

Abstract
A method for the formation of arylnitromethanes is described that employs readily available aryl halides or triflates and small amounts of nitromethane in a dioxane solvent, thereby reducing the hazards associated with this reagent. Specifically, 2–10 equivalents (1–5% v/v) of nitromethane can be employed in comparison to prior work that used nitromethane as solvent (185 equivalents). The present transformation provides high yields at relatively low temperatures and tolerates an array of functionality, including heterocycles and substantial steric encumbrance.
Nitroalkanes comprise a synthetically important class of compounds,1,2 capable of efficient, stereoselective C–C bond forming reactions3,4,5 and subsequent transformation into numerous other functional groups (Scheme 1).6,7 Higher order nitroalkanes have been used to good effect as key intermediates in total synthesis, as demonstrated in recent syntheses of manzamine A8 and (−)-nutlin-3.9 Despite their utility, synthetic access to this class of compounds remains challenging. The generation of arylnitromethanes is particularly challenging.9,10 The inherent limitation in the classical Meyer reaction of metal nitrites11 on benzyl halides arises from the ambident nature of the nitrite nucleophile, resulting in significant quantities of nitrite ester and other byproducts.12 Additionally, even slight steric hindrance on the benzyl halide electrophile (e.g. any ortho-substitution) inhibits the attack of the weak nitrite nucleophile.13
Scheme 1.
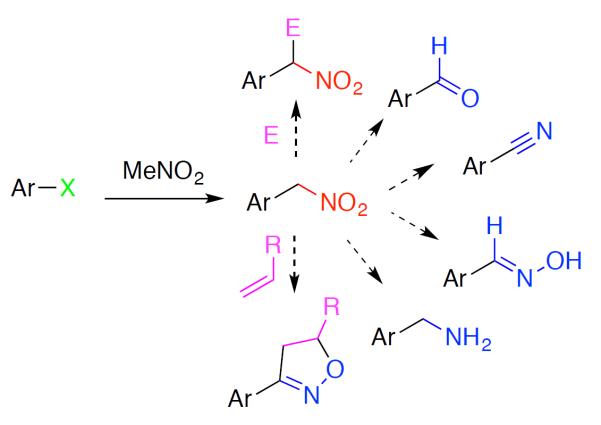
Utility of nitroalkane intermediates.
Encouraged by the success of weak carbon nucleophiles, including higher nitroalkyl congeners,14, 15, 16, 17, 18 as partners in cross-coupling reactions,19 we recently developed a palladium-catalyzed coupling of nitromethane and aryl/heteroaryl halides to afford arylnitromethanes in high yield under mild conditions (Scheme 2).20 Due to its weakly nucleophilic nature, nitromethane was employed as a solvent (0.1 M, 185 equiv) to optimize the yield. Nitromethane is often used as a polar organic solvent in small-scale processes and is stable at ambient temperature and pressure. However, under certain conditions, particularly high pressure and temperature, nitromethane can be explosive.21,22 Caution must also be exercised under basic conditions to avoid formation and isolation of several hazardous species.23,24 Nitronate salts, for example, explode with heat or shock, particularly when dry. Methazonic acid (nitroacetaldehyde oxime) and/or fulminate salts may also be generated, which are highly sensitive compounds and readily detonate with friction, heat, or shock. Despite the routine use of nitromethane as a polar organic solvent, the aforementioned concerns provided ample impetus to reinvestigate the title transformation with the goal of reducing the amount of nitromethane.25 Herein, we report an improved method for the nitromethylation of aryl halides and triflates using 2–10 equivalents of the nitromethane coupling partner (Scheme 2).
Scheme 2.
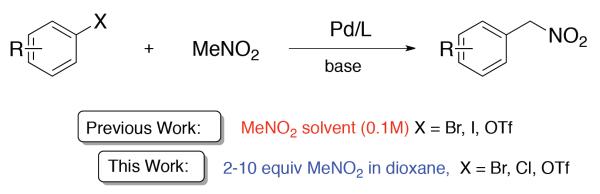
Nitromethylation of aryl halides.
With the goal of finding suitable conditions engendering adequate reactivity while minimizing the amount of nitromethane, a focused parallel microscale experimentation (96 reactions on the 20 μmol halide/100 μL reaction volume scale)26 study was designed building on data from prior examination of the coupling reaction.20 Specifically, the coupling of 4-bromoanisole (1a) with nitromethane (2 equivalents) was examined while varying the ligand, base, and solvent.27 Sufficient reactivity was observed at 70 °C, and several trends emerged. Bases with conjugate acid pKa values close to that of nitromethane (pKa ~10) performed best. Carbonate (pKa ~10) and phosphate (pKa ~13) appeared optimal, while bicarbonate (pKa ~6) yielded only small amounts of product, and tert-butoxide (pKa ~19) led to significant product decomposition. Mild sodium bases (Na2CO3 and NaHCO3) displayed minimal reactivity, likely due to limited solubility. Both THF and dioxane provided improved reactivity relative to CPME. The XPhos, BrettPhos, and CataCXium POMetB ligands all showed comparable reactivity, though examination of the HPLC chromatograms revealed that XPhos possessed the cleanest reaction profile. Verification of the high throughput experimentation (HTE) results (0.020 mmol) and final optimization was performed on bench-scale (0.5 mmol)) reactions (Table 1).
Table 1.
Bench-scale (0.5 mmol) results from selected HTE leads.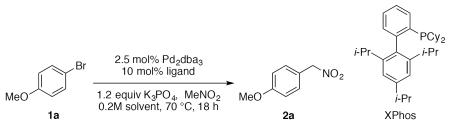
| entry | ligand | solvent | equiv MeNO2 | L:Pd | P:SM at 5 ha | yield (%)b |
|---|---|---|---|---|---|---|
| 1 | cataCXium POMetB |
THF | 2 | 2:1 | 93:7 | 66c |
| 2 | cataCXium POMetB |
1,4-dioxane | 2 | 2:1 | 89:11 | 67c |
| 3 | XPhos | THF | 2 | 2:1 | 67:33 | 42 |
| 4 | XPhos | 1,4-dioxane | 2 | 2:1 | 59:41 | 81 |
| 5 | XPhos | 1,4-dioxane | 5 | 2:1 | 90:10 | 85 |
| 6 | XPhos | 1,4-dioxane | 10 | 2:1 | 90:10 | 92 |
| 7 | XPhos | 1,4-dioxane | 2 | 1.2:1 | 62:38 | 79 |
Determined by GC.
Yield after column chromatography.
9 h reaction time.
Under several conditions, increased starting material consumption was independent from yield due to competing decomposition pathways. Specifically, THF and CataCXium POMetB displayed superior rates in comparison to 1,4-dioxane and XPhos, respectively, yet provided poor yield (entries 1–4).28 The product arylnitromethane 2a was afforded in high yield (81%) with just 2 equivalents of nitromethane using XPhos, dioxane, and K3PO4 (entry 4). As anticipated, increasing the amount of nitromethane to 5 or 10 equivalents increased rates and yields (entries 5 and 6). The ligand to palladium ratio could be decreased to 1.2:1 with minimal affect on reaction efficiency (entry 7).
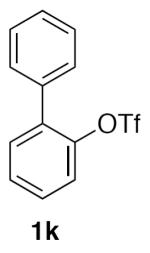
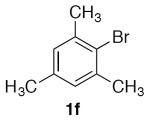
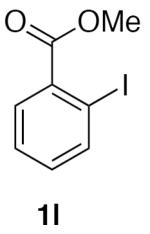
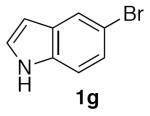
With optimized conditions (Table 2, enties 6–7) secured for the desired transformation, the scope was explored (Table 2). Using 10 equivalents of nitromethane, the coupling proceeds smoothly with electron rich (entries 1, 6) and electron deficient aryl bromides (entries 2–5). Various functional groups, including carbonyls (entries 2–4) and nitro (entry 5), are well tolerated. It is especially noteworthy that acidic ketone 1d did not compete even though coupling of ketones, via the enols, have been reported in other systems15,29,30 at the higher temperature used here compared to the initial report with nitromethane as solvent.20 Bromoindole 1g also provided the nitromethylated heteroarene in high yield (entry 7). Notably, ortho-substitution does not inhibit the reactivity or yield, as determined with a variety of sterically hindered substrates (entries 5, 6, 12, and 13) including the very hindered 2-bromomesitylene 1f. Aryl chlorides proved capable coupling partners (entries 8–10), although more forcing conditions were required and more complex reaction profiles were observed. Sufficient disparity in the reactivity is observed between aryl bromides and chlorides such that chlorides can be carried through to the product (entry 11) providing the opportunity for an additional orthogonal coupling. Biphenyl triflate 1k smoothly afforded the corresponding nitroalkyl product (entry 12), demonstrating the utility of nitromethane in homologating phenolic precursors. In contrast, aryl iodides performed poorly in the transformation, reacting very slowly (entry 13).31 This method is not restricted to nitromethane, being effective with nitroethane14 (Scheme 3) the product of which decomposes far less readily, which accounts for the very high yield (98%).
Table 2.
Scope of the coupling of aryl halides and triflates with 10 equiv nitromethane.
| entry | substrate | yield (%)a | entry | substrate | yield (%)a |
|---|---|---|---|---|---|
| 1 |
|
94 | 8 |
|
77 |
| 2 |
|
77 | 9 |
|
50b |
| 3 |
|
82 | 10 |
|
77b |
| 4 |
|
70 | 11 |
|
86 |
| 5 |
|
82 | 12 |
|
86 |
| 6 |
|
90 | 13 |
|
24 |
| 7 |
|
80 |
Yield after column chromatography.
5 mol% Pd2dba3, 12 mol% XPhos, 80 °C.
Scheme 3.
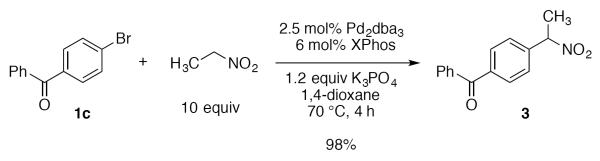
Example of nitroethane coupling.
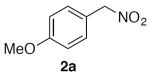
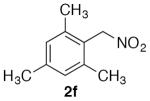
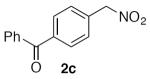
Select examples were exposed to the optimized conditions with just two equivalents of nitromethane and afforded the products in moderate to good yields, albeit requiring longer reaction times (Table 3). Under these coupling conditions, incomplete conversion was observed for several substrates, and increasing the reaction time further resulted in more decomposition of the products.
Table 3.
Coupling of aryl bromides with 2 equiv nitromethane.
Yield after column chromatography.
In conclusion, an improved method for the formation of arylnitromethanes has been discovered that employs readily available aryl halides or triflates and a small excess of nitromethane. The present transformation provides good reactivity at relatively low temperatures and tolerates an array of functionality, including considerable steric encumbrance. Significantly, the amount of nitromethane has been reduced from solvent (100% v/v, 185 equivalents) to 5% (10 equivalents) or 1% (2 equivalents) of the volume utilized in the original protocol, providing a more attractive method for large scale processes. The product arylnitromethanes are produced in high yields and may be readily transformed into the corresponding aldehydes, oximes, and other functionalities.
Experimental Section
Nitromethylation of 4-bromoanisole - Optimization (Table 1)
In a glovebox, an oven-dried microwave vial was charged with Pd2dba3 (11.4 mg, 0.0125 mmol), ligand, and K3PO4 (127 mg, 0.60 mmol). The solvent was added (2.5 mL), followed by nitromethane (2, 5 or 10 equiv) and 4-bromoanisole (62.6 μL, 0.50 mmol). The vial was sealed with a teflon cap, briefly stirred, removed from the glovebox and heated to 70 °C with vigorous stirring for the indicated time. After cooling to rt, the mixture was diluted with CH2Cl2 (20 mL), and washed with 1 M aq HCl, which was then extracted with CH2Cl2 (2 × 20 mL). The combined organics were dried over Na2SO4, filtered, and concentrated. Purification by chromatography (5% EtOAc/Hex) afforded pure 1-methoxy-4-(nitromethyl)benzene as a yellow oil.
General Method for the Nitromethylation of Aryl Halides
In a glovebox, an oven-dried vial was charged with Pd2dba3 (11.4 mg, 0.0125 mmol), XPhos (14.3 mg, 0.030 mmol), K3PO4 (127 mg, 0.60 mmol), and substrate (0.50 mmol). The vial was sealed with a teflon cap, removed from the glovebox, and 1,4-dioxane (2.5 mL) was added, followed by nitromethane (270 μL, 5.0 mmol = General Method A, or 54 μL, 1.0 mmol = General Method B). The mixture was heated to 70 °C with vigorous stirring for the indicated time, then cooled to rt, diluted with CH2Cl2 (15 mL), and washed with 1 M aq HCl (15 mL), which was then extracted with CH2Cl2 (3 × 15 mL). The combined organics were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by chromatography afforded the desired product. All spectral data for known compounds were in good agreement with literature values.20
1-Methoxy-4-(nitromethyl)benzene (2a)
(Table 2, entry 1) General Method A was carried out on 4-bromoanisole with a reaction time of 7 h. Purification by chromatography (5% EtOAc/Hex) afforded the title compound as a pale yellow oil (78.4 mg, 94%): 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 5.38 (s, 2H), 3.83 (s, 3H). (Table 3, entry 1) General Method B was carried out on 4-bromoanisole with a reaction time of 18 h and afforded the title compound after purification as a yellow oil (66.1 mg, 79%).
Ethyl 4-(nitromethyl)benzoate (2b)
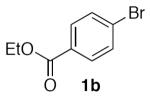
(Table 2, entry 2) General Method A was carried out on ethyl 4-bromobenzoate with a reaction time of 18 h. Purification by chromatography (20% EtOAc/Hex) afforded the title compound as a colorless solid (80.7 mg, 77%): mp: 71–72 °C (lit.20 70–71 °C); 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 5.51 (s, 2H), 4.41 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H).
(4-(Nitromethyl)phenyl)(phenyl)methanone (2c)
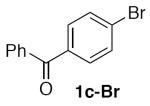
(Table 2, entry 3) General Method A was carried out on 4-bromobenzophenone with a reaction time of 18 h. Purification by chromatography (10 to 20% EtOAc/Hex) afforded the title compound as a colorless solid (99.4 mg, 82%): mp: 86–88 °C (lit.20 88–90 °C); 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.0 Hz, 2H), 7.80–7.83 (m, 2H), 7.62 (tt, J = 7.4, 1.4 Hz, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.51 (t, J = 7.7 Hz, 2H), 5.55 (s, 2H).
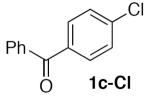
(Table 2, entry 8) General Method A was carried out on 4-chlorobenzophenone with a reaction time of 18 h and afforded the title compound after purification as a light tan solid (93.6 mg, 77%): mp: 85–87 °C.
(Table 3, entry 2) General Method B was carried out on 4-bromobenzophenone with a reaction time of 30 h and afforded the title compound after purification as a tan solid (67.5 mg, 56%): mp: 79–84 °C.
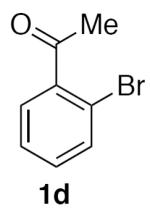
1-(2-(Nitromethyl)phenyl)ethan-1-one (2d)
(Table 2, entry 4) General Method A was carried out on 2'-bromoacetophenone with a reaction time of 18 h. Purification by chromatography (20% EtOAc/Hex) afforded the title compound as a tan solid after trituration with hexanes (62.8 mg, 70%): mp: 76–77 °C (lit.20 87–89 °C); 1H NMR (500 MHz, CDCl3) δ 7.94–7.98 (m, 1H), 7.59–7.62 (m, 2H), 7.38–7.42 (m, 1H), 5.79 (s, 2H), 2.66 (s, 3H).
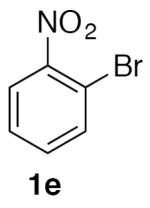
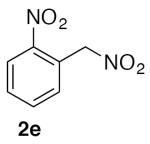
1-Nitro-2-(nitromethyl)benzene (2e)
(Table 2, entry 5) General Method A was carried out on 1-bromo-2-nitrobenzene with a reaction time of 18 h. Purification by chromatography (20% EtOAc/Hex) afforded the title compound as a tan solid after trituration with hexanes (74.5 mg, 82%): mp: 60–63 °C (lit.20 62–64 °C); 1H NMR (500 MHz, CDCl3) δ 8.28 (dd, J = 8.1, 1.3 Hz, 1H), 7.77 (td, J = 7.6, 1.3 Hz, 1H), 7.71 (td, J = 8.0, 1.5 Hz, 1H), 7.52 (dd, J = 7.5, 1.3 Hz, 1H), 5.85 (s, 2H).
(Table 3, entry 3) General Method B was carried out on 1-bromo-2-nitrobenzene with a reaction time of 24 h and afforded the title compound after purification as a tan solid (50.1 mg, 55%): mp: 60–62 °C.
1,3,5-Trimethyl-2-(nitromethyl)benzene (2f)
(Table 2, entry 6) General Method A was carried out on 2-bromomesitylene with a reaction time of 8 h. Purification by chromatography (3% EtOAc/Hex) afforded the title compound as pale needles after trituration with hexanes (80.7 mg, 90%): mp: 71–72 °C (lit.32 70–71 °C); 1H NMR (500 MHz, CDCl3) δ 6.96 (s, 2H), 5.58 (s, 2H), 2.39 (s, 6H), 2.32 (s, 3H).
(Table 3, entry 4) General Method B was carried out on 2-bromomesitylene with a reaction time of 24 h and afforded the title compound after purification as a colorless solid (51.3 mg, 55%): mp: 71–73 °C.
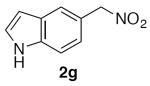
5-(Nitromethyl)-1H-indole (2g)
(Table 2, entry 7) General Method A was carried out on 5-bromoindole with a reaction time of 7 h. Purification by chromatography (15% EtOAc/Hex) afforded the title compound as a tan solid (70.9 mg, 80%): mp: 96–97 °C (lit.20 92–94 °C); 1H NMR (500 MHz, CDCl3) δ 8.35 (bs, 1H), 7.75 (s, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.25–7.29 (m, 2H), 6.59–6.61 (m, 1H), 5.55 (s, 2H).
(Table 3, entry 5) General Method B was carried out on 5-bromoindole with a reaction time of 21 h and afforded the title compound after purification as a tan solid (57.3 mg, 65%): mp: 93–95 °C.
1-Nitro-4-(nitromethyl)benzene (2h)
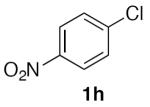
(Table 2, entry 9) General Method A was carried out on 1-chloro-4-nitrobenzene using 5 mol% Pd2dba3 and 12 mol% XPhos at 80 °C with a reaction time of 5 h. Purification by chromatography (12% EtOAc/Hex) afforded the title compound as a yellow solid (45.6 mg, 50%): mp: 82–86 °C (lit.33 84–86 °C); 1H NMR (300 MHz, CDCl3) δ 8.32 (d, J = 8.7 Hz, 2H), 7.68 (d, J = 8.6 Hz, 2H), 5.57 (s, 2H).
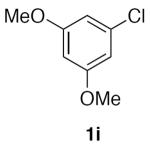
1,3-Dimethoxy-5-(nitromethyl)benzene (2i)
(Table 2, entry 10) General Method A was carried out on 1-chloro-3,5-dimethoxybenzene using 5 mol% Pd2dba3 and 12 mol% XPhos at 80 °C with a reaction time of 5.5 h. Purification by chromatography (15% EtOAc/Hex) afforded the title compound as a tan solid (75.6 mg, 77%): mp: 56–58 °C (lit.34 88 °C); 1H NMR (500 MHz, CDCl3) δ 6.58 (d, J = 2.3 Hz, 2H), 6.52 (t, J = 2.2 Hz, 1H), 5.36 (s, 2H), 3.80 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 161.3, 131.6, 108.0, 101.9, 80.2, 55.6; IR (film) 2916, 2848, 1597, 1551, 1456, 1430, 1372, 1206, 1153, 1066 cm−1; HRMS (CI) calcd for C9H11O2 [M-NO2]+, m/z = 151.0759; found 151.0758.
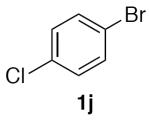
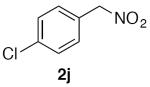
1-Chloro-4-(nitromethyl)benzene (2j)
(Table 2, entry 11) General Method A was carried out on 1-bromo-4-chlorobenzene with a reaction time of 8 h. Purification by chromatography (5% EtOAc/Hex) afforded the title compound as a waxy material that partially melted when handling at room temperature (73.5 mg, 86%): 1H NMR (500 MHz, CDCl3) δ 7.39–7.46 (m, 4H), 5.42 (s, 2H).
(Table 3, entry 6) General Method B was carried out on 1-bromo-4-chlorobenzene with a reaction time of 30 h and afforded the title compound after purification as a low-melting pale solid (39.4 mg, 46%).
2-(Nitromethyl)-1,1'-biphenyl (2k)
(Table 2, entry 12) General Method A was carried out on 1,1'-biphenyl-2-trifluoromethanesulfonate35 with a reaction time of 18 h. Purification by chromatography (3% EtOAc/Hex) afforded the title compound as a colorless oil (92.1 mg, 86%): 1H NMR (300 MHz, CDCl3) δ 7.36–7.56 (m, 7H), 7.28–7.34 (m, 2H), 5.42 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 143.9, 139.8, 131.3, 130.8, 130.1, 129.2, 128.8, 128.3, 128.1, 127.5, 77.5. IR (film) 2920, 2852, 1551, 1481, 1372; HRMS (ESI) calcd for C13H11 [M-NO2]+, m/z = 167.0861; found 167.0858.
Methyl 2-(nitromethyl)benzoate (2l)
(Table 2, entry 13) General Method A was carried out on methyl-2-iodobenzoate with a reaction time of 18 h. Purification by chromatography (8% EtOAc/Hex) afforded the title compound after trituration with hexanes as a colorless solid (23.6 mg, 24%): mp: 66–67 °C (lit.36 64–65 °C); 1H NMR (500 MHz, CDCl3) δ 8.15 (dd, J = 7.7, 1.4 Hz, 1H), 7.62 (td, J = 7.5, 1.4 Hz, 1H), 7.56 (td, J = 7.6, 1.3 Hz, 1H), 7.4 (dd, J = 7.6, 1.0 Hz, 1H), 5.86 (s, 2H), 3.91 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.8, 133.3, 133.2, 131.8, 130.8, 130.4, 130.2, 77.7, 52.6; IR (film) 3024, 2960, 1714, 1569, 1429, 1377, 1285, 1086 cm−1; HRMS (CI) calcd for C9H9O2 [M-NO2]+, m/z = 149.0602; found 149.0597.
(4-(1-Nitroethyl)phenyl)(phenyl)methanone (3)
In a glovebox, an oven-dried vial was charged with Pd2dba3 (11.4 mg, 0.0125 mmol), XPhos (14.3 mg, 0.030 mmol), K3PO4 (127 mg, 0.60 mmol), and 4-bromobenzophenone (130.6 mg, 0.50 mmol). The vial was sealed with a teflon cap, removed from the glovebox, and 1,4-dioxane (2.5 mL) was added, followed by nitroethane (360 μL, 5.0 mmol). The mixture was heated to 70 °C with vigorous stirring for 4 h, then cooled to rt, diluted with CH2Cl2 (15 mL), and washed with 1 M aq HCl (15 mL), which was then extracted with CH2Cl2 (3 × 15 mL). The combined organics were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by chromatography (10% EtOAc/Hex) afforded the title compound as a yellow oil (125.2 mg, 98%): 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.3 Hz, 2H), 7.80 (m, 2H), 7.62 (t, J = 7.6 Hz, 1H), 7.59 (d, J = 8.4 Hz, 2H), 7.50 (t, J = 7.7 Hz, 2H), 5.71 (q, J = 7.0 Hz, 1H), 1.95 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 196.0, 139.5, 139.1, 137.3, 133.0, 130.8, 130.2, 128.6, 127.6, 85.9, 19.7; IR (film) 3058, 2927, 1654, 1546, 1446, 1276, 924 cm−1; HRMS (ESI) calcd for C15H14O [M+H−NO2]+ m/z = 210.1045; found 210.1041.
Supplementary Material
Acknowledgments
We are grateful to the NSF for funding of the High Throughput Laboratory (GOALI CHE-0848460) and NIH (RO1GM087605) for financial support. We thank Accelrys and Virscidian for providing software for the high throughput screening and analysis. Partial instrumentation support was provided by the NIH for MS (1S10RR023444) and NMR (1S10RR022442). We thank Dr. Simon Berritt (Penn Merck HTE Laboratory) and Dr. Spencer Dreher (Merck Research Laboratories) for helpful discussions.
Footnotes
Supporting Information Available General methods, parallel microscale experimentation procedure and data, 1H NMR spectroscopic data for all compounds and 13C NMR for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]
- (2).Ballini R, Palmieri A, Righi P. Tetrahedron. 2007;63:12099–12121. [Google Scholar]
- (3).Luzzio FA. Tetrahedron. 2001;57:915–945. [Google Scholar]
- (4).Marques-Lopez E, Merino P, Tejero T, Herrera RP. Eur. J. Org. Chem. 2009:2401–2420. [Google Scholar]
- (5).Ballini R, Bosica G, Fiorini D, Palmieri A, Petrini M. Chem. Rev. 2005;105:933–971. doi: 10.1021/cr040602r. [DOI] [PubMed] [Google Scholar]
- (6).Seebach D, Colvin EW, Lehr F, Weller T. Chimia. 1979;33:1–18. [Google Scholar]
- (7).Rosini G, Ballini R. Synthesis. 1988:833–847. [Google Scholar]
- (8).Jakubec P, Hawkins A, Felzmann W, Dixon DJ. J. Am. Chem. Soc. 2012;134:17482–17485. doi: 10.1021/ja308826x. [DOI] [PubMed] [Google Scholar]
- (9).Davis TA, Johnston JN. Chem. Sci. 2011;2:1076–1077. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gregor JG, Yoon-Miller SJP, Bechtold NR, Flewelling SA, MacDonald JP, Downey CR, Cohen EA, Pelkey ET. J. Org. Chem. 2011;76:8203–8214. doi: 10.1021/jo2013516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Meyer V, Stüber O. Ber. 1872;5:203–205. [Google Scholar]
- (12).Kornblum N, Smiley RA, Blackwood RK, Iffland DC. J. Am. Chem. Soc. 1955;77:6269–6280. [Google Scholar]
- (13).Kornblum N, Taub B, Ungnade HE. J. Am. Chem. Soc. 1954;76:3209–3211. [Google Scholar]
- (14).For coupling of primary nitroalkanes with aryl halides, see: Vogl EM, Buchwald SL. J. Org. Chem. 2002;67:106–111. doi: 10.1021/jo010953v..
- (15).Fox JM, Huang X, Chieffi A, Buchwald SL. J. Am. Chem. Soc. 2000;122:1360–1370. [Google Scholar]
- (16).Metz AE, Berritt S, Dreher SD, Kozlowski MC. Org. Lett. 2012;14:760–763. doi: 10.1021/ol203303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang M, Zhou J, Kan J, Wang M, Su W, Hong M. Chem. Commun. 2010;46:5455–5457. doi: 10.1039/c0cc01029d. [DOI] [PubMed] [Google Scholar]
- (18).Zhang M, Hu P, Zhou J, Wu G, Huang S, Su W. Org. Lett. 2013;15:1718–1721. doi: 10.1021/ol400507u. [DOI] [PubMed] [Google Scholar]
- (19).Prim D, Campagne J-M, Joseph D, Andrioletti B. Tetrahedron. 2002;58:2041–2075. [Google Scholar]
- (20).Walvoord RR, Berritt S, Kozlowski MC. Org. Lett. 2012;14:4086–4089. doi: 10.1021/ol301713j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McKittrick DS, Irvine RJ, Bergsteinsson I. Ind. Eng. Chem., Anal. Ed. 1938;10:630–631. [Google Scholar]
- (22).Piermarini GJ, Block S, Miller PJ. J. Phys. Chem. 1989;93:457–462. [Google Scholar]
- (23).Markofsky SB. Ullmann's Encyclopedia of Industrial Chemistry, Electronic Release. Wiley-VCH; Weinheim: Oct, 2011. Nitro Compounds, Aliphatic. [Google Scholar]
- (24).Bretherick L. Bretherick's Handbook of Reactive Chemical Hazards. 4th ed. Butterworths; Boston: 1990. [Google Scholar]
- (25).We have not experienced any adverse events in handling the substrates, reaction mixtures, or products via our prior protocol or via the protocol reported herein.
- (26).Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257–9259. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).See Supporting Information for complete HTE data.
- (28).Similarly poor yields were obtained upon halting these reactions earlier.
- (29).Biscoe MR, Buchwald SL. Org. Lett. 2009;11:1773–1775. doi: 10.1021/ol900295u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Culkin DA, Hartwig JF. Acc. Chem. Res. 2003;36:234–245. doi: 10.1021/ar0201106. [DOI] [PubMed] [Google Scholar]
- (31).Per reference 20, aryl iodides couple successfully when using nitromethane as the solvent. The results here are consistent with prior work with aryl iodides when oxidative addition is not rate determining: Wolfe JP, Buchwald SL. J. Org. Chem. 1996;61:1133–1135.
- (32).Grundmann C, Flory K. JustusLiebigs Ann. Chem. 1969;721:91–100. [Google Scholar]
- (33).Bug T, Lemek T, Mayr H. J. Org. Chem. 2004;69:7565–7576. doi: 10.1021/jo048773j. [DOI] [PubMed] [Google Scholar]
- (34).Cameron DW, Hildyard EM. J. Chem. Soc.C. 1968:166–169. [Google Scholar]
- (35).Hara O, Nakamura T, Sato F, Makino K, Hamada Y. Heterocycles. 2006;68:1–4. [Google Scholar]
- (36).Kupchan SM, Wormser HC. J. Org. Chem. 1965;30:3792–3800. doi: 10.1021/jo01022a046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.