

Abstract
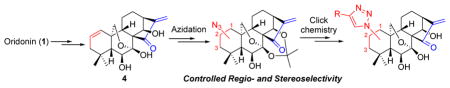
Efficient and concise synthetic approaches have been developed for the rapid and diverse installation of azide functionalities at the C-1, C-2 or C-3 of oridonin (1) with highly controlled regio- and stereoselectivity, while keeping key reactive pharmacophores intact by utilizing unique preactivation strategies based on the common synthon 4. Further functionalization of these azides through click chemistry yielding triazole derivatives successfully provides access to an expanded natural scaffold-based compound library for potential anticancer agents.
Oridonin (1), a highly oxygenated 7,20-epoxy-ent-kaurane-type diterpenoid isolated from the traditional Chinese herb isodon rubescens, is characterized by its densely functionalized and stereochemistry-rich frameworks including an exo-methylene cyclopentanone moiety in the D-ring and a 6-hydroxyl-7-hemiketal group in the B-ring (Fig. 1).1 It has been attracting considerable attention in recent years due to its remarkably unique and safe anticancer pharmacological profile.1e–f In China, oridonin injection is used alone or in combination with other drugs to treat liver cancer and carcinoma of gastric cardia.2 Nevertheless, more extensive clinical applications of oridonin for cancer therapy have been restricted, to a large degree, due to its relatively moderate potency, limited aqueous solubility and bioavailability.3 Consequently, modifications on the scaffold of 1 have become an attractive strategy to create better natural product-like compound libraries. Most previous approaches were attempted by coupling ester appendages to hydroxyl groups of 1.1b,4 To date, little chemical effort has been directed toward modifications of the oridonin Aring to generate diversity, likely owing to the synthetic challenges arising from its structural complexity with multiple reactive functionalities. Recently, we reported a concise synthesis of thiazole-fused oridonin derivatives with enhanced activity and solubility,5 indicating that the rational modifications on oridonin A-ring may have great potential to generate better anticancer agents.
Figure 1.
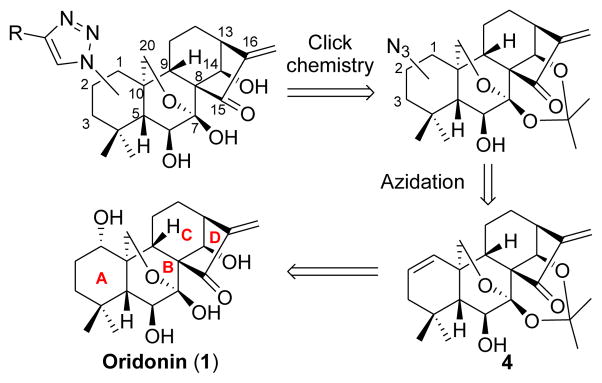
Retrosynthetic analysis of azide- and 1,2,3-triazole-substituted oridonin derivatives.
Click chemistry, a concept initiated by Sharpless,6 has provided a number of nearly perfect “spring-loaded” chemical reactions to elaborate an elegant and selective modification on complex natural products. Particularly, the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC)7 can efficiently afford 1,2,3-triazole scaffolds under mild conditions even in the presence of chemically reactive functionalities. Importantly, the resulting triazole derivatives are fairly stable to metabolic degradation and capable of actively participating in hydrogen bonding and dipole-dipole interactions, providing potential advantages including target binding and cell permeability improvement.8 Herein, we, for the first time, disclose our effort for the efficient synthesis of novel nitrogen-enriched oridonin derivatives with azide and 1,2,3-triazole functionalizations at the C-1, C-2 or C-3 position in a highly regio- and stereo-specific manner.
As illustrated in Fig. 1, our general synthetic strategy to achieve these molecules involves a key and challenging step of azide installation at each of three sites of the Aring with controlled regio- and stereoselectivity, while not destroying other featured functionalities. It is well documented that the presence of the α,β-unsaturated ketone (enone) system in the D-ring of 1 is the main structural determinant for its anticancer activity and destruction of this enone system could counteract its bioactivity.1a–b,d On the other hand, this bioactive enone system is also a chemical electrophilic center susceptible to nucleophilic attack (Michael addition) by various nucleophiles including azide reagents under certain reaction conditions, leading to adducts at the β-position as evidenced in literature.9 Accordingly, a regioselective installation of the azide functionality to the desired sites of the A-ring, but not the enone moiety in the D-ring, is synthetically challenging and essentially required, in developing efficient synthetic protocols. With this goal in mind, we attempted to achieve a differential reactivity of these two sites by activating the functional group of the Aring for a successfully controlled selectivity.
Our synthesis commenced with 1, which is naturally abundant and commercially available (Scheme 1). Protection of 7,14-dihydroxyl of 1 with 2,2-dimethoxypropane10 followed by selective activation of the 1-hydroxyl group with MsCl solely provided mesylate 2 in 74% yield over two steps, which was subsequently subjected to NaN3 in DMF. However, the anticipated substitution reaction at 60 °C failed to give 1-azide 3 but resulted in high recovery of 2. Increasing the reaction temperature to 120 °C produced a complex mixture, presumably owing to the Michael addition to the enone system in the D-ring by azide anions. Therefore, a more reactive electrophilic center deemed necessary to be created at the A-ring. To introduce an allylic alcohol functional moiety to the A-ring, mesylate 2 was chosen as a substrate to undergo an elimination reaction in the presence of Li2CO3 at 110 °C to provide 1-ene 4 in 84% yield, followed by an allylic oxidation with selenium dioxide in refluxing 1,4-dioxane, stereoselectively leading to the 3β-allylic hydroxyl 6 in excellent yield. In this step, the 1-allylic seleninic acid intermediate 5 was only formed from the β-face of the A-ring because of a steric effect of the 7,20-expoxy ring, eventually leading to an installation of the hydroxyl group at the 3β-position in a stereoselective manner that was unambiguously determined through X-ray crystallographic analysis. Initially, allylic alcohol 6 without any preactivation was directly treated with diphenylphosphoryl azide (DPPA)11 and DBU in THF at 0 °C followed by warming up to 60 °C to install an azide group at C-3 for the purpose of atom-economy. Unfortunately, only a diphenyl phosphate intermediate 7, instead of anticipated 3-allylic azide 8, was obtained in 80% yield. Mechanistically, the reactive 3-allylic phosphate of intermediate 7 could not undergo further substitution reaction with the azide anion from the α-face due to the surrounding steric hindrance of C-3α. Accordingly, the 3-allylic alcohol of 6 has to be preactivated in the form of a better leaving group to install the 3-azide with high regioselectivity.
Scheme 1.

Attempts to synthesize 1-azide and 3-azide derivatives 3 and 8
In pursuance of this goal, we intended to convert the 3-allylic alcohol into the 3-allylic chloride for installation of the 3-azide group (Scheme 2). Interestingly, chlorination of 6 with SOCl2 in dry ether at rt exclusively led to 1β-allylic chloride 10 (73%),12 the regio- and stereostructure of which was determined by HMBC and NOESY experiments (Fig. S1 in Supporting Information). In this step, owing to the intrinsic structural feature of the substrate, the in situ generated 3-allylic mesylate 9 underwent an SNi’ reaction with the internal attack of chloride on C-1, instead of the allylic site C-3, leading to the shift of the double bond, in which a stable hexacyclic ring transition state was likely formed from the β-face.13 Subsequent nucleophilic substitution reaction of 10 with NaN3 in dry DMF at rt neatly afforded 1-allylic azide 11 (71%) with inversion of configuration despite steric crowding.14 The high propensity of allylic azides to undergo [3,3]-sigmatropic rearrangement15 suggested a facile route potentially available to 3α-allylic azide 8. Gratifyingly, performing this substitution reaction at 70 °C smoothly redirected the reaction to 3α-azide 8 (74%) with a total control of regio- and stereoselectivity as determined by HMBC and NOESY experiments (Fig. S2 in Supporting Information). These results implied that 1α-azide 11 would be initially produced and then undergo [3,3]-sigmatropic shift to provide 3α-azide 8. Further evidence for this isomerization was also achieved by heating 1α-azide 11 in refluxing CH3CN to solely give 3α-azide 8. This surprisingly high 3α-allylic azide preference is probably ascribed to its less crowded steric environment. As depicted in Scheme 2, the 3-azide group is far away from the bridgehead 20-methylene group, and possesses a more favorable steric surrounding relative to the 1-azide group, leading to this selective thermal isomerization of 1- to 3-type allylic azide.
Scheme 2.

Synthesis of 3-azide-, 1-azide-, 1,2,3-triazole-substituted oridonin derivatives 8, 11, 14, 15, 18 and 19
With key intermediates 11 and 8 in hand, the subsequent click reaction with phenylacetylene catalyzed by CuI in dry CH3CN at rt followed by removal of the acetonide groups with 5% HCl (aq) achieved the corresponding 1,2,3-triazole derivatives 14 and 15 in excellent yields, the structures of which were also confirmed by 1D and 2D NMR experiments (Figs. S3 and S4 in Supporting Information). The stereochemistry of 14 was further unambiguously secured by X-ray crystallographic analysis. As additional proof for selective isomerization of the 1- to 3-type allylic azide, the click reaction of 1-azide 11 in refluxing CH3CN solely provided 3-triazole 13 (70%) instead of 1-triazole 12. To further explore their versatility, triazole derivatives 18 and 19 have also been synthesized in a same fashion with good yields.
Regioselective installation of an azide group at the C-2 of the A-ring also utilized the preactivation strategy to achieve differential reactivity with respect to other reactive functionalities (Scheme 3). After considering various possibilities, the key synthon 4 prepared in Scheme 1 was subjected to epoxidation though m-CPBA-mediated Prilezhaev reaction.16 To our delight, this reaction not only occurred preferentially at the 1-ene rather than the 16-exo-methylene, but also formed 1,2-epoxide ring stereoselectively from the less sterically-hindered β-face to exclusively give 1S,2R-epoxide 20 (86%). Ring-openings of unsymmetrical epoxides by nucleophiles usually lead to non-regioselctive products.17 In our case, azidolysis of epoxide 20 with NaN3 occurred preferentially with attack of azide anion on C-2 due to its less sterically-crowded surrounding relative to C-1. Conducting the reaction in EtOH/H2O (1:1) at 85 °C using NH4Cl as a coordinating salt for 24 h solely afforded 2-azide 21 (51%),18 the structure of which was also unequivocally determined by X-ray crystallographic analysis. In this step, different solvents such as MeOH/H2O, CH3CN/H2O, acetone/H2O or DMF resulted in lower yields than that in aqueous ethanol; extending reaction time decreased the yield; after screening other coordinating salts, LiClO4 proved to be optimal leading to a better yield (60%) than NH4Cl (Table S1 in Supporting Information). The subsequent click reactions of 21 with phenyacetylene or 4-tert-butylphenylacetylene followed by treatment with 5% HCl (aq) successfully generated 2-triazole compounds 24 and 25 in 58% and 80% yields (two steps), respectively.
Scheme 3.

Synthesis of 2-azide-, 1,2,3-triazole-substituted oridonin derivatives 21, 24 and 25
The antiproliferative effects of representative 1,2,3-triazole-substituted analogues against breast cancer cell lines including MCF-7 (ER-positive) and MDA-MB-231 (triple-negative) are summarized in Table 1, indicating that these new compounds with 1,2,3-triazole installed in the A-ring exhibited significantly improved anti-breast cancer activity in comparison with oridonin.
Table 1.
Antiproliferative effects of representative triazole-substituted analogues against human breast cancer cell lines
| compounds | IC50 (μM)a
|
|
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| Oridonin (1) | 6.6 | 29.4 |
| 12 | 0.38 | 0.48 |
| 14 | 2.1 | 1.8 |
| 15 | 2.3 | 1.8 |
| 23 | 2.0 | 1.1 |
| 25 | 1.9 | 3.5 |
Breast cancer cell lines: MCF-7 and MDA-MB-231. Software: MasterPlex ReaderFit 2010, MiraiBio, Inc. Values are mean ± SE of three independent experiments.
In summary, efficient and concise synthetic approaches have been developed for the rapid and diverse installation of azide functionalities at the C-1, C-2 or C-3 position of the oridonin A-ring, while keeping key reactive pharmacophores intact by utilizing unique preactivation strategies based on the common synthon 4. Selective chlorination of 3-allylic alcohol 6 followed by substitution reaction with NaN3 provided 1-azide 11, which further underwent a selective [3,3]-sigmatropic rearrangement to solely afford 3-azide 8; while selective Prilezhaev epoxidation of 1-ene 4 followed by epoxide ring-opening with azide exclusively led to 2-azide 21. By harnessing the intrinsic structural features of the oridonin scaffold, a high degree of regio- and stereoselective control was achieved throughout the whole sequence. Further functionalizations of these azides through click chemistry yielding 1,2,3-triazole derivatives readily provides access to an expanded natural scaffold-based compound library. Intriguingly, these new molecules have demonstrated superior antiproliferative effects against breast cancer cells to oridonin. Our success in the efficient construction of oridonin-based azides or triazoles opens new synthetic avenues to the development of novel potential natural product-like anticancer agents.
Supplementary Material
Acknowledgments
This work was supported by grants P50 CA097007, P30 DA028821, R21 MH093844 from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, Sealy and Smith Foundation grant (to the Sealy Center for Structural Biology and Molecular Biophysics), and John Sealy Memorial Endowment Fund.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. Experimental procedures, characterization data, copies of NMR spectra for new compounds, and X-ray CIF files for compounds 6, 14 and 21. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fujita E, Nagao Y, Kaneko K, Nakazawa S, Kuroda H. Chem Pharm Bull. 1976;24:2118. doi: 10.1248/cpb.24.2118. [DOI] [PubMed] [Google Scholar]; (b) Fujita E, Nagao Y, Kohno T, Matsuda M, Ozaki M. Chem Pharm Bull. 1981;29:3208. doi: 10.1248/cpb.29.3208. [DOI] [PubMed] [Google Scholar]; (c) Yin F, Liang JY, Liu J. J Chin Pharm Univ. 2003;34:302. [Google Scholar]; (d) Sun HD, Huang SX, Han QB. Nat Prod Rep. 2006;23:673. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]; (e) Abelson PH. Science. 1990;247:513. doi: 10.1126/science.2300807. [DOI] [PubMed] [Google Scholar]; (f) Zhang W, Huang Q, Hua Z. Front Biol. 2010;5:540. [Google Scholar]
- 2.(a) Guan YZ, Wei TH. J Med Radiol Technol. 2005;236:43. [Google Scholar]; (b) Wang RL. Chin J Cancer. 1984;8:50. [Google Scholar]; (c) Henan Research Group for Rabdosia Rubescences. Cancer Res Prev Treat. 1976;3:32. [Google Scholar]
- 3.(a) Xu W, Sun J, Zhang T, Ma B, Cui S, Chen D, He Z. Acta Pharmacol Sin. 2006;27:1642. doi: 10.1111/j.1745-7254.2006.00440.x. [DOI] [PubMed] [Google Scholar]; (b) Xu W, Sun J, Zhang T, Ma B, Chen D, He Z. J Shenyang Pharm Univ. 2007;4:220. [Google Scholar]
- 4.(a) Xu J, Yang J, Ran Q, Wang L, Liu J, Wang Z, Wu X, Hua W, Yuan S, Zhang L, Shen M, Ding Y. Bioorg Med Chem Lett. 2008;18:4741. doi: 10.1016/j.bmcl.2008.06.097. [DOI] [PubMed] [Google Scholar]; (b) Wang L, Ran Q, Li D, Yao H, Zhang Y, Yuan S, Zhang L, Shen M, Xu J. Chin J Nat Med. 2011;9:194. [Google Scholar]; (c) Li D, Wang L, Cai H, Zhang Y, Xu J. Molecular. 2012;17:7556. doi: 10.3390/molecules17067556. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang L, Li D, Xu S, Cai H, Yao H, Zhang Y, Jiang J, Xu J. Eur J Med Chem. 2012;52:242. doi: 10.1016/j.ejmech.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 5.Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. J Med Chem. 2013;56:5048. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolb HC, Finn MG, Sharpless KB. Angew Chem. 2001;113:2056. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:2004. [Google Scholar]
- 7.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem. 2002;114:2708. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:2596. [Google Scholar]
- 8.(a) Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Chem Res Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]; (b) Horne WS, Yadav MK, Stout CD, Ghadiri MR. J Am Chem Soc. 2004;126:15366. doi: 10.1021/ja0450408. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Agalave SG, Maujan SR, Pore VS. Chem Asian J. 2011;6:2696. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- 9.(a) Angelini T, Lanari D, Maggi R, Pizzo F, Sartori G, Vaccaro L. Adv Synth Catal. 2012;354:908. [Google Scholar]; (b) Castrica L, Fringuelli F, Gregoli L, Pizzo F, Vaccaro L. J Org Chem. 2006;71:9536. doi: 10.1021/jo061791b. [DOI] [PubMed] [Google Scholar]; (c) Taylor MS, Zalatan DN, Lerchner AM, Jacobsen EN. J Am Chem Soc. 2005;127:1313. doi: 10.1021/ja044999s. [DOI] [PubMed] [Google Scholar]; (d) Guerin DJ, Horstmann TE, Miller SJ. Org Lett. 1999;1:1107. doi: 10.1021/ol9909076. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Cheng Y. Acta Chim Sinica. 1990;48:1185. [Google Scholar]
- 11.(a) Kuliszewska E, Hanbauer M, Hammerschmidt F. Chem Eur J. 2008;14:8603. doi: 10.1002/chem.200800475. [DOI] [PubMed] [Google Scholar]; (b) Kuliszewska E, Hanbauer M, Hammerschmidt F. J Org Chem. 2003;68:3546. [Google Scholar]
- 12.Formation of mesylate followed by treatment with LiCl was also attempted, but failed to give regio- and stereoselective product 10, while resulting in a mixture of several isomers instead.
- 13.(a) Lovchik MA, Goeke A, Fráter G. J Org Chem. 2007;72:2427. doi: 10.1021/jo062264v. [DOI] [PubMed] [Google Scholar]; (b) Gurdeep R. Organic Name Reactions, Reagents and Molecular Rearrangements. KRISHNA Prakashan Media; Meerut, India: 2008. pp. A19–A20. [Google Scholar]
- 14.Indeed, 11 was not very stable and a minor amount of 8 was identified when purified with silica gel column.
- 15.(a) Gagneaux A, Winstein S, Young WG. J Am Chem Soc. 1960;82:5956. [Google Scholar]; (b) Vanderwerf CA, Heasley VL. J Org Chem. 1966;31:3534. [Google Scholar]; (c) Trost BM, Pulley SR. Tetrahedron Lett. 1995;36:8737. [Google Scholar]; (e) Podeschwa MAL, Plettenburg O, ltenbach HJ. Org Biomol Chem. 2003;1:1919. doi: 10.1039/b302622a. [DOI] [PubMed] [Google Scholar]; (f) Chang YK, Lo HJ, Yan TH. Org Lett. 2009;11:4278. doi: 10.1021/ol9016194. [DOI] [PubMed] [Google Scholar]
- 16.McMurry JE, Isser SJ. J Am Chem Soc. 1972;94:7132. [Google Scholar]
- 17.Scriven EFV, Turnbull K. Chem Rev. 1988;88:297. [Google Scholar]
- 18.This ring opening reaction required harsh condition and was still incomplete probably due to 1,3-diaxial interactions in the resulting product 21.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.