

Abstract
BACKGROUND & AIMS
Caspase recruitment domain 9 (CARD9) is an adaptor protein that integrates signals downstream of pattern recognition receptors. CARD9 has been associated with autoinflammatory disorders, and loss-of-function mutations have been associated with chronic mucocutaneous candidiasis, but the role of CARD9 in intestinal inflammation is unknown. We characterized the role of Card9 in mucosal immune responses to intestinal epithelial injury and infection.
METHODS
We induced intestinal inflammation in Card9-null mice by administration of dextran sulfate sodium (DSS) or Citrobacter rodentium. We analyzed body weight, assessed inflammation by histology, and measured levels of cytokines and chemokines using quantitative reverse-transcription polymerase chain reaction and enzyme-linked immunosorbent assay. Cell populations were compared between wild-type and Card9-null mice by flow cytometry analysis.
RESULTS
Colon tissues and mesenteric lymph nodes of Card9-null mice had reduced levels of interleukin (IL)-6, interferon-γ, and T-helper (Th)17 cytokines after administration of DSS, compared with wild-type mice. IL-17A and IL-22 expression were reduced in the recovery phase after DSS administration, coincident with decreased expression of antimicrobial peptides and the chemokine (C-C motif) ligand 20 (Ccl20). Although Card9-null mice had more intestinal fungi based on 18S analysis, their Th17 responses remained defective even when an antifungal agent was administered throughout DSS exposure. Moreover, Card9-null mice had impaired immune responses to C rodentium, characterized by decreased levels of colonic IL-6, IL-17A, IL-22, and regenerating islet-derived 3 gamma (RegIIIγ), as well as fewer IL-22—producing innate lymphoid cells (ILCs) in colon lamina propria.
CONCLUSIONS
The adaptor protein CARD9 coordinates Th17- and innate lymphoid cell-mediated intestinal immune responses after epithelial injury in mice.
Keywords: Colitis, ILC, Inflammatory Response, Mouse Model
The gastrointestinal tract is lined by epithelium, which not only serves as a protective barrier but also assists in nutrient absorption and innate immunity. The mucosa exists indynamic equilibrium with luminal flora and dietary antigens, and immune homeostasis is maintained with the aid of a pre-epithelial layer consisting of protective mucus, antimicrobial peptides (AMPs), and IgA. The intestinal immune system is poised to detect bacterial antigens at the mucosal surface and drive appropriate responses, depending on the nature of the stimulus, to maintain tolerance or initiate immune signaling. In some circumstances, tolerance toward the microbiota is lost, leading to inappropriate immune responses and inflammation. This is the case in inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, and, importantly, an altered composition of the gut microbiota, termed dysbiosis, has been reported in inflammatory bowel disease patients.
Caspase recruitment domain 9 (CARD9) is an adaptor protein that assists in the integration of signals downstream of pattern recognition receptors.1 In particular, CARD9 plays a major role in sensing fungi via several C-type lectins including dectin-1, dectin-2, and mincle. For example, dectin-1 binding by β-glucan, a component of the fungal wall, successively induces the activation of SYK, CARD9, Bcl10, and MALT1, leading to activation of nuclear factor-κB, c-Jun-N-terminal kinase (JNK), and p38 mitogen-activated protein kinase pathways.2,3 Furthermore, CARD9 is required to mount adaptive T-helper (Th)17 responses during fungal infection via dectin-14–6 and during Mycobacteria infection via mincle.7 A loss-of-function mutation in CARD9 was identified in patients with chronic mucocutaneous candidiasis and congenital susceptibility to Candida,4 supporting a role for CARD9 in antifungal innate immunity in human beings.
In addition to its role in sensing fungi, CARD9 is involved in innate immune responses to bacteria and viruses. CARD9 mediates mincle-dependent Mycobacteria sensing and Nod2-dependent p38/JNK signaling,8 enhances Toll-like receptor signaling,9,10 and facilitates microbe-elicited reactive oxygen species production.11 Furthermore, p38 and JNK activation, as well as cytokine production after infection with RNA viruses, relies on CARD9 through Mda5, RIG-I, and MAVS.12 CARD9 is therefore a key adapter protein for innate immunity against a range of microorganisms including intestinal commensals and pathogens.
Although the role of CARD9 in systemic immunity has been explored,1 its function in the gastrointestinal immune system has not yet been investigated. Here, we show that Card9 is a key molecule in intestinal homeostasis and is required to mount appropriate interleukin (IL)-6, IL-17A, interferon (IFN)γ, and IL-22 responses. We show that Card9-null mice are more susceptible to dextran sulfate sodium (DSS)-induced colitis and that recovery is impaired in this model. The enhanced inflammation in Card9-null mice is characterized by defective IFNγ and Th17 cytokine responses, as well as impaired chemokine responses. By using the Th17-dependent Citrobacter rodentium colitis model, we further show that Card9-null mice have fewer colonic Th17 cells and innate lymphoid cells (ILCs) than wild-type mice. As a result, Card9-null mice develop more severe inflammation during C rodentium-induced colitis, indicating that Card9 is important to reestablish homeostasis after epithelial injury.
Materials and Methods
DSS
Mice were fed 3% (wt/vol) DSS (molecular weight, 36,000–50,000; MP Biomedicals, Solon, OH) dissolved in sterile drinking water ad libitum. Animals were monitored daily for weight loss.
C rodentium Infection
Mice were inoculated orally with C rodentium (strain DBS100; American Type Culture Collection, Manassas, VA) as previously described.13,14 Bacteria were grown overnight in Luria broth and resuspended in phosphate-buffered saline (PBS) before infection (0.5 mL/mouse, 1 × 109 colony forming units [CFU] unless otherwise noted). When used, nalidixic acid was administered by intragastric gavage (100 mg/mice) 24 hours before infection with C rodentium. To assess clearance of C rodentium, fecal pellets were homogenized, serially diluted, and plated onto selective MacConkey agar (selective for gram-negative organisms). After overnight culture at 37°C, colonies were counted based on the size and distinctive appearance of C rodentium colonies (pink center with white outer rim), as described previously.
Lymphocyte Isolation and Measurement of Cytokine Production
Lymphocytes were cultured in the presence or absence of anti-CD3 (10 μg/mL) or C rodentium Antigen (50 mg/mL) for 72 hours. Supernatants were analyzed using an enzyme-linked immunosorbent assay (see the Supplementary Materials and Methods section for details).
Ccl20 Assay
Splenic macrophages were isolated using MACs CD11b+ magnetic beads per the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). Cells (1 × 105) were stimulated with 100 ng/mL lipopolysaccharide, 10 μg/mL muramyl dipeptide, and 50 μg/mL tumor necrosis factor (TNF)-α for 4 days, and supernatants were analyzed for chemokine (C-C motif) ligand 20 (Ccl20) production per the manufacturer’s protocol (Quantikine; R&D Systems, Minneapolis, MN).
Laser Injury and Epithelial Permeability Assay
We used a custom-built, 2-photon endomicroscopy system with a side-view probe as previously described.15,16 Micro-wounds were introduced in colonic epithelium via laser, and the epithelial barrier function was assessed 4 days later (see the Supplementary Materials and Methods section for details).
Fungal Translocation
Mesenteric lymph nodes (MLNs) were harvested after DSS recovery (day 12) and 18S ribosomal RNA levels were determined using quantitative reverse-transcription polymerase chain reaction (qRT-PCR) normalized to glyceraldehyde-3-phosphate dehydrogenase (see the Supplementary Materials and Methods section for details).
Mouse Anti-Saccharomyces cerevisiae Antibody Detection
Serum was harvested on days 0, 7, and 12 after DSS exposure as described earlier. Anti–Saccharomyces cerevisiae antibody (ASCA) levels were quantified by an enzyme-linked immunosorbent assay (MyBiosource, San Diego, CA).
Fluconazole Administration
Mice were fed 0.5 mg/mL fluconazole in drinking water (Sigma-Aldrich, St. Louis, MO) 2 days before DSS administration, and everyday thereafter, as previously described.
Lamina Propria Isolation
Colonic and small intestine lamina propria (LP) cells were isolated as previously described with slight modifications (see the Supplementary Materials and Methods section for details).
Statistics
Statistical analyses were performed using the 2-tailed Student t test for unpaired data or by the nonparametric Mann–Whitney test. Differences with a P value less than .05 were considered significant.
Results
Card9-Null Mice Show Impaired Recovery After DSS-Induced Injury
To determine the role of Card9 in mucosal immune responses, we worked with 2 mouse models of epithelial injury. Previous studies have shown that Card9-null mice have normal B-cell, T-cell, and myeloid cell development and function, suggesting that the immune system is intact.1 Upon histologic examination, intestinal epithelial cell distribution appeared normal, including Paneth and goblet cells, and we did not observe spontaneous colitis (data not shown). We therefore decided to induce intestinal injury and inflammation using the DSS colitis model. Mice were killed after receiving 3% DSS in their drinking water for 7 days (acute injury, day 7), or after an additional 5 days in which DSS was discontinued (recovery, day 12). The severity of acute injury (defined by weight loss, colon length, and histologic score) was not statistically different between wild-type and Card9-null mice 7 days after initial DSS exposure (Supplementary Figure 1). However, during recovery, Card9-null mice had greater body weight loss (Figure 1A). Colon shortening, a macroscopic parameter of colitis severity, was more pronounced in Card9-null mice than in wild-type littermates (5.68 ± 0.23 cm vs 6.99 ± 0.30 cm; Figure 1B), and histologic scores were significantly higher in Card9-null mice (Figure 1C and D) 5 days after DSS was discontinued. These deficits during the recovery phase after DSS injury suggest that Card9 may play a role in intestinal healing.
Figure 1.
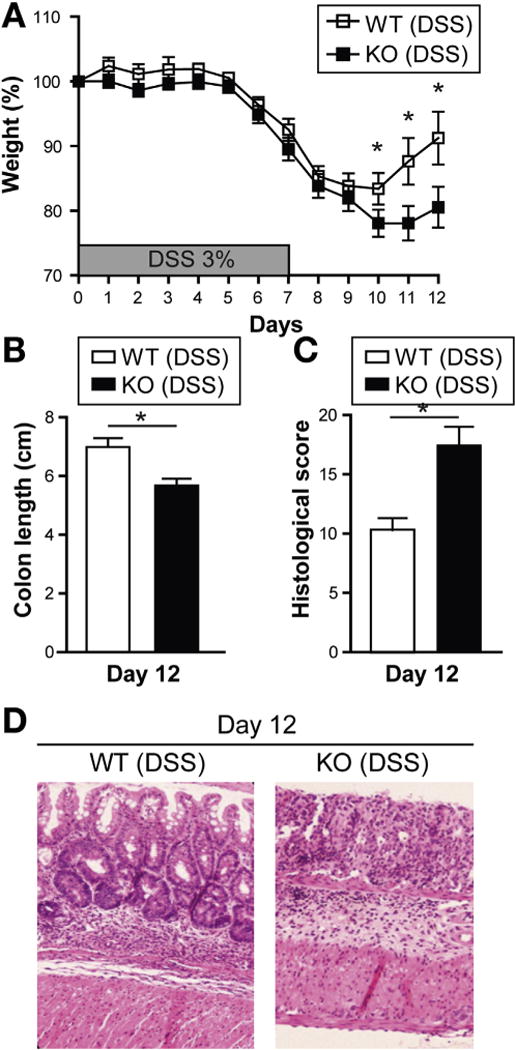
Card9-null mice show impaired recovery after DSS-induced injury. (A) Weight loss in DSS-exposed wild-type (WT) and Card9-null (KO) mice. (B) Colon length of WT and KO mice after administration of DSS. (C) Histologic score of WT and KO mice after administration of DSS. (D) H&E staining of representative cross-sections of medial colon (magnification, 20×). Data are representative of at least 2 independent experiments (N = 5). Error bars represent SEM. *P < .05.
Card9-Null Mice Show Impaired Immune Responses After DSS Administration
To decipher the mechanisms leading to defective recovery from DSS-induced injury in Card9-null mice, we assessed colonic expression levels of both proinflammatory cytokines and cytokines that promote epithelial healing via qRT-PCR (Figure 2A). No significant differences were observed between wild-type and Card9-null mice in the absence of DSS (data not shown). We observed that during acute injury and after discontinuing DSS (recovery), the expression of TNFα, IL-10, and IL-23R were increased similarly in wild-type and Card9-null mice, whereas IL-23, IL-17A, and IFNg were significantly lower in Card9-null mice. The expression of IL-6, IL-17F, IL-21, and IL-22 also were decreased significantly in Card9-null colon compared with wild-type, however, only during recovery after DSS was discontinued (Figure 2A). Systemic IL-6 and IL-17A were similar in wild-type and Card9-null mice after recovery, suggesting that these responses were specific for the intestinal environment (Supplementary Figure 2A). Furthermore, we found decreased IFNγ, IL-17A, IL-21, and IL-22 expression in Card9-null MLNs compared with wild-type, as well as impaired IFNγ and IL-17A secretion after T-cell stimulation (Figure 2B). These results suggest that deficient IL-17 and IL-22 cytokine responses may underlie the defective recovery of Card9-null mice during DSS-induced epithelial injury. Importantly, IL-6 protects the intestinal epithelium from injury by regulating intestinal trefoil factor and/or AMP secretion.18,19 Therefore, the defect in IL-6 expression observed in Card9-null mice also may play a role in impaired epithelial restitution.
Figure 2.
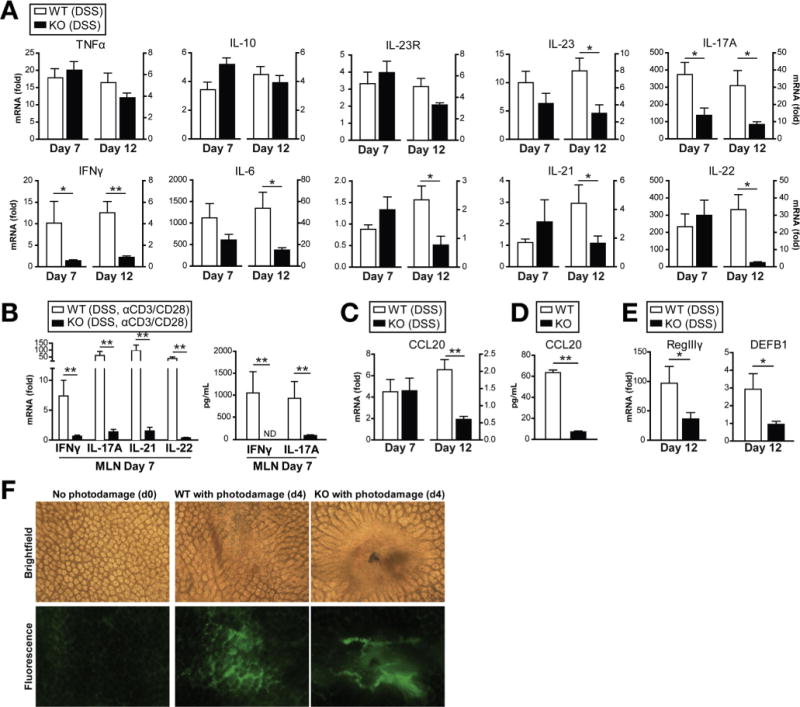
Card9-null mice show impaired immune responses after DSS administration. (A) Cytokine expression in colon of wild-type (WT) and Card9-null (KO) mice as quantified by qRT-PCR. Data are shown as fold changes relative to WT mice without DSS for each time point. (B) Cytokine expression by qRT-PCR (left) and secretion by ELISA (right) in MLN cells after DSS administration and after stimulation with anti-CD3/anti-CD28 antibodies. Data are shown as fold changes relative to WT mice without anti-CD3/anti-CD28. (C) Ccl20 expression in colon of WT and KO mice after DSS administration. Quantification by qRT-PCR. Data are shown as fold changes relative to WT mice without DSS for each time point. (D) Isolated splenic macrophages were stimulated with 100 ng/mL lipopolysaccharide, 10 μg/mL MDP, and 50 μg/mL TNFα for 4 days. Culture supernatants were harvested and analyzed for Ccl20 via enzyme-linked immunosorbent assay. (E) AMP expression in colon of WT and KO mice after recovery from DSS-induced injury. Quantification by qRT-PCR. Data are shown as fold changes relative to WT mice without DSS. (F) Femtosecond laser injuries were introduced into colonic epithelium via a 2-photon endomicroscopy system. Mice were injected with 2 million Daltons fluorescein isothiocyanate dextran before colonoscopy to determine successful photodamage (formation of intravascular clots and extravasation of fluorescent dye after laser exposure). Four days after injury, colons were stained with Evans Blue dye and imaged using bright-field microscopy (top). Residual fluorescein isothiocyanate fluorescence also was assessed using fluorescence microscopy (bottom). Data are representative of at least 2 independent experiments (N = 5). Error bars represent SEM. *P < .05, **P < .01. ND, not detectable.
Chemokines play a major role in recruiting immune cells to sites of injury or infection. Ccl20 is involved in recruitment of Ccr6-expressing cells, including Th17 cells and ILCs, which are the main producers of IL-17 and IL-22 in the gut. In agreement with the cytokine expression results, which showed lower levels of IL-17A, IL-21, and IL-22 in Card9-null colon, expression of the chemokines Ccl20 and monocyte chemoattractant protein-1 (Mcp1) were decreased significantly in Card9-null colon, compared with wild-type, 5 days after DSS was discontinued (Figure 2C and Supplementary Figure 2B). Moreover, when primary macrophages, key Ccl20 secretors, were activated ex vivo, Card9-null macrophages showed impaired production of the Th17-recruiting chemokine (Figure 2D).
To investigate the consequences of reduced IL-17 and IL-22 expression in the intestine of Card9-null mice, we assessed downstream effectors induced by these cytokines. IL-22–mediated signaling acts through STAT3 to regulate mucosal wound healing in intestinal epithelial cells.20 Similar to Card9-null mice, IL-22-null mice develop more severe DSS-induced colitis as measured by weight loss and disease activity, with major differences during recovery.20 IL-22 induces expression of AMPs and the antibacterial protein regenerating islet-derived 3 gamma (RegIIIg) in epithelial cells.21 In line with these data, the expression of RegIIIγ in Card9-null colon was significantly lower than in wild-type mice 5 days after DSS was discontinued (Figure 2E). Moreover, in accordance with the role of IL-17A and IL-17F in the induction of colonic β-defensin (DEFB1) expression,22 β-defensin 1 expression was decreased in Card9-null colon, compared with wild-type, after 5 days of recovery (Figure 2E). Decreased AMP expression in Card9-null mice after recovery may lead to altered responses to intestinal microbiota and suggests one potential mechanism for the observed restitution defect.
To directly assess the defective epithelial restitution in Card9-null mice, we used an in vivo model system to induce epithelial microwounds using laser photodamage. By using 2-photon endomicroscopy, we were able to induce photodamage in the colonic epithelium of wild-type and Card9-null mice and assess epithelial permeability over time. After 4 days, Card9-null epithelium showed more vascular leakage (as detected via fluorescein isothiocyanate dextran) and epithelial permeability (as detected by Evans Blue dye) at the site of injury (Figure 2F). Moreover, although the site of injury was not detected in wild-type mice after 7 days, the site of injury remained identifiable in Card9-null mice at this time point, indicating impaired wound healing (data not shown). Thus, Card9 regulates wound healing, possibly via IL-22-specific functions.
Taken together, these results show that, after DSS exposure, Card9-null mice show impaired epithelial recovery, which is characterized by defective Th17 cytokines, IFNγ, and IL-6 production. The data suggest that repair processes are compromised, which can be explained in part by defective production of restitutive cytokines and AMPs.
Card9 Is Required to Control Fungi in Gut Microbiota
Given the impaired Th17 responses observed in Card9-null mice after DSS injury, we next quantified the level of segmented filamentous bacteria (SFB) present in wild-type and Card9-null mice, because SFB colonization is required for Th17 cell development23,24 (Supplementary Figure 3). We found no difference in SFB levels between wild-type and Card9-null mice in the gut microbiota, ruling out a role for defective SFB colonization in the Th17 phenotype observed in Card9-null mice.
Because Card9 is involved in the immune response to fungi, we also assessed the level of total fungi in the gut microbiota of wild-type and Card9-null mice using 18S analysis (Figure 3A). Interestingly, the level of total fungi was 70% higher in the gut microbiota of Card9-null mice compared with wild-type (Figure 3A). Moreover, greater fungal translocation to the MLN, as measured by 18S qRT-PCR, and higher levels of systemic ASCA were detected in Card9-null mice 5 days after withdrawal of DSS (Figure 3B and C). These data suggest that the gastrointestinal immune system is less efficient in controlling fungi in Card9-null mice. To address whether this alteration in Card9-null mice accounts for enhanced susceptibility to inflammation, we administered the antifungal drug fluconazole before, during, and after DSS exposure. Efficient depletion of the fungi population in the small intestine and colon was validated using qRT-PCR (data not shown). As previously published,17 wild-type mice lost less body weight in the absence of fungi. Similarly, fluconazole-treated Card9-null mice showed less body weight loss than their nontreated counterparts. However, the fluconazole-treated Card9-null mice showed greater weight loss than fluconazole-treated wild-type mice (Figure 3D). Consistent with our observations from mice treated only with DSS (Figure 2B), MLN T cells from fluconazole-treated Card9-null mice produced significantly lower amounts of IL-17A and IFNγ than wild-type mice (Figure 3E). Together, these observations suggest that fungal burden alone does not account for the differences we report.
Figure 3.
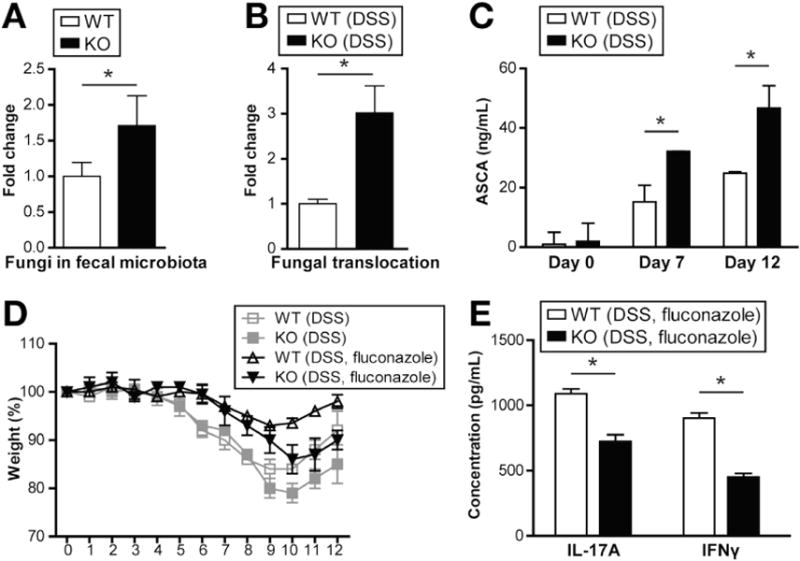
Card9 is required to control fungi in gut microbiota. (A) Basal fungi levels in fecal microbiota of wild-type (WT) and Card9-null (KO) mice were quantified by 18S qRT-PCR, normalized to the threshold cycle value of the 16S ribosomal RNA gene. Data are plotted relative to WT mice. (B) MLNs were harvested 5 days after DSS was discontinued, and 18S fungi levels were determined by qRT-PCR normalized to glyceraldehyde-3-phosphate dehydrogenase. Data are plotted relative to WT mice. (C) Serum ASCA concentrations were quantified on days 0, 7 (acute injury), and 12 (recovery) via enzyme-linked immunosorbent assay. (D) Weight loss in DSS-treated WT and KO mice. Indicated mice were treated with 0.5 mg/mL fluconazole 2 days before, and throughout the 12-day DSS course described earlier. (E) MLN T cells from fluconazole-treated mice (after acute injury) were stimulated with anti-CD3/anti-CD28 for detection of IFNγ and IL-17A by enzyme-linked immunosorbent assay. Data are representative of at least 2 independent experiments (N = 5–6). Error bars represent SEM. *P < .05.
Card9 Is Required to Mount an Appropriate Immune Response Toward C Rodentium
The results acquired in 2 epithelial injury models suggest that Card9 is involved in Th17 responses, and we therefore next used the C rodentium colitis model in which IL-17A, IL-17F, and IL-22 are required for protection against infection.22,25 C rodentium is a gram-negative attaching and effacing bacterial pathogen equivalent to the human enteropathogenic Escherichia coli,26 which infects mouse colon and causes inflammation. We first assessed colonic RegIIIγ, IL-6, IL-17A, and IL-22 expression after C rodentium infection (Figure 4A). As previously reported,25 IL-22, IL-6, and RegIIIγ were induced rapidly, with peak expression around day 5 after infection, whereas IL-17A induction was slower, reaching maximal levels 2 weeks after infection (Figure 4A). In Card9-null mice, the induction of RegIIIγ, IL-6, and Th17 cytokines was lower compared with wild-type mice, suggesting an impaired immune response to C rodentium.
Figure 4.
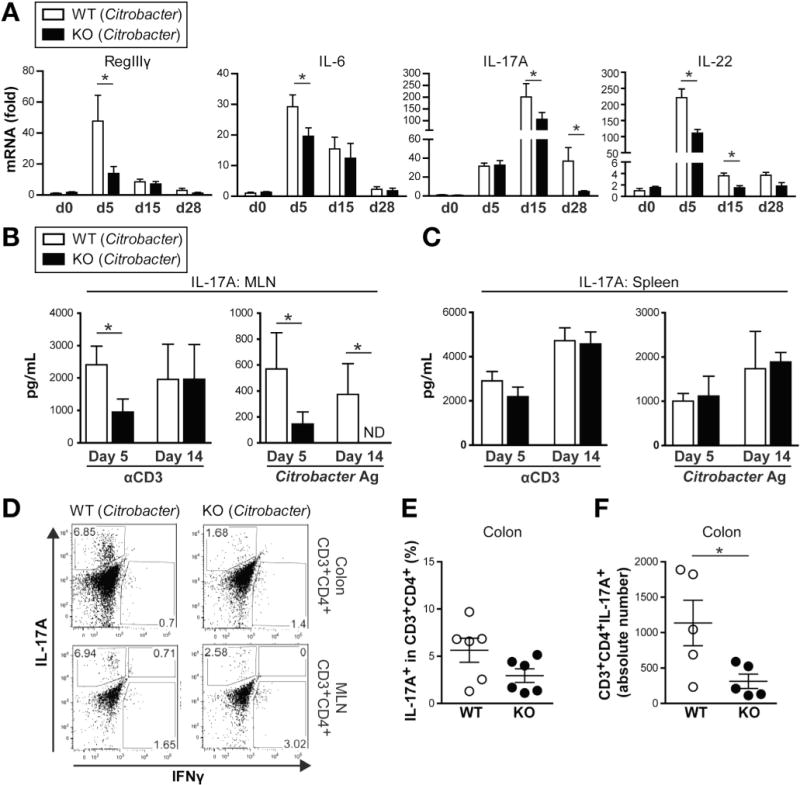
Card9 is required for Th17 and ILC responses in colon after C rodentium infection. (A) Cytokine expression in colon of wild-type (WT) and Card9-null (KO) mice after C rodentium infection, as quantified by qRT-PCR. Data are shown as fold changes relative to uninfected WT mice. (B–C) Secretion of IL-17A by (B) MLN and (C) spleen cells after C rodentium infection. Cells were stimulated with anti-CD3 or C rodentium antigen. ND, not detectable. (D) IL-17A and IFNγ intracellular staining of CD3+CD4+ T cells from colon LP lymphocytes (top) and MLN (bottom). Representative fluorescence-activated cell sorter plots are shown. (E) Proportion of IL-17A+ cells among colon LP lymphocyte CD3+CD4+ T cells. (F) Number of CD3+CD4+IL-17A+ cells in colon LP lymphocytes. Data are representative of at least 2 independent experiments (N = 5). Error bars represent SEM. *P < .05.
We further investigated the immune response elicited by C rodentium by examining total and antigen-specific T-cell cytokine production in MLNs and spleen 5 and 14 days after infection. Isolated MLN lymphocytes were restimulated in vitro with C rodentium antigen or anti-CD3 antibody and, as anticipated, IL-17A production in response to both stimulations was significantly lower in Card9-null mice on day 5 (Figure 4B). However, 2 weeks after infection, the differences between wild-type and Card9-null IL-17A levels persisted only after antigen-specific stimulation (Figure 4B). No significant differences were observed in the spleen, suggesting that T-cell responses are not globally defective but are impaired specifically in cells involved in gut immunity (Figure 4C). Similar results were observed for IFNγ secretion (Supplementary Figure 4A and B).
We next isolated colonic LP cells to assess the frequency and number of Th17 cells in wild-type and Card9-null mice. As shown in Figure 4D–F, Card9-null mice had significantly fewer CD3+CD4+IL-17A+ cells present during C rodentium infection. Given this phenotype in the C rodentium model, we revisited the DSS injury model and likewise observed significantly fewer CD3+CD4+IL-17A+ cells in Card9-null mice in this model (Supplementary Figure 2C and D). Taken together, these data confirm that Card9 is required for Th17-mediated intestinal immune responses.
Card9 Deficiency Is Associated With Decreased ILCs in the Gastrointestinal Tract During C Rodentium Infection
In addition to Th17 cells, ILCs also produce IL-17A and IL-22 in the gut, particularly during C rodentium infection.27–31 Analysis of colon LP cells 5 days after C rodentium infection showed that Card9-null mice had a decreased frequency and number of CD3−CD4+IL-17A+ and CD3−CD4−NKP46+ ILC populations compared with wild-type mice (Figure 5A–F). These results suggest that Card9 is required not only for the development of Th17 cells, but also is important for the generation and/or recruitment of ILCs in the gastrointestinal tract during C rodentium infection.
Figure 5.
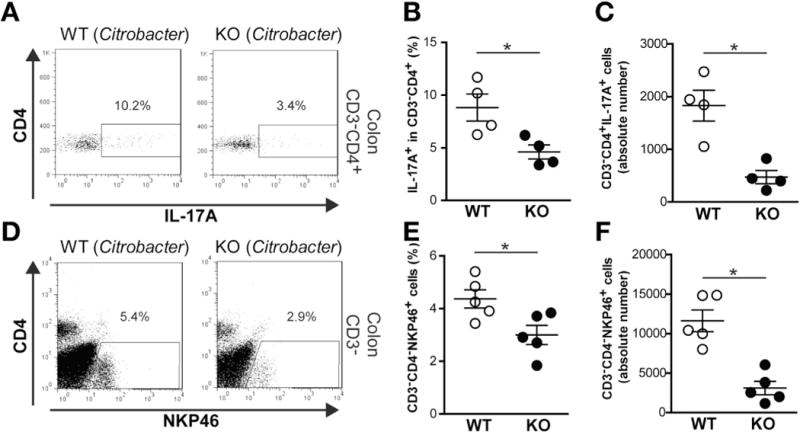
Card9 deficiency is associated with decreased ILCs in the gastrointestinal tract during C rodentium infection. (A) Representative fluorescence-activated cell sorter plots of CD3−CD4+IL-17A+ ILCs in colon LP cells from wild-type (WT) and Card9-null (KO) mice. (B) Proportion and (C) number of CD3−CD4+IL-17A+ ILCs in colon LP. (D) Representative fluorescence-activated cell sorter plots of CD3−CD4−NKP46+ ILCs in colon LP cells. (E) Proportion and (F) number of CD3−CD4−NKP46+ ILCs in colon LP. Data are representative of at least 2 independent experiments (N = 4–6). Error bars represent SEM. *P < .05.
Card9 Is Required to Control C rodentium Infection
Given the observed defect in IL-17A and IL-22 pathways in Card9-null mice during C rodentium–induced colitis, we hypothesized that these mice would be unable to control bacterial propagation and therefore would be more susceptible to infection. As predicted, the levels of C rodentium present in the feces of Card9-null mice were higher than in wild-type mice 7 and 15 days after oral infection with C rodentium (Figure 6A), indicating impairment in bacterial clearance. Complementing these results, we observed increased C rodentium translocation to the spleen 5 days after infection in Card9-null mice compared with wild-type mice (Figure 6B). In addition, the severity of colitis, as assessed by histologic scoring, was higher in Card9-null mice 15 days after infection (Figure 6C and D). Despite increased colitis severity, no mortality was observed and all animals recovered within several weeks.
Figure 6.
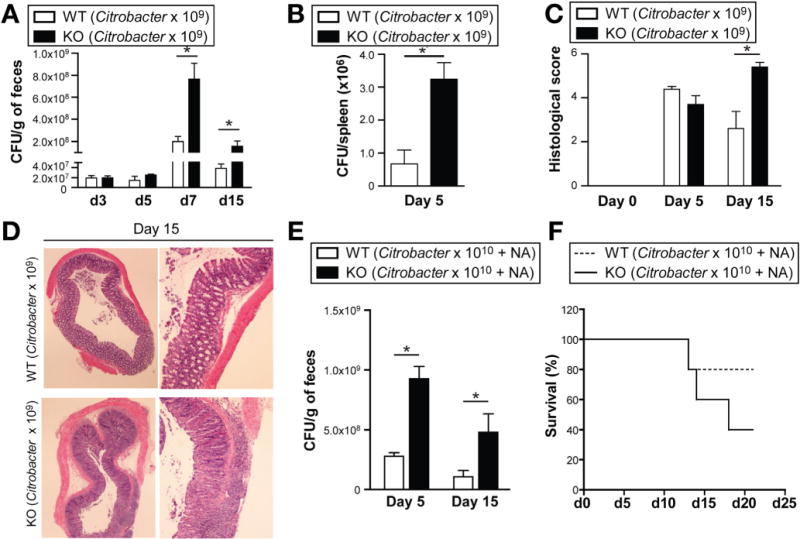
Card9 is required to control C rodentium infection. (A) C rodentium count in feces after infection in wild-type (WT) and Card9-null (KO). (B) C rodentium count in spleen 5 days after infection. (C) Colitis histology score at indicated time points after infection. (D) H&E staining of representative cross-sections of medial colon (magnification, 20x) on day 15 after infection. (E) C rodentium count in feces of mice pretreated with 100 mg nalidixic acid (NA) and infected with 1010 CFU C rodentium. (F) Survival curve of mice pretreated with 100 μg NA and infected with 1010 CFU C rodentium. Data are representative of at least 2 independent experiments (N = 5). Error bars represent SEM. *P < .05.
To better discriminate the altered phenotype in Card9-null mice, we used a modified protocol for C rodentium infection.32 Mice were pretreated with nalidixic acid to disrupt the microbiota and infected with 1 × 1010 CFU C rodentium, a dose 10-fold higher than in the earlier-described experiments. In addition to the expected higher levels of C rodentium in the feces of Card9-null mice (Figure 6E), the Card9-null mice showed an increased mortality rate compared with wild-type mice (Figure 6F), confirming heightened susceptibility to this bacterium.
Discussion
CARD9 is a key adapter molecule that regulates responses to various microbes as well as responses to tissue injury associated with high levels of apoptosis.33 Although the role of CARD9 in systemic immunity against fungi and other microbes has been explored,1 its function in the gastrointestinal tract was unknown before this report. Here, we present evidence that Card9 is important in epithelial injury, ILC, and Th17 responses. Card9-null mice were more susceptible to DSS colitis than wild-type mice as a result of delayed recovery characterized by impaired colonic IFNγ, IL-6, IL-17A, IL-22, and Ccl20 expression. In addition, AMP expression was reduced in Card9-null mice, implicating a defect in the epithelial responses important for healing. By using the C rodentium colitis model, which requires IL-22, IL-17A, and IL-6 for protection,22,25,32 we validated the findings observed in the DSS system. As predicted, Card9-null mice showed impaired expression of IL-6, IL-17A, IL-22, and RegIIIγ in the colon, and fewer Th17 cells and ILCs were detected in the colon LP during C rodentium infection. Based on the insight gained from these 2 models, we propose that Card9 mediates IL-17 and IL-22 signaling pathways, which subsequently regulate intestinal defense mechanisms.
Many pathways function in a coordinated manner to restore homeostasis after epithelial injury, and the data shown here suggest that Card9 may be a novel player in these restitutive processes. After epithelial damage, cytokines and chemokines are secreted to recruit immune cells to the site of injury, whereas necrotic and apoptotic cells release prostaglandin E2 and other growth factors to induce cellular proliferation.34 Wnt5a expression is known to regulate transforming growth factor-β and is required for intestinal epithelial crypt generation, high-lighting the diverse pathways necessary for epithelial restitution.35 In addition, IL-22 also regulates mucosal wound healing,20 and defects in this cytokine exacerbate DSS-induced colitis.36 IL-22 aids in intestinal restitution by inducing expression of epithelial RegIIIγ; as expected, we show here that Card9-null mice express significantly lower levels of RegIIIγ compared with wild-type after recovery from DSS injury. Moreover, Card9-null mice displayed impaired healing after the introduction of epithelial micro-wounds using laser photodamage, high-lighting an inherent defect in restitution. Thus, Card9 expression is required for reestablishing epithelial homeostasis after injury.
ILCs, a main source of IL-22 in both mice and human beings, continuously produce IL-22 to fortify the epithelial barrier.37–39 Because IL-22 messenger RNA levels after acute DSS injury (day 7) were comparable in wild-type and Card9-null mice, but significantly reduced in Card9-null mice after recovery (day 12), we speculate that ILC recruitment and/or proliferation, rather than development, requires Card9 signaling, which will be the subject of future studies. Consistent with this hypothesis, we show that the reduced colonic IL-22 and IL-17 levels in Card9-null mice correlate with a decreased number of CD3−CD4+IL17+ and CD3−CD4+NKp46+ ILCs after epithelial injury. Expression of Ccl20, the ligand for the chemokine receptor Ccr6 that is secreted by myeloid cells to recruit Th17 and ILC populations to the site of injury, is also lower in Card9-null mice after recovery from DSS. Moreover, activated Card9-null macrophages secrete significantly less Ccl20 in response to bacterial ligands in vitro. Together, these data suggest that Card9 may control ILC recruitment to the injury site through induction of Ccl20 by myeloid cells. In addition to ILCs, we show lower absolute numbers of Th17 cells in Card9-null mice after epithelial injury, further suggesting that aberrant Ccl20-mediated recruitment of innate and adaptive immune cells may contribute to enhanced inflammation. Alternatively, Card9 may control ILC and Th17 expansion and function by inducing homeostatic cytokines such as IL-23, which we have shown to be lower in Card9-null mice after recovery from DSS-induced injury.
In addition to defective Th17 cell recruitment, T-cell differentiation also may be compromised in Card9-null mice. IL-6 is required for Th17 cell differentiation in mice,40 and here we show lower levels of IL-6 in Card9-null mice during colitis, which is coincident with a decrease in colonic Th17 cells. Indeed, signals downstream of the Card9 receptors dectin-1 and dectin-2, as well as other pattern recognition receptors, have been shown to direct T-cell differentiation, depending on the receptors that are engaged.41 Recent studies have shown that SFB can induce Th17 differentiation24; however, our data suggest that this mechanism is not skewing our results because both wild-type and Card9-null mice possess similar levels of these bacteria. Therefore, Card9 not only may regulate immune cell recruitment, but also may direct effector T-cell differentiation, both of which are critical during mucosal inflammation.
In addition to bacteria, an abundant and diverse fungal population exists in the gut42 and CARD9 plays a major role in fungal sensing. Mice lacking dectin-1, a Card9-dependent receptor, show increased susceptibility to DSS colitis with an altered response to commensal fungi.17 Similarly, we show here that in the absence of Card9, greater fungal colonization exists in the intestine, suggesting that this adaptor protein plays a role in controlling gut fungi. Consistent with this finding, Card9-null mice have higher systemic ASCA and fungal translocation after DSS injury. However, our experiments using anti-fungal administration suggest that the greater fungal burden does not solely account for the impaired Th17 and IL-22 responses we describe. Therefore, Card9 signaling regulates fungi and, in addition to other Card9-mediated processes, plays a role in intestinal homeostasis.
In conclusion, we have shown that Card9 orchestrates Th17- and ILC-mediated immune responses, which are important for restitution after epithelial injury. The mechanisms by which Card9 regulates these processes will be the subject of future studies.
Supplementary Material
Acknowledgments
Funding
Supported by grants from the French Society of Gastroenterology, the Bettencourt Schueller Foundation, the Ferring Fellowship, and the Arthur Sachs Scholarship (H.S.); The Netherlands Organization for Scientific Research (C.W.); by grants from the National Institutes of Health (DK083756, DK043351, DK086502, and DK060049), the Crohn’s and Colitis Foundation of America, and The Leona M. and Harry B. Helmsley Charitable Trust (R.J.X.).
Editorial assistance was provided by Natalia Nedelsky, as supported by National Institutes of Health grant DK043351 (R.J.X.).
Abbreviations used in this paper
- AMP
antimicrobial peptide
- ASCA
anti–Saccharomyces cerevisiae antibody
- CARD9
caspase recruitment domain 9
- Ccl20
chemokine (C-C motif) ligand 20
- CFU
colony forming unit
- DSS
dextran sulfate sodium
- IFN
interferon
- IL
interleukin
- ILC
innate lymphoid cell
- JNK
c-Jun-N-terminal kinase
- LP
lamina propria
- MLN
mesenteric lymph node
- PBS
phosphate-buffered saline
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- SFB
segmented filamentous bacteria
- Th
T-helper
- TNF
tumor necrosis factor
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Ruland J. CARD9 signaling in the innate immune response. Ann N Y Acad Sci. 2008;1143:35–44. doi: 10.1196/annals.1443.024. [DOI] [PubMed] [Google Scholar]
- 2.Gross O, Gewies A, Finger K, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 3.Hara H, Ishihara C, Takeuchi A, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol. 2007;8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- 4.Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gringhuis SI, Wevers BA, Kaptein TM, et al. Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog. 2011;7:e1001259. doi: 10.1371/journal.ppat.1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 7.Schoenen H, Bodendorfer B, Hitchens K, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010;184:2756–2760. doi: 10.4049/jimmunol.0904013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu YM, Zhang Y, You Y, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 9.Goodridge HS, Shimada T, Wolf AJ, et al. Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J Immunol. 2009;182:1146–1154. doi: 10.4049/jimmunol.182.2.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hara H, Ishihara C, Takeuchi A, et al. Cell type-specific regulation of ITAM-mediated NF-kappaB activation by the adaptors, CARMA1 and CARD9. J Immunol. 2008;181:918–930. doi: 10.4049/jimmunol.181.2.918. [DOI] [PubMed] [Google Scholar]
- 11.Wu W, Hsu YM, Bi L, et al. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat Immunol. 2009;10:1208–1214. doi: 10.1038/ni.1788. [DOI] [PubMed] [Google Scholar]
- 12.Poeck H, Bscheider M, Gross O, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 13.Vallance BA, Deng W, Knodler LA, et al. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun. 2002;70:2070–2081. doi: 10.1128/IAI.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CC, Louie S, McCormick B, et al. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect Immun. 2005;73:5468–5481. doi: 10.1128/IAI.73.9.5468-5481.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim P, Chung E, Yamashita H, et al. In vivo wide-area cellular imaging by side-view endomicroscopy. Nat Methods. 2010;7:303–305. doi: 10.1038/nmeth.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JK, Lee WM, Kim P, et al. Fabrication and operation of GRIN probes for in vivo fluorescence cellular imaging of internal organs in small animals. Nat Protoc. 2012;7:1456–1469. doi: 10.1038/nprot.2012.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iliev ID, Funari VA, Taylor KD, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mashimo H, Wu DC, Podolsky DK, et al. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274:262–265. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 20.Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 22.Ishigame H, Kakuta S, Nagai T, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 26.Schauer DB, Zabel BA, Pedraza IF, et al. Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J Clin Microbiol. 1995;33:2064–2068. doi: 10.1128/jcm.33.8.2064-2068.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cella M, Fuchs A, Vermi W, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanos SL, Bui VL, Mortha A, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Sonnenberg GF, Monticelli LA, Elloso MM, et al. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takatori H, Kanno Y, Watford WT, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geddes K, Rubino SJ, Magalhaes JG, et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med. 2011;17:837–844. doi: 10.1038/nm.2391. [DOI] [PubMed] [Google Scholar]
- 33.Hara H, Saito T. CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 2009;30:234–242. doi: 10.1016/j.it.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12:170–180. doi: 10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- 35.Miyoshi H, Ajima R, Luo CT, et al. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 37.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Sonnenberg GF, Monticelli LA, Alenghat T, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearson C, Uhlig HH, Powrie F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012;33:289–296. doi: 10.1016/j.it.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ott SJ, Kuhbacher T, Musfeldt M, et al. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol. 2008;43:831–841. doi: 10.1080/00365520801935434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.