

Abstract
N-Methyl-d-aspartate (NMDA) receptors mediate excitatory synaptic transmission in the central nervous system and play important roles in synaptic development and plasticity, but also mediate glutamate neurotoxicity. Recently, 2-naphthoic acid (NPA) and its derivatives have been identified as allosteric, noncompetitive NMDA receptor inhibitors. The selectivity of NPA derivatives among NMDA receptor subtypes was mapped structurally to the ligand-binding domain, and was proposed to be mediated by residues on the S1 segment. To delineate the kinetic mechanism by which NPA inhibits NMDA receptor activity, we examined its effects on the NMDA receptor gating reaction. Using whole-cell patch clamping on human embryonic kidney 293 cells expressing recombinant NMDA family of glutamate receptor subunits, GluN1/GluN2A, we found that NPA has a 50% inhibitory effect at 1.9 mM. Further, from one-channel current recordings, we found that 4 mM NPA caused a 62% decrease in open probability by decreasing mean open time 2.5-fold and by increasing mean closed time 2-fold. Kinetic modeling suggested that NPA binding stabilized NMDA receptor closed states and increased the energy barriers toward open states, causing NMDA receptors to dwell longer in pre-open states along the activation pathway. The reaction mechanisms we derived provide quantitative insight into the inhibitory mechanism of NPA and help anticipate its effects on GluN1/GluN2A receptors during both physiologic and pathologic activation modalities.
Introduction
N-Methyl-d-aspartate (NMDA) receptors are glutamate-activated ion channels expressed throughout the central nervous system. These receptors are dimers of heterodimers composed of glycine-binding GluN1 (N1) subunits and glutamate-binding GluN2 (N2) subunits, of which four isoforms (2A–2D) are encoded by separate genes. Each subunit is organized into four distinct domains: an extracellular N-terminal domain, a ligand-binding domain (LBD), a transmembrane pore-forming domain, and an intracellular C-terminal domain (Traynelis et al., 2010). Ca2+ entry through NMDA receptors is required during neuronal development for synapse formation and throughout life for synaptic maintenance and plasticity (Bliss et al., 2003). NMDA receptors are key mediators of glutamate excitotoxicity, and NMDA receptor hyperactivity has been associated with many neurologic conditions, including ischemic stroke, epilepsy, and neurodegenerative disorders (Rothman and Olney, 1986; Lipton and Rosenberg, 1994; Metman et al., 2000).
The different effects of NMDA receptor activity on neuronal survival may correlate with the receptors’ subcellular locations (Petralia et al., 2010). Upon activation by glutamate, depolarizing Na+ and Ca2+ influxes through synaptic NMDA receptors mediate synaptic signal transduction, shape plasticity, and promote neuron survival (Papadia et al., 2008; Lau and Bading, 2009; Léveillé et al., 2010; Bell and Hardingham, 2011). Conversely, activation of extrasynaptic NMDA receptors in pathologic conditions promotes neuronal death by activating separate intracellular pathways (Hardingham and Bading, 2002, 2010; Hardingham et al., 2002; Stark and Bazan, 2011; Kaufman et al., 2012). Although these findings raised hope that NMDA receptor antagonists would be neuroprotective in patients, the clinical trials for stroke and traumatic brain injury have failed so far, due to either low effectiveness or adverse effects caused by completely suppressing NMDA receptor signaling (Albers et al., 1999; Morris et al., 1999; Davis et al., 2000; Muir, 2006). The adverse effects have been attributed to the lack of selectivity of first-generation inhibitors, thus pointing to the need for isoform- or function-selective antagonists (Popescu, 2005; Wyllie et al., 2013). Recently, 2-naphthoic acid (NPA) has been identified as the parent compound of a new family of NMDA receptor allosteric modulators (Costa et al., 2010). Importantly, the compounds in this family have distinct patterns of selectivity at NMDA receptors containing different N2 subunits. Experiments with NMDA receptors containing N2 subunit chimeras suggested that the effect may be mediated by residues located in the LBD, at the interface between the N1 and N2 subunits (Costa et al., 2010), implying that this class of modulators may have a mode of action that is separate from other characterized NMDA receptor allosteric modulators and thus currently unknown. Their ability to discriminate between A/B and C/D subtypes makes NPA derivatives attractive both as possible therapeutics and research tools.
Isoform-specific modulators have tremendous pharmacologic potential (Ogden and Traynelis, 2011). In many brain regions A/B- and C/D-containing receptors are located differentially at synaptic versus extrasynaptic sites, and are thus subject to distinct patterns of agonist exposure (phasic versus tonic) and initiate intracellular cascades with divergent cellular outcomes (survival versus death) (Momiyama, 2000; Hardingham et al., 2002; Brickley et al., 2003; Hardingham and Bading, 2003; Lozovaya et al., 2004; Léveillé et al., 2010). The new features and promising applications of this class of NMDA receptor modulators necessitate a better understanding of their modulatory mechanism. Current models of NMDA receptor activation postulate that after binding agonist, and before populating open states, receptors transition through at least three kinetically resolvable states, which constitute the main activation pathway. Occasionally, receptors escape this pathway by entering desensitized states, and on a seconds-to-minutes timescale they can also change gating mode (Banke and Traynelis, 2003; Popescu and Auerbach, 2003, 2004; Erreger et al., 2005; Schorge et al., 2005; Zhang et al., 2008; Kussius et al., 2009).
To delineate how NPA affects the gating mechanism of NMDA receptors, we examined single-channel currents produced by 2A-containing NMDA receptors in the presence of NPA and in conditions that minimized confounding effects by ambient extracellular ligands. Based on these results, we conclude that NPA-binding reduces NMDA receptor open probability (Po), due to decreased mean open time (MOT) together with increased mean closed time. Kinetic modeling suggests that NPA-binding does not change the number or the sequence of states in the NMDA receptor gating reaction, but influences a specific subset of rate constants and increases the energy barrier for activation and stabilizes closed states. These results provide insight into the inhibitory mechanism of NMDA receptor by NPA and help anticipate the effects of NPA on NMDA receptor responses in both physiologic and pathologic conditions.
Materials and Methods
All methods were similar to those described in detail previously (Popescu and Auerbach, 2003; Kussius et al., 2009; Amico-Ruvio and Popescu, 2010). Briefly, rat GluN1-1a (N1, U08261) and GluN2A (N2A, M91561) subunits were expressed from pcDNA3.1(+) in human embryonic kidney (HEK) 293 cells together with green fluorescent protein.
Whole-Cell Currents.
Whole-cell currents were recorded with intracellular solutions containing (in mM): 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 10 HEPES, and 11 EGTA. Intracellular solutions were adjusted to pH 7.4 (CsOH) and clamped at −70 mV. Clamped cells were perfused with extracellular solutions containing (in mM): 150 NaCl, 2.5 KCl, 0.5 CaCl2, 10 N-(2-hydroxyethyl)piperazine-N′(4-butanesulfonic acid) (HEPBS), 0.01 EDTA, and 0.1 glycine; extracellular solutions were adjusted to pH 8.0 (NaOH). Glutamate (1 mM) and NPA (Sigma-Aldrich, St. Louis, MO) from dimethylsulfoxide (DMSO) stock were added as specified. All extracellular solutions were adjusted to contain 1% DMSO. Currents were measured under two application protocols: either 1) with NPA always present: cells were perfused with extracellular solution containing 4 mM NPA for 5 seconds before switching to a solution containing both 4 mM NPA and 1 mM glutamate; or 2) with NPA only preapplied: cells were perfused with extracellular solution containing 4 mM NPA for 5 seconds before switching to a solution containing 1 mM glutamate and no NPA. Currents were amplified and low-pass filtered at 2 kHz (Axopatch 200B; 4-pole Bessel; Molecular Devices, Sunnyvale, CA), sampled at 5 kHz (Digidata, 1322A; Molecular Devices) and written into digital files with pClamp 10 (Molecular Devices). The data were fitted by using the equation:
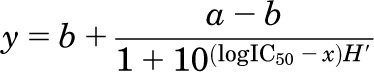 |
where a and b denote the maximum and minimum effects respectively; H is the Hill slope; and IC50 represents the concentration of antagonist that gives a response half-way between a and b. Analyses were done with OriginPro 8.0 software (OriginLab Corporation, Northampton, MA).
Single-Channel Currents.
Single-channel currents were recorded from cell-attached patches with glass electrodes filled with extracellular solution containing (in mM): 150 NaCl, 2.5 KCl, 1 EDTA, 10 HEPBS, 1 glutamate, and 0.1 glycine, as well as 1% DMSO with or without NPA (as control [CTR]), adjusted to pH 8 (NaOH). Channel openings were recorded as sodium influxes after applying +100 mV through the recording pipette. Currents were amplified and low-pass filtered at 10 kHz (Axopatch 200B; 4-pole Bessel), sampled at 40 kHz (PCI-6229, M Series card; National Instruments, Austin, TX) and written into digital files with QuB acquisition software (www.qub.buffalo.edu, University at Buffalo, Buffalo, NY).
Processing and Analyses.
Processing and analyses were done on records with only one active channel and required minimal processing, as described in detail previously (Popescu and Auerbach, 2003; Kussius et al., 2009; Amico-Ruvio and Popescu, 2010). Idealization (segmental K-means) and modeling (maximum interval likelihood) were done in QuB (Qin et al., 1997, 2000) using 12-kHz digitally filtered data, onto which we subsequently imposed a 75 µs resolution. Values for time constants, component areas, and rate constants were calculated by fitting the models indicated to entire single-channel data records.
The minimum number of states necessary to describe the recorded activity was determined by monitoring the increase in log10 likelihood (LL) upon adding closed and then open states, one at a time, to a basal two-state (1C1O) model. For each file, when the increase in the log likelihood value was less than an arbitrary cut-off value of 10 LL units per added state, the model was considered to represent the best fit (Korn and Horn, 1991; Kussius et al., 2009). The model structure was tested by examining changes in LL values upon fitting seven different models simultaneously (global fits) to data contained in three records of (computationally) manageable length. Of the seven models with different pre-open state arrangements, six returned the highest LL values and were indistinguishable. Among these models, one had the same structure as the model in Fig. 4, which was extensively validated for N2A and N2B receptors (Popescu and Auerbach, 2003; Kussius et al., 2009; Amico-Ruvio and Popescu, 2010).
Fig. 4.

Kinetic mechanism of N1/N2A inhibition by NPA. (A) Reaction mechanisms for NPA-free (CTR) and NPA-bound N1/N2A receptors (NPA); rate constants for the steps explicitly incorporated in the model were estimated from fits to one-channel records and are given in s−1 as averages for the records in each data set; blue denotes rate constants that are significantly different. All states represent receptor conformations fully bound with glutamate and glycine. (B) State-occupancy changes calculated from the reaction mechanisms in A. (C) Relative free-energy fluctuations during gating were calculated with the rate constants in A; desensitized states (C4 and C5) are omitted for simplicity; profiles are arbitrarily aligned at O2. C, nonconductive; O, conductive. *,#P < 0.05, Student’s t test, for differences among rate constants (*increase or #decrease).
Rate constants estimated for each transition included in the mode and all other kinetic parameters measured were reported as mean ± S.E.M. for each data set. Significance of differences was evaluated with two-tailed Student’s t tests assuming equal variance; differences were considered significant for P values <0.05 and were expressed as: fold-change = experimental/control or as % change = [(experimental/control) – 1] × 100.
Free-Energy Profiles.
Free-energy profiles were constructed using the rate constants in each model and the relationship ΔΔG0 = −kBTlnKeq, where kB is the Boltzmann constant, T is the absolute temperature, and Keq is the equilibrium constant of the transition considered. Barrier heights were of arbitrary magnitude and are represented as En = ΔG0n + kBT(10 − lnk+n).
Tiered modeling was performed as described previously (Amico-Ruvio et al., 2012). First, the two tiers, which represent gating of NPA-free and NPA-bound receptors, were connected with only one NPA-binding/dissociation step. Within each tier, rate constants that were found to be NPA-independent were fixed at values optimized for CTR. The two tiers were connected sequentially at C3, C2, C1, or O states, and were ranked based on a log likelihood criterion. This process revealed two types of dissociation rate constants (fast and slow) depending on where in the model the connection was placed. Next, we connected the two tiers with multiple NPA-binding/dissociation steps along the activation pathway (C3 through O2). To keep the number of free parameters constant, we scaled the NPA binding/dissociation rate constants to values estimated from the previous step, in which only a single connection between two arms was allowed. Finally, for simulations, glutamate-free and singly glutamate-bound states were added, which were assumed to have NPA affinities similar to the adjacent C3 states.
Simulations.
Simulations of NMDA receptor responses were performed using the tiered model. Responses to square jumps into 1 mM glutamate were evaluated as the sum of time-dependent open state occupancies and were analyzed in a manner similar to experimental macroscopic traces (Popescu et al., 2004). For comparison, the protocols of coapplication/preapplication of NPA and the dose-inhibition curve simulation were similar to those used in our whole-cell experiments. The simulation of physiologic responses was performed with a brief 1-millisecond pulse of 1 mM glutamate. Total charge transfer was estimated by the integral of the open-state occupancies against time. We used previously reported microscopic rates for glutamate binding and dissociation: k+ = 1.7 x 107 M−1s−1 and k− = 60 sec−1, respectively (Popescu et al., 2004).
Results
Inhibitory Effect of NPA on N2A-Containing Receptors.
We recorded currents from HEK293 cells transiently expressing GluN1-1a and GluN2A subunits with the whole-cell patch-clamp method. The extracellular solutions contained maximally effective agonist concentrations (1 mM glutamate and 0.1 mM glycine; each >100-fold EC50); EDTA at 0.01 mM; and had low proton concentration (pH 8, 10 mM HEPBS). Inhibition was measured as decrease in the steady-state current elicited by glutamate following addition of NPA (0–10 mM). The limited solubility of NPA prevented us from investigating higher concentrations. Fitting the data with the Hill equation predicted ∼90% maximal inhibition, with 1.9 mM NPA producing half-maximal inhibition (Fig. 1). This IC50 value is larger than previously measured in Xenopus oocytes (Costa et al., 2010), where 0.1 mM NPA produced 30% inhibition. We attribute this discrepancy to differences in experimental conditions because these previous measurements were done at pH = 7.4, where 2A receptors are tonically ∼50% inhibited (Traynelis and Cull-Candy, 1990) and in the absence of chelators, when trace zinc and magnesium ions further inhibit 2A currents (Nowak et al., 1984; Paoletti et al., 1997).
Fig. 1.

NPA inhibition of steady-state glutamate-elicited N1/N2A receptor currents. (Left) Whole-cell responses were recorded from human embryonic kidney 293 cells expressing N1/N2A receptors. Bars indicate glutamate (1 mM) applications (white) and NPA coapplications (gray). (Right) Levels of equilibrium responses measured after each NPA concentration was normalized to the responses when only glutamate was applied. Circles represent means of normalized values across cells. Line represents the fit of the logistic function to means of normalized responses for each concentration: 0.4 mM (n = 4), 1 mM (n = 4), 2 mM (n = 4), 4 mM (n = 5), 10 mM (n = 4). IC50 is expressed as 95% confidence interval.
Single-Channel Kinetics of NPA-Bound N2A-Containing Receptors.
To investigate the mechanism of NPA inhibition we recorded currents from one-channel cell-attached patches of HEK293 cells expressing N1/N2A receptors with extracellular (pipette) solution containing 4 mM NPA (∼2-fold IC50). Similar to our whole cell recordings, the extracellular solution also contained glutamate (1 mM), glycine (0.1 mM), and EDTA (1 mM) to remove trace divalent cations. We clamped proton concentrations at 10 nM (pH 8) with 10 mM HEPBS (Fig. 2A). As controls, we used a set of recordings obtained in identical conditions and the absence of NPA (CTR) (Fig. 2B). Both data sets included only records that originated from a single active channel, and we processed and analyzed all records as described in detail previously (Kussius et al., 2009).
Fig. 2.
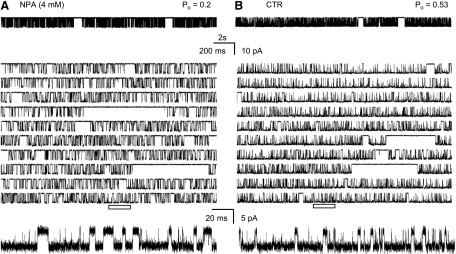
Effects of NPA on single-channel activity of N1/N2A receptors. Traces represent steady-state inward sodium fluxes recorded from cell-attached patches that contained in the recording pipette one active N1/N2A channel: (A) with NPA (4 mM), and (B) without NPA (CTR). For each condition, a 20-second segment is illustrated at two time resolutions in top and middle panels, respectively; bottom panels expand the underlined segment and are displayed filtered, as for analyses, at 12 kHz. All traces represent Na+ currents as downward deflections from a zero-current baseline; Po indicates the open probability calculated for the entire parent record.
We found that 4 mM NPA decreased the average equilibrium open probability (Po) of 2A receptors to 38% of the CTR with no change in the single-channel amplitude (Table 1). Thus, the NPA concentration selected was sufficient to produce a substantial effect on channel gating and had no effect on single-channel conductance. Further, we were able to attribute the decrease in Po to an ∼100% increase in the mean closed time, and an ∼50% decrease in mean open time. Next, we examined in closer detail the mechanism by which these kinetic effects arose.
TABLE 1.
Effects of NPA on average kinetic properties of individual 2A receptors
| Amplitude | Po | MCT | MOT | n | Duration | Events × 106 | |
|---|---|---|---|---|---|---|---|
| pA | ms | ms | min | ||||
| CTR | 10.9 ± 0.8 | 0.53 ± 0.04 | 4.7 ± 0.6 | 5.4 ± 0.7 | 5 | 172 | 2.0 |
| NPA | 10 ± 0.9 | 0.2 ± 0.3* | 9.8 ± 0.8* | 2.5 ± 0.4* | 6 | 117 | 1.0 |
| %Change | −62 | +109 | −54 |
MCT, mean closed time.
P < 0.01, Student’s t test.
NPA-Bound Receptors Had More Frequent Long-Closures and Short-Openings.
The gating mechanism of the 2A-type NMDA receptors has been well characterized kinetically. In the conditions employed in this study, the steady-state gating reaction consists of transitions between five closed and two open states and occasional gating mode shifts. To determine how NPA increased mean closed time and decreased MOT we examined the distribution of closed and open events present in our single-channel records.
Based on a log likelihood criterion, all the one-channel records we obtained with NPA (n = 6) were well described with five closed components (E1 – E5), as we had reported previously for 2A receptors (Kussius et al., 2009). This result indicates that NPA binding did not produce additional closed conformations. Instead, it altered specific closed component areas: E3 and E5 increased ∼2-fold and E4 increased ∼5-fold, at the expense of a 2-fold decrease in the short E1 component area. In addition, the duration of E3 increased 1.6-fold (Fig. 3, A and B). Generally, the short closed components E1–E3 encompass the brief closed events within bursts, and occur along the activation pathway. Therefore, these results strongly imply that NPA can influence receptor activation. Notably, NPA did not change the time constant of the longest closed component (E5), which encompasses the longest desensitized events (Fig. 3, A and B), but increased its area, and the increased abundance of desensitized events was accompanied by ∼50% shorter bursts (0.9 seconds versus 0.4 seconds). Note, however, that the observed increase in the E5 area does not necessarily imply that NPA affected desensitized state(s). Desensitization will occur more frequently simply if the occupancies of adjacent states increase, as will be discussed later (Fig. 4A).
Fig. 3.

NPA increased the areas of specific closed components. (A) Closed intervals observed in two records obtained from N1/N2A receptors in the absence (CTR, 416,413 events) and presence of NPA (4 mM, 1,347,473 events). Probability density functions (gray lines) were calculated by fitting kinetic 5C4O state models (see Fig. 4) to the displayed data; dark lines represent individual exponential components; their time constants [τ, milliseconds (ms)] and areas (a, %) are given as insets. (B) Summary of closed time constants and areas in the two conditions (CTR, n = 5; NPA, n = 6). Average values for CTR are given below each component (in ms). *P < 0.05 (Student’s t test).
In contrast to the specific and robust effects described above for closed events, NPA binding had no significant effects on the durations of any of the four open components. It caused a 3-fold increase in the frequency of the shortest open component, leading to an overall decrease in MOT (Fig. 3, C and D). During activation, NMDA receptors visit two types of open states: first a short ∼0.2 millisecond state, and then a longer state whose duration defines the mode of opening: 2 milliseconds for low, 6 milliseconds for medium, and 11 milliseconds for high modes, respectively (Popescu and Auerbach, 2003). Since neither the durations nor the relative areas of the low, medium, or high open states changed with NPA (Fig. 3D) we conclude that NPA binding did not affect the receptor’s modal transitions. Rather, overall shorter mean open durations reflect a propensity to dwell more often in the shorter of the two open states, regardless of mode.
Together with the observation that NPA did not alter channel conductance, these results exclude the possibility that NPA reduces NMDA receptor currents by blocking the pore or by lengthening desensitized events, and strongly suggest NPA as an allosteric modulator, which primarily affects the NMDA receptor activation.
Kinetic Models of NPA Actions on 2A Receptors.
To interpret the kinetic changes described above, we used an established 5C2O kinetic scheme to fit the sequence of events detected in each single-channel record and averaged the optimized rate constants within each data set. Although this model is a simplification of the complex rearrangements that occur during NMDA receptor gating, it has been shown previously to reproduce accurately key features of its microscopic and macroscopic behaviors (Popescu et al., 2004; Zhang et al., 2008; Kussius et al., 2009). The fitting results showed that NPA significantly changed only three of the 12 rate constants included in the model: kC1-C2, kO1-C1, and kC1-O1. Of these three rate constants, kC1-C2 and kO1-C1 increased 1.4- and 2-fold, respectively, while kC1-O1 decreased 2-fold (Fig. 4A). Thus, this mechanism explains the decrease in current as increased occupancies of states C3 and C2, which reside along the activation pathway (Fig. 4B). Next, we used the reaction mechanisms deduced for CTR and NPA conditions to compare the relative free-energy fluctuations experienced by NMDA receptors in the absence and presence of NPA. We aligned the two profiles arbitrarily at the free-energy level of the long open state O2, based on the observation that openings were of similar durations in both conditions. According to this representation, when NPA is present, albeit at subsaturating concentrations, the free energy barrier to receptor activation is higher, due to more stable preopen states (Fig. 4C). In other words, when NPA is present receptors require more energy to open, and because they dwell longer in preopen states they also have more chances to desensitize.
The description above presents an average picture of receptor behavior in the presence of subsaturating concentrations of NPA and reflects a dynamic mixture of NPA-free and NPA-bound receptors. To more precisely describe the gating of NPA-bound receptors, we expanded the 5C2O model to contain two tiers, one each for NPA-free (top) and NPA-bound (bottom) states. As a first approximation, we connected the tiers through just one NPA association/dissociation step, which we positioned in turn between each of the like-states along the activation pathway. Essentially, the modeling was conducted as described previously (Amico-Ruvio et al., 2012). Along the NPA-free arm, we fixed all rate constants to the values obtained for the CTR data set; whereas along the NPA-bound arm, we fixed only the rate constants that were not significantly changed in the NPA data, to same values as the CTR arm (Fig. 4A). We fitted separately each of the five-tiered models, which differed in the location of the connecting step (C3, C2, C1, O1, and O2), to individual single-channel records in the NPA data set. We observed that the NPA dissociation rate constants estimated with these models fell into two categories: they were slow when tiers were connected through C3 to C2, and they were ∼5-fold faster when tiers were connected C1 through O2. The models with single connection at different states were ranked using a LL criterion (O1>O2>C1>C2>C3). The O1 connected model returned the highest LL (Fig. 5A), which is 41 units larger than that of the second-ranked model, where the connection was at O2. These results suggested that the NPA binding may be state-dependent, with binding and dissociation occurring preferentially from open states. Because this model does not allow NPA binding to closed states, it predicts that the receptor should be insensitive to NPA, if the drug is applied just before but not during stimulation with glutamate.
Fig. 5.

NPA binding kinetics. (A) Tiered model was used to represent the NPA-free (upper) and NPA-bound receptors (lower). Transitions between arms were allowed only between the O1 states. This model was fitted to data obtained at 4 mM NPA (n = 6), and yielded the highest log likelihood. The rate constants (dark color) were fixed to the CTR values obtained as shown in Fig. 4, whereas the rate constants in red were allowed to vary. The association rate constant (kon) and equilibrium dissociation constant (KD) are indicated. The glutamate binding steps used for simulation are shown (gray). (B) Macroscopic responses to a long (5-second) pulse of 1 mM Glu (black line) were simulated, and were compared with whole-cell currents in the absence (black) or presence (red) of 4 mM NPA preapplication (5 seconds).
To test this prediction, we measured glutamate responses from cells that were perfused with NPA-containing solution (4 mM) prior to glutamate application, but not during glutamate exposure. Results showed that NPA reduced NMDA receptor whole-cell currents even when applied exclusively before glutamate (Fig. 5B, whole cell), thus strongly suggesting that: 1) NPA can bind to resting, glutamate-free NMDA receptors, and 2) that NPA remains bound and can encumber glutamate-elicited receptor activation even after excess NPA was washed out. Similarly, our results presented in Fig. 1 show that NPA can also bind to actively gating receptors, since NPA application inhibits steady-state currents produced by saturating concentrations of glutamate.
To gather all this information into a comprehensive model we postulated a mechanism where NPA can bind and dissociate from receptors at any point during gating but the kinetics of binding and dissociation will be different for each state. Based on the results presented above we aimed to limit the number of free parameters computed during statistical fittings, by further proposing that: 1) receptor conformations can be assigned to only two NPA affinity classes: low or high; 2) the change in NPA affinity occurs simultaneously with the C2–C1 transition; and 3) it is primarily due to an ∼5-fold change in dissociation rate. With these assumptions and after fixing NPA-invariant rates to CTR values as described for the previous model, we fitted this extended model individually to single-channel records obtained with NPA. The resulting rate constants are given in Fig. 6A.
Fig. 6.

Kinetic mechanism of N1/N2A inhibition by NPA. (A) Tiered model with transitions allowed at all states except C4 and C5. Rate constants shown in blue are allowed to vary. The on-rates for all closed states were fixed for the values shown, while the off-rates from C1, O1, and O2 (blue) were scaled as 5-fold of those from C2 and C3 (green). This model was fitted to data obtained at 4 mM NPA (n = 6) and also used for simulations. The glutamate binding steps used for simulation are shown (gray). (B) Macroscopic responses to a long (5-second) pulse of 1 mM Glu (black line) were simulated, and agreed well with whole-cell currents as shown in Fig. 5B. (C) The simulated dose-inhibition curve (red) was compared with the whole cell data (IC50 is expressed as 95% confidence interval). Hill slope was 1.0 for simulations and 1.2 for experimental traces.
Experimental Validation for the Proposed Mechanism of NPA Action.
Next, we asked whether the model in Fig. 6A can predict correctly the observed inhibition of glutamate-elicited currents when applied to active receptors (Fig. 1) or when applied to resting receptors (Fig. 5B). The model replicated faithfully the inhibition of steady-state currents illustrated in Fig. 1, and predicted a dose-response curve that was indistinguishable from that calculated from experimentally recorded whole-cell currents (1.5 mM versus 1.9 ± 0.8 mM) (Fig. 6C). Further, currents simulated from NMDA receptor pre-exposed to NPA also reproduced successfully the macroscopic behaviors observed experimentally (Fig. 5B). Based on these results, we propose that a tiered model that includes two NPA affinities (Fig. 6A), although derived on the basis of several simplifying assumptions, captures the necessary detail to account for experimentally observed single-channel and macroscopic behaviors. Last, we used this model to predict how NPA can affect responses from synaptic and extra-synaptic receptors.
Predicted NPA Effects on Synaptic and Extrasynaptic Receptors.
We evaluated the effect of NPA on NMDA receptor synaptic responses by comparing traces simulated with the CTR and the tiered NPA model in Fig. 6B following brief (1 millisecond) pulses of glutamate (1 mM). The resulting traces showed that NPA (4 mM) would inhibit synaptic-like responses from N2A-containing receptors by decreasing the peak current amplitude by 2-fold and also by decreasing the deactivation time ∼2-fold (400 milliseconds versus 200 milliseconds). Together these changes would produce ∼80% decrease in the total charge transfer in response to a single synaptic event (Fig. 7A).
Fig. 7.

Predicted NPA effects on synaptic and extrasynaptic N1/N2A macroscopic responses. (A) Simulations of macroscopic responses to a brief (1-millisecond) pulse of 1 mM Glu (black arrow) (left), simulated traces are normalized to peak and are overlaid for comparison of deactivation time course (right). Red arrow indicates the peak of NMDA receptor response in the presence of 4 mM NPA. (B) Macroscopic responses to a long (5-second) pulse of 1 mM Glu (black line) were simulated with the models in Fig. 4B, and were recorded as whole-cell currents in the absence (black) or presence (red) of 4 mM ambient NPA (right); insets show the same traces normalized to peak for comparison of desensitization time course (left); and normalized to steady-state level for comparison of deactivation time course (right).
We mimicked the effects of NPA on extra-synaptic receptors with the tiered model in Fig. 6B by starting all receptors in glutamate-free states (C0) and setting the NPA concentration to 4 mM. Activation was initiated by switching the glutamate concentration from zero to 0.1 mM, after which receptors were allowed to equilibrate for 5 seconds across all 18 states. The results showed that 4 mM NPA would attenuate both the peak and the steady state currents and that the effect would be slightly larger on the steady state current (Fig. 7B, simulation), but the time course for macroscopic desensitization would be largely unchanged (CTR versus NPA: 2.5 seconds versus 2.0 seconds) (Fig. 7B, simulation, insert). Also, the results predicted ∼2-fold faster deactivation in the presence of 4 mM NPA (CTR versus NPA: 430 milliseconds versus 190 milliseconds) (Fig. 7B, simulation, insert). These results matched closely the whole-cell currents recorded with a similar protocol: for desensitization, the time constants for receptors in the presence and presence of 4 mM NPA are 1.1 ± 0.1 second and 1.2 ± 0.1 second (P = 0.54), respectively; for deactivation, the time constants are 720 ± 50 millisecond and 350 ± 20 millisecond, respectively (P < 0.01) (Fig. 7B). Based on all the results presented we suggest that NPA would inhibit substantially both synaptic and extrasynaptic N2A receptors, with a larger effect on responses to brief synaptic-like stimuli.
Discussion
NMDA receptor hyperactivity has been associated with a variety of central nervous system disorders (Rothman and Olney, 1986; Lipton and Rosenberg, 1994; Metman et al., 2000). Despite efforts to design effective NMDA receptor antagonists, these often failed in clinical trials due to intolerable psychotic effects of completely blocking NMDA receptor signaling (Albers et al., 1999; Morris et al., 1999; Davis et al., 2000). One approach to circumvent this hazard would be to use antagonists of NMDA receptors that are isoform-specific (Ogden and Traynelis, 2011). However, among the clinically evaluated compounds that can presently distinguish among N2 subunits most are 2B receptor selective. In contrast, recently discovered NPA and related compounds have a much wider variety of selectivity among N2 subtypes. Thus, these compounds may serve as valuable tools for studying the functions of different NMDA receptor subtypes and also in clinical applications (Costa et al., 2010). In this study, we delineated the mechanism of NPA inhibition at N2A receptors and inferred their effects on synaptic and extrasynaptic N2A receptors.
We measured the effects of subsaturating concentration of NPA on steady-state currents produced by N2A-type NMDA receptors. Cell-attached single-channel activity was recorded in the presence of saturating concentration of glutamate and glycine and in the virtual absence of extracellular modulators, thus isolating intrinsic effects of NPA on NMDA receptor activity. NPA (4 mM) decreased the Po of N2A receptors ∼2-fold without changing the channel conductance or the gating sequence, but rather by increasing the fraction of longer closures and short opening (Fig. 3). These findings represent definitive proof that NPA modulates NMDA receptor activity with an allosteric mechanism. Fitting the single-channel data to the multistep kinetic model previously deduced for N2A receptors, we found that NPA altered neither the number nor the sequence of the kinetic states considered in the model, but changed significantly a subset of rate constants (Fig. 4A). This result indicated that NPA reduced the gating kinetics of N2A receptors by stabilizing pre-open states visited by receptors during activation (Fig. 4, B and C). Overall, these changes led to increased occupancy of closed states, thus causing a redistribution of receptors among the five closed states considered. We found that in the absence of proton and zinc inhibition and the absence of block by divalent cations, 4 mM NPA caused ∼70% inhibition of N2A currents. These results show that NPA has intrinsic allosteric effects on NMDA receptor gating. In addition, because NPA increased the occupancy of closed states the expectation is that it will also change the apparent sensitivity of N2A receptors to coincident noncompeting ligands, such as protons, polyamines, zinc, or other modulators that work by increasing the relative occupancies of closed, pre-open states. We anticipate that this effect will be reciprocal, and the extent of each interaction will be determined by the relative magnitude of the free-energy change enforced by the contemporaneous ligands.
Further, we determined that NPA binding can occur both before and after glutamate binding (Figs. 1 and 5B). Based on this novel observation, we expanded the minimal kinetic model considered initially (Fig. 4A) to include transitions between NPA-free and NPA-bound states (Fig. 7B). By fitting this model to single-channel data we determined that the change in NPA affinity between closed and open states is ∼5-fold, with NPA dissociation rate being 5-fold faster from open relative to closed receptors. However, given that this state-dependency is relatively weak, with the change in free-energy being on the same scale with those previously reported for other NMDA receptor modulators (Amico-Ruvio et al., 2011, 2012), and also with the difference in free energy between N2A and N2B receptors (Popescu et al., 2004; Amico-Ruvio and Popescu, 2010), we anticipate that the magnitude of the NPA effects will vary substantially with experimental conditions that include such modulators and will also vary with the NMDA receptor isoform considered.
This kinetic model, taken together with previously published kinetic phenotypes of NMDA receptors that carried structure-based mutations allow us to comment on the possible location of NPA-binding on N2A receptors. The structural determinants of NPA binding remain unknown. However structure-activity analyses that predate this work revealed that the NPA-binding site on NMDA receptors can accept molecules that are fairly large, geometrically flat and hydrophobic, and have a negatively charged group (Costa et al., 2012). Conversely, functional measurements combined with NMDA receptor mutagenesis indicated that the NPA effects on NMDA receptors are independent of the receptor’s N-terminal domain but require the N-terminal segment of the glutamate-binding domain (S1). These observations and similarities with known properties of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor modulators, which strengthen the interface between LBDs of separate subunits, led to the hypothesis that the NPA binding site may be located at the interface between LBDs of N1 and N2 subunits. We have recently demonstrated that crosslinking the N1–N2 dimer interface within the LBD of 2A containing receptors dramatically reduces NMDA receptor currents by raising an energy barrier early in the gating reaction and by destabilizing open states (Borschel et al., 2011). While the results we present here do not speak to the location of the NPA binding site, they indicate that the inhibitory mechanism of NPA, which is to stabilize closed receptors (this work), is different from that of mutations that strengthen the interface between LBD of N1 and N2, which is to prevent receptors from opening and to destabilize open states (Borschel et al., 2011).
Similar considerations allow us to propose that NPA has an inhibitory mechanism that is distinct from other allosteric modulators that rely for function on LBDs, such as pregnanolone sulfate (PAS), (E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)-benzoic acid (QNZ46), 3-chloro-4-fluoro-N-[(4-{[2-(phenylcarbonyl)hydrazino]carbonyl}phenyl)methyl]benzene-sulfonamide (TCN201), and 4-(5-(4-bromophenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid (DQP1105) (Bettini et al., 2010; Mosley et al., 2010; Acker et al., 2011; Cameron et al., 2012). Unlike PAS, which primarily increases the occupancy of the longest closed state (desensitized state) by prolonging its duration (Kussius et al., 2009), NPA mainly affected the short closed states located along the activation pathway. In addition, macroscopic data on 2D receptors show that QNZ46 and DQP1105 primarily decrease the equilibrium currents of NMDA receptor under prolonged glutamate stimulus, but does not affect the peak amplitudes significantly (Acker et al., 2011; Hansen and Traynelis, 2011). Assuming the action on N2A receptors is similar in mechanism, the response of QNZ46- or DQP1105-bound NMDA receptors to prolonged stimulus would be quite distinct from that of NPA-bound NMDA receptors, as shown in our study. These considerations further support the argument that the binding site of NPA is different from that of PAS, QNZ46, or DQP1105. More importantly, they indicate that even for inhibitors that target the LBD, separate binding sites can produce distinct inhibitory mechanisms.
At synaptic sites, the NMDA receptors experience controlled exposure to brief glutamate pulses, which have been estimated to reach 1 mM and to last 1 millisecond (Clements, 1996). In contrast, at extrasynaptic sites, and in particular during pathologic conditions, including stroke and certain neurodegenerative disorders, NMDA receptors are exposed to longer lasting glutamate transients. To anticipate how NPA would influence responses from N2A receptors under phasic and tonic glutamate exposure we simulated responses with protocols designed to mimic these. Based on the results, we suggest that: 1) NPA will reduce both the peak amplitude and the deactivation time-constant of synaptic responses mediated by N2A receptors (Fig. 7A), thus considerably decreasing synaptic charge transfer and the time window for synaptic integration; and 2) NPA will reduce the equilibrium response of extrasynaptic or tonically activated N2A receptors (Fig. 6B), thus potentially alleviating the deleterious effects of extrasynaptic calcium influx. Therefore, the mechanism of NPA inhibition we propose here suggests that NPA can have potentially salutary effects in pathologies associated with increased synaptic transmission, as for example in epilepsy, and may also have a neuroprotective effect in pathologies associated with excessive extracellular glutamate, such as ischemia.
In summary, our results indicate that NPA has intrinsic inhibitory activity and it inhibits NMDA receptors with an allosteric mechanism. The kinetic model we developed postulates that NPA binding stabilized closed receptor states that are populated during activation, thus leading to the accumulation of receptors along the activation pathway. This mechanism is clearly distinct from that of other allosteric inhibitors characterized so far, such as zinc, ifenprodil, and pregnanolone sulfate (Kussius et al., 2009; Amico-Ruvio et al., 2011, 2012). In addition, the mechanism we propose predicts that NPA will affect in specific ways responses from synaptic and extrasynaptic N2A-containing receptors, thus providing necessary information in the rational design of therapeutic applications for this class of NMDA receptor inhibitors.
Acknowledgments
The authors thank Eileen M. Kasperek for assistance with molecular biology and cell culture.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CTR
control
- DMSO
dimethylsulfoxide
- DQP1105
4-(5-(4-bromophenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- GluN
NMDA family of glutamate receptor subunits
- HEK
human embryonic kidney
- HEPBS
N-(2-hydroxyethyl)piperazine-N′(4-butanesulfonic acid)
- LBD
ligand-binding domain
- LL
log10 likelihood
- MOT
mean open time
- NMDA
N-methyl-d-aspartate
- NPA
2-naphthoic acid
- PAS
pregnanolone sulfate
- Po
open probability
- TCN201
3-chloro-4-fluoro-N-[(4-{[2-(phenylcarbonyl)hydrazino]carbonyl}phenyl)methyl]benzene-sulfonamide
- QNZ46
(E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)-benzoic acid
Authorship Contributions
Participated in research design: Yu, Popescu.
Conducted experiments: Yu.
Performed data analysis: Yu.
Wrote or contributed to the writing of the manuscript: Yu, Popescu.
Footnotes
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS052669].
References
- Acker T, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, et al. (2011) Mechanism for non-competitive inhibition by novel GluN2C/D NMDA receptor subunit-selective modulators. Mol Pharmacol 80:782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers GW, Clark WM, Atkinson RP, Madden K, Data JL, Whitehouse MJ. (1999) Dose escalation study of the NMDA glycine-site antagonist licostinel in acute ischemic stroke. Stroke 30:508–513 [DOI] [PubMed] [Google Scholar]
- Amico-Ruvio SA, Popescu GK. (2010) Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys J 98:1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico-Ruvio SA, Murthy SE, Smith TP, Popescu GK. (2011) Zinc effects on NMDA receptor gating kinetics. Biophys J 100:1910–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico-Ruvio SA, Paganelli MA, Myers JM, Popescu GK. (2012) Ifenprodil effects on GluN2B-containing glutamate receptors. Mol Pharmacol 82:1074–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Traynelis SF. (2003) Activation of NR1/NR2B NMDA receptors. Nat Neurosci 6:144–152 [DOI] [PubMed] [Google Scholar]
- Bell KF, Hardingham GE. (2011) The influence of synaptic activity on neuronal health. Curr Opin Neurobiol 21:299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, et al. (2010) Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther 335:636–644 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Morris RG. (2003) Introduction. Long-term potentiation and structure of the issue. Philos Trans R Soc Lond B Biol Sci 358:607–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borschel WF, Murthy SE, Kasperek EM, Popescu GK. (2011) NMDA receptor activation requires remodelling of intersubunit contacts within ligand-binding heterodimers. Nat Commun 2:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Misra C, Mok MH, Mishina M, Cull-Candy SG. (2003) NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci 23:4958–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron K, Bartle E, Roark R, Fanelli D, Pham M, Pollard B, Borkowski B, Rhoads S, Kim J, Rocha M, Kahlson M, Kangala M, Gentile L. (2012) Neurosteroid binding to the amino terminal and glutamate binding domains of ionotropic glutamate receptors. Steroids 77:774–779 [DOI] [PubMed] [Google Scholar]
- Clements JD. (1996) Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci 19:163–171 [DOI] [PubMed] [Google Scholar]
- Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Laube B, Jane DE, Monaghan DT. (2012) Structure-activity relationships for allosteric NMDA receptor inhibitors based on 2-naphthoic acid. Neuropharmacology 62:1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Skifter DA, Jane DE, Monaghan DT. (2010) A novel family of negative and positive allosteric modulators of NMDA receptors. J Pharmacol Exp Ther 335:614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, Norris J. (2000) Selfotel in acute ischemic stroke : possible neurotoxic effects of an NMDA antagonist. Stroke 31:347–354 [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563:345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Traynelis SF. (2011) Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors. J Neurosci 31:3650–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. (2002) Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta 1600:148–153 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. (2003) The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26:81–89 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414 [DOI] [PubMed] [Google Scholar]
- Kaufman AM, Milnerwood AJ, Sepers MD, Coquinco A, She K, Wang L, Lee H, Craig AM, Cynader M, Raymond LA. (2012) Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J Neurosci 32:3992–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ and Horn R (1991) Electrophysiology and microinjection; discrimination of kinetic models of ion channel gating, in Methods in Neurosciences Vol 4 (Conn MP ed) pp 428–456, Academic Press, San Diego, CA. [Google Scholar]
- Kussius CL, Kaur N, Popescu GK. (2009) Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci 29:6819–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Bading H. (2009) Synaptic activity-mediated suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition. J Neurosci 29:4420–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA, Puddifoot C, Habel M, Wyllie DJ, Ikonomidou C, et al. (2010) Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci 30:2623–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622 [DOI] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze TSh, Feng B, Monaghan DT, Krishtal OA. (2004) Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’ afterburst EPSC in rat hippocampus. J Physiol 558:451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metman LV, Konitsiotis S, Chase TN. (2000) Pathophysiology of motor response complications in Parkinson’s disease: hypotheses on the why, where, and what. Mov Disord 15:3–8 [DOI] [PubMed] [Google Scholar]
- Momiyama A. (2000) Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. J Physiol 523:621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF, The Selfotel Investigators (1999) Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. J Neurosurg 91:737–743 [DOI] [PubMed] [Google Scholar]
- Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. (2010) Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J Med Chem 53:5476–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW. (2006) Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol 6:53–60 [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465 [DOI] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. (2011) New advances in NMDA receptor pharmacology. Trends Pharmacol Sci 32:726–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. (1997) High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 17:5711–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, et al. (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11:476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L, Stephenson FA, Wenthold RJ. (2010) Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167:68–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G. (2005) Mechanism-based targeting of NMDA receptor functions. Cell Mol Life Sci 62:2100–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G, Auerbach A. (2003) Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci 6:476–483 [DOI] [PubMed] [Google Scholar]
- Popescu G, Auerbach A. (2004) The NMDA receptor gating machine: lessons from single channels. Neuroscientist 10:192–198 [DOI] [PubMed] [Google Scholar]
- Popescu G, Robert A, Howe JR, Auerbach A. (2004) Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 430:790–793 [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. (1997) Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci 264:375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. (2000) A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophys J 79:1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. (1986) Glutamate and the pathophysiology of hypoxic—ischemic brain damage. Ann Neurol 19:105–111 [DOI] [PubMed] [Google Scholar]
- Schorge S, Elenes S, Colquhoun D. (2005) Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J Physiol 569:395–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark DT, Bazan NG. (2011) Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J Neurosci 31:13710–13721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. (1990) Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345:347–350 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Livesey MR, Hardingham GE. (2013) Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology DOI: 10.1016/j.neuropharm.2013.01.016 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Howe JR, Popescu GK. (2008) Distinct gating modes determine the biphasic relaxation of NMDA receptor currents. Nat Neurosci 11:1373–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]