

Abstract
NADPH oxidase is a multi-subunit complex that assembles during phagocytosis to generate reactive oxygen species (ROS). Several components of this complex have been implicated in chronic granulomatous disease and Crohn’s disease, highlighting the importance of ROS in regulating host immune response. In this study, we use genetically deficient mice to elucidate how p40phox, one subunit of the NADPH oxidase complex, functions during intestinal inflammation. We show that p40phox deficiency enhances inflammation in both dextran sulfate sodium-induced and innate immune-mediated murine colitis models. This inflammation is characterized by severe colonic tissue injury, increased proinflammatory cytokines, and increased neutrophil recruitment. We demonstrate that neutrophils are essential during the recovery phase of intestinal inflammation and that p40phox expression is necessary for this restitution. Lastly, using an integrative bioinformatic approach, we show that p40phox deficiency leads to upregulation of chemokine receptor 1 and downregulation of enzymes involved in glycan modifications, including fucosyltransferases and sialyltransferases, during inflammation. We propose that p40phox deficiency enhances intestinal inflammation through the dysregulation of these two pathways in neutrophils.
Introduction
NADPH oxidase is a membrane-bound enzyme complex that generates reactive oxygen species (ROS) critical for pathogen killing upon phagocytosis and for regulating proinflammatory signaling in phagocytic cells. The NADPH oxidase complex is composed of five subunits: p47phox, gp91phox, p22phox, p67phox, and p40phox. Phosphorylation of p47phox induces the activation of the complex by initiating the assembly of its subunits. Once assembled, the complex generates superoxide by transporting electrons from NADPH to phagosomal oxygen (1). The importance of NADPH oxidase in host immune response is illustrated by the association of NADPH oxidase deficiency with chronic granulomatous disease (CGD) (2, 3). CGD is characterized by severe, recurrent bacterial and fungal infections caused by defective respiratory burst function (2, 3). CGD patients often experience gastrointestinal complications, and one third of CGD patients develop intestinal inflammation similar to that observed in Crohn’s disease (CD) (4, 5), highlighting ROS and innate immunity as key components in intestinal homeostasis.
Neutrophils from CD patients are often reported as having decreased respiratory burst function (6–8). Furthermore, genetic studies have associated increased susceptibility for CD with polymorphisms in the genomic region containing NCF4 and NCF2 (9, 10). These genes encode p40phox and p67phox, respectively, supporting a role for NAPDH oxidase dysfunction in promoting intestinal inflammation. Although an association between CD risk and the NCF4 locus has not been found in all meta-analyses of genome-wide association studies (GWAS), this locus has been more specifically associated with the clinical phenotype of ileal CD and recently with perianal disease (10–12). Therefore, the lack of replication is likely due to variation in the representation of ileal vs. colonic CD within the GWAS cohorts (10, 11, 13). Functional studies have confirmed that neutrophils from patients carrying one copy of the NCF4 CD risk allele have impaired ROS production in response to the bacterial peptide fMLP, confirming that impaired p40phox function promotes intestinal inflammation (14). However, in contrast to the observations in human genetics, gp91phox- and p47phox-deficient mice do not exhibit enhanced inflammation during acute dextran sulfate sodium (DSS) colitis (15–17). Thus deciphering how each NADPH oxidase subunit regulates intestinal inflammation is critical.
Here, we show that p40phox-deficient mice have increased susceptibility to DSS-induced colitis and develop severe inflammation, particularly during the recovery phase. Additionally, p40phox-deficient mice are more susceptible to an innate immune model of colitis which is dependent upon anti-CD40 pathway engagement. We demonstrate that neutrophils are essential during the recovery phase of intestinal inflammation and that p40phox expression is necessary for the neutrophil-mediated restitution response. Based on these observations, we developed a bioinformatic approach that integrates analyses of clinical gene expression signatures in CGD patient neutrophils and temporal gene expression profiles during murine DSS colitis (18, 19). Using this method, we were able to identify novel mechanisms and regulators that promote intestinal inflammation in the context of NADPH oxidase deficiency. By applying this analysis to our functional animal model, we propose that p40phox is essential for the resolution of inflammation through downregulation of chemokine receptor 1 (Ccr1) and upregulation of enzymes involved in glycan modifications in neutrophils.
Materials and Methods
Animals
Mice were maintained in specific-pathogen-free facilities at Massachusetts General Hospital (Boston, MA). All animal studies were conducted under protocols approved by the Subcommittee on Research Animal Care (SRAC) at Massachusetts General Hospital. p40phox−/− mice were kindly provided by Phillip T. Hawkins (Babraham Institute, Cambridge, UK). Generation of this knockout line has been previously described and these mice have been backcrossed to the C57BL/6 background (20). Rag1−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained on food and water ad libitum. Mice were used between 7 and 9 weeks of age and strains were age- and gender-matched for each experiment.
DSS colitis
Mice were fed 3% (w/v) dextran sulfate sodium (DSS, MP Biomedicals; MW = 36,000–50,000) dissolved in sterile, distilled drinking water ad libitum. Mice were treated with 3% DSS for 7 days, followed by 5 days of regular drinking water. Animals were monitored daily for weight loss, survival, and disease activity. Disease activity index (DAI) was scored as previously described, based on the average of three parameters: stool consistency (0, 2, 4), fecal blood (0, 2, 4), and percentage weight loss (0–4) (21, 22). Animals that did not survive the full experimental course were not included in body weight and DAI analyses. Colon tissue was harvested at indicated time points for histology, RNA, and flow cytometry analyses.
Anti-CD40 colitis
Acute, innate-mediated colitis was induced as previously described (23). Briefly, Rag1−/− and p40phox−/− × Rag1−/− double knockout (DKO) mice were injected i.p. with 200 μg FGK4.5 anti-CD40 monoclonal antibody (BioXCell, West Lebanon, NH). Age- and sex-matched control mice were treated with a rat IgG2a isotype control, 2A3 (BioXCell). Animals were monitored daily for weight loss and disease activity for 7 days. Disease activity index (DAI) was scored based on the sum of parameters previously detailed including hunching (0–1), wasting (0–1), stool consistency (0–3), proximal colon thickness (0–3), medial colon thickness (0–3), and distal colon thickness (0–3) (24). Colon was harvested for histology 7 days after induction of colitis.
Histology
Colon tissue was fixed in 10% buffered formalin and embedded in paraffin. 5 μm sections were cut and stained with H&E. Longitudinal sections were scored in a blinded fashion using methods previously established in each colitis model, with slight modification (23, 25). Colon tissues from DSS colitis experiments were scored by the following parameters: severity of inflammation (0–3), depth of injury/inflammation (0–3), and crypt damage (0–4). DSS histological scores were multiplied by a factor representing the percentage of tissue involvement: x1 (0–25%), x2 (26–50%), x3 (51–75%), and x4 (76–100%). Thus the maximum colitis score in the DSS model is 40 (25).
Colon tissues from anti-CD40 colitis experiments were prepared in the same manner and scored by the following parameters: epithelial hyperplasia (0–3), goblet cell depletion (0–3), lamina propria infiltrate (0–3), and epithelial cell damage (0–3). Proximal, medial, and distal portions of the colon were individually scored in this model and the colitis score is the sum of all three sections (maximum = 36).
Real-time quantitative PCR
Proximal, medial, and distal colon tissues were harvested at the end of each course of colitis and stored in RNAlater (Ambion, Austin, TX) per the manufacturer’s protocol prior to RNA isolation. RNA was extracted from homogenized tissues using the RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. All RNA samples were reverse-transcribed using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, CA). Using the iQ SYBR Green Supermix (Bio-Rad) for quantitative PCR, mRNA levels were determined using the iCycler with iQ5 Multicolor Real-time PCR Detection System (Bio-Rad). The reaction conditions consisted of 37 cycles of PCR with an annealing temperature of 59°C. The following primers were used: Ccr1 Fwd: aagagcctgaagcagtggaa, Ccr1 Rev: cagattgtagggggtccaga; TNF-α Fwd: GACGTGGAACTGGCAGAAGAG, TNF-α Rev: TTGGTGGTTTGTGAGTGTGAG; IFN-γ Fwd: ATGAACGCTACACACTGCATC, IFN-γ Rev: CCATCCTTTTGCCAGTTCCTC; IL-1β Fwd: GCCCATCCTCTGTGACTCAT, IL-1β Rev: AGGCCACAGGTATTTTGTCG; IL-6 Fwd: GTAGCTATGGTACTCCAGAAGAC, IL-6 Rev: ACGATGATGCACTTGCAGAA; IL-17A Fwd: TTTAACTCCCTTGGCGCAAAA, IL-17A Rev: CTTTCCCTCCGCATTGACAC; i-NOS Fwd: CGTTGGATTTGGAGCAGAAGTG, i-NOS Rev: CATGCAAAATCTCTCCACTGCC; Ccl5 Fwd: ACGTCAAGGAGTATTTCTACAC, Ccl5 Rev: GATGTATTCTTGAACCCACT; Ccl3 Fwd: AGATTCCACGCCAATTCATC, Ccl3 Rev: CTCAAGCCCCTGCTCTACAC; St3gal6 Fwd: gccctttcaaaactgcagag, St3gal6 Rev: tcccaacttcctcttcatgg; St3gal4 Fwd: gcctccaacaagaagcagac, St3gal4 Rev: gatggcaaagtagggaacca; St6gal1 Fwd: ctgcccaaggagaacttcag, St6gal1 Rev: agcgcttttctgtggtgact; Fut4 Fwd: gccattctgctgtgaactga, Fut4 Rev: tgggtgacaggtaaggaagg; Fut9 Fwd: cttcttgacggtgagcacaa, Fut9 Rev: gaatgcttggccataggtgt; HexA Fwd: gggcgtactgctggatacat, HexA Rev: acctccttcacatcctgtgc. The threshold cycle (CT) for each sample was determined for each gene and was normalized to the CT value of the endogenous housekeeping gene GAPDH. Data were calculated using the 2-ΔΔC(T) method (26, 27).
Bacterial translocation
Spleen and mesenteric lymph nodes from DSS-treated mice were weighed and homogenized in 500 μL sterile PBS. Homogenates were plated on tryptic soy broth agar plates and incubated overnight at 37°C before quantification. Data are shown as colony forming units (CFU) per gram of tissue.
Lamina propria isolation
Colonic lamina propria cells were isolated as previously described (28). Briefly, colons were harvested at indicated time points and inverted onto polyethylene tubing (Becton Dickinson, Franklin Lakes, NJ). After being washed in calcium- and magnesium-free PBS, colons were incubated with DTT (Sigma-Aldrich, St. Louis, MO), followed by 30 mM EDTA, all at room temperature. The remaining tissue was further digested using Type IV Collagenase (108 U/mL, Sigma-Aldrich) for 90 minutes. The filtered cells from the digested tissue were then layered on a 45%/72% Percoll (GE Healthcare, Waukesha, WI) gradient and harvested at the interface after centrifugation (650 x g, 15 min) (28).
Flow cytometry
2×105 colonic lamina propria cells were washed in PBS supplemented with 3% FBS (PBS/FBS). Cells were first incubated with 2.4G2 Mouse Fc block in PBS/FBS (BD Pharmingen, San Diego, CA) for 20 minutes at 4°C. Cells were then washed and stained with fluorescent-conjugated antibodies for 20 minutes at 4°C. The following antibodies were purchased from BD Pharmingen and used for our analysis: Ly-6G-FITC, F4/80-PE, and CD11b-APC. The anti-Ccr1 antibody and isotype control were purchased from R&D Systems and used per the manufacturer’s recommendation. Fluorescently labeled lamina propria cells were acquired on a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA) and analyzed using FlowJo Analysis Software (Tree Star, Inc., Ashland, OR).
Neutrophil cytospin
Total lamina propria cells were isolated from mice treated with DSS as described above and Ly-6G+CD11b+F4/80− cells were FACS sorted. 1×105 cells were spun down onto a slide using a Cytospin 3 (ThermoShandon, Waltham, MA) and subjected to Wright’s staining.
Neutrophil depletion
Where indicated, mice were treated with anti-Ly-6G mAb (clone: 1A8, BioXCell) 24 hours prior to DSS or anti-CD40 administration (4 mg/kg body weight i.p.). Age- and sex-matched littermates were injected with PBS. For neutrophil depletion in the DSS model, 24 hours post-depletion, mice were administered 3% DSS for 7 days, followed by regular drinking water for 5 days. Mice received the same dose of anti-Ly-6G every three days during the course of the experiment. For neutrophil depletion in the anti-CD40 model, 24 hours post-depletion, mice were administered anti-CD40 as described above. Mice received the same dose of anti-Ly-6G on day 3 of the 7-day course. Neutrophil depletion efficiency was determined by FACS staining in blood, spleen, bone marrow, and lamina propria.
Bioinformatic analysis
To identify NADPH oxidase-regulated genes underlying pathogenesis in the DSS colitis model, two publicly available datasets were analyzed. The first dataset provides temporal, whole-genome expression profiling during DSS colitis treatment in mice (18). Colon tissue from 12–14-week-old DSS-treated C57BL/6J mice was collected at 0, 2, 4, and 6 days. The second dataset provides global gene expression patterns in polymorphonuclear neutrophils (PMNs) from X-linked chronic granulomatous disease (XCGD) patients and healthy control individuals (19, 29). PMNs were profiled at 0, 90, 180, and 360 minutes after stimulation with IgG and C3bi-coated latex beads to activate phagocytosis. Both datasets were normalized separately using a GCRMA routine in MATLAB. Probes were filtered with low absolute expression values, low entropy, and variance less than the tenth percentile. DSS colitis data were analyzed for differential expression using two-tailed two-sample t test between pairs of data collected from consecutive time points (Supplemental Fig. 1A). 1,283 significant genes were identified with expression change > 2-fold and adjusted p value of < 0.05. We followed a similar strategy for the PMN data and performed a differential-of-differential analysis to identify 124 genes that were significantly differentially expressed in healthy PMNs relative to XCGD PMNs (Supplemental Fig. 1B).
Differentially expressed genes identified in the DSS colitis dataset were clustered into three groups: early responders, mid-to-late responders, and late responders (Supplemental Fig. 2). Each of these groups was then separately analyzed for gene set overlap using the canonical pathway gene sets from mSigDB (30). The differentially expressed genes in the PMN dataset were similarly clustered into two groups: genes that were induced or that were suppressed in the absence of ROS (Supplemental Fig. 3). This was followed by a gene set overlap analysis (Fig. 6C).
Figure 6. An integrative bioinformatic approach identifies neutrophil-specific ROS-dependent genes important during murine colitis.
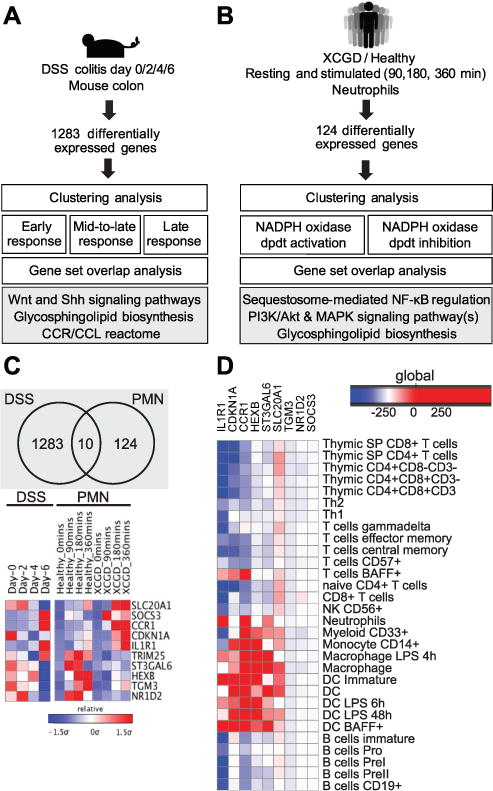
(A) Temporal microarray data from colonic tissues obtained from DSS-treated mice were normalized and analyzed to identify 1,283 differentially expressed genes. Genes enriched in this analysis are involved in the cytokine/chemokine receptor-ligand interactome, glycosphingolipid biosynthesis, and Wnt and sonic hedgehog (Shh) signaling pathways. (B) Temporal profiles of gene expression in neutrophils stimulated for phagocytosis in healthy individuals and XCGD patients were normalized and analyzed to identify 124 differentially expressed genes. Genes involved in PI3K/Akt and MAPK signaling, NF-κB regulation, and glycosphingolipid biosynthesis were enriched in this analysis. (C) A cross-comparison of the two functional analyses (described in A and B) identified 10 genes that are differentially regulated by NAPDH oxidase activity and during DSS colitis. (D) Heatmap displaying human gene expression enrichment scores of the genes identified in (C).
In order to identify NADPH oxidase-dependent genes that were important during DSS-induced colitis, the intersection of the two lists of differentially expressed genes from both DSS and PMN datasets was determined and yielded a set of 10 genes (Fig. 6C).
Enrichment score
Enrichment scores were calculated as previously described (31).
Isolation of lamina propria neutrophils
Lamina propria cells were isolated from WT and p40phox−/− mice as described above. Ly-6G+ cells were isolated via MACS positive selection cell separation kits (Miltenyi Biotec, Auburn, CA). Isolated neutrophils were washed twice with ice-cold PBS and cell pellets were stored at −80°C until RNA was extracted using the RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Quantitative RT-PCR was performed as described above.
In vivo CCR1 antagonist treatment
One hour prior to DSS treatment, WT C57BL/6 mice were injected i.p. with 10 mg/kg body weight of the CCR1 antagonist J113863 (Tocris Bioscience, Bristol, United Kingdom) dissolved in PBS, as described with other CCR1 antagonists (32). Mice were injected once daily thereafter. Assays from treated mice were conducted as described above.
Statistical analysis
Unless otherwise stated, Student’s t test was used to assess the significance of observed differences. A value of p < 0.05 was considered significant. A logrank test was used to assess significance in survival curve comparisons. A two-sided Wilcoxon rank sum test was used to assess significance for DAI scores. The test rejected the null hypothesis at the 5% significance level (*p = 0.0095). All error bars represent standard deviation.
Results
p40phox−/− mice develop severe colitis during the recovery phase of DSS-induced colitis
We first assessed the role of p40phox in intestinal inflammation through the use of an epithelial injury model, consisting of 3% dextran sulfate sodium (DSS) treatment for 7 days followed by 5 days of recovery. p40phox-deficient mice lost significantly more weight than their wild-type (WT) littermates by day 6 during DSS colitis and were unable to recover from this weight loss (Fig. 1A). In addition, p40phox-deficient mice exhibited a higher mortality rate throughout the course of DSS treatment (Fig. 1B). p40phox-deficient mice had significantly more severe disease activity based on a composite index (Fig. 1C) and more severe colonic inflammation on day 12 as shown by a significant decrease in colon length (Fig. 1D) and increase in histology score (Fig. 1E). H&E staining of colon sections revealed inflammation in p40phox−/− mice characterized by epithelial ulceration and leukocyte infiltration (Fig. 1F). Consistent with the inflammation observed in p40phox−/− mice, local TNF-α, IFN-γ, IL-1β, IL-6, iNOS (inducible nitric oxide synthase), and IL-17A levels were significantly higher in p40phox−/− mice than in WT mice on day 12 (Fig. 1G–H). Notably, similar levels of the anti-inflammatory cytokine IL-10 were observed in DSS-treated WT and p40phox−/− colon after 12 days (data not shown). To determine whether NADPH oxidase deficiency affects epithelial permeability during intestinal inflammation, we assessed bacterial translocation to the spleen and mesenteric lymph nodes during DSS colitis. p40phox−/− mice had significantly greater numbers of colony-forming units (CFU) per gram of tissue in both spleen and mesenteric lymph node, suggesting an inability to effectively clear microbes (Fig. 1I). p40phox−/− mice show no spontaneous defect in intestinal permeability, as no bacteria were detected in the peripheral organs without DSS treatment, similar to WT mice (data not shown). Collectively the data demonstrate that p40phox is necessary for the resolution of DSS-induced inflammation and that p40phox deficiency enhances proinflammatory cytokine production during intestinal inflammation.
Figure 1. p40phox−/−mice show increased susceptibility to DSS colitis.
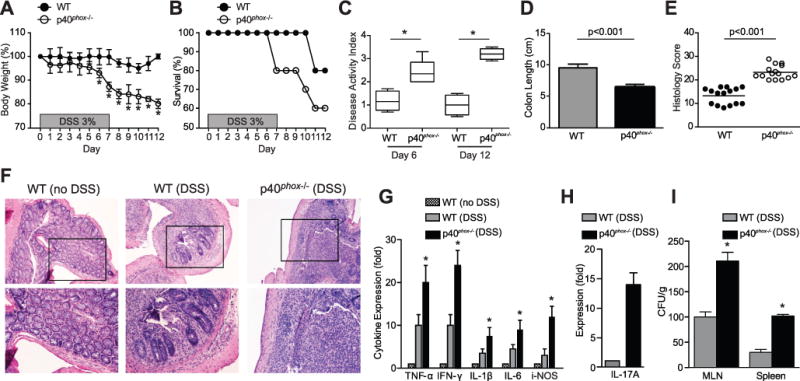
(A) Age- and weight-matched mice were given 3% DSS in drinking water ad libitum for 7 days, followed by 5 days of regular drinking water. Changes in body weight (A) and animal survival (B) were monitored daily (n = 30 per group). Significance was determined using Student’s t test for body weight (*p < 0.01) and logrank test for survival (p = 0.05). (C) Animal weight, stool consistency, and stool blood content were assessed daily to determine disease activity index (DAI). The median DAI scores for days 6 and 12 are shown. Significance was determined by a two-sided Wilcoxon rank sum test; the test rejected the null hypothesis at the 5% significance level (n = 10 per group; *p = 0.0095). (D) Colon length was measured on day 12 (n = 15 per group; WT average = 8.4 ± 0.8 and p40phox−/− average = 6.9 ± 0.5; p < 0.001). (E) Colonic tissue sections were blindly scored for inflammation on day 12. Grading parameters include severity of inflammation, depth of injury, and crypt damage (n = 15 per group; WT average = 14.1 ± 3.7 and p40phox−/− average = 22.7 ± 3.2; p < 0.001). (F) Representative colon sections stained by H&E (top panel: 10X magnification, bottom panel: 20X magnification). (G) Cytokine levels were assessed via qPCR in non-treated WT colon as well as WT and p40phox−/− colon on day 12 after DSS. Fold expression levels shown are relative to WT colon tissue from untreated mice (n = 30 per group; *p < 0.01 comparing WT (DSS) and p40phox−/− (DSS) mice). (H) IL-17A levels were assessed via qPCR in WT and p40phox−/− colon on day 12 after DSS. IL-17A was not detected in the absence of DSS treatment; therefore IL-17A expression fold is relative to the WT DSS condition (n = 30 per group). (I) Bacterial translocation to MLN and spleen was determined on day 12 after DSS administration. The number of colony-forming units per gram of tissue is shown (n = 6 per group; *p < 0.001 comparing WT (DSS) and p40phox−/− (DSS) mice). Unless otherwise noted, significance was determined using Student’s t test. For all panels, data were generated from four independent experiments.
p40phox−/− × Rag1−/− mice develop severe colitis during the recovery phase of DSS-induced colitis
To determine the contribution of the innate immune compartment in this phenotype, we next assessed whether p40phox deficiency also promotes intestinal inflammation in a Rag1-deficient mouse model, which lacks an adaptive immune system. For these experiments, p40phox−/− × Rag1−/− double knockout (DKO) mice were treated with DSS as described above, and resulting phenotypes were compared to Rag1−/− lymphocyte-deficient mice. As shown in Figure 2A, DKO mice lost more weight during DSS treatment than their Rag1−/− counterparts, most notably during the recovery phase (Fig. 2A). In addition, when compared to the weight loss observed in p40phox−/− mice treated with DSS (Fig. 1A), DKO mice lost a comparable percentage at day 12 (p40phox−/− = 20 ± 2.1%; DKO = 22.1 ± 1.2%). Histological and clinical examination demonstrated greater inflammation during DSS in DKO mice than in their Rag1−/− littermates (Fig. 2B–D). The histological score for the DKO group, while significantly greater than the Rag1−/− control cohort, was lower than that seen previously in the lymphocyte-sufficient mice (Fig. 1E), suggesting a potential, albeit secondary, role for lymphocytes in colitis progression. These data indicate that p40phox deficiency renders mice more susceptible to intestinal inflammation, even in the absence of B and T lymphocytes.
Figure 2. p40phox−/−x Rag1−/−mice show increased susceptibility to DSS colitis.
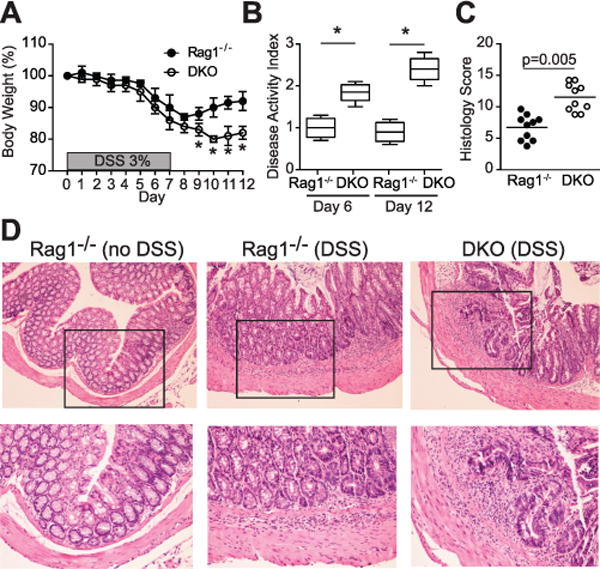
(A) Age- and weight-matched mice were given 3% DSS in drinking water ad libitum for 7 days, followed by 5 days of regular drinking water. Changes in body weight were monitored daily (n = 10 per group; *p < 0.01). (B) Animal weight, stool consistency, and stool blood content were assessed daily to determine DAI. The median DAI scores for days 6 and 12 are shown. Significance was determined by a two-sided Wilcoxon rank sum test; the test rejected the null hypothesis at the 5% significance level (n = 10 per group; *p = 0.0095). (C) Colonic tissues were blindly scored for inflammation on day 12. Grading parameters include severity of inflammation, depth of injury, and crypt damage (n = 10 per group; Rag1−/− average = 6.5 ± 2.4 and DKO average = 12.3 ± 2.6; p = 0.005). (D) Representative colon sections stained by H&E (top panel: 10X magnification, bottom panel: 20X magnification). Unless otherwise noted, significance was determined using Student’s t test. For all panels, data were generated from two independent experiments.
p40phox−/− mice are more susceptible to anti-CD40-induced colitis
In order to define the contribution of p40phox in innate immunity, we next used an anti-CD40 model of colitis. Injection of CD40 monoclonal antibody (mAb) into T and B lymphocyte-deficient Rag1−/− mice induces acute gastrointestinal inflammation and wasting disease within two days and is largely driven by local IL-23 production (23). The histological hallmarks of this colitis model include leukocyte infiltration into the lamina propria, goblet cell depletion, and marked epithelial hyperplasia (23). During the first four days following CD40 mAb treatment, p40phox−/− × Rag1−/− DKO mice lost weight with similar kinetics as their Rag1−/− control littermates. However, DKO mice were unable to recover similarly to their anti-CD40-treated Rag1−/− littermates on days 5–7 (p < 0.01, Fig. 3A). As previously reported, Rag1−/− mice injected with isotype control antibody did not show weight loss (23). In addition to the inability to recover from anti-CD40-induced colitis, DKO mice had a significantly greater disease activity index values as assessed by wasting, stool consistency, and colonic thickness (p < 0.001, Fig. 3B). Similar to the enhanced inflammation of p40phox-deficient mice to DSS, DKO mice had a significantly higher histology score in comparison to Rag1−/− mice (Fig. 3C). Colonic tissues displayed greater inflammation in DKO mice, characterized by polymorphonuclear cell infiltration (Fig. 3D). Together these data demonstrate that p40phox expression is a key component in preserving intestinal homeostasis in an innate immunity model of colitis.
Figure 3. p40phox−/−x Rag1−/−mice show increased susceptibility to anti-CD40-induced colitis.
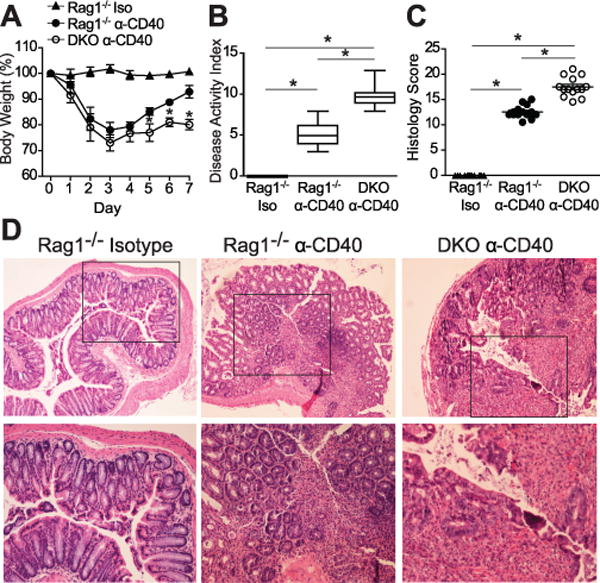
(A) Age- and weight-matched mice were injected i.p. with either anti-CD40 monoclonal depletion antibody (Rag1−/− and DKO mice) or isotype control (Rag1−/− Iso) on day 0. Changes in body weight were monitored daily for 7 days (n = 24 per group; *p < 0.01 between DKO and Rag1−/− mice injected with anti-CD40). (B) Disease activity index was determined on day 7 by scoring hunching, wasting, stool consistency, and colon thickness. Significance was determined by a two-sided Wilcoxon rank sum test; the test rejected the null hypothesis at the 5% significance level (n = 24 per group; *p = 0.0095). (C) Colonic tissue sections were blindly scored for inflammation on day 7. Grading parameters include extent of epithelial hyperplasia, goblet cell depletion, lamina propria infiltration, and epithelial cell damage (n = 14 per group; Rag1−/− average = 12.8 ± 0.5 and DKO average = 17.5 ± 0.5; *p < 0.001). (D) Representative colon sections stained by H&E (top panel: 10X magnification, bottom panel: 20X magnification). Unless otherwise noted, significance was determined using Student’s t test. For all panels, data were generated from four independent experiments.
Neutrophil recruitment in the colon is augmented in p40phox-deficient mice during DSS colitis
Given the exacerbated response of p40phox-deficient animals to murine models of colitis, we next assessed the cellular composition of the colonic lamina propria in WT and p40phox−/− mice after DSS colitis. Flow cytometric analysis revealed a population of Ly-6G+CD11b+F4/80− cells in p40phox-deficient colon whose frequency was significantly greater than their WT counterparts (representative histograms shown in Fig. 4A; WT = 5.4 ± 3%; p40phox−/− = 39.9 ± 5%). Based on cell surface molecule expression, Ly-6G+CD11b+F4/80− cells would be defined as neutrophils; however, in order to definitively categorize this population, the morphology of FACS-sorted Ly-6G+CD11b+F4/80− cells was examined on day 12 after DSS colitis in p40phox−/− mice. Wright’s staining revealed that the sorted Ly-6G+CD11b+F4/80− cells possessed multilobulated nuclei, the hallmark characteristic defining neutrophil morphology (Fig. 4B). Total neutrophil numbers were increased in p40phox lamina propria at day 6 (Fig. 4C; WT = 0.3×105 ± 0.1×105; p40phox−/− = 4.9×105 ± 1×105) and day 12 (Fig. 4C; WT = 1.2×105 ± 0.5×105; p40phox−/− = 10.9×105 ± 2×105). Thus, our data demonstrate enhanced neutrophil recruitment to the colon in p40phox−/− mice following DSS treatment.
Figure 4. Enhanced neutrophil recruitment during DSS colitis in p40phox−/−mice.
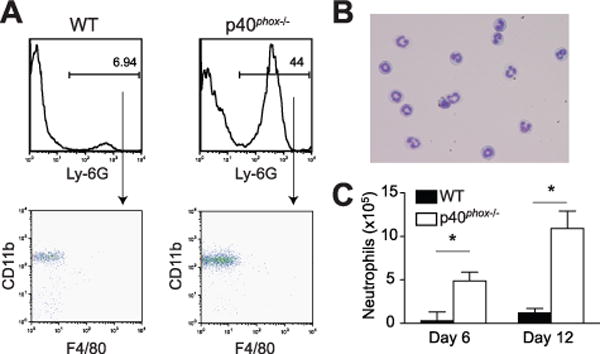
(A) Colonic lamina propria cells were isolated after recovery from DSS (day 12) and stained for flow cytometry analysis. The percentages of Ly-6G+ cells in WT and p40phox−/− LP are shown in the top histograms; CD11b and F4/80 expression of the Ly-6G+ populations are shown in the lower dot plots (n = 10 per group; representative data shown). (B) Cell morphology was assessed in FACS-sorted Ly-6G+CD11b+F4/80− cells from p40phox−/− mice (day 12) using Wright’s staining after Cytospin. Magnification is 40X. (C) The total numbers of Ly-6G+CD11b+F4/80− neutrophils were calculated during acute and recovery phases of DSS colitis. The average of 10 mice per group is displayed (*p < 0.001). Significance was determined using Student’s t test. For all panels, data were generated from three independent experiments.
p40phox-sufficient neutrophils play a protective role in intestinal inflammation
Given the increased susceptibility to DSS during the recovery phase and the enhanced neutrophil infiltration in p40phox−/− mice, we next addressed the role of neutrophils in intestinal inflammation in the context of p40phox deficiency. Neutrophils are responsible for initiating early responses in inflammation by aiding in pathogen-killing ROS generation, epithelial imprinting, and immune cell recruitment (33, 34). During the resolution phase of inflammation, however, neutrophils promote wound healing via heterotypic cell interactions with the epithelium, inducing the expression of enzymes that facilitate the production of anti-inflammatory lipid mediators and mucins (35–38).
To complement our genetic model involving NAPDH oxidase-deficient neutrophils and to determine whether neutrophils contribute to intestinal homeostasis, we next depleted neutrophils during DSS inflammation using a Ly-6G-specific depletion antibody (39). We monitored body weight and survival during DSS administration (7 days), followed by a water recovery period (5 days) (Fig. 5). Interestingly, WT mice that had been depleted of neutrophils prior to DSS treatment lost significantly more weight than their neutrophil-intact counterparts and had a 100% mortality rate by day 10 (Fig. 5A, 5C), indicating a protective role for neutrophils during DSS colitis. In contrast, p40phox−/− mice exhibited no significant difference in body weight loss with or without neutrophils (Fig. 5B, 5D), demonstrating that the protective role of neutrophils is dependent on p40phox expression. We next assessed the role of neutrophils in the anti-CD40 model and found that 100% of the neutrophil-depleted mice did not survive past day 4, indicating that neutrophils are also necessary for recovery in this colitis model (Fig. 5E–H). Together these observations indicate that neutrophils are critical for resolution of intestinal inflammation and that p40phox expression is necessary for this process.
Figure 5. Ly-6G+neutrophils are necessary for recovery from DSS and anti-CD40 colitis.
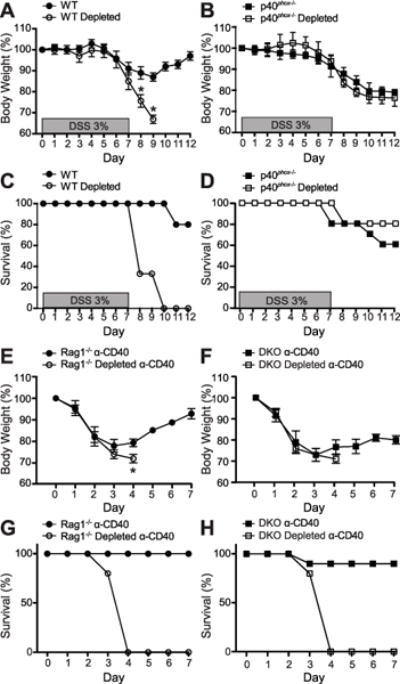
DSS colitis: Age- and weight-matched mice were either pre-treated with PBS or Ly-6G neutrophil depletion antibody prior to DSS administration. Changes in body weight (A and B) and animal survival (C and D) were monitored daily (n = 8 per group). Significance was determined using Student’s t test for body weight (*p < 0.01) and logrank test for survival (p < 0.001 for panel C; p = 0.399 for panel D). Anti-CD40 colitis: Age- and weight-matched mice were either pre-treated with PBS or Ly-6G neutrophil depletion antibody prior to anti-CD40 administration. Changes in body weight (E and F) and animal survival (G and H) were monitored daily (n = 5 per group). Significance was determined using Student’s t test for body weight (*p < 0.01) and logrank test for survival (p = 0.0002 for panel G; p = 0.0043 for panel H). For all panels, data were generated from two independent experiments.
NAPDH oxidase activity regulates expression of chemokine receptor 1 and enzymes involved in glycan modifications
We next developed an integrative bioinformatic approach to identify novel NADPH oxidase-dependent genes and pathways important during intestinal inflammation, particularly in the neutrophil compartment (Fig. 6, Supplemental Fig. 1). We first assessed time course gene expression patterns from whole murine colon from C57BL/6 WT mice treated with DSS (18). After normalization and analysis of the microarray data, we identified 1,283 differentially expressed genes (Fig. 2). The majority of the early-responding genes (day 0–2) were suppressed by day 2 and were largely enriched for sonic hedgehog and Wnt signaling pathways (Fig. 6A, Supplemental Fig. 2). The group of mid-to-late-responding genes (day 2–6) were enriched for the glycosphingolipid biosynthesis pathway (Fig. 6A, Supplemental Fig. 2). Eight of 26 genes in the glycosphingolipid biosynthesis pathway, including fucosyltransferases and sialyltransferases Fut2, Fut9, Fut4, St3gal4, St3gal6, Abo, B3galt5, and B3gnt5, were significantly differentially downregulated during this acute DSS phase (Supplemental Fig. 2). Finally, the late-responding genes showed significant enrichment for the cytokine-cytokine receptor interactome (Fig. 6A, Supplemental Fig. 2). Notably, these genes included chemotactic receptors and ligands such as Ccr1, Ccl2, and Ccr2. Thus, the analysis of temporal gene expression profiles in murine colon during DSS highlights important roles for hedgehog and Wnt signaling pathways, glycosphingolipid biosynthesis, and the activation of the chemokine reactome.
Having established that neutrophils and p40phox play key roles in intestinal homeostasis, we next analyzed data from a study chronicling global gene expression patterns in stimulated polymorphonuclear leukocytes (PMNs) isolated from healthy controls and X-linked chronic granulomatous disease (XCGD) patients. 124 differentially expressed genes were identified in our analysis (19, 29). We found that genes induced during phagocytosis in XCGD neutrophils were enriched for members of the NF-κB, PI3K/Akt, and MAPK signaling pathways (Fig. 6B, Supplemental Fig. 3). Similarly, the expression of several NF-κB-dependent proinflammatory cytokines, including IL-6, was enhanced in p40phox−/− mice during DSS colitis (Fig. 1G). Notably, HexA and HexB, two genes highly upregulated in healthy patients but not induced during phagocytosis in XCGD PMNs, belong to the glycosphingolipid biosynthesis pathway, a network highlighted in our DSS microarray analysis (Supplemental Fig. 2, 3).
Cross-comparison of these two functional analyses highlighted 10 genes regulated both by NADPH oxidase activity in human PMNs and by intestinal inflammation in the DSS colitis model: CCR1, ST3GAL6, SLC20A1, SOCS3, CDKN1A, IL1R1, TRIM25, HEXB, TGM3, and NR1D2 (Fig. 6C). Most of these genes have enriched expression in neutrophils and myeloid cells, suggesting their relevance in the context of p40phox, which is also highly expressed in these cell types (Fig. 6D). Among them, CCR1, a neutrophil chemokine receptor, has been previously implicated in murine intestinal inflammation (32, 40). Quantitative PCR confirmed that Ccr1 expression in colonic tissue from DSS-challenged mice is more highly induced in p40phox−/− mice than in WT controls (Fig. 7A). This induction of Ccr1 expression is largely neutrophil-specific, as we detected higher levels of Ccr1 expression in isolated p40phox−/− lamina propria neutrophils compared to WT neutrophils during the acute phase of DSS colitis (Fig. 7B). Moreover, p40phox−/− neutrophils isolated from the lamina propria expressed significantly higher levels of cell surface Ccr1 protein as detected by flow cytometry (Fig. 7C). Notably, no difference in expression was observed in the Ccr1 ligands Ccl3 and Ccl5 in WT and p40phox−/− colon during DSS colitis, suggesting an inherent increased capacity for p40phox−/− neutrophils to migrate (Fig. 7D). Thus, enhanced Ccr1 expression upon decreased NAPDH oxidase activity may contribute to increased neutrophilic recruitment during DSS colitis.
Figure 7. p40phoxregulates neutrophilCcr1expression and this pathway is important for intestinal inflammation restitution.
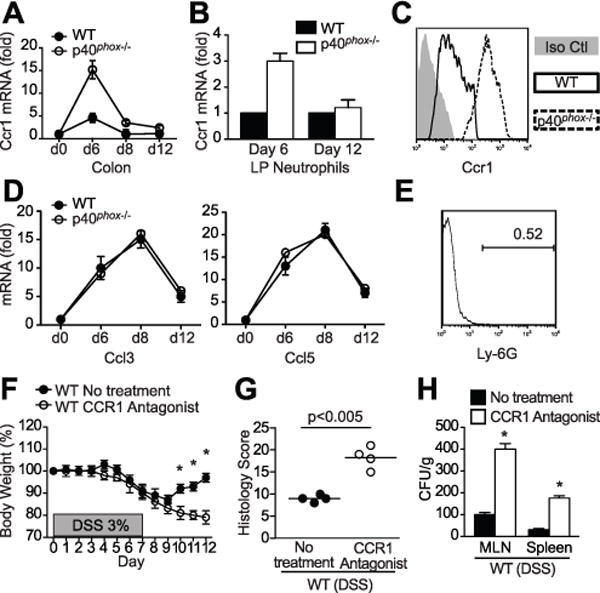
(A) Ccr1 transcript levels were assessed via qPCR in WT and p40phox−/− colon on days 0, 6, 8, and 12 during the DSS course (7 days 3% DSS followed by 5 days of water). Fold expression changes are relative to day 0. Data were generated from four independent experiments (n = 12 per group). (B) Ccr1 transcript levels were assessed via qPCR in lamina-propria isolated Ly-6G+ neutrophils from WT and p40phox−/− mice on days 6 and 12 during DSS colitis. Fold expression change is relative to Ccr1 expression in isolated WT neutrophils. Data were generated from four independent experiments (n = 5 per group). (C) Lamina propria cells isolated 6 days after 3% DSS treatment were stained with both Ly-6G and Ccr1 antibodies and analyzed by flow cytometry. The histogram shows an overlay of rat IgG2B isotype control (grey) and Ccr1 staining on Ly-6G+ cells from WT (solid line) and p40phox−/− (dotted line) mice. Data were generated from two independent experiments (n = 5 per group; representative data shown). (D) qPCR was run for Ccl3 and Ccl5, two Ccr1 ligands, in colon tissue from WT and p40phox−/− mice during DSS colitis. Fold expression changes are relative to day 0. Data were generated from four independent experiments (n = 12 per group). (E, F, G, and H) WT mice were treated with the CCR1 antagonist J113863 (10 mg/kg body weight) 24 hours prior to DSS administration and every day thereafter. LP Ly-6G+ cell frequency was assessed in CCR1 antagonist-treated mice (a representative histogram is shown) (E), body weights were monitored daily (F), colon histology was scored on day 12 (G), and bacterial translocation was quantified on day 12 (H). For panels E-H, data were generated from two independent experiments (n = 4 per group). Significance was determined using Student’s t test (*p < 0.001).
Together the bioinformatic and DSS data suggest that Ccr1 expression is regulated by ROS and that neutrophils are important for resolving inflammation. In order to directly examine the role of Ccr1 in DSS colitis, we next blocked Ccr1 using a molecular antagonist prior to DSS treatment in WT mice. As predicted, neutrophils were not recruited efficiently to the colon (Fig. 7E; compare to Fig. 4A). However, WT mice that received the CCR1 antagonist experienced significantly greater weight loss, more severe inflammation, and increased bacterial translocation compared to non-treated animals (Fig. 7F–H). Together these data suggest that Ccr1 expression and subsequent neutrophil recruitment is required for DSS recovery, a process that is dysregulated in the absence of NADPH oxidase.
The glycosphingolipid biosynthesis pathway was highlighted in our analysis of both DSS and XCGD datasets, suggesting that this pathway may be important in neutrophils during inflammation. Within this pathway, the overlap specifically highlighted HexB and St3gal6, enzymes that are involved in the synthesis and termination of carbohydrate chains and that modify glycolipids and glycoproteins. We confirmed in our DSS model that St3gal6 is downregulated during acute colitis; however, as inflammation resolved during the recovery period, St3gal6 transcript levels increased and were significantly higher in WT mice compared to p40phox−/− mice (Fig. 6C, 8A) (18). St3gal6 is a transferase involved in the biosynthesis of carbohydrates, particularly through sialylation, and has been implicated in diseases characterized by aberrant mucus production (41–43). In addition to St3gal6, analysis of the DSS time course dataset highlighted two additional sialyltransferases (St3gal4 and St6gal1) and two fucosyltransferases (Fut4 and Fut9) that are downregulated during acute colitis and are also involved in glycan modifications. We investigated the expression of these genes during DSS treatment and observed that each gene was expressed at significantly lower levels in p40phox−/− mice during colitis and, unlike WT mice, expression of these transferases remained low after the recovery phase at day 12 (Fig. 8A). Lastly, HexA, the most differentially expressed gene between XCGD patients and healthy controls in the human PMN dataset, was significantly lower in p40phox−/− mice compared to WT mice during DSS colitis (Fig. 8A). Together, our data show that NADPH oxidase deficiency induces a downregulation of genes involved in post-translational modifications of glycoproteins and glycolipids. These findings highlight the value of combining integrative genomic approaches with mouse models of disease.
Figure 8. p40phoxregulates expression of enzymes involved in glycan modifications.
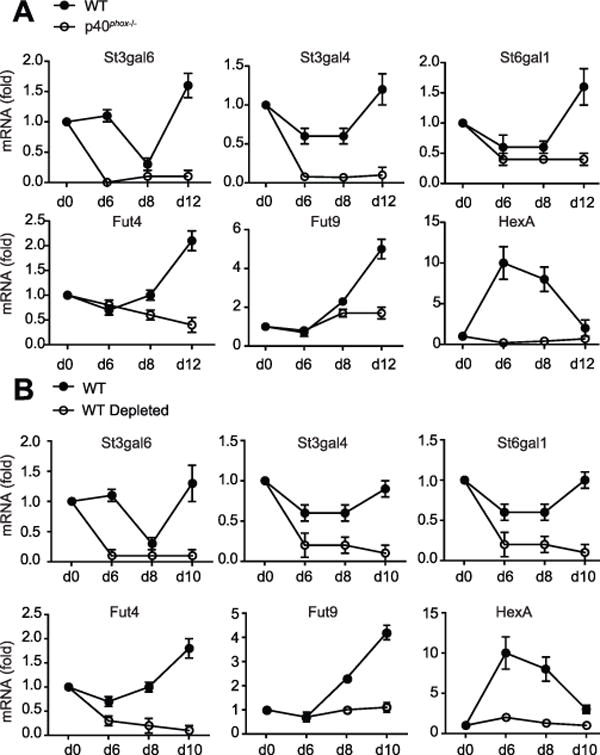
(A) Expression of enzyme members of the glycosphingolipid biosynthesis pathway were assessed via qPCR in WT and p40phox−/− colon on days 0, 6, 8, and 12 during the DSS course described above. Fold expression changes at each time point are relative to day 0. Data were generated from four independent experiments (n = 12 per group). (B) Expression of enzyme members of the glycosphingolipid biosynthesis pathway were assessed via qPCR in WT and WT Ly-6G+ neutrophil-depleted colon on days 0, 6, 8, and 10 during the DSS course described above. Fold expression changes at each time point are relative to day 0. Data were generated from two independent experiments (n = 8 per group).
These data confirm our bioinformatic predictions, in which we propose a set of ROS-dependent genes important during inflammation. However, based on the human dataset, we can only surmise that neutrophils are the relevant cell type driving this phenotype. Therefore, we next investigated the expression levels of the target genes in colon harvested from neutrophil-depleted WT mice after DSS. Expression of all six glycan-modifying proteins was significantly diminished in neutrophil-depleted WT mice compared to the non-depleted WT cohort (Fig. 8B). In addition, no differences in expression were detected in p40phox−/− mice with or without neutrophils (data not shown). These studies demonstrate that p40phox-intact neutrophils are required for appropriate expression of genes important for glycan post-translational modifications during DSS colitis.
Discussion
Mutations that impair NADPH oxidase function have been linked to CGD (2, 3), and recent genetic studies have identified associations between ileal CD and genomic regions containing NADPH oxidase genes (9–11). We investigated the role of the p40phox subunit in intestinal homeostasis using gene-targeted mice and demonstrated that mice lacking the p40phox subunit of NADPH oxidase are more susceptible to DSS-induced colitis. In addition, p40phox-deficient mice showed enhanced neutrophil infiltration, a hallmark of both inflammatory bowel disease (IBD) and CGD (34). We report the first evidence that NAPDH oxidase deficiency is crucial for the resolution phase, rather than the acute phase, of inflammation. Previous studies of DSS colitis that used genetically deficient mice for other NADPH oxidase subunits have reported different outcomes in their models (15–17). Krieglstein et al. demonstrated that the absence of p47phox had no effect on colitis severity and colonic myeloperoxidase production after 7 days of 3% DSS treatment. Furthermore, through the use of an iNOS inhibitor, the investigators showed that in the absence of p47phox and iNOS, animals were protected from DSS colitis (16). On the other hand, Bao et al. reported less colonic tissue damage and reduced myeloperoxidase levels after 7 days of 2.5% DSS in gp91phox-deficient mice compared to WT (15). Although these studies have reported that susceptibility to DSS colitis is not affected by p47phox deficiency and is lessened in the absence of gp91phox, we hasten to note that the recovery phase of inflammation was not investigated in these studies (15–17).
In this study, we show that the absence of p40phox in Rag1−/− mice enhances susceptibility to anti-CD40-induced colitis, suggesting that ROS in the innate compartment is essential during intestinal inflammation. However, in p40phox−/− × Rag1−/− DKO mice, DSS-induced inflammation was less severe than in p40phox−/− mice, implying a contribution from lymphocytes. Given that NAPDH oxidase activity has recently been shown to play a role in T cell differentiation and activation, p40phox-deficient mice may have altered T cell responses in addition to the neutrophil phenotype. p47phox deficiency has been associated with impaired T regulatory cell induction (44, 45) and increased IL-23 and TH17 response (44, 45). Indeed, we observed 50% fewer peripheral T regulatory cells in p40phox−/− mice than in WT mice (data not shown). While our data highlight an important role for p40phox in neutrophils, we recognize that NAPDH oxidase activity may affect other components of the inflammatory response.
We demonstrate that neutrophil infiltration in p40phox−/− mice is coincident with greater inflammation during DSS challenge. However, despite greater numbers of recruited lamina propria neutrophils in p40phox−/− mice during DSS, the ROS-deficient phagocytes are unable to control infiltrating bacteria, as demonstrated by greater bacterial translocation to peripheral lymph nodes. This inability to efficiently kill microbes during DSS colitis may contribute to the exacerbated inflammation described in this NADPH oxidase-deficient model. During inflammation, ROS produced by neutrophils are thought to not only assist in anti-bacterial clearance but also to damage surrounding tissue (33, 34). However, mice deficient in the neutrophil chemokine Cxcl1 fail to recruit neutrophils during DSS colitis and show more severe symptoms, highlighting a role for neutrophils in restoring mucosal barrier integrity (46). In order to address whether neutrophils are necessary for recovery from DSS- and/or anti-CD40-induced colitis, as well as to establish whether p40phox−/− neutrophils are pathogenic, we performed in vivo neutrophil depletion experiments. Depletion of WT neutrophils increased disease severity and induced 100% mortality by day 10 during DSS colitis and by day 4 during anti-CD40 colitis, suggesting that neutrophils are important in the resolution of intestinal inflammation. Neutrophil depletion in p40phox−/− mice treated with DSS, however, induced no significant differences in weight loss and inflammation compared to their p40phox−/− neutrophil-replete counterparts, suggesting that an inflammatory threshold had already been met in the absence of ROS. Previous reports using the RP-3 monoclonal antibody suggested that neutrophil depletion in WT rats suppresses DSS colitis; however, this antibody targeted the Gr1 antigen which consists of both Ly-6G and Ly-6C molecules and therefore depleted not only neutrophils but also a subset of macrophages. Thus, we propose that the observed differences may be due to the depletion of several immune populations in previous studies and/or the kinetics of antibody administration (47). Our experimental design, in which a Ly-6G neutrophil-specific depletion antibody was repeatedly injected throughout the DSS course, differs from one-time depletion studies in which new neutrophils may be generated and recruited to assist in the resolution of inflammation at later stages of DSS. Using another anti-Gr1 antibody (RB6-8C5), Qualls et al. demonstrated protection from DSS at early time points, which was lost by day 7 of treatment (48). Thus we extend these reports by describing a role for neutrophils during the resolution phase of intestinal inflammation.
To identify disease-relevant pathways mediated by p40phox, we developed an integrative approach by combining functional studies of genetically deficient mice and murine models of IBD with cell-specific data extracted from clinical microarray studies of CGD. Bioinformatic analysis of the XCGD human PMN dataset revealed a signature in NFκB signaling, suggesting that NAPDH oxidase deficiency induces hyperactivity of this pathway. Data obtained from our murine model confirmed that proinflammatory cytokine expression was indeed higher in p40phox−/− mice than in WT mice during DSS colitis, correlating with the degree of inflammation in the colon. Thus, our model mimics human disease and demonstrates a role for p40phox in the regulation of proinflammatory mediators.
We identified a subset of genes whose expression is affected both by impaired NADPH oxidase activity in XCGD neutrophils and by inflammation in mouse colon. Given our in vivo data implicating aberrant neutrophil recruitment, the neutrophil cell-specific analysis was key to unraveling the underlying mechanisms of increased colitis severity in p40phox-deficient mice. Validating the findings from our computational analysis, we confirmed that during early DSS colitis, induction of the neutrophil chemokine receptor Ccr1 was significantly higher in p40phox−/− colon compared to WT. Analysis of isolated lamina propria cells confirmed that in the context of NADPH oxidase deficiency, Ccr1 expression on neutrophils is significantly higher, despite equal levels of Ccl3 and Ccl5 chemoattractants present in the colon. As Ccr1 plays a key role in neutrophil infiltration (32, 40), its upregulation in p40phox−/− neutrophils likely accounts for the observed enhanced neutrophil recruitment in p40phox−/− colon during DSS. However, despite their greater recruitment, p40phox−/− neutrophils are unable to resolve intestinal inflammation. By using a Ccr1 antagonist in WT mice, we were able to block neutrophil recruitment to the colon during DSS colitis. Interestingly, we observed greater disease susceptibility in this setting, in agreement with a key role for neutrophils during healing. These data, as predicted by bioinformatic analyses, show not only that Ccr1 is regulated by p40phox but moreover that neutrophils are necessary during the restitution phase of DSS.
Our integrated bioinformatic analysis further identified NADPH oxidase activity as a novel regulator of the enzymes involved in the modification of glycan structures during intestinal inflammation. Six genes identified through our integrative genomics approach (St3gal6, St3gal4, St6gal1, Fut4, Fut9, and HexA) were significantly impaired in p40phox−/− mice during acute and recovery phases of DSS colitis. Moreover, by assessing these genes in neutrophil-depleted WT mice, we were able to demonstrate that p40phox-intact neutrophils are required for their expression during DSS inflammation. Sialyltransferases and fucosyltransferases such as St3gal4, St6gal1, Fut4, and Fut9 are key regulators of leukocyte trafficking through selectin ligand formation. For example, St3gal4 promotes neutrophil adhesion (49, 50), while St6gal1 deficiency has been reported to induce neutrophilic inflammation (51). Thus, decreased expression of sialyltransferases and fucosyltransferases in the absence of p40phox may contribute to aberrant neutrophil trafficking. Furthermore, alterations in mucosal thickness and mucin structure have been reported in IBD and other mucosal disorders due to aberrant glycosylation (41–43, 52–57). St3gal6, St3gal4, Fut4, and Fut9 have been implicated in intestinal mucin modification and their impaired expression in p40phox−/− mice may compromise the protective mucosal barrier integrity, resulting in enhanced inflammation (37, 58). Moreover, FUT4 contributes to epithelial wound repair during airway inflammation (59, 60), suggesting that its decreased expression in p40phox deficiency may contribute to impaired resolution of inflammation. Together, our observations indicate that p40phox deficiency leads to alterations in the expression of enzymes modifying glycan structures, which may induce aberrant intestinal neutrophil recruitment and impaired wound healing.
By combining functional studies in genetically engineered murine models with computational analysis, we have demonstrated that p40phox deficiency induces an aberrant neutrophilic inflammation in response to intestinal injury, likely due to alterations in the mechanisms controlling leukocyte recruitment and wound healing.
Supplementary Material
Acknowledgments
We thank Bret Morin for technical support and Natalia Nedelsky for review of the manuscript and helpful discussions.
This work was supported by National Institutes of Health Grants DK060049, DK086502, DK043351, and AI062773 (to R.J.X.), DK52510 (to C.T.), and The Leona M. and Harry B. Helmsley Charitable Trust (to R.J.X.). H.S. was supported by grants from the French Society of Gastroenterology, the Bettencourt Schueller Foundation, the Philippe Foundation, and the Arthur Sachs Scholarship.
References
- 1.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 2.de Oliveira-Junior EB, Bustamante J, Newburger PE, Condino-Neto A. The human NADPH oxidase: primary and secondary defects impairing the respiratory burst function and the microbicidal ability of phagocytes. Scand J Immunol. 2011;73:420–427. doi: 10.1111/j.1365-3083.2010.02501.x. [DOI] [PubMed] [Google Scholar]
- 3.Dinauer MC. Chronic granulomatous disease and other disorders of phagocyte function. Hematology Am Soc Hematol Educ Program. 2005:89–95. doi: 10.1182/asheducation-2005.1.89. [DOI] [PubMed] [Google Scholar]
- 4.Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, Hilligoss DM, Malech HL, Gallin JI, Holland SM. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114:462–468. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 5.Rahman FZ, Marks DJ, Hayee BH, Smith AM, Bloom SL, Segal AW. Phagocyte dysfunction and inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1443–1452. doi: 10.1002/ibd.20449. [DOI] [PubMed] [Google Scholar]
- 6.Curran FT, Allan RN, Keighley MR. Superoxide production by Crohn’s disease neutrophils. Gut. 1991;32:399–402. doi: 10.1136/gut.32.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gionchetti P, Campieri M, Guarnieri C, Belluzzi A, Brignola C, Bertinelli E, Ferretti M, Miglioli M, Barbara L. Respiratory burst of circulating polymorphonuclear leukocytes and plasma elastase levels in patients with inflammatory bowel disease in remission. Dig Dis Sci. 1994;39:550–554. doi: 10.1007/BF02088341. [DOI] [PubMed] [Google Scholar]
- 8.Hayee B, Rahman FZ, Tempero J, McCartney S, Bloom SL, Segal AW, Smith AM. The neutrophil respiratory burst and bacterial digestion in Crohn’s disease. Dig Dis Sci. 2010;56:1482–1488. doi: 10.1007/s10620-010-1426-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R, Lam GY, Hu P, Murchie R, Sherlock M, Gana JC, Russell RK, Glogauer M, Duerr RH, Cho JH, Lees CW, Satsangi J, Wilson DC, Paterson AD, Griffiths AM, Silverberg MS, Brumell JH. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2011 doi: 10.1136/gutjnl-2011-300078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun. 2008;9:561–565. doi: 10.1038/gene.2008.49. [DOI] [PubMed] [Google Scholar]
- 12.Eglinton TW, Roberts R, Pearson J, Barclay M, Merriman TR, Frizelle FA, Gearry RB. Clinical and genetic risk factors for perianal Crohn’s disease in a population-based cohort. Am J Gastroenterol. 2012;107:589–596. doi: 10.1038/ajg.2011.437. [DOI] [PubMed] [Google Scholar]
- 13.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D’Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak-Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D’Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somasundaram R, Deuring JJ, van der Woude CJ, Peppelenbosch MP, Fuhler GM. Linking risk conferring mutations in NCF4 to functional consequences in Crohn’s disease. Gut. 2011;61:1097. doi: 10.1136/gutjnl-2011-301344. [DOI] [PubMed] [Google Scholar]
- 15.Bao S, Carr ED, Xu YH, Hunt NH. Gp91(phox) contributes to the development of experimental inflammatory bowel disease. Immunol Cell Biol. 2011;89:853–860. doi: 10.1038/icb.2011.4. [DOI] [PubMed] [Google Scholar]
- 16.Krieglstein CF, Cerwinka WH, Laroux FS, Salter JW, Russell JM, Schuermann G, Grisham MB, Ross CR, Granger DN. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med. 2001;194:1207–1218. doi: 10.1084/jem.194.9.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori M, Stokes KY, Vowinkel T, Watanabe N, Elrod JW, Harris NR, Lefer DJ, Hibi T, Granger DN. Colonic blood flow responses in experimental colitis: time course and underlying mechanisms. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1024–1029. doi: 10.1152/ajpgi.00247.2005. [DOI] [PubMed] [Google Scholar]
- 18.Fang K, Bruce M, Pattillo CB, Zhang S, Stone R, 2nd, Clifford J, Kevil CG. Temporal genomewide expression profiling of DSS colitis reveals novel inflammatory and angiogenesis genes similar to ulcerative colitis. Physiol Genomics. 2010;43:43–56. doi: 10.1152/physiolgenomics.00138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, DeLeo FR. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol. 2004;172:636–643. doi: 10.4049/jimmunol.172.1.636. [DOI] [PubMed] [Google Scholar]
- 20.Ellson CD, Davidson K, Ferguson GJ, O’Connor R, Stephens LR, Hawkins PT. Neutrophils from p40phox−/− mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med. 2006;203:1927–1937. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 22.Murthy SN, Cooper HS, Shim H, Shah RS, Ibrahim SA, Sedergran DJ. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38:1722–1734. doi: 10.1007/BF01303184. [DOI] [PubMed] [Google Scholar]
- 23.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 24.Manocha M, Rietdijk S, Laouar A, Liao G, Bhan A, Borst J, Terhorst C, Manjunath N. Blocking CD27-CD70 costimulatory pathway suppresses experimental colitis. J Immunol. 2009;183:270–276. doi: 10.4049/jimmunol.0802424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krieglstein CF, Anthoni C, Cerwinka WH, Stokes KY, Russell J, Grisham MB, Granger DN. Role of blood- and tissue-associated inducible nitric-oxide synthase in colonic inflammation. Am J Pathol. 2007;170:490–496. doi: 10.2353/ajpath.2007.060594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 28.Sidhu M, Cotoner CA, Guleng B, Arihiro S, Chang S, Duncan KW, Ajami AM, Chau M, Reinecker HC. Small molecule tyrosine kinase inhibitors for the treatment of intestinal inflammation. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. 2002;99:6901–6906. doi: 10.1073/pnas.092148299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood. 2010;115:5376–5384. doi: 10.1182/blood-2010-01-263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morteau O, Castagliuolo I, Mykoniatis A, Zacks J, Wlk M, Lu B, Pothoulakis C, Gerard NP, Gerard C. Genetic deficiency in the chemokine receptor CCR1 protects against acute Clostridium difficile toxin A enteritis in mice. Gastroenterology. 2002;122:725–733. doi: 10.1053/gast.2002.31873. [DOI] [PubMed] [Google Scholar]
- 33.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 34.Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. 2007;42:787–798. doi: 10.1007/s00535-007-2096-y. [DOI] [PubMed] [Google Scholar]
- 35.Calder PC. Immunomodulation by omega-3 fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2007;77:327–335. doi: 10.1016/j.plefa.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol. 2010;184:4062–4068. doi: 10.4049/jimmunol.0903002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGuckin MA, Linden SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 38.Norling LV, Serhan CN. Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J Intern Med. 2010;268:15–24. doi: 10.1111/j.1365-2796.2010.02235.x. [DOI] [PubMed] [Google Scholar]
- 39.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 40.Ajuebor MN, Hogaboam CM, Kunkel SL, Proudfoot AE, Wallace JL. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J Immunol. 2001;166:552–558. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- 41.Groux-Degroote S, Krzewinski-Recchi MA, Cazet A, Vincent A, Lehoux S, Lafitte JJ, Van Seuningen I, Delannoy P. IL-6 and IL-8 increase the expression of glycosyltransferases and sulfotransferases involved in the biosynthesis of sialylated and/or sulfated Lewisx epitopes in the human bronchial mucosa. Biochem J. 2008;410:213–223. doi: 10.1042/BJ20070958. [DOI] [PubMed] [Google Scholar]
- 42.Fischer AJ, Goss KL, Scheetz TE, Wohlford-Lenane CL, Snyder JM, McCray PB., Jr Differential gene expression in human conducting airway surface epithelia and submucosal glands. Am J Respir Cell Mol Biol. 2009;40:189–199. doi: 10.1165/rcmb.2008-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nozaki K, Ogawa M, Williams JA, Lafleur BJ, Ng V, Drapkin RI, Mills JC, Konieczny SF, Nomura S, Goldenring JR. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology. 2008;134:511–522. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, Kuijpers TW, Holmdahl R, van Kooten C, Gelderman KA. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, Mathews CE. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. 2010;185:5247–5258. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shea-Donohue T, Thomas K, Cody MJ, Aiping Z, Detolla LJ, Kopydlowski KM, Fukata M, Lira SA, Vogel SN. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 2008;14:117–124. doi: 10.1177/1753425908088724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Natsui M, Kawasaki K, Takizawa H, Hayashi SI, Matsuda Y, Sugimura K, Seki K, Narisawa R, Sendo F, Asakura H. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J Gastroenterol Hepatol. 1997;12:801–808. doi: 10.1111/j.1440-1746.1997.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 48.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 49.Ellies LG, Sperandio M, Underhill GH, Yousif J, Smith M, Priatel JJ, Kansas GS, Ley K, Marth JD. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 50.Frommhold D, Ludwig A, Bixel MG, Zarbock A, Babushkina I, Weissinger M, Cauwenberghs S, Ellies LG, Marth JD, Beck-Sickinger AG, Sixt M, Lange-Sperandio B, Zernecke A, Brandt E, Weber C, Vestweber D, Ley K, Sperandio M. Sialyltransferase ST3Gal-IV controls CXCR2-mediated firm leukocyte arrest during inflammation. J Exp Med. 2008;205:1435–1446. doi: 10.1084/jem.20070846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nasirikenari M, Segal BH, Ostberg JR, Urbasic A, Lau JT. Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal I. Blood. 2006;108:3397–3405. doi: 10.1182/blood-2006-04-014779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carnoy C, Ramphal R, Scharfman A, Lo-Guidice JM, Houdret N, Klein A, Galabert C, Lamblin G, Roussel P. Altered carbohydrate composition of salivary mucins from patients with cystic fibrosis and the adhesion of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 1993;9:323–334. doi: 10.1165/ajrcmb/9.3.323. [DOI] [PubMed] [Google Scholar]
- 53.Davril M, Degroote S, Humbert P, Galabert C, Dumur V, Lafitte JJ, Lamblin G, Roussel P. The sialylation of bronchial mucins secreted by patients suffering from cystic fibrosis or from chronic bronchitis is related to the severity of airway infection. Glycobiology. 1999;9:311–321. doi: 10.1093/glycob/9.3.311. [DOI] [PubMed] [Google Scholar]
- 54.Lamblin G, Lhermitte M, Lafitte JJ, Filliat M, Degand P, Roussel P. Comparative study of bronchial mucins isolated from the sputum of patients suffering from cystic fibrosis or other chronic bronchial diseases author’s transl. Bull Eur Physiopathol Respir. 1977;13:175–190. [PubMed] [Google Scholar]
- 55.Lo-Guidice JM, Wieruszeski JM, Lemoine J, Verbert A, Roussel P, Lamblin G. Sialylation and sulfation of the carbohydrate chains in respiratory mucins from a patient with cystic fibrosis. J Biol Chem. 1994;269:18794–18813. [PubMed] [Google Scholar]
- 56.Rhim AD, Stoykova LI, Trindade AJ, Glick MC, Scanlin TF. Altered terminal glycosylation and the pathophysiology of CF lung disease. J Cyst Fibros. 2004;3(Suppl 2):95–96. doi: 10.1016/j.jcf.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 57.Zhang X, Kiechle FL. Review: Glycosphingolipids in health and disease. Ann Clin Lab Sci. 2004;34:3–13. [PubMed] [Google Scholar]
- 58.Einerhand AW, I, Renes B, Makkink MK, van der Sluis M, Buller HA, Dekker J. Role of mucins in inflammatory bowel disease: important lessons from experimental models. Eur J Gastroenterol Hepatol. 2002;14:757–765. doi: 10.1097/00042737-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Allahverdian S, Wang A, Singhera GK, Wong BW, Dorscheid DR. Sialyl Lewis X modification of the epidermal growth factor receptor regulates receptor function during airway epithelial wound repair. Clin Exp Allergy. 2010;40:607–618. doi: 10.1111/j.1365-2222.2010.03455.x. [DOI] [PubMed] [Google Scholar]
- 60.Allahverdian S, Wojcik KR, Dorscheid DR. Airway epithelial wound repair: role of carbohydrate sialyl Lewisx. Am J Physiol Lung Cell Mol Physiol. 2006;291:L828–836. doi: 10.1152/ajplung.00120.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.