

Abstract
Coronary heart disease (CHD) is the leading cause of death worldwide. Mitochondrial genetic determinant for the development of CHD remains poorly explored. We report there the clinical, genetic, molecular and biochemical characterization of a four-generation Chinese family with maternally inherited CHD. Thirteen of 32 adult members in this family exhibited variable severity and age-at-onset of CHD. Mutational analysis of their mitochondrial genomes identified the tRNAThr 15927G>A mutation belonging to the Eastern Asian haplogroup B5. The anticipated destabilization of a highly conserved base-pairing (28C-42G) by the 15927G>A mutation affects structure and function of tRNAThr. Northern analysis revealed ≈80% decrease in the steady-state level of tRNAThr in the mutant cell lines carrying the 15927G>A mutation. The 15927G>A mutation changed the conformation of tRNAThr, as suggested by slower electrophoretic mobility of mutated tRNA with respect to the wild-type molecule. In addition, ∼39% reduction in aminoacylated efficiency of tRNAThr was observed in mutant cells derived from this Chinese family. An in vivo mitochondrial protein labeling analysis showed ∼53% reduction in the rate of mitochondrial translation in mutant cells. The impaired mitochondrial protein synthesis leads to defects in overall respiratory capacity or malate/glutamate-promoted respiration or succinate/glycerol-3-phosphate-promoted respiration, or N,N,N′,N′-tetramethyl-pphenylenediamine/ascorbate-promoted respiration in mutant cells. An increasing production of reactive oxygen species was observed in the mutant cells carrying the 15927G>A mutation. These results provide the direct evidence that the tRNAThr 15927G>A mutation is associated with CHD. Our findings may provide new insights into pathophysiology and intervention targets of this disorder.
INTRODUCTION
Cardiovascular diseases are the biggest cause of deaths worldwide. Cardiovascular diseases include high blood pressure, coronary heart disease (CHD), cardiomyopathy, heart failure and stroke. In particular, CHD annually results in 502 000 deaths in USA, and more than 700 000 deaths in China (1,2). CHD is a common complex disorder, which can be caused by single gene or multifactorial conditions resulting from interactions between environmental and inherited risk factors (3–5). Efforts to identify genetic determinants of CHD have been directed primarily on nuclear genomes (6). Genome-wide association studies in the population of European and Asian ancestries have identified several genetic loci that are associated with the risk of CHD (7–10). Especially, a SNP, rs6903956 in C6orf105 on chromosome 6p24 is associated with susceptibility to CHD in the Chinese population (11). However, the mitochondrial genetic determinant for the development of CHD remains poorly explored. Mitochondrial dysfunction has been implicated to contribute to a wide range of cardiovascular disorders such as cardiomyopathy and hypertension (12–15). In particular, mutations in mitochondrial DNA (mtDNA) have been associated with hypertension (15,16). Of these, the 4291T>C mutation in the tRNAIle gene has been associated with a cluster of metabolic defects, including hypertension, hypercholesterolemia and hypomagnesemia (17). Furthermore, the tRNAIle 4263A>G, tRNAMet 4435A>G mutations and the 4401A>G mutation in the junction of tRNAMet and tRNAGln genes have been associated with hypertension in several Chinese pedigrees (18–20).
To investigate whether mitochondrial genetic defects contribute to the pathogenesis of CHD, we have initiated a systematic and extended mutational screening of mtDNA in a large cohort of CHD subjects in the Chinese population. One four-generation Han Chinese family (Fig. 1) with maternally transmitted CHD was ascertained at the Cardiac Clinic of Anzhen Hospital, Beijing, China. Thirteen of the 32 adult members in this family exhibited the variable severity and age-at-onset of CHD. Mutational analysis of their mitochondrial genomes identified the tRNAThr 15927G>A mutation belonging to the Eastern Asian haplogroup B5 in this Chinese family (21). This 15927G>A mutation is localized at the highly conserved base in the anticodon stem, corresponding to conventional position 42 of the tRNAThr (22,23). Thus, it is anticipated that the 15927G>A mutation alters the structure and function of tRNAThr, thereby causing the mitochondrial dysfunction necessary for the development of CHD. Functional significance of the 15927G>A mutation was further evaluated by examining the steady-state levels and aminoacylation capacities of mitochondrial tRNAs including tRNAThr by using lymphoblastoid cell lines derived from five affected matrilineal relatives carrying the 15927G>A mutation and from three control individuals lacking the mtDNA mutation. These cell lines were further assessed for the effects of the 15927G>A mutation on mitochondrial protein synthesis, endogenous respiration and substrate-dependent respiration as well as the production of reactive oxygen species (ROS).
Figure 1.

A Han Chinese pedigree with CHD. Affected individuals are indicated by filled symbols. An arrowhead denotes proband (BJH15-III-7).
RESULTS
Clinical presentation
The proband (BJH15-III-7) developed CHD at the age of 40 years. He presented to the Cardiology Clinic of Anzhen Hospital for further clinical and biochemical evaluations at the age of 55 years. He was diagnosed as having ischemia (extent of CAD based on extent of angiographic coronary was narrowed >85%), hypertension (160/95 mm Hg) and hypercholesterolemia [total cholesterol (TC) = 220 mg/dl and low-density lipoprotein cholesterol (LDL-C)>160 mg/dl). He did not have other clinical abnormalities, including diabetes mellitus, vision and hearing impairments, and renal and neurological disorders. As shown in Figure 1 and Table 1, a clinical and biochemical evaluation of other 31 members in this family revealed a high prevalence of CHD, hypertension and hyperlipidemia. Of other 12 matrilineal relatives, 7 members suffered from only CHD, 1 subject (III-6) had CHD with hypercholesterolemia and 1 individual (II-3) exhibited CHD with hypertension and hypercholesterolemia. All affected fathers with CHD, except the subject II-3 who married with the affected subject II-4, never transmitted the trait to their offsprings, whereas all affected mothers transmitted the trait to their offsprings. These features are the maternal transmissions of CHD, suggesting the mitochondrial involvement. In addition, the individual II-4, who was the wife of II-3 but not a sibling of II-2 an II-6, suffered from CHD at the age of 70 years. A corresponding mitochondrial DNA mutation by the subject II-4 would be introduced independently into the pedigree, causing CHD of subjects III-12 and III-13. Thus, the subjects II-4, III-12, III-13, IV-7 and IV-8 represented a different maternal pedigree.
Table 1.
Summary of clinical and biochemical data for some members in one Han Chinese Pedigree (BJH15)
| Subjects | Gender | Age of test (years) | Age of onset (years) | ECG | Ectent of CAD narrow (%) | Systolic BP (mmHg) | Diastolic BP (mmHg) | TC (mg/dl) | LDL (mg/dl) | Smoker |
|---|---|---|---|---|---|---|---|---|---|---|
| II-2 | F | 78 | N | 115 | 85 | 175 | 110 | No | ||
| II-3 | M | 60 | 60 | ischemia | 50 | 150 | 95 | 220 | 145 | Yes |
| II-4 | F | 76 | 70 | ischemia | 50 | 135 | 85 | 190 | 100 | No |
| II-5 | M | 80 | N | 150 | 90 | 230 | 140 | No | ||
| II-6 | F | 76 | 62 | ischemia | 50 | 138 | 85 | 170 | 98 | No |
| III-1 | M | 62 | N | 130 | 90 | 198 | 115 | Yes | ||
| III-2 | F | 59 | 53 | ischemia | 50 | 140 | 80 | 178 | 96 | No |
| III-3 | M | 63 | N | 150 | 95 | 188 | 100 | Yes | ||
| III-4 | F | 58 | N | 135 | 85 | 170 | 92 | No | ||
| III-5 | M | 60 | N | 125 | 75 | 175 | 100 | No | ||
| III-6 | F | 56 | 45 | ischemia | 55 | 140 | 85 | 210 | 140 | No |
| III-7 | M | 55 | 40 | ischemia | 85 | 160 | 95 | 220 | 160 | Yes |
| III-8 | F | 53 | N | 135 | 80 | 250 | 160 | No | ||
| III-9 | M | 51 | 49 | ischemia | 50 | 125 | 80 | 180 | 86 | Yes |
| III-10 | F | 46 | N | 130 | 80 | 187 | 108 | No | ||
| III-11 | M | 55 | N | 135 | 75 | 190 | 110 | No | ||
| III-12 | F | 53 | 52 | ischemia | 65 | 140 | 80 | 170 | 95 | No |
| III-13 | M | 50 | 40 | ischemia | 70 | 135 | 80 | 180 | 110 | Yes |
| III-14 | F | 49 | N | 140 | 85 | 195 | 100 | No | ||
| III-15 | M | 50 | 45 | ischemia | 50 | 130 | 85 | 170 | 110 | Yes |
| III-16 | F | 50 | N | 145 | 90 | 185 | 105 | No | ||
| IV-1 | M | 40 | 35 | ischemia | 50 | 125 | 80 | 180 | 108 | No |
| IV-2 | M | 42 | 28 | ischemia | 55 | 125 | 70 | 190 | 110 | Yes |
| IV-3 | M | 38 | 30 | ischemia | 60 | 120 | 80 | 188 | 106 | No |
| IV-4 | F | 29 | N | 55 | 120 | 80 | 160 | 88 | No | |
| IV-5 | F | 26 | N | 120 | 80 | 140 | 80 | No | ||
| IV-6 | M | 32 | N | 120 | 75 | 160 | 96 | Yes | ||
| IV-7 | M | 26 | N | 125 | 80 | 160 | 108 | Yes | ||
| IV-8 | M | 22 | N | 120 | 75 | 175 | 100 | Yes | ||
| IV-9 | F | 24 | N | 110 | 65 | 180 | 110 | No | ||
| IV-10 | M | 25 | N | 125 | 75 | 168 | 110 | No |
F, female; M, male; N, electrocardiography (ECG) was normal; Ectent of CAD narrow (%) was determined by Coronary angiography; BP, blood pressure; TC, total cholesterol; LDL, lipoprotein cholesterol.
Mutational analysis of mitochondrial genome
Southern blot analysis showed no evidence of mitochondrial DNA deletion or duplication among 32 members of this family (data not shown). We then performed a PCR amplification of fragments spanning their mitochondrial genomes and subsequent DNA sequence analysis in six matrilineal relatives (II-2, II-6, III-2, III-7, III-9 and IV-3) who were offsprings of I-2, and subject II-4 with two of her offsprings (III-12 and III-13). As shown in Supplementary Material, Table, these subjects carried the identical tRNAThr 15927G>A mutation and distinct sets of polymorphisms (31 identical variants of each 43 variants between III-7 and III-12) belonging to the Eastern Asian haplogroup B5 on their maternal lineages (21,24). The 15927G>A mutation, as shown in Figure 2, locates at the fourth base in the anticodon stem, corresponding to conventional position 42 of the tRNAThr (22,23). A guanine at this position is a conserved base in sequenced threonine tRNA from bacteria to human mitochondria (22,23). It was anticipated that the 15927G>A mutation destabilizes a very conservative base pairing (28C-42G) on the anticodon stem of this tRNAThr, thus causing a failure in tRNA metabolism. To determine whether the 15927G>A mutation is homoplasmy in cells, the fragments spanning the tRNAThr gene were PCR amplified and subsequently digested with HpaII as the mutation disrupted the site for this restriction enzyme. As shown in Figure 2C, there was no detectable wild-type DNA in all-available matrilineal relatives, indicating that the 15927G>A mutation was present in homoplasmy in these matrilineal relatives but absent in other members in this family. In addition, allele frequency analysis showed that 3 of 80 genetically unrelated individuals with CHD and 2 of 262 Chinese control subjects who were free from CHD carried the 15927G>A mutation. Further genetic and clinical evaluation of 5 subjects (3 with CHD and 2 controls) carrying the mutation showed that only one Chinese family (BJH45) exhibited a maternal transmission of CHD (supplementary Material, Figure). Entire mtDNA sequence analysis of proband BJH45 III-2 was shown in the supplementary Material, Table.
Figure 2.
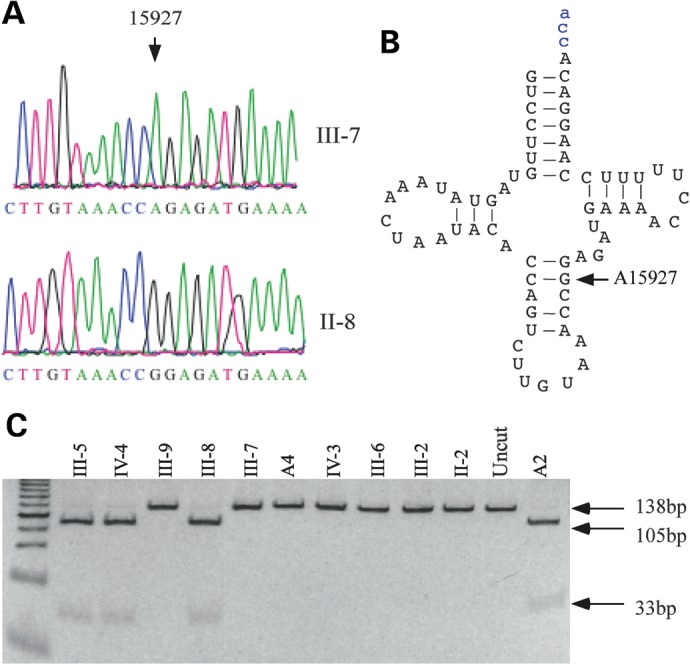
Identification and qualification of the 15927G>A mutation in the mitochondrial tRNAThr gene. (A) Partial sequence chromatograms of tRNAThr gene from an affected individual (III-7) and a married-in-control (II-8). An arrow indicates the location of the base changes at position 15927. (B) The location of the 15927G>A mutation in the mitochondrial tRNAThr. Cloverleaf structure of human mitochondrial tRNAThr is derived from Suzuki et al. (22). Arrow indicates the position of the 15927G>A mutation. (C) Quantification of 15927G>A mutation in the tRNAThr gene of mutants and controls derived from the Chinese families. PCR products amplified from total DNA isolated from whole blood of subjects were digested with HpaII and analyzed by electrophoresis in a 7% polyacrymide gel stained with ethidium bromide.
Marked decrease in the levels of mitochondrial tRNAThr
To examine whether the 15927G>A mutation affects the tRNAThr metabolism, we subjected mitochondrial RNAs from lymphoblastoid cell lines to Northern blots and hybridized them with digoxigenin (DIG)-labeled oligodeoxynucleotide probes for tRNAThr and four other tRNAs (23,25). As shown in Figure 3A, the amount of tRNAThr in five mutant cell lines carrying the 15927G>A mutation were markedly decreased, compared with those in three control cell lines lacking the mtDNA mutation. For comparison, the average levels of tRNAThr in various control and mutant cell lines were normalized to the average levels in the same cell lines for tRNALeu(CUN), tRNALys, tRNASer(AGY) and tRNAHis, respectively. As shown in Figure 3B, the average levels of tRNAThr in the five mutant cell lines were 20, 18.2, 18.2 and 20% of the average values of three controls after normalization to tRNALeu(CUN), tRNALys, tRNASer(AGY) and tRNAHis, respectively.
Figure 3.

Northern-blot analysis of mitochondrial tRNA. (A) Equal amounts (2 μg) of total mtRNA samples from the various cell lines were electrophoresed through a denaturing polyacrylamide gel, were electroblotted and were hybridized with DIG-labeled oligonucleotide probes specific for the tRNAThr, tRNALys, tRNALeu(CUN), tRNASer(AGY) and tRNAHis, respectively. (B) Quantification of mitochondrial tRNA levels. Average relative tRNAThr content per cell, normalized to the average content per cell of tRNALys, tRNALeu(CUN), tRNASer(AGY) and tRNAHis in cells derived from five affected subjects carrying the 15927G>A mutation and three Chinese controls (A6, IV-4 and III-8) lacking the mutation. The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent determinations of each tRNA content in each cell line and three determinations of the content of reference tRNA marker in each cell line. The error bars indicate two standard errors of the mean (SEM). P indicates the significance, according to the t-test, of the difference between mutant mean and control mean.
Altered aminoacylation of tRNAThr
The aminoacylation capacities of tRNAThr, tRNALeu(CUN), tRNALys, and tRNASer(AGY) in control and mutant cell lines were examined by the use of electrophoresis in an acid polyacrylamide/urea gel system to separate uncharged tRNA species from the corresponding charged tRNA, electroblotting and hybridizing with above tRNA probes (26). As shown in Figure 4, the upper band represented the charged tRNA, and the lower band was uncharged tRNA. Electrophoretic patterns showed that either charged or uncharged tRNAThr in cell lines carrying the 15927G>A mutation migrated much slower than those of cell lines lacking this mutation. However, there were no obvious differences in electrophoretic mobility of tRNALeu(CUN), tRNALys and tRNASer(AGY) between the cell lines carrying the 15927G>A mutation and cell lines lacking this mutation. Notably, the efficiencies of aminoacylated tRNAThr in these mutant cell lines reflected 39% reduction, ranged from 35 to 43%, relative to the average control values (P = 0.005). However, the levels of aminoacylation in tRNALeu(CUN), tRNALys and tRNASer(AGY) in mutant cell lines were comparable with those in the control cell lines.
Figure 4.

In vivo aminoacylation assay for mitochondrial tRNA. (A) Equal amounts (2 µg) of total mitochondrial RNA purified from various cell lines under acid conditions were treated with electrophoresis at 4°C through an acid (pH 5.1) 10% polyacrylamide /7 m urea gel, electroblotted onto a positively charged nylon membrane and hybridized with DIG-labeled oligonucleotide probes specific for mitochondrial tRNAThr. The blots were then stripped and rehybridized with DIG-labeled probes for tRNALys, tRNALeu(CUN) and tRNASer(AGY), respectively. (B) In vivo aminoacylated proportion of tRNAThr in the mutant and controls. The calculations were based on three independent determinations. The error bars indicate two SEs.
Mitochondrial protein synthesis defect
To examine whether the failure in tRNA metabolism caused by the 15927G>A mutation impairs mitochondrial translation, cell lines derived from five affected matrilineal relatives carrying the 15927G>A mutation and three controls were labeled for 30 min with [35S] methionine–[35S] cysteine in methionine-free regular DMEM medium in the presence of 100 µg/ml of emetine to inhibit cytosolic protein synthesis (27). Figure 5A shows typical electrophoretic patterns of the mitochondrial translation products of the mutant and control cell lines. Patterns of the mtDNA-encoded polypeptides of the cells carrying the 15927G>A mutation were qualitatively identical to those of the control cells, in terms of electrophoretic mobility of the various polypeptides. However, cell lines carrying the 15927G>A mutation trended to a decrease in the total rate of labeling of the mitochondrial translation products relative to those of the control cell lines. Figure 5B shows a quantification of the results of a large number of labeling experiments and electrophoretic runs, which were carried out using Image-Quant program analysis of appropriate exposures of the fluorograms and normalization to data obtained for the 143B.TK– sample. In fact, the overall rates of labeling of the mitochondrial translation products in the cell lines derived from five affected individuals carrying the 15927G>A mutation ranged from 23 to 69%, with an average of 47% (P = 0.005) relative to the mean value measured in the control cell lines.
Figure 5.

Mitochondrial translation assay. (A) Electrophoretic patterns of the mitochondrial translation products of lymphoblastoid cell lines and of 143B.TK– cells labeled for 30 min with [35S]methionine in the presence of 100 µg/ml of emetine. Samples containing equal amounts of total cellular protein (30 µg) were run in SDS/polyacrylamide gradient gels. COI, COII and COIII indicate subunits I, II and III of cytochrome c oxidase; ND1, ND2, ND3, ND4, ND4L, ND5 and ND6, subunits 1, 2, 3, 4, 4L, 5 and 6 of the respiratory chain reduced nicotinamide-adenine dinucleotide dehydrogenase; A6 and A8, subunits 6 and 8 of the H+-ATPase; and CYTb, apocytochromeb. (B) Quantification of the rates of the mitochondrial translation labeling. The rates of mitochondrial protein labeling, as detailed elsewhere (26), were expressed as percentages of the value for 143B.TK– in each gel, with error bars representing 2 SEMs. A total of three independent labeling experiments and three electrophoretic analyses of each labeled preparation were performed on cell lines. Graph details and symbols are explained in the legend to Figure 3.
Respiration deficiency
The endogenous respiration rates of cell lines derived from five affected individuals carrying the15927G>A mutation and three controls were measured by determining the O2 consumption rate in intact cells, as described previously (28). As shown in Figure 6A, the rate of total O2 consumption in the cell lines derived from five affected individuals varied from 59 to 79%, with an average of 67% (P = 0.001) relative to the mean value measured in the control cell lines.
Figure 6.

Respiration assays. (A) Average rates of endogenous O2 consumption per cell measured in different cell lines are shown, with error bars representing two SE. A total of four to six determinations were made on each of lymphoblastoid cell lines. (B) Polarographic analysis of O2 consumption in digitonin-permeabilized cells of the various cell lines using different substrates and inhibitors. The activities of the various components of the respiratory chain were investigated by measuring on 1 × 107 digitonin-permeabilized cells the respiration dependent on malate/glutamate, on succinate/G3P and on TMPD/ascorbate. A total of three to six determinations were made on each of the lymphoblastoid cell lines. Graph details and symbols are explained in the legend to Figure 3. mal/glu, malate/glutamate-dependent respiration; succ/G-3-P, succinate/G3P-dependent respiration; and asc/TMPD, TMPD/ascorbate-dependent respiration. Graph details and symbols are explained in the legend to Figure 3.
To investigate which of the enzyme complexes of the respiratory chain was affected in the mutant cell lines, O2 consumption measurements were carried out on digitonin-permeabilized cells, using different substrates and inhibitors (29). As shown in Figure 6B, in the cell lines derived from five affected individuals, the rate of malate/glutamate-driven respiration, which depends on the activities of NADH: ubiquinone oxidoreductase (Complex I), ubiquinol–cytochrome c reductase (Complex III) and cytochrome c oxidase (Complex IV), but usually reflects the rate-limiting activity of Complex I, was very significantly decreased, relative to the average rate in the three control cell lines, by 54–77% (65% on the average; P = 0.001). Similarly, the rate of succinate/glycerol-3-phosphate (G3P)-driven respiration, which depends on the activities of Complexes III and IV, but usually reflects the rate-limiting activity of Complex III, was significantly affected in the mutant cell lines, relative to the average rate in the control cell lines, by 62–81% (70% on the average; P = 0.0001). Furthermore, the rate of N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD)/ascorbate-driven respiration, which reflects the activity of Complex IV, exhibited a 63–76% reduction in Complex IV activity (69% on the average; P = 0.001) in the mutant cell lines relative to the average rate in the control cell lines.
ROS production increases
To investigate whether the mitochondrial dysfunction increases the production of ROS, we measured the levels of the ROS generation in the vital cells derived from five affected matrilineal relatives carrying the 15927G>A mutation and three control individuals lacking the mutation using flow cytometry under normal and H2O2 stimulation (30,31). Geometric mean intensity was recorded to measure the rate of ROS of each sample. The ratio of geometric mean intensity between unstimulated and stimulated with H2O2 in each cell line was calculated to delineate the reaction upon increasing level of ROS under oxidative stress. As shown in Figure 7, the levels of ROS generation in the lymphoblastoid cell lines derived from five affected individuals carrying the mtDNA mutation ranged from 117 and 123%, with an average 122% (P = 0.0001) of the mean value measured in the control cell lines.
Figure 7.

The ROS production assays. The rates of production in ROS from five affected matrilineal relatives and three control individuals were analyzed by BD-LSR II flow cytometer system with or without H2O2 stimulation. The relative ratio of intensity (stimulated versus unstimulated with H2O2) was calculated. The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Figure 3.
DISCUSSION
The present study provides the first direct evidence that mitochondrial genetic defect is associated with CHD. Adult matrilineal relatives in one Chinese kindred exhibited a syndrome including CHD, hypertension and hyperlipidemia. In particular, CHD is transmitted on the maternal lineage with a pattern, indicating mitochondrial inheritance. mtDNA analysis of their maternal lineage identified a homoplasmy 15927G>A mutation at a highly conserved nucleotide (G42) of tRNAThr, where the position is important for the stability and identity of tRNA (22). The anticipated destabilization of base-pairing (28C-42G) by the 15927G>A mutation may affect secondary structure and function of this tRNA, as in the cases of tRNAIle 4300A>G and tRNALeu(UUR) 3273T>C mutations (32,33). The 15927G>A mutation changed the conformation of tRNAThr, as suggested by slower electrophoretic mobility of mutated tRNA with respect to the wild-type molecule. However, the aminoacylation level of the tRNAThr was not impaired, but the steady-state level of the tRNA was reduced 44% in lymphoblastoid cell lines derived from the Chinese control subjects carrying the 15927G>A mutation (23). The mutant tRNAThr may be metabolically less stable and more subject to degradation, thereby lowering the level of the tRNA, as in the case of 3243A>G mutation in the tRNALeu(UUR) (25,34). However, an ∼70% decrease in the level of tRNA responsible for the significant respiratory defects was proposed as the threshold level that produces a clinical phenotype associated with a mitochondrial tRNA mutation (34,35). Thus, the relatively mild mitochondrial dysfunction caused by the 15927G>A mutation suggests that this mutation is by itself insufficient to produce a clinical phenotype (23).
In the present investigation, the tRNAThr carrying the 15927G>A mutation was charged to a lesser extent by the mitochondrial threonyl-tRNA synthetase, thereby altering aminoacylation. An ∼39% reduction in aminoacylated efficiency of tRNAThr in mutant cells derived from this Chinese family with CHD apparently aggravates a failure in tRNA metabolism associated with the 15927G>A mutation, thereby yielding an ∼80% decrease in the steady-state level of tRNAThr in mutant cell lines. This reduced level of tRNA is below the threshold level that leads to a clinical phenotype (34–36). A shortage of tRNAThr was responsible for ∼53% decrease in the rate of mitochondrial protein synthesis in the mutant cells, which is the proposed threshold level to support a normal respiratory phenotype (37). The impaired mitochondrial protein synthesis apparently leads to defects in overall respiratory capacity or malate/glutamate-promoted respiration or succinate/glycerol-3-phosphate-promoted respiration, or N,N,N′,N′-tetramethyl-p-phenylenediamine/ascorbate-promoted respiration in mutant cells (25,36). Furthermore, the reduced mitochondrial protein synthesis may also result in the decrease in the activity of Complex V. Moreover, altered activities of Complexes I and III can lead to more electrons leakage from electron transport chain, and in turn, increase the generation of ROS (31,38). Here, an increase in ROS production was detected in cell lines derived from affected matrilineal relatives carrying the 15927G>A mutation. The increasing generation of ROS can damage to DNA, lipids, proteins and membranes. As a result, these biochemical defects can cause dysfunction or apoptosis in cardiac muscle cells, thereby producing the clinical phenotype. Therefore, these mitochondrial dysfunctions contribute to the pathogenesis of matrilineal CHD (12,39–41).
The homoplasmic nature of 15927G>A mutation in the tRNAThr gene hints to mild nature of mutation, evidenced by a mild mitochondrial dysfunction observed in mutant cells (23). These suggest that the 15927G>A mutation in the tRNAThr gene is the inherited risk factors necessary for the development of CHD. The nuclear modifier genes, environmental and epigenetic factors, as well as personal life styles may also contribute to the development of CHD in these subjects carrying the mtDNA mutation (42,43). In particular, defects in nuclear modifier genes can worsen mitochondrial dysfunctions caused by the primary pathogenic mtDNA mutations, thereby modulating the phenotypic manifestation of primary mtDNA mutations (37,44). Indeed, genes encoding the mitochondrial tRNA synthetase or modifying enzyme can modulate the phenotypic variability of mitochondrial tRNA or 12S rRNA mutations (44,45). The coexistence of three tRNA mutations contributed to high penetrance of hypertension in two Chinese families (46). Despite harboring the 15927G>A mutation belonging to haplogroup B5b (21,23), more drastic defects in tRNA metabolism in this family with CHD than those in other Chinese families strongly indicates the involvement of nuclear modifier genes required for tRNA metabolism in the development of CHD. Potential defects in nuclear genes may worsen mitochondrial dysfunction caused by the 15927G>A mutation, thereby producing the clinical phenotype. In particular, the tissue specificity of 15927G>A mutation in the tRNAThr in the Chinese family is likely attributed to tissue-specific tRNA metabolism or the involvement of nuclear modifier genes.
In summary, our findings demonstrate that mitochondrial dysfunction caused by the tRNAThr 15927G>A mutation is associated with CHD. The 15927G>A mutation altered tRNA metabolism, thereby impairing mitochondrial translation, respiration and increasing the production of ROS. Possible defects in nuclear modifiers may worsen the mitochondrial dysfunction caused by the 15927G>A mutation. Subsequently, mitochondrial dysfunctions and longstanding increase of ROS in cardiovascular cells may lead to the development of CHD. Thus, our findings may provide new insights into the understanding of pathophysiology and valuable information for management and treatment of CHD.
MATERIALS AND METHODS
Subjects
One four-generation Han Chinese family (BJH15) (Fig. 1) and 80 genetically unrelated subjects with CHD were ascertained at the Cardiac Clinics of Beijing Anzhen Hospital, Beijing, China. A total of 262 Han Chinese control subjects were obtained from a panel of unaffected Han Chinese individuals from the same area. Informed consent, blood samples and clinical evaluations were obtained from all participating family members, under protocols approved by the ethics committee of the Anzhen Hospital and Zhejiang University.
Risk factors assessment
Risk factors were considered as hypertension, hyperlipidemia, diabetes mellitus, cigarette smoking and family history for CHD. Blood pressure: a physician measured the systolic and diastolic blood pressures of subjects using a mercury column sphygmomanometer and a standard protocol. The first and the fifth Korotkoff sounds were taken as indicators of systolic and diastolic blood pressure, respectively. The average of three such systolic and diastolic blood pressure readings was taken as the examination blood pressure. Hypertension was defined according to the recommendation of the Joint National Committee on Detection, Evaluation and Treatment of High Blood Pressure (JNC VI) (47) as a systolic blood pressure of 140 mmHg or higher and/or a diastolic blood pressure of 90 mmHg or greater. Diabetes mellitus was defined as hyperglycemia, requiring antidiabetic drugs or fasting blood sugar over 126 g/dl (American Diabetes Association, the Expert Committee on the diagnosis and classification of diabetes mellitus, 1999). Hyperlipidemia was defined as plasma LDL-C>130 mg/dl or TC >200 mg/dl or using lipid-lowering drugs at the time of investigation according to The Third Report of the National Cholesterol Education Program guidelines. (Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Cholesterol in Adults, 2001). Patients reporting cigarette use during the year prior to examination were considered as smokers.
Mutational analysis of mitochondrial genome
Genomic DNA was isolated from whole blood of all participants for this investigation using Puregene DNA Isolation Kits (Gentra Systems, Minneapolis, MN, USA). Southern blot analysis of mitochondrial genomes of 32 members of this Chinese family was carried out as detailed elsewhere (48). The entire mitochondrial genomes of six matrilineal relatives (II-2, II-6, III-2, III-7, III-9 and IV-3) who were offsprings of I-2, and subject II-4 with two of her offsprings (III-12 and III-13) in this Chinese family were PCR amplified in 24 overlapping fragments by use of sets of the light-strand and the heavy strand oligonucleotide primers, as described elsewhere (49). Each fragment was purified and subsequently analyzed by direct sequencing in an ABI 3700 automated DNA sequencer using the Big Dye Terminator Cycle sequencing reaction kit. The resultant sequence data were compared with the updated consensus Cambridge sequence (GenBank accession number: NC_012920) (24). The presence and degree of the 15927G>A mutation for members of this family, 80 genetically unrelated individuals with CHD and control subjects were performed as detailed elsewhere (23).
Cell cultures
Lymphoblastoid cell lines were immortalized by transformation with the Epstein-Barr virus, as described elsewhere (50). Cell lines derived from five affected individuals (II-2, III-2, III-6, III-9 and IV-3) carrying the 15927G>A mutation and two married-in-controls (III-8 and IV-4) in this Chinese family, and a genetically unrelated control subject (A6) were grown in RPMI 1640 medium (Invitrogen), supplemented with 10% fetal bovine serum (FBS).
Mitochondrial tRNA northern analysis
Total mitochondrial RNA were obtained using TOTALLY RNATM kit (Ambion) from mitochondria isolated from lymphoblastoid cell lines (∼4.0 × 108 cells), as described previously (51). Five micrograms of total mitochondrial RNA were electrophoresed through a 10% polyacrylamide/7 m urea gel in Tris-borate-EDTA buffer (TBE) (after heating the sample at 65°C for 10 min), and then electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis with oligodeoxynucleotide probes. For the detection of mitochondrial tRNAs, the following non-radioactive DIG-labeled oligodeoxynucleotides specific for tRNAThr, tRNALeu(CUN), tRNALys, tRNAHis and tRNASer(AGY) were detailed elsewhere (23,44). DIG-labeled oligodeoxynucleotides were generated by using DIG oligonucleotide Tailing kit (Roche). The hybridization was carried out as detailed elsewhere (23,36,44). Quantification of density in each band was made as detailed previously (23,36,44).
Mitochondrial tRNA aminoacylation analysis
Total mitochondrial RNAs were isolated as above but under acid condition. The total mitochondrial RNA (2 µg) was electrophoresed at 4°C through an acid 10% polyacrylamide/7 m urea gel in a 0.1 m sodium acetate (pH 5.0) to separate the charged and uncharged tRNA, as detailed elsewhere (23,26). Then RNAs were electro-blotted onto a positively charged membrane (Roche) and hybridized sequentially with the specific tRNA probes as above.
Analysis of mitochondrial protein synthesis
Pulse-labeling of the cell lines for 30 min with [35S]methionine–[35S]cysteine in methionine-free DMEM in the presence of emetine, electrophoretic analysis of the translation products and quantification of radioactivity in the whole-electrophoretic patterns or in individual well-resolved bands were carried out as detailed previously (27).
O2 consumption measurements
Rates of O2 consumption in intact cells were determined with a YSI 5300 oxygraph (Yellow Springs Instruments) on samples of 5 × 106 cells in 1.5 ml of a special DMEM medium lacking glucose, supplemented with 10% dialyzed FBS (28). Polargraphic analysis of digitonin-permeabilized cells, using different respiratory substrates and inhibitors, to test the activity of the various respiratory complexes was carried out as detailed previously (29).
ROS measurements
ROS measurements were performed following the procedures detailed elsewhere (30,31). Briefly, approximate 2 × 106 cells of each cell line were harvested, resuspended in PBS supplemented with 100 µm of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and then incubated at 37°C for 20 min. After washing with PBS twice, cells were resuspended in PBS in the presence of 2 mm freshly prepared H2O2 and 2% FBS and then incubated at room temperature for another 5 min. Cells were further washed with PBS and resuspended with 1 ml of PBS with 0.5% paraformaldehyde. Samples with or without H2O2 stimulation were analyzed by BD-LSR II flow cytometer system (Beckton Dickson, Inc.), with an excitation at 488 nm and emission at 529 nm. A total of 10 000 events were analyzed in each sample.
Statistical analysis
Statistical analysis was performed by the unpaired, two-tailed Student's t-test contained in Microsoft Office Excel (version 2007). P indicates the significance, according to the t-test, of the difference between mutant mean and control mean. Differences were considered significant at a P < 0.05.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
The authors are grateful for the support of the members of the Chinese family. This work was supported by NIH grant RO1DC07696 and from an NIH research grant (206-08) from the Bureau of Science and Technology, Wenzhou Municipal Government, China to M.X.G.
Supplementary Material
REFERENCES
- 1.Lopez A.D., Mathers C.D., Ezzati M., Jamison D.T., Murray C.J. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X.H., Lu Z.L., Liu L. Coronary heart disease in China. Heart. 2008;94:1126–1131. doi: 10.1136/hrt.2007.132423. [DOI] [PubMed] [Google Scholar]
- 3.Sing C.F., Stengård J.H., Kardia S.L. Genes, environment, and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2003;23:1190–1196. doi: 10.1161/01.ATV.0000075081.51227.86. [DOI] [PubMed] [Google Scholar]
- 4.Wilson P.W., D'Agostino R.B., Levy D., Belanger A.M., Silbershatz H., Kannel W.B. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 5.Khot U.N., Khot M.B., Bajzer C.T., Sapp S.K., Ohman E.M., Brener S.J., Ellis S.G., Lincoff A.M., Topol E.J. Prevalence of conventional risk factors in patients with coronary heart disease. J. Am. Med. Assoc. 2003;290:898–904. doi: 10.1001/jama.290.7.898. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q. Molecular genetics of coronary artery disease. Curr. Opin. Cardiol. 2005;20:182–188. doi: 10.1097/01.hco.0000160373.77190.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peden J.F., Farrall M. Thirty-five common variants for coronary artery disease: the fruits of much collaborative labour. Hum. Mol. Genet. 2011;20:R198–R205. doi: 10.1093/hmg/ddr384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trégouët D.A., Konig I.R., Erdmann J., Munteanu A., Braund P.S., Hall A.S., Grosshennig A., Linsel-Nitschke P., Perret C., DeSuremain M., et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat. Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 9.Schunkert H., König I.R., Kathiresan S., Reilly M.P., Assimes T.L., Holm H., Preuss M., Stewart A.F., Barbalic M., Gieger C., et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peden J.F., Hopewell J.C., Saleheen D., Chambers J.C., Hager J., Soranzo N., Collins R., Danesh J., Elliott P., Farrall M. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 11.Wang F., Xu C.Q., He Q., Cai J.P., Li X.C., Wang D., Xiong X., Liao Y.H., Zeng Q.T., Yang Y.Z., et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat. Genet. 2011;43:345–349. doi: 10.1038/ng.783. [DOI] [PubMed] [Google Scholar]
- 12.Nisoli E., Clementi E., Carruba M.O., Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ. Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 13.Ballinger S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005;38:1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Davidson S.M., Duchen M.R. Endothelial mitochondria: contributing to vascular function and disease. Circ. Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 15.Marian A.J. Mitochondrial genetics and human systemic hypertension. Circ. Res. 2011;108:784–786. doi: 10.1161/CIRCRESAHA.111.242768. [DOI] [PubMed] [Google Scholar]
- 16.Liu C., Yang Q., Hwang S.J., Sun F., Johnson A.D., Shirihai O.S., Vasan R.S., Levy D., Schwartz F. Association of genetic variation in the mitochondrial genome with blood pressure and metabolic traits. Hypertension. 2012;60:949–956. doi: 10.1161/HYPERTENSIONAHA.112.196519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson F.H., Hariri A., Farhi A., Zhao H., Petersen K.F., Toka H.R., Nelson-Williams C., Raja K.M., Kashgarian M., Shulman G.I., Scheinman S.J., Lifton R.P. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li R., Liu Y., Li Z., Yang L., Wang S., Guan M.X. Failures in mitochondrial trnamet and trnagln metabolism caused by the novel 4401A>G mutation are involved in essential hypertension in a han chinese family. Hypertension. 2009;54:329–337. doi: 10.1161/HYPERTENSIONAHA.109.129270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li R., Liu Y., Li Z., Yang L., Wang S., Guan M.X. The mitochondrial transfer RNAMet 4435A>G mutation is associated with maternally hypertension in a Chinese pedigree. Hypertension. 2009;53:1083–1090. doi: 10.1161/HYPERTENSIONAHA.109.128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S.W., Li R.H., Fettermann A., Li Z.B., Qian Y.P., Liu Y.Q., Wang X.J., Zhou A., Mo J.Q., Yang L., et al. Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ. Res. 2011;108:862–870. doi: 10.1161/CIRCRESAHA.110.231811. [DOI] [PubMed] [Google Scholar]
- 21.Kong Q.P., Bandelt H.J., Sun C., Yao Y.G., Salas A., Achilli A., Wang C.Y., Zhong L., Zhu C.L., Wu S.F., Torroni A., Zhang Y.P. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum. Mol. Genet. 2006;15:2076–2086. doi: 10.1093/hmg/ddl130. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T., Nagao A., Suzuki T. Human mitochondrial tRNAs: Biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011;45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Lu J., Zhu Y., Yang A., Yang L., Li R., Chen B., Qian Y., Tang X., Wang J., Zhang X., Guan M.X. Mitochondrial tRNAThr 15927G>A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA A1555G mutation in four Chinese families. Pharmacogenet. Genom. 2008;18:1059–1070. doi: 10.1097/FPC.0b013e3283131661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 25.Li R., Guan M.X. Human mitochondrial leucyl-tRNAsynthetase corrects mitochondrial dysfunctions due to the MELAS and diabetes associated tRNALeu(UUR) A3243G mutation. Mol. Cell Biol. 2010;30:2147–2154. doi: 10.1128/MCB.01614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Enríquez J.A., Attardi G. Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 1996;264:183–196. doi: 10.1016/s0076-6879(96)64019-1. [DOI] [PubMed] [Google Scholar]
- 27.Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- 28.King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 29.Hofhaus G., Shakeley R.M., Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- 30.Mahfouz R., Sharma R., Lachner J., Aziz N., Agarwal A. Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil. Steril. 2008;92:819–827. doi: 10.1016/j.fertnstert.2008.05.087. [DOI] [PubMed] [Google Scholar]
- 31.Qian Y., Zhou X., Liang M., Qu J., Guan M.X. The altered activity of complex III may contribute to the high penetrance of Leber's hereditary optic neuropathy in a Chinese family carrying the ND4 G11778A mutation. Mitochondrion. 2011;11:871–877. doi: 10.1016/j.mito.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Taylor R.W., Giordano C., Davidson M.M., d'Amati G., Bain H., Hayes C.M., Leonard H., Barron M.J., Casali C., Santorelli F.M., et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003;41:1786–1796. doi: 10.1016/s0735-1097(03)00300-0. [DOI] [PubMed] [Google Scholar]
- 33.Campos Y., Gamez J., Garcia A., Andreu A.L., Rubio J.C., Martin M.A., del Hoyo P., Navarro C., Cervera C., Garesse R., Arenas J. A new mtDNA mutation in the tRNALeu(UUR) gene associated with ocular myopathy. Neuromusc. Disord. 2001;11:477–480. doi: 10.1016/s0960-8966(00)00223-6. [DOI] [PubMed] [Google Scholar]
- 34.Chomyn A., Enriquez J.A., Micol V., Fernandez-Silva P., Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 2000;275:19198–19209. doi: 10.1074/jbc.M908734199. [DOI] [PubMed] [Google Scholar]
- 35.Guan M.X., Enriquez J.A., Fischel-Ghodsian N., Puranam R., Lin C.P., Marion M.A., Attardi G. The deafness-associated mtDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase ND6 subunit gene expression. Mol. Cell Biol. 1998;18:5868–5879. doi: 10.1128/mcb.18.10.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R.A., Guan M.X. Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 2004;32:867–877. doi: 10.1093/nar/gkh226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan M.X., Fischel-Ghodsian N., Attardi G. Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 2001;10:573–580. doi: 10.1093/hmg/10.6.573. [DOI] [PubMed] [Google Scholar]
- 38.Lenaz G., Baracca A., Carelli V., D'Aurelio M., Sgarbi G., Solaini G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim. Biophys. Acta. 2004;1658:89–94. doi: 10.1016/j.bbabio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 39.Postnov Y.V., Orlov S.N., Budnikov Y.Y., Doroschuk A.D., Postnov A.Y. Mitochondrial energy conversion disturbance with decrease in ATP production as a source of systemic arterial hypertension. Pathophysiology. 2007;14:195–204. doi: 10.1016/j.pathophys.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Addabbo F., Montagnani M., Goligorsky M.S. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galluzzi L., Kepp O., Trojel-Hansen C., Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2007;111:1198–1207. doi: 10.1161/CIRCRESAHA.112.268946. [DOI] [PubMed] [Google Scholar]
- 42.Vasan R.S., Beiser A., Seshadri S., Larson M.G., Kannel W.B., Agostino R.B., Levy D. Residual lifetime risk for developing hypertension in (2002). Residual lifetime risk for developing hypertension in middle-aged women and men: the Framingham Heart Study. JAMA. 2002;287:1003–1010. doi: 10.1001/jama.287.8.1003. [DOI] [PubMed] [Google Scholar]
- 43.Djousse L., Driver J.A., Gaziano J.M. Relation between modifiable lifestyle factors and lifetime risk of heart failure. JAMA. 2009;302:394–400. doi: 10.1001/jama.2009.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan M.X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E., et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perli E., Giordano C., Tuppen H.A., Montopoli M., Montanari A., Orlandi M., Pisano A., Catanzaro D., Caparrotta L., Musumeci B., et al. Isoleucyl-tRNA synthetase levels modulate the penetrance of a homoplasmic m.4277T>C mitochondrial tRNAIle mutation causing hypertrophic cardiomyopathy. Hum. Mol. Genet. 2012;21:85–100. doi: 10.1093/hmg/ddr440. [DOI] [PubMed] [Google Scholar]
- 46.Qiu Q., Li R., Jiang P., Xue L., Lu Y., Song Y., Han J., Lu Z., Zhi S., Mo J.Q., Guan M.X. Mitochondrial tRNA mutations are associated with maternally inherited hypertension in two Han Chinese pedigrees. Hum. Mutat. 2012;33:1285–1293. doi: 10.1002/humu.22109. [DOI] [PubMed] [Google Scholar]
- 47.Guidelines Subcommittee. 1999. World Health Organization-International Society of Hypertension Guidelines for the Management of Hypertension. Guidelines Subcommittee. J. Hypertens. 1999;17:151–183. [PubMed] [Google Scholar]
- 48.Guan M.X., Fischel-Ghodsian N., Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 1996;5:963–972. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- 49.Rieder M.J., Taylor S.L., Tobe V.O., Nickerson D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller G., Lipman M. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl Acad. Sci. USA. 1973;70:190–194. doi: 10.1073/pnas.70.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King M.P., Attardi G. Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 1993;1268:10228–10237. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.