

Abstract
Background
Clinical trial and epidemiological data support that the cardiovascular effects of estrogen are complex, including a mixture of both potentially beneficial and harmful effects. In animal models, estrogen protects females from vascular injury and inhibits atherosclerosis. These effects are mediated by estrogen receptors (ERs), which when bound to estrogen can bind to DNA to directly regulate transcription. ERs can also activate several cellular kinases by inducing a “rapid” non-nuclear signaling cascade. However, the biologic significance of this rapid signaling pathway has been unclear.
Methods and Results
Here, we develop a novel transgenic mouse in which rapid signaling is blocked by over-expression of a peptide that prevents ERs from interacting with the scaffold protein, striatin (the Disrupting Peptide Mouse, DPM). Microarray analysis of ex vivo-treated mouse aortas demonstrates that rapid ER signaling plays an important role in estrogen-mediated gene regulatory responses. Disruption of ER-striatin interactions also eliminates the ability of estrogen to stimulate cultured endothelial cell migration and to inhibit cultured vascular smooth muscle cell growth. The importance of these findings is underscored by in vivo experiments demonstrating loss of estrogen-mediated protection against vascular injury in the DPM mouse following carotid artery wire injury.
Conclusions
Taken together, these results support that rapid, non-nuclear ER signaling contributes to the transcriptional regulatory functions of ER, and is essential for many of the vasoprotective effects of estrogen. These findings also identify the rapid ER signaling pathway as a potential target for the development of novel therapeutic agents.
Keywords: cardiovascular diseases, hormones, molecular biology, signal transduction
INTRODUCTION
Epidemiologic studies demonstrate approximately two-fold lower rates of cardiovascular disease (CVD) in age-matched premenopausal women compared to men, but a near equalization of CVD risk in women after menopause, suggesting that endogenous estrogen protects women against CVD. Clinical trials of postmenopausal estrogen therapy however demonstrate mixed CVD effects. These findings underscore the complexity of the cardiovascular effects of estrogen, and support the need for a better understanding of the molecular mechanisms by which estrogen regulates cardiovascular biology. In vivo animal-based studies, including those in the mouse, have shown that estrogen protects against both vascular injury (reducing medial hyperplasia and vascular smooth muscle cell growth) and atherosclerosis 1–5. In cell culture, 17β estradiol (E2) inhibits vascular smooth muscle cell (VSMC) proliferation 6, 7. E2 also promotes the growth and migration of vascular endothelial cells, an essential aspect of vascular healing 8–11.
The cardiovascular functions of estrogen depend on estrogen receptors alpha and beta (ERα and ERβ, for review see 3–5, 12, 13). Mouse knock out studies have shown that ERα is required for estrogen-dependent protection from vascular injury 1 and from atherosclerosis 2, and that ERβ is important for E2-mediated vasodilation, protection from age-related hypertension, and proper VSMC ion channel functioning 14. Estrogen receptors are transcription factors that, when bound by E2, bind to estrogen response elements (EREs), and activate or repress target gene transcription. ER can also be recruited to chromatin through interaction with other transcription factors, such as SP1, NFκB and AP-1 15, 16. We and others have shown that ERs, in the presence of estrogen, regulate many genes important for vascular biology 17–23.
In addition to their chromatin binding functions, agonist-bound ERs also form signaling complexes in calveolae on the inner plasma membrane 12, 16, 24–27 . Interactions between membrane-bound ER and mediator proteins (including calveolin-1, shc, ras, striatin, MNAR and Gαi) mediate the activation of several important cellular kinases, including c-Src, PI3-kinase, Akt, and the ERK1/2, JNK and p38 MAP kinases, which regulate the activity of numerous downstream effectors. These pathways have been variously referred to as “rapid,” “non-canonical,” “non-nuclear” or “membrane-initiated” ER signaling. For simplicity, we will refer to these effects of ER as the “rapid” pathway, and to the functions of ER when bound to chromatin as the “genomic” pathway.
The overall functional importance of rapid ER signaling in the vascular gene regulatory and atheroprotective responses to estrogen has been largely unknown, due to the absence of models for selective abrogation of this ER signaling pathway. We have previously shown that the adapter protein striatin interacts directly with ERα and that complex formation between ERα and striatin is required for the rapid signaling functions of ER. We previously identified the binding site within ERα that mediates striatin binding, and demonstrated that overexpression of this peptide (amino acids 176–253 of ERα) disrupts ER/striatin complex formation, and blocks MAPK, AKT and eNOS activation by E2 in cultured cells, but does not prevent direct gene activation through ER binding to EREs 28. Here we develop a novel transgenic mouse that expresses this disrupting peptide (Disrupting Peptide Mouse; DPM), and use this model to examine the functions of rapid signaling in mediating both the gene regulatory and physiological functions of estrogen in vascular cells and tissues.
METHODS
Vectors and adenoviruses
pCMV-ER176-253-FLAG was created by cloning human ERα amino acids 176–253, previously shown to disrupt ERα-striatin binding, in frame with the FLAG epitope tag in a CMV-driven expression plasmid (pCMV-Tag, Agilent). Adenovirus expressing ER176-253-FLAG was generated by inserting PCR-amplified ER176-253-FLAG sequences into a CMV promoter-containing pShuttle vector (Clontech), with virus production using the Agilent Ad-Easy system. The striatin-shRNA vector was created by inserting the striatin short-hairpin RNA sequences; forward: GATCCCCCAAGGTCGACAACTACTCAttcaagagaTGAGTAGTTGTCGACCTTGTTTTTGGAAA, and reverse: AGCTTTTCCAAAAACAAGGTCGACAACTACTCAtctcttgaaTGAGTAGTTGTCGACCTTGGGG, into pSUPER (OligoEngine). The eNOS-luciferase reporter was created by inserting the eNOS promoter sequences (from -1621) upstream of luciferase in the pGL3-basic reporter vector (Promega). The L7RH-β-galactosidase control reporter 29, CMV-ERα expression vector 30, and Ad-GFP infection control vector , 31 have been described previously.
Transgenic DPM Mouse Model
Ten C57Bl/6 females (3–4 weeks old, from Jackson labs) were each administered 5 I.U of pregnant mare serum (PMS) by IP injection, followed by 5 I.U of human chorionic gonatropin 48 hours later. They were then mated with B6D2F1 stud males. The following day, one-cell embryos were harvested from the donor females and injected with the pCMV-ER176-253-FLAG vector at a concentration of 5ng/μl, and cultured in drops of KSOM overnight at 37 °C and 5% CO2. Five oviduct transfers of 30 two-cell embryos each were performed using 0.5-day pseudopregnant C57Bl/6 foster females. Pups were genotyped from tail biopsies using PCR with T3 and T7 primers flanking the insert. The integrity of the insert sequences was also confirmed using the T3 and ER3615 (CCTGTTTTTATCAATGGTGCACTG) or T7 and ER3463 (CGCTAGTGTGCAGTGTGCAATGAC) insert-specific primer sets (data not shown). These studies used heterozygous (TG+/TG-) mice derived from BL6 x DPM heterozygote matings. The colony used for all experiments was founded by mouse DPM2 (from Fig. 1).
Figure 1.
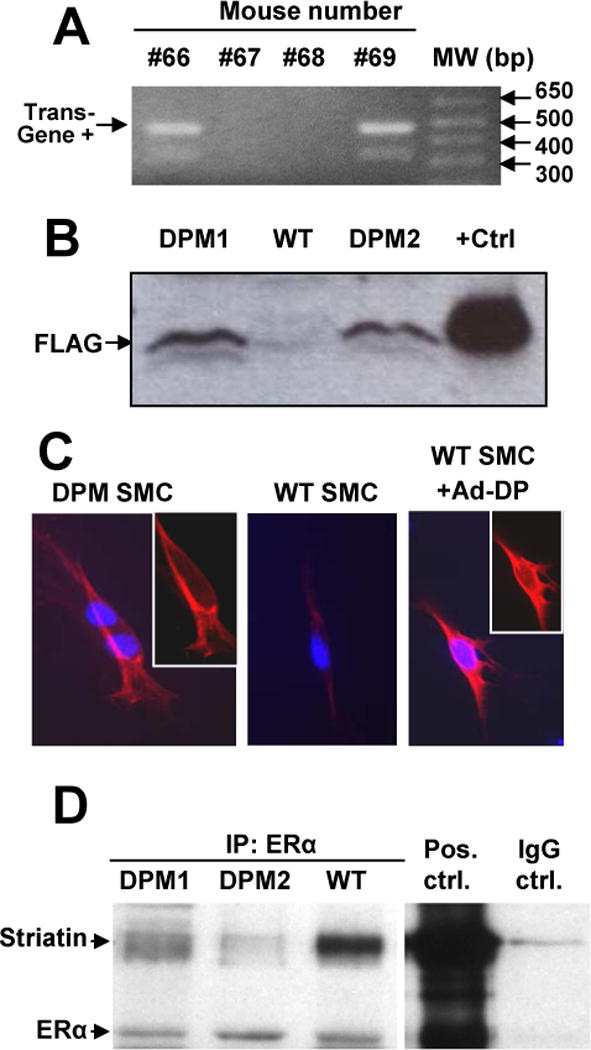
Interactions between ERα and striatin are blocked in a novel ERα176-253 Disrupting Peptide Mouse (DPM). (A) Tail DNA was amplified using primers flanking the ERα176-253-FLAG insert, yielding an ~480 bp transgene product – mice #66 and #69 were positive for the transgene. (B) The ERα176-253-FLAG peptide, as detected by anti-FLAG Western blot, is expressed in DPM but not WT liver cells. “+Ctrl”: Cos-1 cells transfected with an ERα176-253-FLAG expression plasmid. (C) Immunostaining for the disrupting peptide ERα176-253-FLAG (red) in mouse smooth muscle cells. Insets: same image without overlay of blue DAPI nuclear stain. (D) Lysates from liver samples of two different DPM mice, and one WT mouse were immunoprecipitated with anti-ERα antibody, before Western blotting with antibodies to Striatin or ERα. “Pos. ctrl”: positive control immunoprecipitation with both anti-ERα and anti-striatin antibodies from Cos-1 cells overexpressing ERα. “IgG ctrl.” Negative control immunoprecipitation with non-immune IgG antibody.
Cell lines and culture methods
Aortic smooth muscle cells (AoSMCs) were cultured from aortic explants from WT and DPM mice, as per 32, and grown in phenol red-free DMEM with 10% charcoal-stripped bovine growth serum (stripped BGS). EAhy926 cells (a human umbilical vein endothelial cell hybrid line, a kind gift of C.J. Edgell, UNC, Chapel Hill) were stably transfected with the striatin-shRNA vector or control empty vector (pSUPER). 24 hours after transfection, cells were placed in selective media with puromycin at 5 μg/ml (Sigma) for 2 to 3 weeks. Single colonies were selected from 96 well plates and maintained in the presence of 2 μg/ml puromycin. Low passage (<10) BAECs were cultured in DMEM with 10% FBS, and HUVEC cells in M199, 10% BGS, 50 μg/ml ECGS (Biomedical Technologies) and 0.1 mg/ml heparin. For Fig. 2B, BAECs were transfected with striatin-shRNA vector or vector alone by electroporation, followed by 1.5μg/ml puromycin selection for 48h, then a change to serum free medium with puromycin for 24h, before treatment with E2 or FBS. Lung endothelial cells were isolated from WT and DPM mice by mixing anti-CD31 coated beads with a lung cell suspension. The sorted cells were growing in DMEM + 10% BGS + Heparin + ECGS. When the cells were 60–75% confluent, a second sorting was performed using an anti-ICAM-2 antibody. The doubly-sorted cells were then grown as above, and endothelial cell purity was checked by eNOS immunostaining. To examine rapid E2 signaling, the cells were changed to low serum medium (1% with no ECGS) for 16 hours, and to serum free medium for 6 hours, and treated with 10 nM E2 or vehicle for 20 min before harvest. To measure eNOS protein induction, cells at ~70% confluence were treated with 10 nM E2 or vehicle for 16 hours in serum-free medium, and the medium supplemented with low, 2%, serum for an additional 8 hours. This supplementation was necessary because we found that lung ECs became visibly sick when starved of serum for greater than 16 hrs.
Figure 2.
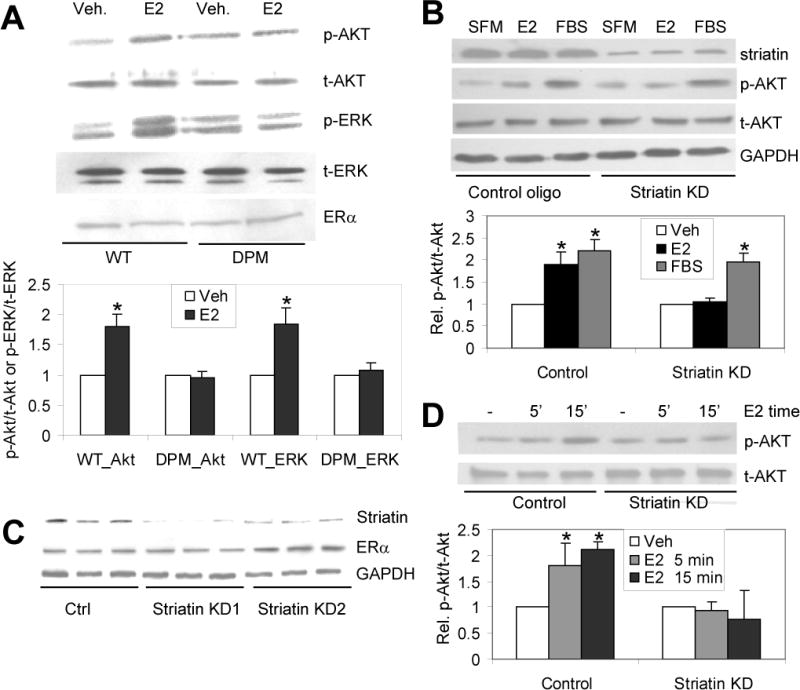
Rapid signaling is lost in cells from DPM mice, and in primary and immortalized cells lacking striatin. (A) Primary lung endothelial cells from WT and DPM mice were progressively starved for serum over ~12 hours, before treatment with 10 nM E2 or vehicle for 20 minutes. The total cell lysates were immunoblotted with the indicated antibodies. (B) Primary bovine aortic endothelial cells (BAECs) were cultured in DMEM supplemented with 10% FBS and striatin expression was knocked down (KD) via transient shRNA transfection. Cells were grown for 24 hrs in serum-free media then treated with vehicle (SFM), 10nM E2 or 10%FBS for 20 min, and immunoblotted with the indicated antibodies. (C) Western blot showing the reduction of striatin protein in stably transfected EAhy926 knock down cell lines (StriatinKD 1 & 2) but not in an empty-vector control cell line (Ctrl). Shown are results from 3 separate cultures derived from 2 independent clones for each cell line. Lower panels: immunoblots for ERα (showing ERα protein levels are unaffected by striatin shRNAs) and for GAPDH (loading control). (D) Stably-transfected striatin knock down or empty vector control EAhy926 cells were cultured in DMEM supplemented with 10% FBS, followed by treating with SFM for 24 h, followed by treatment with 10 nM E2 for 5 or 15 minutes. Cell lysates were immunoblotted for the indicated antibodies. Experiments were done in triplicate (for B and D) or quadruplicate (for A). Bars: standard deviation. “*”: p. <.05. Veh = Vehicle treated; E2- 17β-estradiol treated.
Co-Immunoprecipitation and immunodetection methods
Co-immunoprecipitation assays in Fig. 1 and Supplemental Fig. 4 were performed using anti-ERα (H184, Santa Cruz Biotechnologies) or anti-ERβ (Affinity BioReagents) antibodies, essentially as described in 28. For Western blots, the following additional antibodies were used: anti-FLAG M2 (Sigma), anti-GAPDH (Calbiochem), HC20 anti-ERα (Santa Cruz Biotechnologies) anti-striatin and anti-eNOS (BD Bioscience), with visualization using anti-mouse- or anti-rabbit-HRP secondary antibody (GE Healthcare UK LTD) and ECL (Amersham Pharmacia). For Fig. 1C, cultured primary AoSMCs were fixed and stained with M2 antibody (1:500), with fluorescent detection using donkey anti-mouse Cy3 (1:500, Jackson Immunoresearch). Cells were counter-stained with DAPI for visualization of the nucleus. COS-1 or cultured AoSMCs transfected with ER176-253-FLAG or ERα expression vectors were used as positive controls in Fig. 1D and 1C.
AoSMC growth assays
AoSMCs from passage 7 to 10 were switched to media with 0.5% BGS for 18 hr, treated with 100 nM β-estradiol (E2, Sigma) or EtOH vehicle for 30 minutes and then with 20ng/ml PDGF (InVitrogen) or 100 mM acetic acid, 0.1% BSA vehicle for 24 hrs before RNA was harvested using the RNeasy plus kit (Qiagen). cDNA was prepared using the Super Script VILO kit (InVitrogen), and qRT-PCR performed using QuantiTect SYBR Green (Qiagen) and the primers: Pcna: forward CTAGCCATGGGCGTGAAC, reverse GAATACTAGTGCTAAGGTGTCTGCAT, and Gapdh: forward CACTGAAGGGCATCTTGG, reverse CATTGTCATACCAGGAAATGAG.
Migration assays
P1 BAEC or HUVEC cells were grown to ~85% confluence in 6-well Biocoat plate (BD Biosciences), and infected with infected with GFP (control) or ERα176-253-FLAG disrupting peptide adenovirus for 24 hrs in SFM. A single scratch “wound” was made with a plastic p200 tip. Cells were rinsed to remove non-adhered cells, and incubated for 20hrs in media containing 10 nM E2, 1 μM ICI-182,780 (ICI, TOCRIS Bioscience) or EtOH vehicle, or 10% FBS. Cells were fixed in paraformaldehyde and DAPI-stained. Cell migration was measured as the number of DAPI-stained nuclei that had entered the scratch area (as demarcated by reference marks, using photographs at time 0 versus 20 hrs after scratching).
eNOS expression assays
Stable EAhy926 striatin knock down or control cell lines were co-transfected with the eNOS-luciferase reporter, L7RH-β-galactosidase control reporter 29, and CMV-ERα expression 30 vectors using Polyfect transfection reagent (Qiagen). After 24 hrs, the media was replaced with serum-free media (SFM) for 24 hrs, and cells were treated with 10 nM E2 and/or 100 nM ICI, as indicated, for 24 hours before cell lysis. eNOS luciferase assay (Promega) counts were normalized by dividing by β-gal assay (Applied Biosystems) counts. Results for each cell line were then normalized to the vehicle-alone control (SFM).
Carotid artery injury model
The mouse carotid injury model used in this study has been described in detail previously 1 (see timeline in Fig. 6A). Briefly, WT and DPM mice aged 8 to 12 weeks were ovariectomized at day -14, and randomly selected to receive either placebo pellets (-E2) or 17β-estradiol-containing pellets (+E2; 0.25mg/60-day release pellet, Innovative Research) at day -7. At day 0, the mice were anesthetized with inhaled isoflurane and the left common carotid artery was denuded of its endothelium by intraluminal passage of a wire. At the same time, osmotic minipumps were implanted subcutaneously to release bromodeoxyuridine (BrdU; 25 mg/kg/day). At day 14, mice were sacrificed for tissue and blood harvest. Both carotid arteries were harvested after perfusion fixation with 10% formalin, and embedded in paraffin. Parallel sections were subjected to standard hematoxylin-eosin and elastin staining, to measure medial area (the area between the internal and external elastic laminae), and to immunostaining with anti-BrdU Pure (BD Biosciences) to measure BrdU positive dividing cells in the same region, as previously described 1. All staining and subsequent analysis was done in a fully blinded fashion. Medial area was measured as the area within the innermost and outermost elastic lamina in magnification 20 images of elastin stained sections, using SPOT basic and ImagePro Plus software. Because an initial analysis indicated no evident or significant differences between +E2 and –E2 groups of uninjured arteries from the same strain (WT or DPM), these pairs of groups were combined in the analysis presented.
Figure 6.
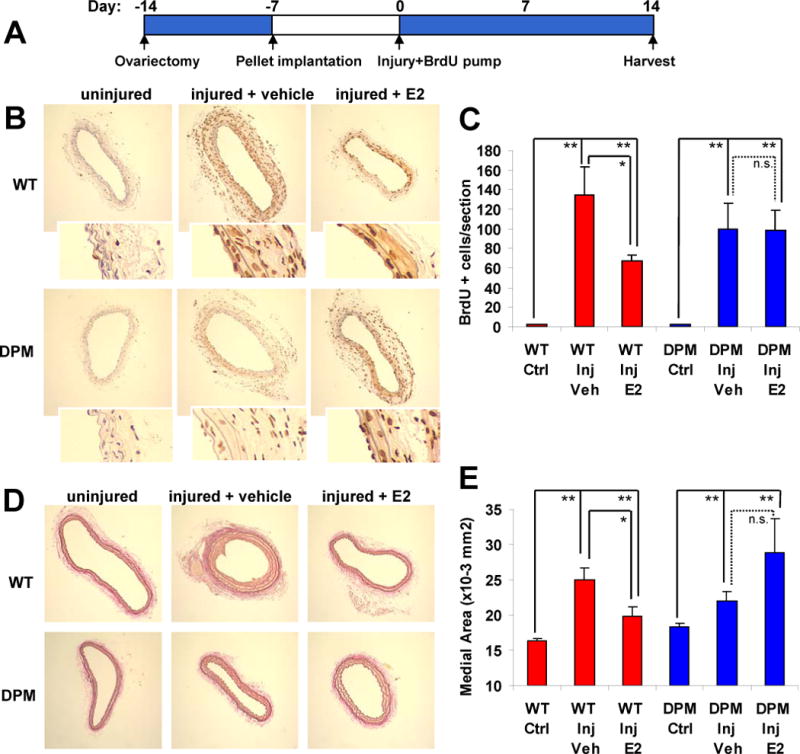
The ability of estrogen to protect against carotid arterial injury is lost in DPM mice. (A) Time course of the carotid artery wire injury model. 11 to 12 mice were used for each condition (see Supplemental Table 4 for weight measurements and other control parameters). (B) Representative anti-BrdU stained carotid artery sections, showing the effect of E2 on SMC proliferation in WT versus DPM mice. Insets: 4x higher magnification to show that BrdU stain is localized to cell nuclei. (C) Quantitation of BrdU positive cells per section. (D) Representative elastin stained sections showing the effect of estrogen on medial thickness in DPM versus WT mice. (E) Quantitation of medial thickness measurements. Bars: standard error of the mean. “*”: p<.05, “**”: p<.01.=, “n.s.”: no significant difference.
Microarray expression analysis
Aortas from WT and DPM mice were surgically removed from 10–12 week old female mice that had been overiectomized 7 days prior, and treated ex vivo for 4 hours with either 10nM estradiol or ethanol vehicle (control). Four aortas were used for each group, with two groups for each +E2 condition (WT and DPM) and three groups for each +Veh condition. RNA from each group was extracted from frozen tissue with TRIzol (InVitrogen), purified using the RNeasy kit (Qiagen), and reverse transcribed to cDNA using Superscript (InVitrogen). Labeling and hybridization to Mouse Genome 430A 2.0 microarrays (Affymetrix, Santa Clara, Ca.) was done as per the Dana Farber core facility protocol (http://chip.dfci.harvard.edu/). Raw microarray data was normalized using the GCRMA method, and differential expression analyzed using Limma with array quality weighting 33, 34. Three contrasts were performed: DPM_E2-DPM_Veh (estrogen response in DPM), WT_E2-WT_Veh (estrogen response in WT), and (DPM_E2-DPM_Veh)-(WT_E2-WT_Veh) (differential estrogen response between DPM and WT). For each contrast, we selected genes having expression differences of greater than 1.25 fold, with p values of <.02. Raw and processed data is available on GEO under accession number GSE35558.
Transcription factor binding site analysis
Promoter sequences, from -1000 to +200 bp, for the 48 top differentially E2 up-regulated genes and top 24 differentially down-regulated genes (DPM_E2/V / WT_E2/V, p<.01, fold change >1.25) were acquired from the UCSC genome browser table lookup function (mouse mm9 genome). As a control, non-regulated set, we used -1000 to +200 bp promoter sequences from the 3935 genes on the array that were expressed at least 1.1-fold greater than array background in all conditions and that showed less than 1.1-fold change E2/Veh in both WT and DPM aortas. We then performed Storm analysis as part of the CREAD package 35 to identify matches to the 585 transcription factor binding site weight matrices in the TRANSFAC database (courtesy of BIOBASE) with a functional depth of >.85. The significance of over- or under-representation of binding sites in foreground versus background sets was assessed using two-tailed binomial tests, corrected for multiple testing using the Benjamini-Hochberg method.
Statistical analyses
Statistical analyses (for all but the expression array and TFBS studies described above) were done using SigmaStat and Excel software. Data from each experiment was first examined using a two-way ANOVA to confirm significant deviation from the null hypothesis. Comparisons between pairs of conditions were then made using two-tailed t-tests, and adjusted for multiple testing using the Benjamini-Hochberg method.
RESULTS
Transgenic DPM mice carrying an expression construct for the FLAG tagged ERα176-253 ER/striatin disrupting peptide were identified by PCR-based genotypic analysis of tail DNA (Fig. 1A). ERα176-253-FLAG protein expression in liver was confirmed by Western blot against the FLAG epitope (Fig. 1B). DPM mice are born in Mendelian ratios, grow normally, are viable and fertile, and appear to be developmentally normal. By immunoflourescence, the peptide was found to be expressed throughout the cytoplasm and nucleus of cultured vascular smooth muscle cells isolated from the aortae of the transgenic mouse (Fig. 1C). The localization and expression levels observed were similar to those seen in smooth muscle cells infected with an adenovirus bearing the ERα176-253-FLAG expression construct (Fig. 1C, right panel). Expression of the ERα176-253 peptide blocked the ability of striatin to interact with ERα in co-immunoprecipitation assays in cells and tissues of the DPM mouse (Fig. 1D and data not shown).
Previously we found that expression of the ERα176-253-FLAG peptide in EAhy926 endothelial cells blocked the E2-induced rapid phosphorylation of Akt and eNOS (28). Here we find that expression of the ERα176-253 in the DPM mouse also functionally blocks rapid signaling through ERα, as evidenced by the E2-induced phosphorylation of Akt and ERK in WT- but not DPM-derived primary lung endothelial cells (Fig. 2A). To confirm that loss of rapid, E2-induced Akt phosphorylation resulted from loss of striatin binding, we transiently knocked down striatin in primary bovine aortic endothelial cells (BAECs; average striatin protein levels reduced 68%, data not shown). We found that transient reduction of striatin expression in BAECs also resulted in loss of rapid phosphorylation of Akt (Fig. 2B). Next, to facilitate studies of long-term estrogen responses in a transfectable cell model, we generated stable striatin knock down and empty-vector control EAhy926 cells (Fig. 2C), and showed that the striatin knock down cells also lose E2-mediated induction of Akt phosphorylation (Fig. 2D).
In addition to activating eNOS in endothelial cells via rapid ER signaling, E2 also upregulates expression of the eNOS gene via a transcriptional mechanism 36, 37. To test whether rapid signaling was important for this upregulation, we examined the effects of E2 treatment on eNOS protein levels in the striatin knock-down versus control EAHy926 cells. We found that the E2-dependent increase in endogenous eNOS protein abundance observed in control cells was lost in the striatin knock down cells (Fig. 3A). Striatin knock down did not, however, prevent the induction of eNOS protein levels by fetal bovine serum (FBS). We also found that E2 activated an eNOS promoter-luciferase construct in control cell lines (an effect that was blocked by the ER antagonist ICI182780), but not in the striatin knock down cell lines (Fig 3B). In addition, while primary lung endothelial cells isolated from WT mice showed E2-dependent induction of eNOS protein, this induction was lost in lung ECs from DPM mice (Fig. 3C). Taken together, these data indicate that, for eNOS and perhaps other endogenous ER target genes, rapid ER signaling modulates the gene regulatory effects of E2.
Figure 3.
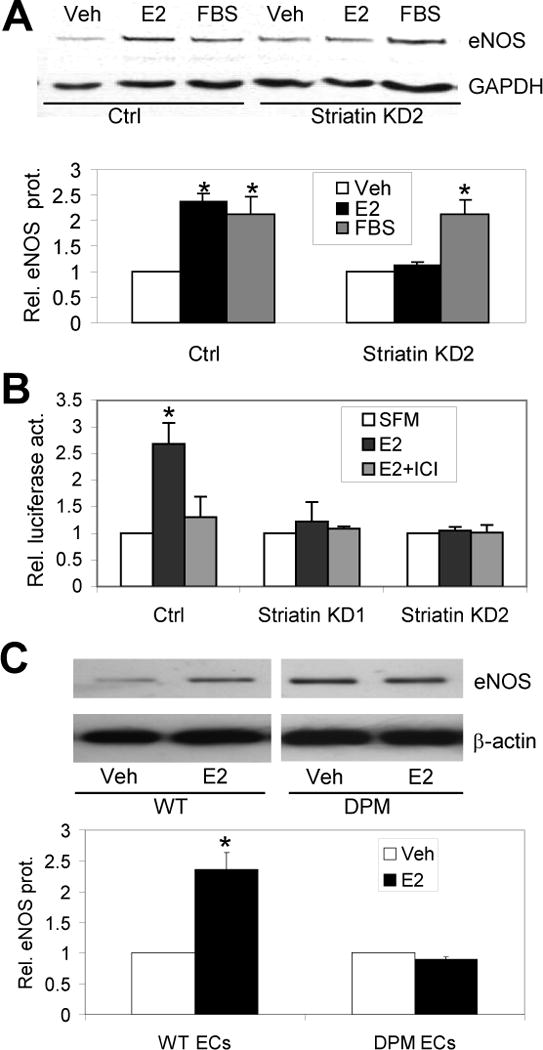
Rapid signaling through striatin is required for transcriptional upregulation of eNOS by E2. (A) Western blot showing eNOS protein levels, relative to control GAPDH (with quantitation below), in control or stably transfected striatin knock down EAhy926 endothelial cell lines treated with serum free media and vehicle (Veh), 10 nM E2 (E2) or 10% FBS for 24 hrs. (B) E2 increases activity of an eNOS-promoter luciferase reporter construct in transiently-transfected control EAhy926 cells, and this effect is blocked by the ER antagonist ICI182780 (ICI). E2 had no effect on the eNOS promoter construct in stably transfected cells with striatin knock-down. (C) Primary lung endothelial cells from WT and DPM mice were treated with vehicle or 10 nM E2 for 24 hrs, before Western blotting as in (A). Relative eNOS/β-actin levels were normalized to WT and DPM vehicle controls. Experiments were done in triplicate (for B and C) or quadruplicate (for A). Bars: standard error of the mean (for A); standard deviation (for B and C). *; p<.05 relative to Veh or SFM controls.
To more broadly test the hypothesis that rapid signaling is important for the transcriptional response of vascular tissues to estrogen, we performed expression microarray analysis in an aortic explant model. Briefly, female wild type (WT) or DPM mice were ovariectomized, and aortas harvested at day 7 and treated ex vivo with E2 or vehicle for 4 hrs (in two to three groups of 4 mice per condition), before isolation of mRNA for expression array analysis. As in our prior studies 22, we chose a 4 hour time point for this experiment in order to identify genes that were likely to be direct targets of ER signaling, rather than targets of factors whose transcription was altered in first-wave responses. We used an aortic explant model in order to precisely control the timing and concentration of E2 in an intact vascular tissue. We found 243 genes that were significantly regulated by E2 treatment in WT aortas (165 upregulated and 78 downregulated, p<=.02 and fold-change >=1.25, Fig. 4A, left), and 179 genes were regulated by E2 treatment in DPM aortas (140 upregulated and 39 downregulated, Fig. 4A, middle). Interestingly, the overlap between these sets was low, with only 27 genes being regulated in the same direction by E2 in both WT and DPM aortas (Fig. 4A, right).
Figure 4.
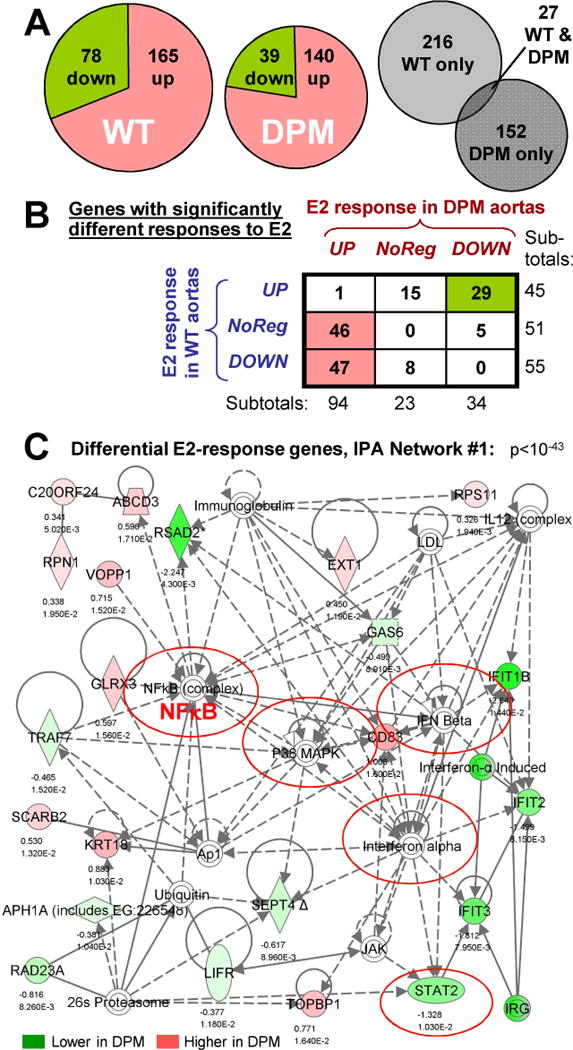
Identification of differentially E2-responsive genes in DPM versus WT by expression microarray. (A) Left & center: Pie charts showing the fraction of up- versus down-regulated genes amongst the 243 or 179 significantly E2-regulated genes in WT or DPM aortas (p<.02, fold-change > 1.25). Right: Venn diagram showing the small fraction of genes that are significantly regulated, in the same direction, by E2 in both WT and DPM mice. (B) Classification of the 151 genes showing significant differential E2 response in DPM versus WT mouse aortas. Rows: response in WT; Columns: response in DPM. (C) Top IPA network for differentially E2-regulated genes between WT and DPM. Solid lines: direct interactions. Dotted lines: indirect interactions. Arrows: regulatory action of one protein on another’s activity or expression. Lines without arrows: protein-protein interactions. Loops: self-regulatory of homo-multimerization events. Genes differentially up- or down-regulated by E2, DPM vs WT, are shown in red and green, respectively, with log2 fold change and p-value below them. Complexes of multiple proteins are indicated by concentric circles. Nodes connected to many differentially-regulated genes indicate proteins that may be involved in the coordinate regulation of these genes.
Next, we compared E2 responses in DPM mice (DPM_E2/DPM_Veh) to those in WT mice (WT_E2/WT_Veh), and found that 151 genes showed significantly different E2 response in DPM versus WT mice. 101 of these showed higher E2-induced expression in DPM mice, while 50 showed higher E2-induced expression in WT mice (see Supplemental Table 1). Interestingly, as described further below and in the Discussion, many of these genes are of possible importance in vascular development, function and disease (shaded in gray in Supplemental table 1).We classified the E2 responses for each differentially-regulated gene as up-regulated (UP), down-regulated (DOWN) or non-regulated (NR) in each strain individually (WT or DPM), setting a fold change of less than 1.1 as NR (see Supplemental Table 1 for fold-change values). Strikingly, the results indicate that about 50% of these differentially regulated genes showed opposing regulatory effects of E2 in the two mouse lines (e.g. up in WT and down in DPM, or vice versa, Fig. 4B). Almost all of the remaining differential-response genes showed non-regulation in one strain and either up or down regulation in the other. Interestingly, most of the differentially regulated genes (94 out of 151) were either non-regulated or down-regulated by E2 in WT mice, and became upregulated by E2 in the DPM mice (see Discussion)
Ingenuity Pathway Analysis (IPA) of these differentially-regulated genes indicated that they are significantly associated with cardiovascular disease and development, cholesterol metabolism, HIF1 signaling, and endocrine system development and disorders (Supplemental Table 2). We also looked at IPA networks, which can suggest common regulatory mechanisms for subgroups of differentially regulated genes by showing the shortest physical or regulatory connections amongst these genes and between these genes and cellular kinases, transcription factors and other proteins. Interestingly, many of the differentially-regulated genes in these networks clustered around transcription factors that cooperate with ER at its binding sites, or that are known or potential targets for regulation by the kinases activated by rapid signaling (including NFκB (and the regulatory subunit of the IκB kinase, IKBKG), Myc, Stat2, p53, HNF4α, HNF1α and FoxE1), or around nuclear hormones and their receptors (including progesterone, AR, GR/NR3C1 and RARαred circles in Fig. 4C and Supplemental Figures 1 through 3). Equally strikingly, most of the other differentially regulated genes in these networks were centered around kinases that are activated by rapid signaling, growth factors and their receptors, and mediators of inflammatory responses (including p38 MAPK, PI3K, Akt and Jnk; BMP6, PDGF BB, VEGF and the VEGF-receptor FLT1; and IFNα, IFNβ, prostaglandin E2 and TNF). These observations are consistent with the hypothesis that the gene regulatory effects of rapid signaling proceed through modification of transcription factors by PI3K, Akt and MAPKs, and suggest the involvement of both previously-implicated and novel transcription factors (see Discussion).
One possible explanation for the differential gene regulatory effects of E2 in WT versus DPM mice is that rapid signaling alters the activity of transcription factors which bind to consensus sequences in regulated promoters. To test this possibility, we searched the promoter regions (-1000 to +200 bp) of the top 55 differentially upregulated genes (DPM_E2/DPM_Veh > WT_E2/WT_Veh, p<.01, fold-change >1.25) or the top 28 differentially downregulated genes (DPM_E2/DPM_Veh < WT_E2/WT_Veh) for matches to 585 transcription factor binding site (TFBS) consensus matrices using Storm 35. The site frequencies for this foreground data were then compared to site frequencies in a background set composed of the promoters of 3935 genes that were detectably expressed in aortas but were not regulated by E2 in either WT or DPM aortas. Consensus elements that were significantly over- or under-represented in each group are shown in Supplemental Table 3. In the promoters of genes that were differentially upregulated by E2 in DPM aortas relative to WT, we found 27 TFBSes significantly over-represented (including Stat4 and Stat5a sites) and 22 TFBSes significantly under-represented (including SP1 sites). In the promoters of genes that are differentially downregulated by E2 in DPM compared to WT, we found five over-represented TFBSes (including CEBP and Jun sites) and four under-represented TFBSes (including AHR-HIF and p53).
The importance of rapid signaling on E2-dependent gene regulation suggests that rapid signaling might also be important for the physiological responses of vascular cells and tissues to estrogen. Prior cell culture studies have shown that estrogen can inhibit the growth of cultured smooth muscle cells – providing one likely cellular mechanism by which estrogen inhibits vascular remodeling in vivo6, 7. Consistent with those studies, we found that E2 significantly inhibited PDGF-stimulated proliferation of cultured mouse aortic smooth muscle cells (AoSMCs) derived from WT mice, (p<.01). However, E2 had no significant anti-proliferative effect on DPM-derived AoSMCs (with proliferation measured by PCNA mRNA or cell count, Figures 5A and 5B).
Figure 5.
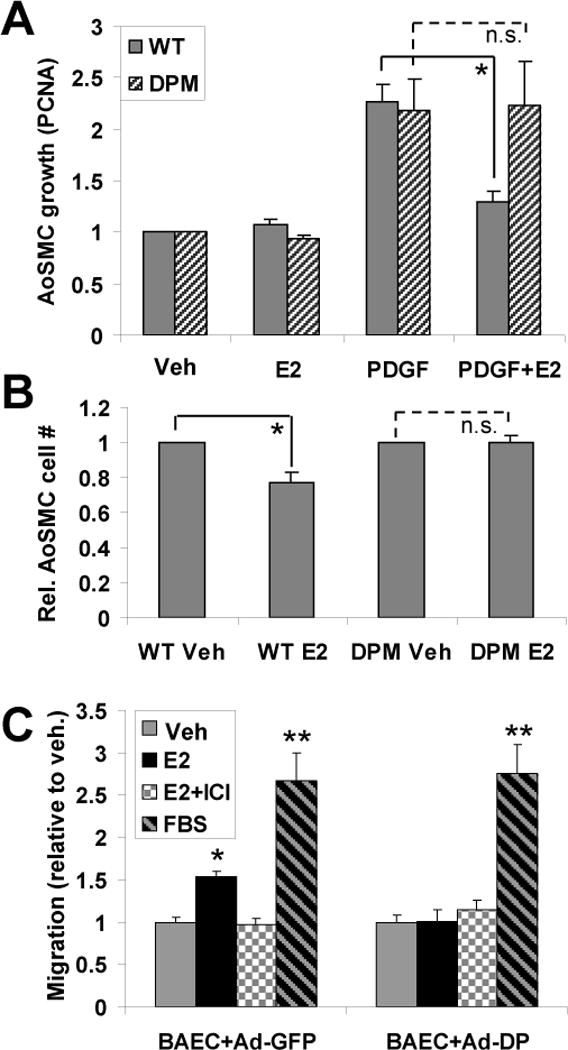
The ERα176-253 striatin-blocking peptide eliminates the effect of E2 on SMC growth and endothelial cell migration. (A) Cultured aortic smooth muscle cells (AoSMCs) from WT and DPM mice were treated with E2 and/or PDGF as indicated. Cell growth relative to untreated controls was measured by quantitative RT-PCR for the DNA-replication sliding clamp protein, PCNA, whose expression is tightly coupled to cell growth 7, with normalization to GAPDH. Each experiment was repeated four times, with two technical replicates per experiment. (B) As in (A), but with cell growth after 2 days measured by counting of trypsin-released cells using a hemocytometer. The experiment was repeated twice with three technical replicates, and data were normalized to the vehicle control value for WT or for DPM. (C) BAECs were infected with control adenovirus containing GFP (Ad-GFP) or ERα176-253-FLAG peptide (Ad-DP) expression vectors, and assayed for migration under the indicated conditions in a scratch-wound assay. Each experiment was repeated three times, with four technical replicates. Bars: standard error of the mean. “**”: p<.001 (relative to EtOH control), “*”: p<.02, “n.s.”: no significant difference.
E2 also stimulates migration of vascular endothelial cells 9–11, providing a second potential mechanism by which estrogen may protect against vascular injury. To examine the role of rapid signaling in this response, we infected bovine aortic endothelial cells (BAECs) with adenovirus carrying either an ERα176-253 expression plasmid or a control GFP expression plasmid. We found that control BAECs showed significant migration to E2 that was blocked by the estrogen antagonist ICI (Fig. 5C, “BAEC+Ad-GFP” control set). By contrast, expression of ERα176-253 in BAECs blocked E2-stimulated migration (Fig. 5C, “BAEC+Ad-DP”). It, however, did not block migration stimulated by FBS. Similar results were also seen in human umbilical vein endothelial cells (HUVECs), where E2-induced migration was seen in Ad-GFP infected cells but not in Ad-DP infected cells (1.59-fold vs. 1.00-fold, N=4, p<.008).
Finally, in our prior studies, we showed that E2 can inhibit VSMC proliferation and protect against carotid artery injury, in vivo, in wild-type but not in whole body ERα knock out mice 1. To determine whether rapid signaling plays a role in E2-mediated vascular protection, we compared wild type and DPM mice in the same carotid artery injury model (Fig. 6A). Strikingly, we found E2 significantly decreased injury-induced proliferation of smooth muscle cells in WT mice (as evidenced by a decreased number of BrdU positive cells), but had no significant effect in the DPM mice (Fig. 6B, with quantification in Fig. 6C). Similarly, whereas estrogen treatment significantly inhibited injury-induced increases in medial area in the WT mice, this effect was lost in DPM mice (Figs. 6D and 6E).
DISCUSSION
Here we have used a novel model in which rapid ER signaling is inhibited by preventing striatin-ER interaction to show that rapid signaling plays an important role in mediating the transcriptional responses of aortic cells to E2, and is also required for several effects of estrogen on vascular cells and tissues, in vitro and in vivo. With respect to transcription, we find that the transcriptional response of mouse aortas to E2 is greatly altered by the specific loss of rapid signaling through ER. This is consistent with previous studies showing a role for rapid signaling in the regulation of specific genes in various systems 16, 24. It is also consistent with a recent study in human MCF7 breast cancer cells which found that approximately one quarter of the genes that are regulated by E2 are also regulated by estrogen dendrimer conjugates (EDCs) which cannot enter the nucleus and, thus, only activate the rapid pathway 38. Taken together, these observations indicate that rapid signaling plays a major role in vascular gene regulatory responses to estrogen.
About half of the differentially-regulated genes showed opposite regulation by E2 in DPM versus WT mice, suggesting that rapid ER signaling may counter the regulatory effects of genomic signaling on gene expression, thus converting a normally stimulatory E2 response to a repressive response and vice versa. Such an effect could arise via rapid pathway kinases modifying, and altering the functions of, ER, ER co-regulators, and/or transcription factors that cooperate with ER. For instance, MAPKs, which are stimulated by rapid signaling, phosphorylate and activate ERα and ERβ 39, 40 and E2 has also been shown to promote the activation of Steroid Receptor Coactivator 3 by phosphorylation 41. Furthermore, 93 out of 151 differentially regulated genes went from down-regulated or non-regulated in WT to up-regulated in DPM, indicating that rapid signaling pathways may be particularly important for blocking activation and/or mediating repression through ER and E2.
Since mouse aortae are primarily composed of smooth muscle cells, the changes in mRNA levels we observe are likely to stem from regulatory effects in SMCs. This is likely to be one reason why eNOS induction by E2 in WT aortas was not as great as that observed in endothelial cells in culture. It is possible, however, that particularly strong E2-up-regulatory events in the minority of aortic endothelial or adventitial cells could also be detected in our expression array analysis.
The simplest regulatory model for rapid signaling in E2 transcriptional response would be that transcription factors that are modified by rapid signaling kinases bind to DNA to activate or repress a distinct set of endogenous E2 target genes, which is independent from the set of genes regulated by ER binding to chromatin. If this were the case though, we would expect that most differentially-regulated genes would fall into the class of regulated in WT aortas and non-regulated in DPM mice. Surprisingly, though, relatively few genes had this characteristic (23 out of 151, Fig. 4B). This indicates that the relationship between rapid signaling through membrane-bound ER and genomic signaling through chromatin-bound ER is much more complex, with the E2 response of most endogenous target genes determined by some combination of both ER functions. Indeed, one of the largest single classes (46 genes) was non-regulated in WT and became upregulated in DPM. These genes may represent targets at which rapid signaling normally functions to prevent a gene activation response mediated by ER binding to chromatin.
Through pathway and literature analysis, we found that many of the genes showing differential E2 responses in DPM versus WT aortas were associated with vascular function and disease. This suggests that the gene regulatory effects of rapid signaling might be important in mediating the physiological effects we observe for rapid signaling (promoting E2-dependent endothelial cell migration and inhibiting VSMC growth – both in vitro and in vivo). Differentially-regulated genes that might play a role in these processes include: CNOT7, a protein involved in controlling cell proliferation that is upregulated in smooth muscle cells with contact 42, FLT1, a VEGF-receptor that is important for angiogenesis and is also upregulated in cancer, ischemia and inflammatory disease 43, 44, and LTBP2, an extracellular TGFβ binding protein with roles in modulating the elasticity of the extracellular matrix 45. In addition, several interferon-activated genes, including Ifit1, 2 and 3 and Rsad2, were upregulated in WT and downregulated in DPM, and these genes cluster around IFN nodes in IPA network 1. This suggests that rapid signaling promotes upregulation and counters downregulation of many interferon-responsive genes. We also found three differentially-regulated genes that are involved in the metabolism of estrogen and other steroid hormones: HSD17B8/H2-Ke6, HSD17B10 and UGT1A1. Additional details for other genes of possible importance in mediating the vascular effects of rapid signaling are provided in Supplemental Table 1 (genes highlighted in gray).
Interestingly, several of the TFBSes that we found to be enriched or under-represented in differentially E2-responsive promoters (DPM vs. WT) are known targets of rapid pathway signaling kinases (including SP1, STATs 4, 5 & 6, C/EBP, Elk1 and VJUN – which is related to the c-Jun component of AP-1) 16. Several were also found to be foci in top IPA networks (including STATs HNF4, p53 and myc), indicating that the relative enrichment of these TFBSes may contribute to the coordinate regulation of groups of genes by rapid signaling. Notably, there is considerable overlap between the differentially enriched transcription factors associated with rapid pathway-specific transcriptional responses and transcription factors that can cooperate with or recruit ER to chromatin (including SP1, Jun/AP-1, Stat5 and C/EBP) 16. This highlights the interconnectedness between rapid signaling to transcription factors and the genomic effects of ER binding to chromatin (at EREs or at cooperating transcription factor binding sites), and is consistent with the observation that relatively few genes were uniquely regulated by rapid signaling (e.g. regulated by E2 in wild-type aortas and non-regulated in DPM aortas).
Factors whose sites are over-represented on differentially down-regulated promoters (those with decreased E2-dependent transcription in DPM relative to WT), such as STAT4 and STAT5a, may normally function to activate transcription via rapid signaling, while sites over-represented on differentially up-regulated promoters (such as C/EBP) may normally repress transcription in response to rapid signaling. Conversely, factors whose sites are under-represented in differentially regulated promoters may serve to counteract rapid pathway effects. For instance the under-enrichment of SP1 sites in differentially up-regulated promoters suggests that SP1 (perhaps through its ability to recruit ER to chromatin 16) may block gene repression through rapid ER signaling.
Strikingly, expression of the disrupting peptide in DPM mice eliminated the protective effect of E2 in a carotid artery injury model, blocking the E2-mediated decrease in medial thickening and the number of BrdU positive dividing cells (two measures of VSMC growth and proliferation in response to injury). These observations fit well with recent findings from a complementary model, in which selective activation of rapid signaling by EDCs was found to inhibit smooth muscle cell growth and enhance endothelial cell regrowth, in a similar arterial injury model, as efficiently as E2 46. The consistent interpretation between both of these model systems (selective inhibition resulting in loss of E2 protection and selective activation resulting in protection similar to E2) indicate that rapid signaling through ERs is both necessary and sufficient for some of the most important vasoprotective effects of estrogen. We also find that abrogation of rapid signaling through ER blocks two positive cellular responses to estrogen: eliminating both E2-mediated inhibition of cultured VSMC growth and E2-mediated stimulation of endothelial cell migration, suggesting cellular mechanisms by which rapid signaling may mediate the protective effects of E2 in vascular injury. It is worth noting that these cell culture studies employed a single 10 nM concentration of E2 in the absence of growth factors from serum, similar to many prior studies of estrogen function. It will be important, however, for future studies to consider the effects of lower nM E2 concentrations (more typical of circulating estrogen levels in non-pregnant animals) alone or in combination with growth hormones that can also influence ER activity 15, 16.
We have previously shown that whole body ERα knock out mice lose the protective effects of E2 in the same carotid artery injury model used here 1. Considered together with our current observations in DPM mice, this suggests that the protection from vascular injury afforded by E2 may require rapid signaling specifically through ERα. It is important to note, however, that vascular tissues also express ERβ, and we find that striatin interacts to a similar extent with ERβ and ERα in co-immunoprecipitation experiments, and even directly as purified proteins (Supplemental Fig. 4). Thus, while the loss of E2-dependent protection from vascular injury in DPM mice may be largely mediated through ERα, either or both of ERα or ERβ may mediate other effects that result from inhibition of rapid signaling, including alterations in E2-dependent gene regulation and loss of E2-stimulated EC migration.
Considered all together, these data indicate that rapid signaling through ER is of critical importance in mediating the vascular protective effects of estrogen. As such, we anticipate that an increased understanding of the mechanisms of rapid signaling, and its effects on both cell physiology and gene regulation, could facilitate the development of novel pathway-selective ERα activators as therapies to promote vascular health.
Supplementary Material
Acknowledgments
FUNDING SOURCES
This research was supported by an NIH NHLBI grant, R01-HL056069, to R.H.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None.
References
- 1.Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res. 2002;90:1087–1092. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- 2.Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta-estradiol’s atheroprotective effects on lesion size in Apoe-/- mice. J Clin Invest. 2001;107:333–340. doi: 10.1172/JCI11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang XP, Reckelhoff JF. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hypertens. 2011;20:133–138. doi: 10.1097/MNH.0b013e3283431921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 5.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 6.Bhalla RC, Toth KF, Bhatty RA, Thompson LP, Sharma RV. Estrogen reduces proliferation and agonist-induced calcium increase in coronary artery smooth muscle cells. Am J Physiol. 1997;272:H1996–2003. doi: 10.1152/ajpheart.1997.272.4.H1996. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura Y, Suzuki T, Miki Y, Tazawa C, Senzaki K, Moriya T, Saito H, Ishibashi T, Takahashi S, Yamada S, Sasano H. Estrogen receptors in atherosclerotic human aorta: inhibition of human vascular smooth muscle cell proliferation by estrogens. Mol Cell Endocrinol. 2004;219:17–26. doi: 10.1016/j.mce.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Arnal JF, Fontaine C, Billon-Gales A, Favre J, Laurell H, Lenfant F, Gourdy P. Estrogen receptors and endothelium. Arterioscler Thromb Vasc Biol. 2010;30:1506–1512. doi: 10.1161/ATVBAHA.109.191221. [DOI] [PubMed] [Google Scholar]
- 9.Razandi M, Pedram A, Levin ER. Estrogen signals to the preservation of endothelial cell form and function. J Biol Chem. 2000;275:38540–38546. doi: 10.1074/jbc.M007555200. [DOI] [PubMed] [Google Scholar]
- 10.Geraldes P, Sirois MG, Bernatchez PN, Tanguay JF. Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: role of p38 and p42/44 mitogen-activated protein kinase. Arterioscler Thromb Vasc Biol. 2002;22:1585–1590. doi: 10.1161/01.atv.0000035393.11854.6a. [DOI] [PubMed] [Google Scholar]
- 11.Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK, Schnaper HW. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. 1995;91:755–763. doi: 10.1161/01.cir.91.3.755. [DOI] [PubMed] [Google Scholar]
- 12.Shaul PW. Novel role of estrogen receptors in vascular endothelium. Semin Perinatol. 2000;24:70–74. doi: 10.1016/s0146-0005(00)80060-9. [DOI] [PubMed] [Google Scholar]
- 13.Xing D, Nozell S, Chen YF, Hage F, Oparil S. Estrogen and mechanisms of vascular protection. Arterioscler Thromb Vasc Biol. 2009;29:289–295. doi: 10.1161/ATVBAHA.108.182279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- 15.Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7:497–508. doi: 10.2174/138920206779315737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 17.Ihionkhan CE, Chambliss KL, Gibson LL, Hahner LD, Mendelsohn ME, Shaul PW. Estrogen causes dynamic alterations in endothelial estrogen receptor expression. Circ Res. 2002;91:814–820. doi: 10.1161/01.res.0000038304.62046.4c. [DOI] [PubMed] [Google Scholar]
- 18.Koike H, Karas RH, Baur WE, O’Donnell TF, Jr, Mendelsohn ME. Differential-display polymerase chain reaction identifies nucleophosmin as an estrogen-regulated gene in human vascular smooth muscle cells. J Vasc Surg. 1996;23:477–482. doi: 10.1016/s0741-5214(96)80014-0. [DOI] [PubMed] [Google Scholar]
- 19.Nuedling S, Karas RH, Mendelsohn ME, Katzenellenbogen JA, Katzenellenbogen BS, Meyer R, Vetter H, Grohe C. Activation of estrogen receptor beta is a prerequisite for estrogen-dependent upregulation of nitric oxide synthases in neonatal rat cardiac myocytes. FEBS Lett. 2001;502:103–108. doi: 10.1016/s0014-5793(01)02675-8. [DOI] [PubMed] [Google Scholar]
- 20.Kazi AA, Molitoris KH, Koos RD. Estrogen rapidly activates the PI3K/AKT pathway and hypoxia-inducible factor 1 and induces vascular endothelial growth factor A expression in luminal epithelial cells of the rat uterus. Biol Reprod. 2009;81:378–387. doi: 10.1095/biolreprod.109.076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedersen SH, Nielsen LB, Pedersen NG, Nilas L, Ottesen B. Hormone therapy modulates ET(A) mRNA expression in the aorta of ovariectomised New Zealand White rabbits. Gynecol Endocrinol. 2009;25:175–182. doi: 10.1080/09513590802549833. [DOI] [PubMed] [Google Scholar]
- 22.Schnoes KK, Jaffe IZ, Iyer L, Dabreo A, Aronovitz M, Newfell B, Hansen U, Rosano G, Mendelsohn ME. Rapid recruitment of temporally distinct vascular gene sets by estrogen. Mol Endocrinol. 2008;22:2544–2556. doi: 10.1210/me.2008-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol. 2007;21:1281–1296. doi: 10.1210/me.2006-0497. [DOI] [PubMed] [Google Scholar]
- 24.Meyer MR, Haas E, Prossnitz ER, Barton M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol Cell Endocrinol. 2009;308:9–16. doi: 10.1016/j.mce.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin ER. Membrane oestrogen receptor alpha signalling to cell functions. J Physiol. 2009;587:5019–5023. doi: 10.1113/jphysiol.2009.177097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–14743. doi: 10.1074/jbc.R110.191791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendelsohn ME. Nongenomic, ER-mediated activation of endothelial nitric oxide synthase: how does it work? What does it mean? Circ Res. 2000;87:956–960. doi: 10.1161/01.res.87.11.956. [DOI] [PubMed] [Google Scholar]
- 28.Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci USA. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnerot C, Rocancourt D, Briand P, Grimber G, Nicolas JF. A beta-galactosidase hybrid protein targeted to nuclei as a marker for developmental studies. Proc Natl Acad Sci USA. 1987;84:6795–6799. doi: 10.1073/pnas.84.19.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karas RH, Gauer EA, Bieber HE, Baur WE, Mendelsohn ME. Growth factor activation of the estrogen receptor in vascular cells occurs via a mitogen-activated protein kinase-independent pathway. J Clin Invest. 1998;101:2851–2861. doi: 10.1172/JCI1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leon RP, Hedlund T, Meech SJ, Li S, Schaack J, Hunger SP, Duke RC, DeGregori J. Adenoviral-mediated gene transfer in lymphocytes. Proc Natl Acad Sci USA. 1998;95:13159–13164. doi: 10.1073/pnas.95.22.13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. 1994;89:1943–1950. doi: 10.1161/01.cir.89.5.1943. [DOI] [PubMed] [Google Scholar]
- 33.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004:3. doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 34.Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schones DE, Smith AD, Zhang MQ. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. doi: 10.1186/1471-2105-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H, Grohe C. 17 Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc Res. 1999;43:666–674. doi: 10.1016/s0008-6363(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 37.Tan E, Gurjar MV, Sharma RV, Bhalla RC. Estrogen receptor-alpha gene transfer into bovine aortic endothelial cells induces eNOS gene expression and inhibits cell migration. Cardiovasc Res. 1999;43:788–797. doi: 10.1016/s0008-6363(99)00159-5. [DOI] [PubMed] [Google Scholar]
- 38.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay A, Tremblay GB, Labrie F, Giguere V. Ligand-independent recruitment of SRC-1 to estrogen receptor beta through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–519. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 41.Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Bogdan JA, Adams-Burton C, Pedicord DL, Sukovich DA, Benfield PA, Corjay MH, Stoltenborg JK, Dicker IB. Human carbon catabolite repressor protein (CCR4)-associative factor 1: cloning, expression and characterization of its interaction with the B-cell translocation protein BTG1. Biochem J. 1998;336(Pt 2):471–481. doi: 10.1042/bj3360471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 44.Parenti A, Brogelli L, Filippi S, Donnini S, Ledda F. Effect of hypoxia and endothelial loss on vascular smooth muscle cell responsiveness to VEGF-A: role of flt-1/VEGF-receptor-1. Cardiovasc Res. 2002;55:201–212. doi: 10.1016/s0008-6363(02)00326-7. [DOI] [PubMed] [Google Scholar]
- 45.Hirai M, Horiguchi M, Ohbayashi T, Kita T, Chien KR, Nakamura T. Latent TGF-beta-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. Embo J. 2007;26:3283–3295. doi: 10.1038/sj.emboj.7601768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.