Baka populations, despite their different H. pylori infection rates. However, the relatively small number of individuals sampled and the non-randomized design of the study preclude solid conclusions about the associations between H. pylori and pathology as observed by endoscopy.
In total, 191 H. pylori isolates were cultured from all biopsies, and seven MLST housekeeping gene fragments [38] were sequenced from each isolate. An alignment of the concatenated sequences (3,406 bp) revealed 113 distinct haplotypes. Ten haplotypes were shared among more than one individual, the most common of which was found in five individuals, including three Baka Pygmies and two non-Baka individuals. Isolates from antrum and corpus differed in 31 individuals, although most of these strain pairs varied only slightly. There was only one pair of highly divergent strains, which is a clear indication of a mixed infection with unrelated strains. Finding identical haplotypes in multiple individuals indicates that transmission is frequent, relative to mutation and recombination rates, especially considering the numbers of strain pairs from single individuals that differed slightly in their haplotype sequences.
Population structure and phylogeography
Bayesian clustering using both the “no admixture model” and the “linkage model” of STRUCTURE [39] assigned most haplotypes to the populations hpNEAfrica (67; 59.3%) or hpAfrica1 (44; 38.9%) (Figure 2A). Two strains isolated from one non-Baka individual were assigned to hpEurope. hpAfrica2 was not isolated nor did any isolate belong to a novel, Pygmy-specific population. The occurrence of hpAfrica1 strains is not surprising because this population has been isolated repeatedly from Bantu speakers [9]. However, isolating hpNEAfrica in Cameroon was unexpected because it has previously only been found in Afro-Asiatic and Nilo-Saharan speakers [8].
Figure 2. Bayesian population assignments using STRUCTURE 2.0.

(A) DISTRUCT plot of the population assignment of Cameroonian H. pylori haplotypes in a global reference dataset as determined by the “no admixture model” (K = 6) and the “linkage model” (K = 6). Each bacterial isolate is depicted by a thin vertical line, which is divided into K colored segments representing the membership coefficients in each cluster. Black lines separate isolates of different populations/subpopulations. Populations are labeled above the figure, subpopulations below the figure. Cameroonian non-Baka and Baka H. pylori haplotypes are shown on the right side of the plot. (B) DISTRUCT plots of the subpopulation assignment of H. pylori hpAfrica1 haplotypes using the “no admixture model” (K = 2, K = 3). Strains were grouped together according to their geographical source. (C) DISTRUCT plot of the population structure of H. pylori hpNEAfrica haplotypes according to the “no admixture model” (K = 2). Strains were grouped together according to their geographical source.
The proportions of Baka and non-Baka in our study that were infected with hpNEAfrica or hpAfrica1 strains were very similar (Table 1), as were the relative levels of nucleotide diversity within hpNEAfrica and hpAfrica1 haplotypes from both communities (Table 2). Intra- and inter-population pair-wise haplotype differences did not reject a null hypothesis of panmixia (p(Ks) = 0.12) between Baka and non-Baka populations.
Table 2. Genetic diversity of hpAfrica1 and hpNEAfrica haplotypes isolated from Baka Pygmies and non-Baka agriculturalists.
| H. pylori population | Ethnicity | N | h | S | k | π (SD) |
| hpAfrica1 | Baka | 7 | 6 | 183 | 76.29 | 0.02240 (0.00318) |
| non-Baka | 40 | 38 | 336 | 81.22 | 0.02385 (0.00082) | |
| hpNEAfrica | Baka | 10 | 8 | 233 | 80.80 | 0.02372 (0.00202) |
| non-Baka | 70 | 60 | 440 | 76.59 | 0.02249 (0.00064) |
N, total number of sequences; h, number of haplotypes; S, Number of polymorphic (segregating) sites; k, average number of nucleotide differences; π, nucleotide diversity; SD, standard deviation.
Using a composite-likelihood approach to estimate population demographic parameters under an isolation with migration model (Jaatha 2.0) [40], we estimated the time to population split of H. pylori between Baka and non-Baka using explicit demographic models of constant population size and exponential expansion (see Methods for details). The composite likelihood for model 1 (−197.4695; constant population size in Baka) was higher than for scenario 2 (−197.5009; exponential growth in both populations), but estimates for time to population split and effective population size (Table 3) were very similar. Gene flow into the Baka population was higher when Baka bacterial population size was assumed to be constant, although confidence limits for both estimates were widely overlapping. Allowing for gene flow since divergence, we inferred a median date for the split between the Baka and non-Baka H. pylori of 2,110 and 2,406 years for constant size and expansion models respectively, with combined confidence limits of 856–3,973 years. These analyses indicate that the modern infections of Baka by H. pylori began no earlier than 4,000 years ago, soon after Baka came into contact with non-Baka agriculturalists (∼3–6 kya).
Table 3. Population demographic parameters for H. pylori from Baka Pygmies and non-Baka agriculturalists derived using an isolation-with-migration model and assuming two demographically explicit scenarios.
| Theta (θ) | Timing (τ) | Gene flow (m) | Growth (α) | Recombination (rho) | ||||
| Model 1: Constant Baka population size, exponential growth of non-Baka population | ||||||||
| θ 1 (non-Baka) | τ | T (years) | Baka→non-Baka | non-Baka→Baka | α 1 non-Baka | α 2 Baka (fixed) | rho | |
| Median | 13.78 | 0.0006 | 2,110 | 0.2145 | 0.0781 | 0.0023 | 0 | 0.0033 |
| c.i. L | 13.76 | 0.0003 | 856 | 0.0292 | 0.0047 | 0.0007 | 0 | 0.0003 |
| c.i. U | 13.87 | 0.0012 | 3,981 | 0.6325 | 0.2662 | 0.0062 | 0 | 0.0476 |
| Model 2: Exponential growth of both Baka and non-Baka populations | ||||||||
| θ 1 (non-Baka) | τ | T (years) | Baka→non-Baka | non-Baka→Baka | α 1 non-Baka | α 2 Baka | rho | |
| Median | 13.78 | 0.0007 | 2,406 | 0.4373 | 0.0250 | 0.0008 | 0.0012 | 0.0023 |
| c.i. L | 13.71 | 0.0003 | 983 | 0.0502 | 0.0054 | 0.0004 | 0.0003 | 0.0002 |
| c.i. U | 13.89 | 0.0012 | 3,973 | 0.9352 | 0.0910 | 0.0016 | 0.0042 | 0.0018 |
Population parameter theta (θ) = 4N1μ, where N1 is the effective population size of the reference population (non-Baka) and μ is the mutation rate. t is the timing parameter in coalescent units, and T the time to split between Baka and non-Baka (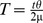 ). m is the fraction of the population replaced by migrants per generation:
). m is the fraction of the population replaced by migrants per generation:  of population 1 (non-Baka) are replaced per generation and
of population 1 (non-Baka) are replaced per generation and 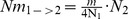 of population 2 (Baka) are replaced per generation. The θ estimated by the software refers only to population 1. c.i., confidence interval; L, lower; U, upper.
of population 2 (Baka) are replaced per generation. The θ estimated by the software refers only to population 1. c.i., confidence interval; L, lower; U, upper.
We also analyzed our Cameroon sample together with other African haplotypes previously assigned to hpAfrica1 or hpNEAfrica, respectively. The most consistent results were obtained for a K = 2 scenario within hpAfrica1, wherein 43 haplotypes from Cameroon were assigned to hspWAfrica and only one to hspSAfrica (Figure 2B). Interestingly, at K = 3, most Cameroonian haplotypes formed a novel subpopulation, hspCAfrica, together with three haplotypes from Angola. These analyses also provide evidence for genetic structure within hpNEAfrica, for which a K = 2 hypothesis was most likely. All haplotypes from Ethiopia (51), Somalia (2) and isolates from Sudan (42/63) and Algeria (1/2) clustered together in an ‘East African’ subpopulation, hspEastNEA, whereas all isolates from Cameroon (67), Nigeria (8) and some isolates from Sudan (21/63) and Algeria (1/2) formed a second ‘Central/West African’ subpopulation which we call hspCentralNEA (Figure 2C).
ClonalFrame was used to further elucidate the substructure of both the hpAfrica1 and hpNEAfrica populations. The resulting genealogies (Figures S2 and S3) highlight the interspersed distribution of Baka H. pylori isolates among the non-Baka strains from Cameroon, without Baka-specific groupings. Both phylogenies comprised several clades, all supported in over 80% of the posterior sample, but corresponding only roughly to the subpopulations within hpAfrica1 and hpNEAfrica.
H. pylori infection among Baka Pygmies is recent
We were intrigued by the low frequency with which the Baka were infected by H. pylori, which could further corroborate the hypothesis of recent acquisition by the Baka. In order to investigate these observations quantitatively, we implemented an age-structured SI model, building on an existing model of H. pylori infection developed by Rupnow et al. [41]. In concordance with observed data, the model assumes that infection is life-long once acquired. Unlike in the original model, we did not further subdivide the compartment of infected individuals by severity or pathology. The model population was divided into three age groups: children, youths and adults; and we stipulated that a fraction of individuals are born non-susceptible, in keeping with observations that show that prevalence never attains 100%, even in highly infectious settings [42], and consistent with the implementation of the existing model [41]. Our model includes the following assumptions: 1) The Baka population is both stable and stationary, that is, both the total population size and the fraction in each age class are constant, allowing us to estimate constant birth and age-specific death rates. The assumption of a stable population is consistent with our coalescent simulations above, that show only very slight differences in splitting times between models of constant population size and population expansion (Table 3). 2) Infection does not affect all-cause mortality. This was demonstrated in a recent study [43]. 3) Inter-age class transmission is governed by the characteristics of the less-transmissive partner. This is conservative, but represents an increase in transmission from the Rupnow et al. model [41], which assumes that inter-age class transmission is negligible. 4) The population is closed, with no migration or transmission from external sources. This is unlikely in reality, given the close proximity of agriculturalist communities; however, since the Baka are quite reclusive, we consider it a reasonable simplifying assumption. Additionally, since the Baka have only recently (3–6 kya) come into secondary contact with agriculturalists [33], [34], the assumption would likely hold true for Baka populations prior to this time. 5) Survival decreases linearly within age classes. As did the original model, we also assume that transmission is entirely person-to-person. We then parameterized the model according to our best estimates for demographic and transmission parameters among Pygmies (Table 4). Allowing for inter-age class transmission yielded nine transmission constants (β) rather than the three intra-class coefficients used by Rupnow et al. [41].
Table 4. Base scenario model parameters determined empirically for Baka Pygmies and Cameroon.
| Model parameter | Baka Pygmies | Cameroon (countrywide) | ||
| Value | Reference | Value | Reference | |
| Population size (N) | 40,000 | [35] | 200,000 | [41] |
| Life expectancy (Le) (years) | 16.6 | [52] | 51.0 | WHO1 |
| Survivorship at age 5 (S5) | 0.7 | estimated | 0.846 | WHO |
| Survivorship at age 15 (S15) | 0.4 | [52] | 0.823 | WHO |
| Fraction born immune (NS) | 0.15 | [42] | 0.15 | [42] |
| Transmission coefficients | All derived from [41] | All derived from [41] | ||
| βCC | 0.0000183 | 0.0000183 | ||
| βCY | 0.00000367 | 0.00000367 | ||
| βCA | 0.0000004 | 0.0000004 | ||
| βYC | 0.00000367 | 0.00000367 | ||
| βYY | 0.00000367 | 0.00000367 | ||
| βYA | 0.0000004 | 0.0000004 | ||
| βAC | 0.0000004 | 0.0000004 | ||
| βAY | 0.0000004 | 0.0000004 | ||
| βAA | 0.0000004 | 0.0000004 | ||
WHO, 2012.
WHO Global Observatory Health Data Repository. World Health Organization, Geneva, Switzerland. http://apps.who.int/gho/data/. Last accessed on 11/02/2012.
N: For the Cameroon-wide simulation to validate the model, a population of 200,000 was adopted, following Rupnow et al. [41].
Le: For the Baka, the Migliano et al. estimate for the Aka was used [52].
S5: Fraction of individuals that reach five years of age. For the Baka, no estimate was available, so the base scenario assumes that half of mortality that will occur by age 15 takes place by age 5, in keeping with typically high infant mortality in developing-world contexts.
S15: Fraction of individuals that reach fifteen years of age. For the Baka, the Migliano et al. estimate for the Aka was used [52].
NS: Fraction born non-susceptible, i.e., the fraction of individuals that cannot become infected “for physiologic, physical, or immunologic reasons” [41]. Estimated from observed maximum prevalences in the developing world.
βXY: = Transmission coefficients, with X and Y in {C,Y, A}, where these stand for child, youth and adult age classes, respectively, yielding nine coefficients in all. This represents the probability of transmission from class X to class Y, and is derived from Rupnow et al. [41], as described in the text.
The model thus specified indicates a low equilibrium prevalence for the Baka of ∼10.2% under the assumptions of the base scenario. This is close to what we observed, and suggests that the time required to establish a stable equilibrium at this prevalence is considerably shorter than the estimated age of the most recent common ancestor of H. pylori haplotypes from Baka and non-Baka - under the base scenario the estimated time to equilibrium was 1,164 years. We tested the sensitivity of this model by varying the parameter estimates individually across a wide range of values, encompassing plausible ranges of demographic variation estimated from the literature or prior experience with the Baka (Figure 3). Equilibrium infection rates among adults remained low (<40%) over the ranges of plausible variation for individual parameters. Moreover, infection was nearly or entirely eliminated in the population for some values within these ranges for most parameters, suggesting that the Baka are near a limit for viable maintenance of population-level H. pylori infection. The sensitivity analysis indicated that the most crucial parameter was population size (Figure 3A). In fact, the observed infection rate of 20.8% is achieved at equilibrium in the model with a small population increase, to N = 43,000 from the 40,000 assumed in the base scenario. Mean life expectancy also appears to have a strong but non-linear effect on infection, with equilibrium prevalence falling to zero over intermediate values (Figure 3C). Time to equilibrium increased rapidly as equilibrium infection rates approached zero, decreasing the meaningfulness of this statistic at particularly low infection rates. However, within the range of uncertainty, time to equilibrium is reached in less than 1,000 years for those scenarios where the equilibrium infection rate is equal to or exceeds the value we observed in the Baka.
Figure 3. Sensitivity analysis of equilibrium values for H. pylori infection (primary y-axes) and time to equilibrium (secondary y-axes) in Baka Pygmies for six demographic and transmission parameters (panels A–F).

For each panel, adults are in green, youths in red, children in blue. The dashed vertical line shows the value of the parameter adopted in the base scenario; the two solid vertical lines delimit plausible ranges of variation in this population, where these can be estimated from the literature. The dotted black line illustrates the time in years required for the prevalence in adults to approach to within 1% of the equilibrium value.
Increasing the population size to 200,000 and applying model parameters that are typical of Cameroon (Table 4) yielded infection frequencies that are consistent with non-Baka populations in Cameroon (Figure 4), and other parts of the developing world [42]. This supports the validity of the model and is consistent with the idea that human population sizes were fairly large during the 100,000 years that they have been infected by H. pylori [4].
Figure 4. Model predicted prevalence under the base scenario versus observed H. pylori prevalences by age in Cameroon.

The model-predicted prevalence rises in steps with the three age classes (i.e., child, youth, adult).
As with all epidemiological models, our results should be interpreted with care. Following Rupnow et al. [41], we incorporated a frequency-dependent transmission parameter (rather than a density-dependent parameter). This is a common assumption for directly-transmitted diseases, but the estimated values from the original model might be inaccurate when applied to substantially different population sizes. Moreover, it is possible that another model structure might better capture the complex reality underlying H. pylori transmission, such as a model incorporating the possibility of environmental transmission. Although the utility of the original model in various contexts [41], [44], [45] suggests that it captures many of the most important aspects of transmission, further empirical work on the importance of environmental transmission in sub-Saharan Africa would allow us to refine our model accordingly. There is also uncertainty in parameter estimates. For example, the proportion of non-susceptible individuals can vary across populations as a function of innate immunity, lifestyle, contact patterns and/or factors such as atrophic gastritis which eliminates infection [42]. Our results rely on the accuracy of demographic and transmission estimates for the Baka, which in many cases are undefined or highly unusual for human populations. Further work verifying the accuracy of these estimates would be useful.
Discussion
Based on the >100 kyr long association between H. pylori and man and our recent findings that the San hunter-gatherers of southern Africa were the original hosts of hpAfrica2 [4], we expected to identify unique H. pylori lineages that have long been associated with West African Baka Pygmies. Our results did not identify such lineages. Instead, H. pylori haplotypes from Baka belong to the hpAfrica1 or hpNEAfrica populations, which are also prevalent among other Africans (Figure 1A). Surprisingly, we also found that Baka were only rarely infected with H. pylori, significantly less than global frequencies in developing countries [6], [7], including their agriculturalist neighbors.
Phylogeography
Under a K = 3 scenario, we identified a novel subpopulation within hpAfrica1, designated hspCAfrica, which because it comprises haplotypes from Cameroon and Angola, provides support for a Bantu agriculturalist migration along the western coast of Central Africa [30], [31] (see also Figure S1). Interestingly, we did not detect this population among 91 previously isolated hpAfrica1 strains from Namibia and South Africa, which were designated hspSAfrica and hspWAfrica (Figure 2B). This suggests that the western Bantu migration that brought hspCAfrica to Angola was not able to cross the Namib Desert and progress as far south as present-day South Africa. This agrees with the hypothesis that the Bantu who originally colonized South Africa were not part of this western migration, and instead brought hpAfrica1 to South Africa via the alternative eastern coastal route [31], [32]. We would therefore expect to find both hspSAfrica and hspWAfrica in high frequency among eastern African populations. Investigating the frequency of hpAfrica1 throughout south-central Africa could potentially shed even further light on the enigmatic Bantu migrations.
We also defined two subpopulations of hpNEAfrica, hspEastNEA and hspCentralNEA, which are distributed across the Sahel from Nigeria to Somalia. Sudan contains both subpopulations, suggesting that this subdivision may have originated there. Alternatively, the presence of both populations in Sudan may represent more recent, secondary migrations. The existence of hspCentralNEA in Cameroon, Nigeria and Algeria may correspond to the Holocene expansions that carried Nilo-Saharan languages from northeast Africa into the Sahara. We note however, that most of the Cameroonians from whom we isolated hpNEAfrica were Bantu speakers who presumably originated in West Africa. This situation resembles the frequent isolation of hpEurope and hpAfrica1 in South America, where they have largely displaced the ancestral hspAmerind bacteria [9], and the isolation of multiple populations of H. pylori from Australian aboriginals in addition to their ancestral hpSahul bacteria [10]. We also found that phylogenetic reconstructions of both hpAfrica1 (Figure S2) and hpNEAfrica (Figure S3) revealed greater detail about population structure than did Bayesian clustering. These showed a more gradual or gradient-like transition from one population/clade to another. Together, this evidence suggests that H. pylori is a sensitive indicator of human population admixture. Further genetic studies on populations in Cameroon may provide insights on local colonization processes, and the origins and diffusion of agriculturalist communities.
Gene flow and contraction
The strong similarity of haplotypes from Baka Pygmies and non-Baka agriculturalists and the absence of significant differentiation between them justifies treating these H. pylori as a single metapopulation. This inference is plausible because Baka are in daily contact with their Bantu agriculturalist neighbors [46], and these populations exchange a low number of human migrants per generation [25]. Although we also inferred high gene flow between Baka and non-Baka populations [47], phylogenetic analyses suggest that Baka strains were derived from non-Baka ancestors. Under a hypothesis of acquisition of H. pylori by one group from another, we would expect that strains isolated from the donor population would occupy ancestral positions closer to the base of a subclade and that they would form closely related clusters, some strains of which would be passed on to the recipient population. This is precisely what we observe in both hpAfrica1 and hpNEAfrica phylogenies (Figures S2 and S3) - groupings among non-Baka strains, with one of the strains belonging to a Baka. We interpret these results, as well as the unusually low equilibrium prevalence of H. pylori among Baka, as reflecting the acquisition of H. pylori by the Baka via secondary contact with their non-Baka agriculturalist neighbors. This most likely occurred around 2,500 years ago with an upper bound of 3,973 years (Table 3). This age estimate for the Cameroon H. pylori metapopulation is in complete agreement with a hypothesis of recent secondary contact because Bantu expansions have been dated by archaeology to 3–5 kyr [34] and by human population genetics to ∼6 kyr [33]. Furthermore, the number of distinctly different and unrelated haplotypes found among the Baka is also suggestive of several independent agriculturalist to hunter-gatherer transmission events.
Factors influencing H. pylori infection rate
According to our H. pylori-specific transmission model, the levels of infection observed in Baka can be achieved in a short timeframe after introduction into a naïve population, and are reproducible within the range of uncertainty in parameter estimates. The infection rate of 10.2% predicted by the model under the base scenario was slightly lower than the observed rate, possibly implying that the latter is influenced by continuously introduced infection from non-Baka agriculturalists. Regular and ongoing contact or cross-migration between Baka and agriculturalists would also imply a higher effective population size, which would increase the observed infection rate relative to that implied by the closed model. Conversely, we note that our samples were from Baka individuals with gastric complaints, which might overestimate the population prevalence of infection in the Baka (an effect that might be even more pronounced for non-Baka, who were sampled from among hospital patients).
Possibly the most striking finding from the simulations was that even small fluctuations in population size can result in the extinction of population-wide infection. This observation may explain the apparent absence of ancient bacterial lineages among Baka. While there are currently no estimates on the prehistoric census size of San or Pygmies, archaeological evidence suggests that San occupied a much wider territory than Pygmies [48]. From molecular demographic reconstructions, we know that prehistoric hunter-gatherer populations were larger [49], denser [50] and more constant in size [47] than in the present day, but they suffered declines during the Holocene at the advent of Neolithic agriculture. The Baka population size is now ∼40,000 [35], which is less than a fifth of the 208,000 extant San speakers [51]. Thus, the San, being less affected by the rise of agriculture, were able to retain their ancient H. pylori lineages, while any that existed among the Baka may have died out leaving an uninfected population which could acquire novel lineages from their non-Baka neighbors on secondary contact. The postulated ancient lineages might be found by more extensive sampling, and/or by sampling H. pylori from Eastern Pygmies. Alternatively, ancient haplotypes may never have existed and Baka were first infected with H. pylori in recent times.
A second interesting observation was that the short life expectancy among Pygmies [52] contributes to maintenance of endemic H. pylori infection. Transmission of H. pylori is particularly high during childhood [1], which in turn results in higher prevalence where life expectancy is low and children make up a larger proportion of the population. Our simulations indicate that the expected prevalence drops with increasing life expectancy - though it increases with age class, as individuals, once infected, remain infected. Intriguingly, prevalence rises once more when life expectancy exceeds 40 years, despite lower rates of transmission in adults. This is probably because longer-lived adults can potentially transmit infections for much longer periods. However, these insights were obtained by varying single parameters while holding other variables constant at base-scenario levels; simultaneous fluctuation in several parameter values would result in a wider range of estimated endemic prevalence values.
Apart from violations of model assumptions, other explanations could potentially also account for the low prevalence of H. pylori among the Baka, including their hunter-gatherer diet based on forest products and the medicinal use of rain-forest plants with antibiotic properties [53]–[55]. A low prevalence of H. pylori in Java, Malaysia and Tanzania has also been attributed to local diet [56] but those epidemiological data have not been considered in light of a demographic transmission model as presented here [57]. Finally, genetic or innate immunity that is specific to the Baka may also have played a role.
Materials and Methods
Study area, sample selection, and ethics statement
We undertook field collection of gastric biopsies at two settlements in the rainforests of the East Province of Cameroon. Yokadouma and Abong Mbang are towns of more than 20,000 inhabitants that lie about 250 km apart. The Bantu populations are engaged mainly in primary agriculture (hereafter referred to as agriculturalists), while the Baka are hunter-gatherer communities that live in the forests surrounding both settlements. Study participants were selected from among individuals complaining of gastric pain. Esophagogastroduodenoscopy was performed by a certified gastroenterologist (A.M.) with written informed consent at Yokadouma and Abong Mbang Hospitals under ethics certificate 0002/ERCC/CBNO2 from the Cameroon Bioethics Initiative (CAMBIN), administrative authorization 631.19-09 from the Cameroon Ministry of Public Health (Secretariat General, Division of Health Operations Research), and with permission of the ethics committee of the Charité Hospital in Berlin, Germany (ethics certificate EA1/071/07). Among agriculturalists, samples were taken from patients at their respective district hospitals, whereas Baka - who were unlikely to attend the hospital - were recruited directly from their communes by an NGO worker familiar with the area. Baka communes were provided minor food incentives (e.g., rice) for participation, which did not depend on the number of participants from each village. Non-participation or refusal was rare among approached subjects, both at the hospital and in the field.
H. pylori samples and culture
Gastric biopsies were taken from 178 Cameroonians belonging to 25 different ethnic groups (Table 1). All individuals were endoscopically evaluated for gastroduodenal pathology, histological evaluation was not performed. Biopsies were obtained from both the antrum and corpus of the stomachs of 176 people, while only single biopsies from the corpus were obtained from two individuals. All biopsies were immersed in PBS (phosphate buffered saline) and immediately frozen in liquid nitrogen, kept at −80°C and couriered to the Hannover Medical School. H. pylori was cultivated by inoculation of the biopsies on blood agar plates (Columbia-Agar-Basis II, Oxoid, Wesel) supplemented with 10% horse blood (Oxoid), and the antibiotics vancomycin (10 mg/l), polymyxin B (2500 U/l), amphotericin B (4 mg/l) and trimethoprim (5 mg/l). Plates were incubated under microaerobic conditions (5% O2, 10% CO2, 85% N2) for 3 to 5 days at 37°C. In case of lack of bacterial growth, the plates were incubated for longer periods of time. Since concomitant infection with several different H. pylori strains has been previously observed [2], strains were purified by single colony isolation from one colony from each of the antrum and corpus biopsies, resulting in a total of 191 isolates. To ensure optimal efficiency of cultures, the entire biopsies were used for culture, and antrum and corpus biopsies were cultured by independent investigators on separate days, with overall excellent agreement between results.
Molecular genetics
Genomic DNA of bacterial strains was isolated using the QIAamp DNA Mini Kit (Qiagen). Seven housekeeping gene fragments (atpA, efp, mutY, ppa, trpC, ureI, yphC) were amplified by PCR. Primer sequences and primer combinations can be found at http://pubmlst.org/helicobacter. PCR was performed under the following conditions: initial denaturation of 5 min at 94°C, 30 cycles of 1 min at 94°C, 1 min of annealing, 1 min at 72°C, and a final extension of 7 min at 72°C. The annealing temperature was either 57°C (atpA, trpC), 55°C (yphC), 53°C (efp, mutY), or 51°C (ppa, ureI). PCR products were purified using the QIAquick 96 PCR Purification Kit (Qiagen). PCR amplicons were sequenced bidirectionally using the BigDye Terminator v1.1 Cycle Sequencing Kit and the 3130xl Genetic Analyzer (Applied Biosystems). Sequence data were analyzed using BioNumerics v6.01 (Applied Maths NV, Sint-Martens-Latem, Belgium).
Diversity and structure
Data graphing and statistical analyses were performed with Origin 8.0 (OriginLab, USA) and SigmaStat 3.5 (SYSTAT, USA). Population genetic diversity indices were calculated using DnaSP v5 [58].
A test of population subdivision was applied to evaluate whether Baka and non-Baka H. pylori populations were panmictic [59]. This statistic is particularly effective in detecting population structure in the presence of high recombination, as is the case for H. pylori. The test generated a random distribution for the Ks statistic (sum of the mean number of pairwise differences for each population weighted by population) using a Monte Carlo approach that randomized genotypes between the two populations using 10,000 simulations. The null hypothesis of panmixia is not rejected if p(Ks)>0.05.
We performed Bayesian population assignments on the 113 Cameroonian H. pylori haplotypes along with another 792 previously published global reference haplotypes [8]–[10]. The “no admixture model” and “linkage model” of the program STRUCTURE 2.0 [39] were used to assign the Cameroonian isolates to one of the known modern H. pylori populations [8]–[10] and to explore their level of reciprocal admixture, respectively. In order to determine the number of bacterial populations (K) within the dataset ten independent STRUCTURE runs of 50,000 iterations following a burn-in period of 25,000 iterations for each K value were analyzed and compared for consistency. To reveal subpopulation structure subsequent analyses were performed separately for hpAfrica1 and hpNEAfrica isolates with ten independent STRUCTURE runs for each K value. The Cameroonian isolates assigned to hpAfrica1 (n = 44) were further analyzed together with 93 hspSAfrica and 73 hspWAfrica reference isolates. The Cameroonian isolates assigned to hpNEAfrica (n = 67) were further analyzed together with 126 hpNEAfrica reference isolates. The latter include 65 previously published haplotypes [8], [9] as well as 61 unpublished haplotypes obtained from individuals in Sudan in 2004 and 2005. Culture and sequence analysis of the Sudanese isolates were performed as described above for the samples from Cameroon. The STRUCTURE runs yielding the highest model log-likelihood were graphically displayed using the program DISTRUCT [60].
The software ClonalFrame v1.1 [61] was used to determine ancestral relationships among strains within the hpAfrica1 and hpNEAfrica populations. ClonalFrame reconstructs phylogenetic genealogies while accounting for horizontal gene transfer and its value in consistently reconstructing H. pylori phylogenies has been demonstrated previously [4], [10]. The program was run with 100,000 MCMC iterations with a thinning interval of 100 after an initial burn-in phase of 50,000 iterations. Ten independent ClonalFrame runs were performed. Using Treefinder [62] an 80% consensus tree of all independent ClonalFrame runs was computed. Three hpAfrica2 haplotypes were used as an outgroup to root both hpAfrica1 and hpNEAfrica trees.
Gene flow and coalescence
We used the software Jaatha 2.0 [40] to estimate the time to population split and the level of gene flow between Baka and non-Baka H. pylori populations. This coalescence-based software uses the observed joint allele frequency spectrum as a summary statistic to estimate the composite likelihood of posterior parameters in user-defined demographic models. This flexibility allows the user to control for between-population differences in demographic history by explicitly simulating either constant or expanding populations. Furthermore, Jaatha is also able to model recombination within the coalescent framework, controlling for horizontal gene transfer, and is particularly robust in estimating the time to population split in cases of recent population divergence [40], as may be likely among the studied populations. We ran two models: the first assuming that the non-Baka bacterial population underwent exponential growth and Baka remained constant in size; and the second with both populations evolving under exponential growth. These two settings approximate well the possible demographic histories of the two bacterial populations based on previous knowledge of host demographic history. As a first step, we used wide prior values (see Table S2) to explore the parameter probability space for the best starting point for a refined estimate of the posterior parameters. The probability space was divided into a number of equal blocks in which coalescent simulations using the software ms [63] were performed. Since we simulated sets of 6 and 7 parameters for Baka constant and expansion models, probability space was divided into 36 and 37 blocks, respectively, for each of which 200 simulations were performed using the range of prior parameter values. The joint allele frequency spectra were divided into 23 independent Poisson-distributed values and fitted using a generalized linear model. A score proportional to the likelihood was calculated for each parameter set and those with the two highest scores were used as starting points in the final search. Then, a search of 10,000 simulations was repeated 200 times for the two best starting points with five independent seeds. The 10 best parameter combinations for each of these 10 simulations were then subjected to a further simulation step of 100,000 iterations from which the composite likelihood of each parameter combination was calculated. The distribution of posterior parameters was used to estimate confidence intervals. The times to population split in years were calculated using a mutation rate (μ = 2.09×10−6), which is the average of the three estimates presented in Morelli et al. [64].
Transmission model
To estimate the length of time needed to reach equilibrium H. pylori infection among Baka Pygmies, we applied a simple age-structured transmission model, modified from Rupnow et al. [41]. The model assumes a standard Susceptible-Infected (SI) structure dividing the model population into three age groups, namely, children (0–5 years old), youths (5–15 yo) and adults (15+ yo). For a description of model parameters see Table 4.
We constructed a base scenario using the best available estimates for the demographic and transmission parameters: life expectancy (Le), population size (N), survivorship at age 5 (S5), survivorship at age 15 (S15), and fraction of individuals born into the non-susceptible class (NS). Since demographic variables for Western Pygmies are largely uncharacterized [65], we used parameters from a Pygmy population adjacent to the Baka (the Aka) [52] where available; during sensitivity analysis we included the entire range of plausible parameter variation, estimated from the literature where possible. In contrast to the model by Rupnow et al. [41], we allowed for inter-age class transmission. To obtain approximate values for the resulting nine transmission constants (β), we divided those obtained for the prior model - i.e., for intra-age class transmission - by three, and assumed that transmission between age groups was governed by the characteristics of the less-transmissive group. To test the validity of the β-values thus specified, we applied them to a simulation using a population of 200,000 (following Rupnow et al. [41]) and demographic parameters for Cameroon. Because the results fit well with prior observations of age-specific prevalence (see Figure 4), we retained these values for the base scenario.
To test the sensitivity of results to changes in input parameters, we varied each parameter incrementally, while holding all others at base scenario values. When varying transmission constants, we assumed that the relative strength of transmission among age groups remains constant. Simulations were run until the system reached equilibrium with a time step (dt) of 0.25 years, using the Runge-Kutta 4 integration method in iThink 7.0.3 (High Performance Systems, Inc., Hanover, NH).
Data deposition
This publication made use of the Helicobacter pylori Multi Locus Sequence Typing website (http://pubmlst.org/helicobacter/) developed by Keith Jolley and sited at the University of Oxford [66]. The development of this site has been funded by the Wellcome Trust and European Union. Each new isolate from Cameroon and Sudan was assigned an ID number (1663–1836).
Supporting Information
Major human population events in recent African prehistory. This map summarises the potential major human demographic events as inferred from [4], [20], [25], [27], [28], [33], [47], [67]. Ancestral hunter-gatherer populations split into extant populations, beginning with Northern and Southern San (32–47 kya) and Western and Eastern Pygmies ∼20 kya. The Nilo-Saharan speakers originated in northeast Africa from where they migrated both north-westward and south-eastward. Their north-westward migration was favored by a climatic change from dry to more humid conditions during the humid Holocene period (6–9 kya). Bantu-speaking agriculturalist populations expanded from their homeland in what is present-day Nigeria/Cameroon, beginning ∼6 kya, in two independent waves along the western and eastern flanks of Africa into southern Africa. They thus spread their languages and agriculture through most of sub-Saharan Africa within the last 5 kyr.
(TIF)
Phylogenetic relationships among hpAfrica1 isolates. Phylogenetic relationships among hpAfrica1 isolates as determined by ClonalFrame. The haplotypes are colored according to their geographical source and symbols (triangles, circle) specify the respective subpopulation determined by STRUCTURE. Cameroonian haplotypes from non-Baka (brown color) or Baka (pink color) are marked. The hpAfrica2 strains used as outgroup to root the tree are separately indicated. The number of isolates is shown in brackets.
(PDF)
Phylogenetic relationships among hpNEAfrica isolates. Phylogenetic relationships among hpNEAfrica isolates as determined by ClonalFrame. The color-code corresponds to the geographical source. Cameroonian haplotypes isolated from non-Baka (brown color) or Baka (pink color) are indicated. One haplotype was isolated from both non-Baka and Baka individuals (dark-green color). The hpAfrica2 strains used as outgroup to root the tree are separately indicated. The number of isolates is shown in brackets.
(PDF)
Frequencies of endoscopy findings in Baka Pygmy and non-Baka agriculturalist study participants.
(DOC)
Prior parameters used in coalescent simulations with Jaatha 2.0 and their values.
(DOC)
Acknowledgments
We thank Jessika Schulze, Birgit Brenneke and Friederike Kops for excellent technical assistance with culturing of H. pylori strains and MLST sequencing, and Yousra Mirghani and Hatim Mudawi for help with obtaining H. pylori from Sudan. We also thank Marcia Rupnow and Julie Parsonnet for very useful input which informed the design of our transmission model. We thank all participants, especially the Baka people, without their participation and commitment this study would not have been possible.
Funding Statement
This work was supported by a special grant from the Max-Planck Gesellschaft zur Förderung der Wissenschaften, ERA-NET PathoGenoMics projects HELDIVNET (0313930B) and HELDIVPAT (0315905A) from the German Ministry of Education and Research (BMBF), and grant SFB900/A1 from the German Research Foundation (DFG). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kusters JG, van Vliet AH, Kuipers EJ (2006) Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19: 449–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwarz S, Morelli G, Kusecek B, Manica A, Balloux F, et al. (2008) Horizontal versus familial transmission of Helicobacter pylori . PLoS Pathog 4: e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Magalhaes Queiroz DM, Luzza F (2006) Epidemiology of Helicobacter pylori infection. Helicobacter 11 Suppl 1: 1–5. [DOI] [PubMed] [Google Scholar]
- 4. Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, et al. (2012) Age of the association between Helicobacter pylori and man. PLoS Pathog 8: e1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ndip RN, Malange Takang AE, Ojongokpoko JE, Luma HN, Malongue A, et al. (2008) Helicobacter pylori isolates recovered from gastric biopsies of patients with gastro-duodenal pathologies in Cameroon: current status of antibiogram. Trop Med Int Health 13: 848–854. [DOI] [PubMed] [Google Scholar]
- 6. Guisset M, Coton T, Rey P, Debonne JM (1997) Helicobacter pylori infection in developing countries. Med Trop 57: 77–82. [PubMed] [Google Scholar]
- 7. Frenck RW Jr, Clemens J (2003) Helicobacter in the developing world. Microbes Infect 5: 705–713. [DOI] [PubMed] [Google Scholar]
- 8. Linz B, Balloux F, Moodley Y, Hua L, Manica A, et al. (2007) An African origin for the intimate association between humans and Helicobacter pylori . Nature 445: 915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, et al. (2003) Traces of human migrations in Helicobacter pylori populations. Science 299: 1582–1585. [DOI] [PubMed] [Google Scholar]
- 10. Moodley Y, Linz B, Yamaoka Y, Windsor HM, Breurec S, et al. (2009) The peopling of the Pacific from a bacterial perspective. Science 323: 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wirth T, Wang X, Linz B, Novick RP, Lum JK, et al. (2004) Distinguishing human ethnic groups by means of sequences from Helicobacter pylori: lessons from Ladakh. Proc Natl Acad Sci U S A 101: 4746–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tay CY, Mitchell H, Dong Q, Goh KL, Dawes IW, et al. (2009) Population structure of Helicobacter pylori among ethnic groups in Malaysia: recent acquisition of the bacterium by the Malay population. BMC Microbiol 9: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Breurec S, Guillard B, Hem S, Brisse S, Dieye FB, et al. (2011) Evolutionary history of Helicobacter pylori sequences reflect past human migrations in Southeast Asia. PLoS One 6: e22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, et al. (2011) Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol 11: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delport W, Cunningham M, Olivier B, Preisig O, van der Merwe SW (2006) A population genetics pedigree perspective on the transmission of Helicobacter pylori . Genetics 174: 2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Behar DM, Villems R, Soodyall H, Blue-Smith J, Pereira L, et al. (2008) The dawn of human matrilineal diversity. Am J Hum Genet 82: 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonder MK, Mortensen HM, Reed FA, de Sousa A, Tishkoff SA (2007) Whole-mtDNA genome sequence analysis of ancient African lineages. Mol Biol Evol 24: 757–768. [DOI] [PubMed] [Google Scholar]
- 19. Hellenthal G, Auton A, Falush D (2008) Inferring human colonization history using a copying model. PLoS Genet 4: e1000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, et al. (2009) The genetic structure and history of Africans and African Americans. Science 324: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhivotovsky LA, Rosenberg NA, Feldman MW (2003) Features of evolution and expansion of modern humans, inferred from genomewide microsatellite markers. Am J Hum Genet 72: 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, et al. (2010) Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, et al. (2008) Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451: 998–1003. [DOI] [PubMed] [Google Scholar]
- 24. Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, et al. (2008) Worldwide human relationships inferred from genome-wide patterns of variation. Science 319: 1100–1104. [DOI] [PubMed] [Google Scholar]
- 25. Patin E, Laval G, Barreiro LB, Salas A, Semino O, et al. (2009) Inferring the demographic history of African farmers and pygmy hunter-gatherers using a multilocus resequencing data set. PLoS Genet 5: e1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quintana-Murci L, Quach H, Harmant C, Luca F, Massonnet B, et al. (2008) Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter-gatherers and Bantu-speaking farmers. Proc Natl Acad Sci U S A 105: 1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlebusch CM, Lombard M, Soodyall H (2013) MtDNA control region variation affirms diversity and deep sub-structure in populations from southern Africa. BMC Evol Biol 13: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Drake NA, Blench RM, Armitage SJ, Bristow CS, White KH (2011) Ancient watercourses and biogeography of the Sahara explain the peopling of the desert. Proc Natl Acad Sci U S A 108: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillipson DW (2005) African Archeology. Cambridge: Cambridge University Press. 389 p.
- 30.Newman JL (1995) The Peopling of Africa: A Geographic Interpretation. New Haven: Yale University Press. 235 p.
- 31. Pakendorf B, Bostoen K, de Filippo C (2011) Molecular perspectives on the Bantu expansion: a synthesis. Language Dynam Change 1: 50–88. [Google Scholar]
- 32. Ehret C (2001) Bantu expansions: re-envisioning a central problem of early African history. Int J Afr Hist Stud 34: 5–41. [Google Scholar]
- 33. Montano V, Ferri G, Marcari V, Batini C, Anyaele O, et al. (2011) The Bantu expansion revisited: a new analysis of Y chromosome variation in Central Western Africa. Mol Ecol 20: 2693–2708. [DOI] [PubMed] [Google Scholar]
- 34. Schoenbrun D (2001) Representing the Bantu Expansions: What's at Stake? Int J Afr Hist Stud 34: 1–4. [Google Scholar]
- 35.Bahuchet S (2011) Languages of African rainforest « pygmy » hunter-gatherers: language shifts without cultural admixture. In: Guldemann T, McConvell P, Rhodes R, editors. Historical linguistics and hunter-gatherers populations in global perspective. Available: http://hal.archives-ouvertes.fr/docs/00/54/82/07/PDF/Bahuchet_2006-Leipzig_Version1.pdf Accessed July 19, 2013.
- 36. Palmer DD, Watson KR, Allen MJ (1994) Helicobacter pylori infection and peptic ulcer disease in Cameroon, West Africa. J Clin Gastroenterol 18: 162–164. [DOI] [PubMed] [Google Scholar]
- 37. Ndip RN, Malange AE, Akoachere JF, MacKay WG, Titanji VP, et al. (2004) Helicobacter pylori antigens in the faeces of asymptomatic children in the Buea and Limbe health districts of Cameroon: a pilot study. Trop Med Int Health 9: 1036–1040. [DOI] [PubMed] [Google Scholar]
- 38. Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, et al. (1999) Recombination and clonal groupings within Helicobacter pylori from different geographic regions. Mol Microbiol 32: 459–470. [DOI] [PubMed] [Google Scholar]
- 39. Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naduvilezhath L, Rose LE, Metzler D (2011) Jaatha: a fast composite-likelihood approach to estimate demographic parameters. Mol Ecol 20: 2709–2723. [DOI] [PubMed] [Google Scholar]
- 41. Rupnow MF, Shachter RD, Owens DK, Parsonnet J (2000) A dynamic transmission model for predicting trends in Helicobacter pylori and associated diseases in the United States. Emerg Infect Dis 6: 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pounder RE, Ng D (1995) The prevalence of Helicobacter pylori infection in different countries. Aliment Pharmacol Ther 9 Suppl 2: 33–39. [PubMed] [Google Scholar]
- 43. Chen Y, Segers S, Blaser MJ (2013) Association between Helicobacter pylori and mortality in the NHANES III study. Gut 62: 1262–9 doi:10.1136/gutjnl-2012-303018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rupnow MF, Shachter RD, Owens DK, Parsonnet J (2001) Quantifying the population impact of a prophylactic Helicobacter pylori vaccine. Vaccine 20: 879–885. [DOI] [PubMed] [Google Scholar]
- 45. Rupnow MF, Chang AH, Shachter RD, Owens DK, Parsonnet J (2009) Cost-effectiveness of a potential prophylactic Helicobacter pylori vaccine in the United States. J Infect Dis 200: 1311–1317. [DOI] [PubMed] [Google Scholar]
- 46.Ndameu B (2003) Case Study 7: Cameroon - Boumba Bek - Protected areas and indigenous peoples: The paradox of conservation and survival of the Baka in Moloundou region (south-east Cameroon). In: Nelson J, Hossack L, editors. Indigenous Peoples and Protected Areas in Africa: From Principles to Practice. Moreton-in-Marsh: Forest Peoples Programme. pp. 215–241.
- 47. Batini C, Lopes J, Behar DM, Calafell F, Jorde LB, et al. (2011) Insights into the demographic history of African Pygmies from complete mitochondrial genomes. Mol Biol Evol 28: 1099–1110. [DOI] [PubMed] [Google Scholar]
- 48.Dornan S (1925) Pygmies & Bushmen of the Kalahari. London: C. Struik. 318 p.
- 49. Excoffier L, Schneider S (1999) Why hunter-gatherer populations do not show signs of pleistocene demographic expansions. Proc Natl Acad Sci U S A 96: 10597–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hamilton MJ, Milne BT, Walker RS, Burger O, Brown JH (2007) The complex structure of hunter-gatherer social networks. Proc Biol Sci 274: 2195–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trevilla L (2009) Ethnologue: Languages of the World. Dallas: SIL International. 1248 p.
- 52. Migliano AB, Vinicius L, Lahr MM (2007) Life history trade-offs explain the evolution of human pygmies. Proc Natl Acad Sci U S A 104: 20216–20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kuete V (2010) Potential of Cameroonian plants and derived products against microbial infections: a review. Planta Med 76: 1479–1491. [DOI] [PubMed] [Google Scholar]
- 54. Brusotti G, Cesari I, Frassa G, Grisoli P, Dacarro C, et al. (2011) Antimicrobial properties of stem bark extracts from Phyllanthus muellerianus (Kuntze) Excell. J Ethnopharmacol 135: 797–800. [DOI] [PubMed] [Google Scholar]
- 55. Assob JC, Kamga HL, Nsagha DS, Njunda AL, Nde PF, et al. (2011) Antimicrobial and toxicological activities of five medicinal plant species from Cameroon traditional medicine. BMC Complement Altern Med 11: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Graham DY, Yamaoka Y, Malaty HM (2007) Thoughts about populations with unexpected low prevalences of Helicobacter pylori infection. Trans R Soc Trop Med Hyg 101: 849–851. [DOI] [PubMed] [Google Scholar]
- 57. Farag TH, Stoltzfus RJ, Khalfan SS, Tielsch JM (2007) Unexpectedly low prevalence of Helicobacter pylori infection among pregnant women on Pemba Island, Zanzibar. Trans R Soc Trop Med Hyg 101: 915–922. [DOI] [PubMed] [Google Scholar]
- 58. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- 59. Hudson RR, Boos DD, Kaplan NL (1992) A statistical test for detecting geographic subdivision. Mol Biol Evol 9: 138–151. [DOI] [PubMed] [Google Scholar]
- 60. Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138. [Google Scholar]
- 61. Didelot X, Falush D (2007) Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Hudson RR (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- 64. Morelli G, Didelot X, Kusecek B, Schwarz S, Bahlawane C, et al. (2010) Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet 6: e1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Becker NS, Verdu P, Hewlett B, Pavard S (2010) Can life history trade-offs explain the evolution of short stature in human pygmies? A response to Migliano et al. (2007). Hum Biol 82: 17–27. [DOI] [PubMed] [Google Scholar]
- 66. Jolley KA, Maiden MC (2010) BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Campbell MC, Tishkoff SA (2010) The evolution of human genetic and phenotypic variation in Africa. Curr Biol 20: R166–R173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Major human population events in recent African prehistory. This map summarises the potential major human demographic events as inferred from [4], [20], [25], [27], [28], [33], [47], [67]. Ancestral hunter-gatherer populations split into extant populations, beginning with Northern and Southern San (32–47 kya) and Western and Eastern Pygmies ∼20 kya. The Nilo-Saharan speakers originated in northeast Africa from where they migrated both north-westward and south-eastward. Their north-westward migration was favored by a climatic change from dry to more humid conditions during the humid Holocene period (6–9 kya). Bantu-speaking agriculturalist populations expanded from their homeland in what is present-day Nigeria/Cameroon, beginning ∼6 kya, in two independent waves along the western and eastern flanks of Africa into southern Africa. They thus spread their languages and agriculture through most of sub-Saharan Africa within the last 5 kyr.
(TIF)
Phylogenetic relationships among hpAfrica1 isolates. Phylogenetic relationships among hpAfrica1 isolates as determined by ClonalFrame. The haplotypes are colored according to their geographical source and symbols (triangles, circle) specify the respective subpopulation determined by STRUCTURE. Cameroonian haplotypes from non-Baka (brown color) or Baka (pink color) are marked. The hpAfrica2 strains used as outgroup to root the tree are separately indicated. The number of isolates is shown in brackets.
(PDF)
Phylogenetic relationships among hpNEAfrica isolates. Phylogenetic relationships among hpNEAfrica isolates as determined by ClonalFrame. The color-code corresponds to the geographical source. Cameroonian haplotypes isolated from non-Baka (brown color) or Baka (pink color) are indicated. One haplotype was isolated from both non-Baka and Baka individuals (dark-green color). The hpAfrica2 strains used as outgroup to root the tree are separately indicated. The number of isolates is shown in brackets.
(PDF)
Frequencies of endoscopy findings in Baka Pygmy and non-Baka agriculturalist study participants.
(DOC)
Prior parameters used in coalescent simulations with Jaatha 2.0 and their values.
(DOC)