

Abstract
Multiple sclerosis (MS) is the most common autoimmune disease of the central nervous system (CNS). It is characterized by the infiltration of autoreactive immune cells into the CNS, which target the myelin sheath, leading to the loss of neuronal function. Although it is accepted that MS is a multifactorial disorder with both genetic and environmental factors influencing its development and course, the molecular pathogenesis of MS has not yet been fully elucidated. Here, we studied the longitudinal gene expression profiles of whole-blood RNA from a cohort of 195 MS patients and 66 healthy controls. We analyzed these transcriptomes at both the individual transcript and the biological pathway level. We found 62 transcripts to be significantly up-regulated in MS patients; the expression of 11 of these genes was counter-regulated by interferon treatment, suggesting partial restoration of a ‘healthy’ gene expression profile. Global pathway analyses linked the proteasome and Wnt signaling to MS disease processes. Since genotypes from a subset of individuals were available, we were able to identify expression quantitative trait loci (eQTL), a number of which involved two genes of the MS gene signature. However, all these eQTL were also present in healthy controls. This study highlights the challenge posed by analyzing transcripts from whole blood and how these can be mitigated by using large, well-characterized cohorts of patients with longitudinal follow-up and multi-modality measurements.
INTRODUCTION
Multiple sclerosis (MS) is a debilitating disease of the central nervous system (CNS), affecting primarily young adults, with a prevalence of about 100 per 100 000 in northern Europeans and their descendants (1). Disease pathogenesis is thought to be mediated by autoreactive T-cells and B-cells, although innate immune mechanisms have also been implicated (2). Pathogenic immune processes lead to a breakdown of the blood–brain barrier, enabling increased access to the CNS of immune cells, which target the myelin sheath of axons. MS is a multifactorial disorder with both genetic and environmental factors influencing its development and course (3). Genome-wide, more than 60 loci have been identified that influence MS risk, and among these, the HLA locus has the strongest effect (4).
The commonly used disease-modifying treatments (DMTs) interferon (IFN) beta and glatiramer acetate are believed to modulate the immune response, reduce new inflammatory lesions in the CNS and partially protect against progression of disability. However, patients vary considerably in their responsiveness to these therapies, and for any individual patient, the natural history of MS is extremely heterogeneous, varying from a benign condition to a devastating and rapidly incapacitating disease. For these reasons, a better characterization of patients is much needed to ultimately understand the diversity of disease presentation. Recent studies in neurodegenerative disorders and autoimmune diseases (5–8) suggest that gene expression changes in blood mirror pathologic processes in the CNS. Blood transcriptomics have also been used to study therapeutic response to treatment with different drugs, toxins and infections in different diseases (9–11). Several microarray-based gene expression studies have used whole blood or peripheral blood mononuclear cells (PBMCs) to investigate de-regulated patterns of gene expression in MS patients (12–31). Unfortunately, owing to small sample sizes and disease heterogeneity, reproducibility across studies has been limited. In this study, we set out to assess gene expression profiles in whole blood in a well-characterized longitudinal cohort comprising 195 MS patients and 66 healthy controls. We followed a multi-analytical approach to identify both individual transcripts and biological pathways implicated in MS pathogenesis as well as in response to therapeutic drugs. In addition, we integrated the transcriptomes with available genome-wide genetic variants in order to determine expression quantitative trait loci (eQTL) (32,33).
RESULTS
We performed whole-blood transcriptional profiling in 195 MS patients at the time of enrollment (baseline), and after 1 and 2 years of follow-up. We also profiled 66 healthy individuals at two different time points (1 year apart) as controls (see Supplementary Material, Fig. S1, for a description of the analytical strategy). After stringent quality control, 397 arrays were analyzed as a discovery set, and an independent set of 229 arrays were analyzed for validation. Details of the cohort are provided in Table 1. The quality of microarray data was further assessed by analyzing a set of 48 transcripts in 44 random samples by an independent technology (NanoString). The correlation between the expression values as determined by the two techniques was high (range 0.76–0.88; Supplementary Material, Fig. S2), indicating the reliability of the array data set.
Table 1.
Major characteristics of the study cohorta
| Data set | Discovery (397 arrays) |
Replication (229 arrays) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Condition | MS (n = 120) |
Ctrl (n = 41) |
MS (n = 75) |
Ctrl (n = 25) |
||||||
| Visit | BL | F/U Y1 | F/U Y2 | BL | F/U Y1 | BL | F/U Y1 | F/U Y2 | BL | F/U Y1 |
| CIS/RR/SP (%) | 17/77/6 | 14/78/8 | 13/80/7 | NA | NA | 25/69/5 | 15/82/3 | 11/87/2 | NA | NA |
| Median disease duration (range) | 6 (0–45) | 7 (1–46) | 8 (2–47) | NA | NA | 5 (0–34) | 5 (1–35) | 6 (2–27) | NA | NA |
| Median EDSS (range) | 1.5 (0–7) | 2 (0–7) | 2 (0–7) | NA | NA | 1.5 (0–5) | 2 (0–6) | 2 (0–6) | NA | NA |
| Median age (range) | 43 (22–66) | 44 (23–67) | 45 (24–66) | 46 (26–66) | 46 (27–64) | 45 (23–61) | 46 (23–62) | 46 (25–61) | 42 (27–61) | 43 (31–62) |
| % Female | 71 | 71 | 70 | 74 | 76 | 66 | 64 | 65 | 59 | 50 |
| Number of subjects untreated/IFN-treated | 54/57 | 42/67 | 37/61 | 38/NA | 41/NA | 21/38 | 20/41 | 15/48 | 22/NA | 24/NA |
MS, multiple sclerosis patients; Ctrl, healthy controls; BL, baseline; F/U Y1/Y2, follow-up year 1 or 2; CIS, clinically isolated syndrome; RR, relapsing-remitting; SP, secondary progressive; EDSS, Expanded Disability Status Scale; IFN, interferon; NA, not applicable.
aPatients' characteristics were determined at the start of the study.
Global gene expression in IFN-treated patients (n = 58) was compared with that of untreated MS subjects (n = 62) in the discovery data set (Table 2). We determined the gene expression differences between untreated and treated patients at each of three yearly time points and computed the union of these differences as differentially regulated genes. As expected, IFN treatment was associated with broad gene expression changes: 749 genes were differentially expressed at a false discovery rate (FDR) of 1%. Increasing FDR stringency to 0.01% decreased the number of genes to 262, of which 260 (99%) were also found differentially expressed in the replication data set (Supplementary Material, Table S1). Among the most significantly and strongly differentially expressed genes were EPSTI1 (epithelial stromal interaction 1), OAS3 (2′5′-oligoadenylate synthetase 3), IFI44L (interferon-induced protein 44-like) and RSAD2 (radical S-adenosyl methionine domain containing 2). Notably, 45% of the reported 260 genes are known IFN-responsive genes as recorded in the Interferome database (http://www.interferome.org/, last accessed date on Spring, 2012), indicating that a robust signature can be reliably detected from whole-blood total RNA. Furthermore, the identified signature clearly discriminated treated from untreated subjects (Fig. 1 and Supplementary Material, Fig. S3).
Table 2.
Characteristics of MS patients, split by treatmenta
| Data set | Discovery (318 arrays) |
Replication (183 arrays) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | None (n = 62) |
IFN (n = 58) |
None (n = 27) |
IFN (n = 48) |
||||||||
| Visit | BL | F/U Y1 | F/U Y2 | BL | F/U Y1 | F/U Y2 | BL | F/U Y1 | F/U Y2 | BL | F/U Y1 | F/U Y2 |
| CIS/RR/SP (%) | 26/65/9 | 26/60/14 | 27/68/8 | 9/88/4 | 6/90/4 | 5/89/7 | 57/33/10 | 25/70/5 | 27/73/0 | 8/89/3 | 10/88/2 | 6/92/2 |
| Median disease duration (range) | 5.5 (0–32) | 7 (1–33) | 8 (2–32) | 7 (0–45) | 7 (1–46) | 8 (2–47) | 1 (0–22) | 2 (1–35) | 4 (2–14) | 7 (0–34) | 6 (1–35) | 7 (2–27) |
| Median EDSS (range) | 1 (0–6.5) | 2 (0–6.5) | 2 (0–6) | 1.5 (0–7) | 2 (0–7) | 2 (0–7) | 1 (0–5) | 1.5 (0–5.5) | 2 (0–5) | 1.5 (0–4) | 2.5 (0–6) | 2 (0–6) |
| Median age (range) | 43 (22–63) | 47 (27–64) | 48 (28–64) | 43 (23–66) | 43 (23–67) | 44 (24–66) | 45 (24–59) | 47 (25–62) | 46 (26–61) | 46 (23–61) | 45 (23–60) | 46 (25–61) |
| % Female | 76 | 74 | 70 | 67 | 69 | 70 | 57 | 60 | 73 | 71 | 66 | 63 |
| Number of subjects | 54 | 42 | 37 | 57 | 67 | 61 | 21 | 20 | 15 | 38 | 41 | 48 |
| Median time on IFN (range) (days) | NA | NA | NA | 737 (2–3511) | 849 (44–3895) | 1227 (50–4239) | NA | NA | NA | 956 (1–4171) | 1081 (16–4535) | 1233 (92–4902) |
BL, baseline; F/U Y1/Y2, follow-up year 1 or 2; CIS, clinically isolated syndrome; RR, relapsing–remitting; SP, secondary progressive; EDSS, Expanded Disability Status Scale; IFN, interferon; NA, not applicable.
aPatients' characteristics were determined at the start of the study.
Figure 1.

PCA of untreated and IFN-treated patients by the expression of IFN signature genes in the discovery (A) and replication (B) data sets. On the x-axis, principal component (PC) 2 is plotted, on the y-axis PC 3 and on the z-axis PC 4. IFN-treated patients are displayed in orange, untreated patients in blue. As indicated by the colored ellipses, these principal components split samples into two groups, corresponding to whether subjects were treated or not.
We then turned to comparing gene expression changes in untreated MS patients versus healthy controls. Because gene expression patterns were relatively stable across the three time points, we adopted a cross-sectional analysis strategy. MS patients were compared at each time point with all data points available for controls. The two time points for controls were treated as replicates, assuming that in the absence of disease processes, the expression of the majority of genes would not change significantly within a year (correlation coefficients between the two time points for controls ranged between 92 and 99%; data not shown). As in the IFN analysis, differentially expressed genes were determined as the union of gene expression differences observed for each of the three time points.
In contrast with the transcriptional responses observed for IFN treatment, gene expression differences between untreated cases and controls were much more subtle, with a maximal expression difference of 1.46-fold. Taking advantage of our large sample size and two-tiered approach, we applied an FDR cut-off of 1% in the discovery data set to increase the list of potential hits. Out of 79 differentially expressed transcripts, 62 (78%) were confirmed in the replication data set (Supplementary Material, Table S2). Of interest, 11 out of 51 overlapping transcripts (22%) were also differentially expressed at nominally significant P-values in a publicly available, independent data set [ANZgene data (18,23)]. Furthermore, the overall concordance in differentially expressed transcripts identified by our microarrays and by the ANZgene data was significantly higher than expected (at an FDR of 0.05, both data sets shared 22 up-regulated genes, 18 more than expected by chance, P-value by chi-square test: 2.2 × 10−16), supporting the validity of our findings. Interestingly, 11 of the 62 differentially expressed genes (18%, all up-regulated in MS) were consistently down-regulated by IFN treatment (nominal P-value ≤ 0.05 in at least five of the six studied time points in both discovery and replication data sets), suggesting that DMT counteracts potentially pathogenic gene expression patterns in MS patients. Table 3 lists these 11 genes, some of which encode for proteins with known immune functions (S100A11, LST1, FCGRT, GMFG). Also, the transcription of genes involved in proteasome function (PSMA7), Wnt signaling (CSNK2B) and oxidative phosphorylation (COX4I1) was affected by IFN treatment. In addition, IFN counter-regulated the expression of PARK7 (also known as DJ-1), a gene which has previously been reported to be up-regulated in MS (34–36). Seven of the 11 genes (64%) were also found to be significantly down-regulated in at least 50% of tested publicly available data sets assessing MS patients before and after the start of IFN treatment (IFN data: see Materials and Methods). Of note, these seven validated genes included two MS genes that were replicated in the ANZgene data (PARK7 and COX4I1). Interestingly, 24% of the genes we found to be significantly up-regulated in MS in the ANZgene data also showed counter-regulation in at least 50% of the IFN data, suggesting that one means by which IFN treatment shows therapeutic benefit might be restoration of ‘healthy’ gene expression.
Table 3.
MS signature genes counter-regulated by interferon treatmenta
| Gene symbol | Fold change (P-value) |
Gene name | Gene function | Validated | |
|---|---|---|---|---|---|
| MS versus Ctrl | IFN versus untreated | ||||
| PARK7 | 1.239 (2 × 10−7) | 0.873 (0.001) | Parkinson protein 7 | Mitochondrial oxidative stress, TRAIL-induced cell death (65); mutated form causes early-onset Parkinson disease (66); previously linked to MS (34–36) | Yes |
| CSNK2B | 1.207 (9 × 10−8) | 0.895 (0.002) | Casein kinase 2, beta polypeptide | Anti-viral response (67), Wnt signaling (68); susceptibility region for rheumatoid arthritis (69) | No |
| USE1 | 1.163 (2 × 10−7) | 0.943 (0.039) | Unconventional SNARE in the ER 1 homolog | Localizes to the endoplasmatic reticulum (ER) and Golgi | Yes |
| S100A11 | 1.201 (2 × 10−7) | 0.919 (0.008) | S100 calcium binding protein A11 | Interleukin signaling (70) | No |
| LST1 | 1.275 (1 × 10−7) | 0.874 (0.002) | Leukocyte-specific transcript 1 | Myeloid transmembrane protein, expression increased in rheumatoid arthritis and up-regulated by LPS, IFN-gamma and bacterial infection (71) | Yes |
| FCGRT | 1.126 (1 × 10−5) | 0.905 (2 × 10−5) | Fc fragment of IgG, receptor, transporter, alpha | Binds to, and increases the stability of, IgG in serum (72) | Yes |
| GMFG | 1.160 (1 × 10−4) | 0.908 (0.007) | Glia maturation factor, gamma | Hematopoietic cell development (73) | Yes |
| MRFAP1 | 1.134 (6 × 10−7) | 0.938 (0.007) | Morf4 family associated protein 1 | Uncertain | No |
| COX4I1 | 1.225 (3 × 10−9) | 0.914 (0.006) | Cytochrome c oxidase subunit IV isoform 1 | Oxidative phosphorylation | Yes |
| C19orf43 | 1.159 (4 × 10−6) | 0.941 (0.033) | Open reading frame 43 (Chr 19) | Uncertain | Yes |
| PSMA7 | 1.129 (3 × 10−6) | 0.947 (0.019) | Proteasome subunit, alpha type, 7 | Component of proteasome; among others, functions in cell cycle and viral replication (74) | No |
aCtrl, control; IFN, interferon; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; Wnt, wingless-related MMTV integration site; LPS, lipopolysaccharide. Validated, also found to be down-regulated in publicly available data sets measuring gene expression before and after administration of IFN (see Materials and Methods).
Despite modest differences in expression levels, the identified MS signature is discriminatory in unsupervised hierarchical clustering (Fig. 2). The heatmap shows a uniform cluster of MS patients (group A) as well as several smaller uniform clusters of controls, an observation that stands in the replication set. Of note, MS cases who do not belong to group A, rather clustered with the controls (group B), indicating that gene expression changes evoked by the disease are much more heterogeneous and complex than those induced by IFN.
Figure 2.

Unsupervised hierarchical clustering of MS patients and healthy controls according to the expression of MS signature genes in the discovery (A) and the replication (B) data sets. The rows are different genes, the columns reflect different experiments. The colored bar above the heatmap identifies patients (orange) and controls (grey). Two subgroups of MS patients, group A with a stronger signature and group B, emerge. Blue depicts low expression and yellow high expression. Hierarchical clustering was performed using Euclidean distance and average clustering.
We used the Gene Enrichment Profiler tool (37) to assess the expression of transcripts in the MS signature in each of 126 normal cell types and tissues. This analysis revealed that the identified signature contains genes strongly expressed in myeloid cells, T-cells and other blood cell types, whereas brain and other tissues show a relative depletion for those genes (Supplementary Material, Fig. S4). Although this is not unexpected given the origin of the samples, it demonstrates that we detect the coordinated expression of transcripts in these cell types, thus providing support for our analysis. Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis revealed that the MS signature is enriched in energy-generating, metabolic and degradative processes as well as transport (Supplementary Material, Table S3). These signatures of cellular activation, also found in another large study (18), might reflect the sizeable fraction of activated T-cells in MS samples.
We next used a systems biology approach in which expression profiles were integrated with protein interaction networks, and heuristic searches were conducted to identify modules—groups of proteins involved in the same biological function—enriched in differentially expressed genes. We used the list of transcripts differentially expressed between MS (untreated) and healthy controls as input to the jActive modules plugin (38) of the network visualization software Cytoscape (39). We found 52 statistically significant networks, out of which 43 modules were constituted of more than 10 proteins (Supplementary Material, Table S4). GO enrichment analysis of these modules revealed that they were overwhelmingly representing immune pathways (such as wound healing, T-cell receptor signaling, B-cell activation). As immune processes are known to play a role in MS, this finding indicates that our analysis approach identifies non-random selections of genes involved in disease pathogenesis. Additional cellular processes that were among the most significantly enriched categories were insulin and transforming growth factor β receptor signaling, cell cycle and transcriptional regulation, apoptosis as well as vesicular transport. Similarly, KEGG enrichment analysis of these modules yielded cell cycle and cancer as well as immune categories, including innate immune functions such as phagocytosis and complement and coagulation cascades. Interestingly, we identified several networks representing cell adhesion and transendothelial migration pathways, all mechanisms likely involved in MS pathogenesis. One of the most significant networks was enriched in immune and cell cycle-related pathways, highlighting these processes as active players of disease pathogenesis. Detailed information on all significant networks is provided in Supplementary Material, Tables S5–S7. One of the most significant networks, enriched in immune and cell cycle-related pathways, is shown in Supplementary Material, Figure S5.
Because genome-wide single-nucleotide polymorphism (SNP) genotypes were available for 59 untreated patients and 28 healthy controls from the discovery set as well as for 27 cases and 25 controls from the replication set (40), we finally used the gene expression data to identify eQTL, i.e. SNPs associated with differential gene expression. We identified 178 transcripts with high variance (see Materials and Methods) across all cases and controls (separately) at multiple time points. We found 103 cis-eQTL shared between MS cases and controls, showing significant P-values in both the discovery (Bonferroni-corrected P-values) and the replication (nominal P-values) data sets (Supplementary Material, Table S8). Only 29% of these have been reported before. Of note, 50% of the significant eQTL were located within 60 000 bp of the transcriptional start site of the regulated genes (Fig. 3). This strongly suggests that the reported associations are potentially functional. Two genes from the MS signature were among identified novel eQTL: rs3173833 was associated with the expression of TMEM176B, rs7806458, rs10952287 and rs2072443 with the expression of both TMEM176B and TMEM176A (Fig. 4). Since these two genes are located next to each other, it is likely that their expression is co-regulated; indeed, a recent study observed matching expression patterns of the murine homologs (41). The associated SNPs most likely represent one haplotype [pairwise linkage disequilibrium (LD) ranges between 0.8 and 1].
Figure 3.
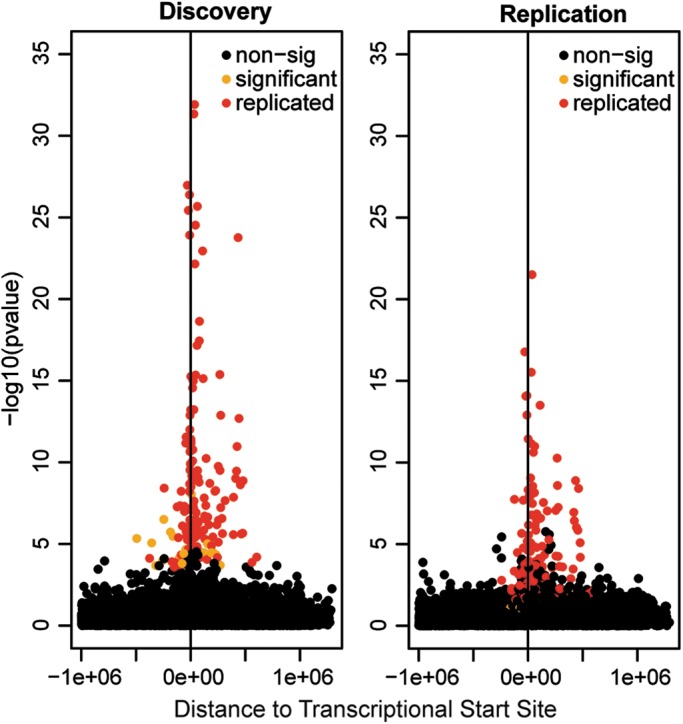
Location of general cis-eQTL. Association P-values in discovery (A) and replication (B) data sets are plotted against the distance of each cis-SNP from the transcription start of studied transcripts. SNPs that were found to be significant in both discovery and replication data sets (replicated) are displayed in red, those with significant P-values only in the discovery data set (significant) are displayed in orange, all non-significant SNPs (non-sig) are shown in black.
Figure 4.

Genetic make-up of MS gene cis-eQTL. (A and B) Manhattan plots of association P-values of all studied SNPs and the expression of one of the MS-associated genes with replicated cis-eQTL, TMEM176A, encoded on chromosome 7, in the discovery (A) and the replication (B) data sets. Each chromosome is displayed in a different color. Note the pronounced peak of association P-values on chromosome 7, on which TMEM176A is encoded, especially in the discovery data set. (C and D) Log2-transformed expression values for TMEM176A in dependence of the most significantly associated SNP, rs7806458 (genotype), in the discovery (C) and the replication (D) data sets in both MS patients (MS) and controls (CTRL). (E) University of California at Santa Cruz (UCSC) genome browser-based visualization of the genetic locus comprising TMEM176A, TMEM176B and the SNPs associated with their expression (shared: rs7806458, rs10952287, rs2072443; TMEM176B only: rs3173833), marked in red in the upper track. In addition to UCSC genes, RefSeqGenes and Human mRNA tracks, location of the probes on the analyzed microarray (‘Core PS’, Affy Exon Array) as well as the Encode Integrated Regulation tracks ‘Layered H3K27Ac’, ‘DNase Clusters’ and ‘Txn Factor ChIP’ are shown. Also, Vista Enhancers (‘HMR-Conserved Non-coding Human Enhancers’) and transcription factor binding sites (‘HMR Conserved Transcription Factor Binding Sites’, TFBS conserved) are displayed. Common SNPs (as of dbSNP v135) are shown in the bottom track.
We also investigated whether any eQTL were specific for either MS or healthy controls, but did not find consistent eQTL differences in the discovery and replication data sets (data not shown).
DISCUSSION
We here present the largest longitudinal gene expression study in MS to date. Although several smaller expression profile experiments in MS have been reported (12–28), independent validation is missing and replication across studies is either minimal or null. Owing to the large number of samples analyzed here, we were not only able to identify an MS signature despite modest effect sizes (fold expression changes); but we could also split our data into two separate sets, thus providing a means for replication. The identified MS signature from untreated subjects differentiated cases from controls reasonably well in unsupervised clustering (Fig. 2). A distinct cluster of MS samples was observed (group A), whereas the remaining MS subjects appeared more heterogeneous and partly intertwined with controls (group B). This type of aggregation has been observed before (27) and could not be explained by any of the assessed clinical parameters [disease course, gender, EDSS, number of copies of the risk allele DRB1*15:01, disease duration or age (data not shown)] and may reflect underlying etiological heterogeneity. Recently, a large cross-sectional study reported the clustering of MS patients into two groups by virtue of gene expression profiles derived from PBMCs; these two groups differed in the expression level of 98 probes as well as by clinical disease activity (28). With expression profiling from only one time point being available, the authors could not comment on whether a given patient would always belong to the same MS subgroup or could switch groups (28). To follow up on this finding, we defined two MS subgroups either based on the previously described 98 probe-signature or based on the inherent structure of our data. Although most patients clustered with the same subgroup at all three time points, a fraction of patients changed groups, depending on the method used to define the two subgroups (data not shown). When considering only patients who remained in the same subgroup over time, we were not able to identify significant differences in clinical activity between groups by survival analysis (data not shown). However, it is possible that the smaller sample size in our study may have restricted our power to detect differences.
Interestingly, both KEGG and pathway analyses of the MS gene expression signature identified over-representation of elements of the proteasome. These results are supported by previous findings implicating the proteasome in MS pathogenesis (42–44) and suggest that the proteasome might be a relevant target to decrease overt disease activity. Likewise, both KEGG and pathway analyses hint to a role for the SNARE complex, whose assembly has recently been linked to proteasome function and neurodegeneration (45). Two of the modules in the pathway analysis revealed the involvement of Wnt (wingless-related MMTV integration site) signaling in MS. Notably, the unfiltered GO and KEGG categories enriched in the MS gene signature (Supplementary Material, Tables S3 and S9) significantly overlap with a recently published categorical analysis of the transcriptional response to Wnt1 in cultured human neural progenitor cells (46). Previous reports have also associated the Wnt pathway with MS susceptibility (47) and particularly, with de/remyelination (48,49). Altogether, this body of data supports the Wnt pathway as a player in MS pathogenesis, a fact that warrants a closer look at this critical developmental pathway.
Surprisingly, we find several indications for the involvement of neuronal processes in the pathogenesis of MS in our data set. This finding has to be interpreted cautiously as the analyzed transcriptomes were acquired from blood, not from neuronal cells. Further studies are needed to determine whether these signatures in whole blood truly represent processes in the CNS.
No significant changes in longitudinal gene expression were identified that could discriminate the two groups (data not shown), suggesting that yearly measurements may not be frequent enough to capture the true dynamics of gene expression. Alternatively, the duration of the study (2 years) was not sufficient to detect the cumulative impact of disease processes on blood gene expression.
Our data on the transcriptional consequences of IFN treatment are in accordance with previous gene expression studies (reviewed in 50). By applying a very stringent FDR cut-off (FDR < 10−4), we identified an IFN gene expression signature that clearly differentiated treated from untreated patients (Fig. 1). Interestingly, we observed that a subset of patients within the treated group showed a stronger signature. Although this observation was replicated in the validation data set, no correlation was found with any of the assessed clinical parameters (IFN formula, days on treatment, disease duration, EDSS, disease course, number of copies of the risk allele DRB1*15:01, age, gender, principle component 1; data not shown) that could explain the sample aggregation. Since many treated patients do not show this strong signature at all assessed time points, the stronger response to IFN might rather reflect some variability in the reaction to injection than a specific attribute of affected individuals.
Although we identified many cis-eQTL with very significant P-values (Supplementary Material, Table S8), we did not find evidence of trans-eQTL. This might be related to the fact that, for the latter analysis, correction for multiple testing is much more extensive (genome-wide versus SNPs in the same genomic region only), thus increasing the chance of eliminating true discoveries (type II error). Also, no eQTL specific for MS patients or healthy controls were identified. One possibility for the lack of disease-specific eQTL might be the limiting sample size.
In summary, gene expression differences between MS patients and controls were modest, possibly due to both disease heterogeneity and the choice of research specimen (whole blood). Analysis of a large data set allowed us to overcome these natural limitations, and a number of significant de-regulated genes could be detected. A proportion of transcripts up-regulated in untreated patients were counter-regulated by IFN treatment, suggesting a set of possible effectors for this first-line therapy in MS.
MATERIALS AND METHODS
Samples and consent
This study was approved by the University of California at San Francisco (UCSF) Committee on Human Research. The EPIC study enrolled 500 MS patients and 500 controls in 2004 and has been following them on a yearly basis until now. At each visit, subjects are assessed by neurological examination as well as neuro-imaging, and blood samples are collected. Blood samples from 120 MS patients (at three consecutive years) and 41 healthy controls (at two time points) participating in the EPIC study were selected at random as discovery data set. Another set of 75 MS patients (at three consecutive years) and 25 healthy controls (at two time points) were selected at random as the replication data set. Major characteristics of the study cohort are given in Table 1.
RNA preparation and microarrays
Blood was drawn into PAXgene collection tubes and RNA was isolated using the PAXgene Blood RNA kit (Qiagen, Valencia, CA, USA), following the manufacturer's instructions except for an additional washing step before RNA elution. DNA was digested on columns using the RNase-free DNase Set (Qiagen). RNA quality control, labeling and hybridization onto Affymetrix Human Exon 1.0 ST Arrays (Affymetrix, Inc., Santa Clara, CA, USA) were performed by the core facility of Duke Institute for Genome Sciences and Policies. The raw data are available at Gene Expression Omnibus, accession number GSE41850.
NanoString data generation and analysis
To test the reliability of the microarray data, we assessed the expression profiles from a subset of 20 patients at different time points (44 samples in total) using an independent technique, NanoString nCounter® assays (NanoString Technologies, Seattle, WA, USA). RNA from selected samples was re-extracted and sent to the Oncogenomics Core Facility of the University of Miami. After quality control was performed [Agilent 2100 Bioanalyzer (Agilent Technology, Santa Clara, CA, USA) analysis; RIN ranging between 7.9 and 9.7], the expression of 48 selected genes and 3 housekeeping genes was assessed. Data were analyzed in R. One sample did not pass quality control and was excluded. Data were log2-transformed before being normalized in two steps. We first used assay internal positive controls to calculate a normalization factor for each sample (by dividing the median of all counts of the positive controls by their sum) and then normalized data by the expression of housekeeping genes (GAPDH, PPP1CA, HPRT1). This was accomplished by determining the average counts of all housekeeping genes for all samples and then computing a normalization factor for each sample (obtained by dividing the median of these averages by the average counts of the housekeeping genes of each sample). Sixteen samples were run in replicates, and the correlation coefficients for these samples ranged from 0.99 to 1 (data not shown), supporting that the data were highly reliable. Comparing the NanoString data with the same samples assessed by microarrays also yielded correlation coefficients ranging from 0.76 to 0.88 (Supplementary Material, Fig. S2), supporting the good quality of the expression arrays.
Genotyping
The genotypes on this data set have been published before in the context of a genome-wide association study (GWAS) (40). Arrayed SNPs were re-annotated to hg19 using the liftOver tool of the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgLiftOver, last accessed date on February, 2012) and the R package biomaRt (51).
Microarray data normalization and determination of differentially regulated genes
Microarray data were processed in R using the R package ‘aroma.affymetrix’ (www.aroma-project.org). Thirteen outliers were detected by both principal component analysis (PCA) and the package ‘arrayQualityMetrics’ (52) and were thus excluded from further analysis. For the comparison of IFN-treated and untreated patients, all arrays of IFN-treated and untreated patients were processed together (i.e. background-corrected and normalized). Similarly, for the comparison of untreated patients and controls, all arrays of untreated patients and controls were processed together in a separate analysis. Data were background-corrected (‘RMAbackgroundCorrection’) and quantile normalized before core probe sets were summarized to transcript level, using a custom-made cdf file. This cdf file excludes all probes known to span SNPs, which could interfere with hybridization and thus introduce noise (53). Data were log2-transformed and filtered for variance using the ‘genefilter’ package [probe sets showing a difference between the 10 and 90% quantiles >0.7 (IFN analysis) or 0.6 (MS versus controls) were further analyzed]. Probe sets were annotated to genes using biomaRt. In the discovery data set, differentially expressed genes were identified by applying stringent FDR-corrected P-value filters; these genes were then tested for validation in the replication data set.
The approach for the cross-sectional data analysis is depicted in Supplementary Material, Figure S2. In brief, expression profiles of two groups (IFN-treated versus -untreated, MS patients versus controls) were compared with each other for each of the three available time points (baseline, follow-up year 1 and follow-up year 2). For controls, the two available time points were considered biological replicates and were averaged; these averaged expression profiles were then used for comparison with untreated MS patients for all three time points. Differential expression analysis was performed for each time point using the package ‘limma’ (54) [including gender as a covariate in the linear model in the MS versus controls comparison, as gender-specific gene expression has recently been reported in MS (21)]. Thus, three lists of genes were generated, defining differentially expressed genes as genes passing an FDR cut-off at either of the three time points. The union of these significant genes was then assessed in the replication data set. We considered as differentially expressed only those transcripts that passed FDR in the discovery data set and that reached a nominal P-value of ≤0.05 at any of the three tested time points in the replication data set. In addition to statistical significance, transcripts had to show differential regulation in the same direction (up- or down-regulated) at all three time points in both the discovery and replication data sets to be considered further.
Enrichment analysis
Analyses for the enrichment of GO and the KEGG categories were performed in R, using the Bioconductor packages ‘GO.db’, ‘KEGG.db’ and ‘GOstats’ (55), conditioning parent terms on their child terms. To assess the relative enrichment of identified genes in different cell types and tissues, we used Gene Enrichment Profiler (http://xavierlab2.mgh.harvard.edu/EnrichmentProfiler/, last accessed date on January, 2012) (37), which displays both gene expression and enrichment levels across normal tissues, reflecting the degree of specificity of a gene for a certain tissue.
In silico gene expression analysis
To validate and compare findings in our data set with previously published data, the following data sets were downloaded from GEO using the R package ‘GEOquery’ (56): GSE26104, GSE19285, GSE10655, GSE24427, GSE33464 (IFN-treated versus -untreated MS patients; IFN data) and GSE17048 (untreated MS patients versus controls: ANZgene data). Probes were assigned to genes using either contributor's annotation or, preferably, the R package ‘biomaRt’. Down-regulation of genes of interest by interferon was assessed by comparing baseline (untreated) samples with samples collected after the first injection (GSE19285), around 1 month (GSE24427, GSE10655, GSE33464) or 3 months (GSE26104) of IFN treatment using a paired, one-tailed t-test. Significance of differential expression of the genes in the identified MS signature in the ANZgene was determined using a one-tailed t-test. Global differential expression analysis with the ANZgene data was performed following our analysis strategy (unspecific filtering of probes, limma, FDR correction).
Pathway/network analysis
For network analysis, nominal P-values of all tested genes for all time points and both batches (in total six different P-values per gene) were loaded into Cytoscape (39). Using the protein–protein interaction network from the Human Protein Reference Database (HPRD; http://www.hprd.org/, last accessed date on Spring, 2012) (57–59), the plugin jActive modules (38) was run to identify interaction networks of proteins whose genes are differentially expressed in MS. jActive modules were run using the following parameters: the maximum number of modules was set to 1000, the overlap threshold between modules to 0.2 and the search depth to 2. This analysis yielded 52 significant (score >3) networks, out of which 43 were constituted of more than 10 proteins. These networks were analyzed for GO (biological processes, database downloaded on 27 March 2012) and KEGG (database downloaded on 28 March 2012) enrichments using the Cytoscape plugin ClueGO (60), assessing only categories with at least five proteins and applying fusion of similar GO terms. The Mosaic analysis was run using standard settings (61), mapping proteins in the module to GOSlim categories, using HUGO Gene Nomenclature Committee identifiers.
eQTL analysis
Genotypes were pruned using PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/, last accessed date on May, 2012) (62), excluding samples or SNPs with >10% missing data as well as SNPs with a minor allelic frequency of <0.1, being in higher LD than 0.9 (r2; assessing SNPs pair-wise in a window of 50 SNPs, before moving the window by 5 SNPs) or deviating from the Hardy–Weinberg equilibrium with a P-value <0.001.
Different sets of genes were studied in the eQTL analysis. First, we tested genes that were found to be in the MS gene expression signature. Then, we determined consistent but variably expressed genes separately in cases and controls by filtering for variance using the ‘genefilter’ R package. In cases, we filtered for genes with a difference between the 10 and 90% quantile of >1.2; in controls, this difference had to be >1.1. Only genes that passed the filter at all time points in both cases and controls were studied. In addition, genes had to be located on autosomes and the corresponding probe sets had to have unambiguous annotation (i.e. for example, probe sets aligning to multiple members of a gene family were excluded). Using these criteria, 179 genes were selected for eQTL analysis.
Gene expression levels of all time points were averaged before eQTL analysis was performed in PLINK (–assoc) and evaluated in R. In order to assess whether MS patients and controls differ in any eQTL, PLINK was run with the –gxe flag, testing the same genes. We defined cis-SNPs as SNPs located within 1 Mb upstream or downstream of the analyzed gene, trans-SNPs as all other SNPs. P-values were adjusted for the number of tests by Bonferroni correction. In order to be reported as a genetic interaction, eQTL had to be consistent, i.e. to show a significant (≤0.05) adjusted P-value in the discovery data set and a significant nominal P-value in the replication data set. We determined whether identified eQTL had been reported before by querying the GWAS catalog (http://www.genome.gov/gwastudies/, last accessed date on May, 2012) (63) and the seeQTL database (http://www.bios.unc.edu/research/genomic_software/seeQTL/, last accessed date on May, 2012) (64).
SUPPLEMENTARY MATERIAL
FUNDING
Recruitment of study participants, clinical and MRI assessment and genome scan genotyping were originally funded by GlaxoSmithKline. S.E.B. is a Harry Weaver Neuroscience Scholar of the US National MS Society (NMSS). D.N. is a fellow from the Deutsche Forschungsgemeinschaft (DFG) Research Grant Program. This work was supported by the NMSS (NMSS RG4051A2 to S.E.B.) and National Institutes of Health (R01 NS 026799 to S.L.H. and J.R.O.).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Pam Qualley, Refujia Gomez, Robin R. Lincoln and Hourieh Mousavi for patient recruitment and sample management. We thank Henrik Bengtsson (UCSF) for the generation of the custom-made cdf file.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Rosati G. The prevalence of multiple sclerosis in the world: an update. Neurol. Sci. 2001;22:117–139. doi: 10.1007/s100720170011. [DOI] [PubMed] [Google Scholar]
- 2.Sospedra M., Martin R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Oksenberg J.R., Baranzini S.E. Multiple sclerosis genetics—is the glass half full, or half empty? Nat. Rev. Neurol. 2010;6:429–437. doi: 10.1038/nrneurol.2010.91. [DOI] [PubMed] [Google Scholar]
- 4.Haines J.L., Ter-Minassian M., Bazyk A., Gusella J.F., Kim D.J., Terwedow H., Pericak-Vance M.A., Rimmler J.B., Haynes C.S., Roses A.D., et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat. Genet. 1996;13:469–471. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 5.Nagasaka Y., Dillner K., Ebise H., Teramoto R., Nakagawa H., Lilius L., Axelman K., Forsell C., Ito A., Winblad B., et al. A unique gene expression signature discriminates familial Alzheimer's disease mutation carriers from their wild-type siblings. Proc. Natl Acad. Sci. USA. 2005;102:14854–14859. doi: 10.1073/pnas.0504178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsen N.J., Moore J.H., Aune T.M. Gene expression signatures for autoimmune disease in peripheral blood mononuclear cells. Arthritis Res. Ther. 2004;6:120–128. doi: 10.1186/ar1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang Y., Gilbert D.L., Glauser T.A., Hershey A.D., Sharp F.R. Blood gene expression profiling of neurologic diseases: a pilot microarray study. Arch. Neurol. 2005;62:210–215. doi: 10.1001/archneur.62.2.210. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y., Xu H., Du X., Lit L., Walker W., Lu A., Ran R., Gregg J.P., Reilly M., Pancioli A., et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J. Cereb. Blood Flow Metab. 2006;26:1089–1102. doi: 10.1038/sj.jcbfm.9600264. [DOI] [PubMed] [Google Scholar]
- 9.Pereira E., Tamia-Ferreira M.C., Cardoso R.S., Mello S.S., Sakamoto-Hojo E.T., Passos G.A., Donadi E.A. Immunosuppressive therapy modulates T lymphocyte gene expression in patients with systemic lupus erythematosus. Immunology. 2004;113:99–105. doi: 10.1111/j.1365-2567.2004.01929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rotger M., Dang K.K., Fellay J., Heinzen E.L., Feng S., Descombes P., Shianna K.V., Ge D., Gunthard H.F., Goldstein D.B., et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. 2010;6:e1000781. doi: 10.1371/journal.ppat.1000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt S., Rainer J., Riml S., Ploner C., Jesacher S., Achmuller C., Presul E., Skvortsov S., Crazzolara R., Fiegl M., et al. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood. 2006;107:2061–2069. doi: 10.1182/blood-2005-07-2853. [DOI] [PubMed] [Google Scholar]
- 12.Achiron A., Gurevich M., Friedman N., Kaminski N., Mandel M. Blood transcriptional signatures of multiple sclerosis: unique gene expression of disease activity. Ann. Neurol. 2004;55:410–417. doi: 10.1002/ana.20008. [DOI] [PubMed] [Google Scholar]
- 13.Achiron A., Feldman A., Mandel M., Gurevich M. Impaired expression of peripheral blood apoptotic-related gene transcripts in acute multiple sclerosis relapse. Ann. NY Acad. Sci. 2007;1107:155–167. doi: 10.1196/annals.1381.017. [DOI] [PubMed] [Google Scholar]
- 14.Arthur A.T., Armati P.J., Bye C., Heard R.N., Stewart G.J., Pollard J.D., Booth D.R. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med. Genet. 2008;9:17. doi: 10.1186/1471-2350-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bomprezzi R., Ringner M., Kim S., Bittner M.L., Khan J., Chen Y., Elkahloun A., Yu A., Bielekova B., Meltzer P.S., et al. Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum. Mol. Genet. 2003;12:2191–2199. doi: 10.1093/hmg/ddg221. [DOI] [PubMed] [Google Scholar]
- 16.Booth D.R., Arthur A.T., Teutsch S.M., Bye C., Rubio J., Armati P.J., Pollard J.D., Heard R.N., Stewart G.J. Gene expression and genotyping studies implicate the interleukin 7 receptor in the pathogenesis of primary progressive multiple sclerosis. J. Mol. Med. (Berl.) 2005;83:822–830. doi: 10.1007/s00109-005-0684-y. [DOI] [PubMed] [Google Scholar]
- 17.Brynedal B., Khademi M., Wallstrom E., Hillert J., Olsson T., Duvefelt K. Gene expression profiling in multiple sclerosis: a disease of the central nervous system, but with relapses triggered in the periphery? Neurobiol. Dis. 2010;37:613–621. doi: 10.1016/j.nbd.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 18.Gandhi K.S., McKay F.C., Cox M., Riveros C., Armstrong N., Heard R.N., Vucic S., Williams D.W., Stankovich J., Brown M., et al. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 2010;19:2134–2143. doi: 10.1093/hmg/ddq090. [DOI] [PubMed] [Google Scholar]
- 19.Iglesias A.H., Camelo S., Hwang D., Villanueva R., Stephanopoulos G., Dangond F. Microarray detection of E2F pathway activation and other targets in multiple sclerosis peripheral blood mononuclear cells. J. Neuroimmunol. 2004;150:163–177. doi: 10.1016/j.jneuroim.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Martinelli-Boneschi F., Fenoglio C., Brambilla P., Sorosina M., Giacalone G., Esposito F., Serpente M., Cantoni C., Ridolfi E., Rodegher M., et al. MicroRNA and mRNA expression profile screening in multiple sclerosis patients to unravel novel pathogenic steps and identify potential biomarkers. Neurosci. Lett. 2012;508:4–8. doi: 10.1016/j.neulet.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Menon R., Di Dario M., Cordiglieri C., Musio S., La Mantia L., Milanese C., Di Stefano A.L., Crabbio M., Franciotta D., Bergamaschi R., et al. Gender-based blood transcriptomes and interactomes in multiple sclerosis: involvement of SP1 dependent gene transcription. J. Autoimmun. 2012;38:J144–J155. doi: 10.1016/j.jaut.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Ramanathan M., Weinstock-Guttman B., Nguyen L.T., Badgett D., Miller C., Patrick K., Brownscheidle C., Jacobs L. In vivo gene expression revealed by cDNA arrays: the pattern in relapsing-remitting multiple sclerosis patients compared with normal subjects. J. Neuroimmunol. 2001;116:213–219. doi: 10.1016/s0165-5728(01)00308-3. [DOI] [PubMed] [Google Scholar]
- 23.Riveros C., Mellor D., Gandhi K.S., McKay F.C., Cox M.B., Berretta R., Vaezpour S.Y., Inostroza-Ponta M., Broadley S.A., Heard R.N., et al. A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS One. 2010;5:e14176. doi: 10.1371/journal.pone.0014176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkijarvi S., Kuusisto H., Paalavuo R., Levula M., Airla N., Lehtimaki T., Kaprio J., Koskenvuo M., Elovaara I. Gene expression profiles in Finnish twins with multiple sclerosis. BMC Med. Genet. 2006;7:11. doi: 10.1186/1471-2350-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satoh J., Nakanishi M., Koike F., Miyake S., Yamamoto T., Kawai M., Kikuchi S., Nomura K., Yokoyama K., Ota K., et al. Microarray analysis identifies an aberrant expression of apoptosis and DNA damage-regulatory genes in multiple sclerosis. Neurobiol. Dis. 2005;18:537–550. doi: 10.1016/j.nbd.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Tian Y., Apperson M.L., Ander B.P., Liu D., Stomova B.S., Jickling G.C., Enriquez R., Agius M.A., Sharp F.R. Differences in exon expression and alternatively spliced genes in blood of multiple sclerosis compared to healthy control subjects. J. Neuroimmunol. 2011;230:124–129. doi: 10.1016/j.jneuroim.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 27.van Baarsen L.G., van der Pouw Kraan T.C., Kragt J.J., Baggen J.M., Rustenburg F., Hooper T., Meilof J.F., Fero M.J., Dijkstra C.D., Polman C.H., et al. A subtype of multiple sclerosis defined by an activated immune defense program. Genes Immun. 2006;7:522–531. doi: 10.1038/sj.gene.6364324. [DOI] [PubMed] [Google Scholar]
- 28.Ottoboni L., Keenan B.T., Tamayo P., Kuchroo M., Mesirov J.P., Buckle G.J., Khoury S.J., Hafler D.A., Weiner H.L., De Jager P.L. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Sci. Transl. Med. 2012;4:153ra131. doi: 10.1126/scitranslmed.3004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achiron A., Feldman A., Magalashvili D., Dolev M., Gurevich M. Suppressed RNA-polymerase 1 pathway is associated with benign multiple sclerosis. PLoS One. 2012;7:e46871. doi: 10.1371/journal.pone.0046871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gurevich M., Achiron A. The switch between relapse and remission in multiple sclerosis: continuous inflammatory response balanced by Th1 suppression and neurotrophic factors. J. Neuroimmunol. 2012;252:83–88. doi: 10.1016/j.jneuroim.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Cox M.B., Bowden N.A., Scott R.J., Lechner-Scott J. Altered expression of the plasminogen activation pathway in peripheral blood mononuclear cells in multiple sclerosis: possible pathomechanism of matrix metalloproteinase activation. Mult. Scler. 2013 doi: 10.1177/1352458513475493. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Cheung V.G., Spielman R.S. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat. Rev. Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lev N., Ickowicz D., Barhum Y., Blondheim N., Melamed E., Offen D. Experimental encephalomyelitis induces changes in DJ-1: implications for oxidative stress in multiple sclerosis. Antioxid. Redox Signal. 2006;8:1987–1995. doi: 10.1089/ars.2006.8.1987. [DOI] [PubMed] [Google Scholar]
- 35.Hirotani M., Maita C., Niino M., Iguchi-Ariga S., Hamada S., Ariga H., Sasaki H. Correlation between DJ-1 levels in the cerebrospinal fluid and the progression of disabilities in multiple sclerosis patients. Mult. Scler. 2008;14:1056–1060. doi: 10.1177/1352458508093616. [DOI] [PubMed] [Google Scholar]
- 36.van Horssen J., Drexhage J.A., Flor T., Gerritsen W., van der Valk P., de Vries H.E. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic. Biol. Med. 2010;49:1283–1289. doi: 10.1016/j.freeradbiomed.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 37.Benita Y., Cao Z., Giallourakis C., Li C., Gardet A., Xavier R.J. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood. 2010;115:5376–5384. doi: 10.1182/blood-2010-01-263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ideker T., Ozier O., Schwikowski B., Siegel A.F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl. 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 39.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baranzini S.E., Wang J., Gibson R.A., Galwey N., Naegelin Y., Barkhof F., Radue E.W., Lindberg R.L., Uitdehaag B.M., Johnson M.R., et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009;18:767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Condamine T., Le Texier L., Howie D., Lavault A., Hill M., Halary F., Cobbold S., Waldmann H., Cuturi M.C., Chiffoleau E. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J. Leukoc. Biol. 2010;88:507–515. doi: 10.1189/jlb.1109738. [DOI] [PubMed] [Google Scholar]
- 42.Fissolo N., Kraus M., Reich M., Ayturan M., Overkleeft H., Driessen C., Weissert R. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 2008;38:2401–2411. doi: 10.1002/eji.200838413. [DOI] [PubMed] [Google Scholar]
- 43.Mayo I., Arribas J., Villoslada P., Alvarez DoForno R., Rodriguez-Vilarino S., Montalban X., De Sagarra M.R., Castano J.G. The proteasome is a major autoantigen in multiple sclerosis. Brain. 2002;125:2658–2667. doi: 10.1093/brain/awf274. [DOI] [PubMed] [Google Scholar]
- 44.Mishto M., Bellavista E., Ligorio C., Textoris-Taube K., Santoro A., Giordano M., D'Alfonso S., Listi F., Nacmias B., Cellini E., et al. Immunoproteasome LMP2 60HH variant alters MBP epitope generation and reduces the risk to develop multiple sclerosis in Italian female population. PLoS One. 2010;5:e9287. doi: 10.1371/journal.pone.0009287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma M., Burre J., Sudhof T.C. Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci. Transl. Med. 2012;4:147ra113. doi: 10.1126/scitranslmed.3004028. [DOI] [PubMed] [Google Scholar]
- 46.Wexler E.M., Rosen E., Lu D., Osborn G.E., Martin E., Raybould H., Geschwind D.H. Genome-wide analysis of a Wnt1-regulated transcriptional network implicates neurodegenerative pathways. Sci. Signal. 2011;4:ra65. doi: 10.1126/scisignal.2002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galimberti D., Macmurray J., Scalabrini D., Fenoglio C., De Riz M., Comi C., Comings D., Cortini F., Villa C., Serpente M., et al. GSK3beta genetic variability in patients with multiple sclerosis. Neurosci. Lett. 2011;497:46–48. doi: 10.1016/j.neulet.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 48.Fancy S.P., Harrington E.P., Yuen T.J., Silbereis J.C., Zhao C., Baranzini S.E., Bruce C.C., Otero J.J., Huang E.J., Nusse R., et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fancy S.P., Baranzini S.E., Zhao C., Yuk D.I., Irvine K.A., Kaing S., Sanai N., Franklin R.J., Rowitch D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goertsches R.H., Zettl U.K., Hecker M. Sieving treatment biomarkers from blood gene-expression profiles: a pharmacogenomic update on two types of multiple sclerosis therapy. Pharmacogenomics. 2011;12:423–432. doi: 10.2217/pgs.10.190. [DOI] [PubMed] [Google Scholar]
- 51.Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 52.Kauffmann A., Gentleman R., Huber W. arrayQualityMetrics—a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415–416. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan S., Zhang W., Bleibel W.K., Cox N.J., Dolan M.E. SNPinProbe_1.0: a database for filtering out probes in the Affymetrix GeneChip human exon 1.0 ST array potentially affected by SNPs. Bioinformation. 2008;2:469–470. doi: 10.6026/97320630002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 55.Falcon S., Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 56.Davis S., Meltzer P.S. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and Bioconductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 57.Mishra G.R., Suresh M., Kumaran K., Kannabiran N., Suresh S., Bala P., Shivakumar K., Anuradha N., Reddy R., Raghavan T.M., et al. Human protein reference database – 2006 update. Nucleic Acids Res. 2006;34:D411–D414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peri S., Navarro J.D., Amanchy R., Kristiansen T.Z., Jonnalagadda C.K., Surendranath V., Niranjan V., Muthusamy B., Gandhi T.K., Gronborg M., et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prasad T.S., Kandasamy K., Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol. Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 60.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.H., Pages F., Trajanoski Z., Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang C., Hanspers K., Kuchinsky A., Salomonis N., Xu D., Pico A.R. Mosaic: making biological sense of complex networks. Bioinformatics. 2012;28:1943–1944. doi: 10.1093/bioinformatics/bts278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xia K., Shabalin A.A., Huang S., Madar V., Zhou Y.H., Wang W., Zou F., Sun W., Sullivan P.F., Wright F.A. seeQTL: a searchable database for human eQTLs. Bioinformatics. 2012;28:451–452. doi: 10.1093/bioinformatics/btr678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saiki S., Sato S., Hattori N. Molecular pathogenesis of Parkinson's disease: update. J. Neurol. Neurosurg. Psychiatry. 2012;83:430–436. doi: 10.1136/jnnp-2011-301205. [DOI] [PubMed] [Google Scholar]
- 66.Bonifati V., Dekker M.C., Vanacore N., Fabbrini G., Squitieri F., Marconi R., Antonini A., Brustenghi P., Dalla Libera A., De Mari M., et al. Autosomal recessive early onset parkinsonism is linked to three loci: PARK2, PARK6, and PARK7. Neurol. Sci. 2002;23(Suppl. 2):S59–S60. doi: 10.1007/s100720200069. [DOI] [PubMed] [Google Scholar]
- 67.Sun Z., Ren H., Liu Y., Teeling J.L., Gu J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 2011;85:1036–1047. doi: 10.1128/JVI.01734-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dominguez I., Sonenshein G.E., Seldin D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell. Mol. Life Sci. 2009;66:1850–1857. doi: 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harney S.M., Vilarino-Guell C., Adamopoulos I.E., Sims A.M., Lawrence R.W., Cardon L.R., Newton J.L., Meisel C., Pointon J.J., Darke C., et al. Fine mapping of the MHC Class III region demonstrates association of AIF1 and rheumatoid arthritis. Rheumatology (Oxford) 2008;47:1761–1767. doi: 10.1093/rheumatology/ken376. [DOI] [PubMed] [Google Scholar]
- 70.Bin L., Howell M.D., Kim B.E., Hall C.F., Streib J.E., Leung D.Y. Inhibition of S100A11 gene expression impairs keratinocyte response against vaccinia virus through downregulation of the IL-10 receptor 2 chain. J. Allergy Clin. Immunol. 2009;124:270–277. doi: 10.1016/j.jaci.2009.05.002. 277.e271. [DOI] [PubMed] [Google Scholar]
- 71.Mulcahy H., O'Rourke K.P., Adams C., Molloy M.G., O'Gara F. LST1 and NCR3 expression in autoimmune inflammation and in response to IFN-gamma, LPS and microbial infection. Immunogenetics. 2006;57:893–903. doi: 10.1007/s00251-005-0057-2. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki T., Ishii-Watabe A., Tada M., Kobayashi T., Kanayasu-Toyoda T., Kawanishi T., Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J. Immunol. 2010;184:1968–1976. doi: 10.4049/jimmunol.0903296. [DOI] [PubMed] [Google Scholar]
- 73.Shi Y., Chen L., Liotta L.A., Wan H.H., Rodgers G.P. Glia maturation factor gamma (GMFG): a cytokine-responsive protein during hematopoietic lineage development and its functional genomics analysis. Genomics Proteomics Bioinformatics. 2006;4:145–155. doi: 10.1016/S1672-0229(06)60027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du H., Huang X., Wang S., Wu Y., Xu W., Li M. PSMA7, a potential biomarker of diseases. Protein Pept. Lett. 2009;16:486–489. doi: 10.2174/092986609788167824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.