

Abstract
Understanding the migration patterns of invasive organisms is of paramount importance to predict and prevent their further spread. Previous attempts at reconstructing the entire history of the sudden oak death (SOD) epidemic in California were limited by: (1) incomplete sampling; (2) the inability to include infestations caused by a single genotype of the pathogen; (3) collapsing of non-spatially contiguous yet genetically similar samples into large meta-samples that confounded the coalescent analyses. Here, we employ an intensive sampling coverage of 832 isolates of Phytopthora ramorum (the causative agent of SOD) from 60 California forests, genotyped at nine microsatellite loci, to reconstruct its invasion. By using age of infestation as a constraint on coalescent analyses, by dividing genetically indistinguishable meta-populations into highly-resolved sets of spatially contiguous populations, and by using Bruvo genetic distances for most analyses, we reconstruct the entire history of the epidemic and convincingly show infected nursery plants are the original source for the entire California epidemic. Results indicate that multiple human-mediated introductions occurred in most counties and that further disease sources were represented by large wild infestations. The study also identifies minor introductions, some of them relatively recent, linked to infected ornamental plants. Finally, using archival isolates collected soon after the discovery of the pathogen in California, we corroborate that the epidemic is likely to have resulted form 3 to 4 core founder individuals evolved from a single genotype. This is probably the most complete reconstruction ever completed for an invasion by an exotic forest pathogen, and the approach here described may be useful for the reconstruction of invasions by any clonally reproducing organism with a relatively limited natural dispersal range.
Electronic supplementary material
The online version of this article (doi:10.1007/s10530-013-0453-8) contains supplementary material, which is available to authorized users.
Keywords: Emergent forest disease, Genetic epidemiology, Microsatellite, Coalescent analysis, Network analysis
Introduction
Since the time-scale of most biological invasions is so short, population genetic inference based upon the neutral theory of molecular evolution is typically limited (at least for eukaryotes) to questions such as determining the geographic origin of the invasion, testing for bottlenecks, and testing for hybridization with native species (Fitzpatrick et al. 2012). Viruses and pathogenic microbes (e.g. bacteria, phytoplasmas, and some fungi) with high mutation rates and short generation times may be exceptions to this rule and indeed genetic data are increasingly being used to trace disease transmission pathways. For example, the high-rate of nucleotide sequence evolution of RNA viruses permits detailed phylogenetic reconstructions of disease transmission pathways even within the short time-scale of epidemics. Disease outbreaks studied in this way include foot-and-mouth disease (Cottam et al. 2008), HIV (Leitner and Albert 1999), hepatitis C (Bracho et al. 2005), Ebolavirus (Walsh et al. 2005) and SARS coronavirus (Liu et al. 2005). Recent analytical advances are also beginning to permit the combining of genetic data with traditional epidemiological data (e.g. the tracing of contacts, timing and spatial proximity of infections). Within the field of disease ecology, the use of genetic data and their integration with information regarding the timing, spread and incidence of disease outbreaks has been more limited. This may be because of differences in specialization between field pathologists and molecular biologists (Milgroom and Peever 2003), because of greater difficulty in obtaining detailed epidemiological data, and because basic knowledge about the population biology and genetics of many economically important pathogens is lacking (Fletcher et al. 2006; Goss et al. 2009b). Although newly emergent plant pathogens are often typified by reduced genetic diversity due to strong bottlenecks experienced during intercontinental transport combined with the strictly clonal reproduction occurring when a single mating type is introduced (Engelbrecht et al. 2004; Garbelotto 2008; Gonthier and Garbelotto 2011), strong directional selection processes may occur as introduced pathogens colonize new habitats (Goss et al. 2009b; Grünwald and Goss 2011; Paoletti et al. 2006). Consequently, these pathogens experience local diversification that may be tracked at highly-variable neutral markers as novel genotypes emerge. This pattern of clonal evolution permits the identification of particular genotypes, which may be traced to infer the origins, relationships and transmission routes of epidemics (Cummings and Relman 2002; Goss et al. 2009b). Such “microbial forensics” is made possible by the increasingly easy development of polymorphic markers and by the advent of coalescent algorithms that permit detailed assessment of population genetic parameters (e.g. effective population size and migration rates) over time (Grünwald and Goss 2011). The power of these approaches can be enhanced by the integration of historical and epidemiological data (Milgroom and Peever 2003).
The sudden oak death (SOD) pathogen Phytopthora ramorum (Straminopila: Oomycetes, Peronosporales) (Werres et al. 2001) evolves in a clonal, stepwise manner characterized by the local evolution of novel genotypes from introduced founders (Ivors et al. 2006; Mascheretti et al. 2008, 2009; Prospero et al. 2007, 2009). P. ramorum has reached epidemic levels in the native coastal forests of California and southern Oregon (Rizzo and Garbelotto 2003), causing lethal trunk lesions on tanoak (Lithocarpus densiflora), coast live oak (Quercus agrifolia), Californian black oak (Quercus kelloggii), and possibly Canyon live oak (Quercus chrysolepis). The pathogen also causes a more progressive die-back called “ramorum blight” in over 100 species of wild and ornamental plants (Davidson et al. 2003; Grünwald et al. 2008; Rizzo et al. 2005). SOD symptoms were first noticed in 1994–1995 in Californian oaks and tanoaks (Garbelotto et al. 2001; Rizzo et al. 2002) and in Rhododendron and Viburnum plants in European nurseries (Werres et al. 2001). Subsequent studies showed that the two most relevant infectious substrates in California forests are provided by bay laurel (Umbellularia californica) and tanoak leaves, while oaks and tanoak boles appear to be non-infectious (Reviewed in Rizzo and Garbelotto 2003).
Genetic data suggest that P. ramorum is an exotic pathogen in Europe and in North America, introduced in the nursery trade of both continents from unknown origins at least four times independently—as indicated by the presence of four clonal lineages: EU1, EU2, NA1, and NA2 (reviewed in Garbelotto and Hayden 2012). NA1 is also responsible for the natural infestation in North American forests, while EU1, NA1, and NA2 genotypes have all been isolated only from nurseries in North America (Goss et al. 2011; Hansen et al. 2003; Ivors et al. 2004). Forest infestations are present in a single county in Southern Oregon (Curry County), and in 14 contiguous counties in California from Humboldt to Monterey, spanning over 700 km of mostly coastal habitat (Fig. 1; also see www.sodmap.org). The EU2 lineage has been recently discovered in formal gardens of Northern Ireland and Northwestern Great Britain (Van Poucke et al. 2012). Indeed, the four lineages probably originated from geographically isolated populations that have been clonally reproducing for almost 200,000 years and have a reduced ability to reproduce sexually (Goss et al. 2009b).
Fig. 1.
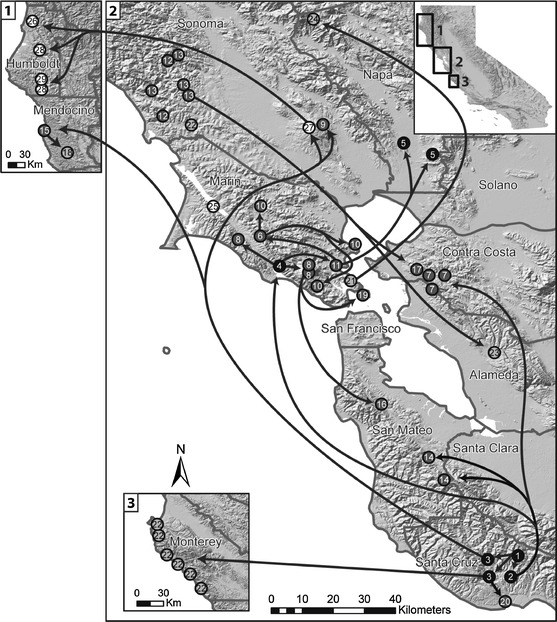
Map showing the major locations of the forests from which P. ramorum isolates were gathered and also representing the overall range of distribution of P. ramorum in California. Transmission routes supported by coalescent analysis are shown. Populations are shaded according to their likely membership in four genetic clusters: black with white text = cluster 1; dark gray with white text = cluster 3; light gray with black text = cluster 4, white with black text = cluster 2. Numbers refer to final populations collapsed into the same population if genetically undistinguishable within each county (Supplemental Table 1)
Epidemiological (Davidson et al. 2005) and population genetic studies (Mascheretti et al. 2008, 2009; Prospero et al. 2004, 2007) indicate that although wild populations are genetically structured due to limited natural dispersal abilities, there may be widespread long-distance gene flow due to human mediated transport. Human mediated transport through the movement of infected ornamental plants among nurseries across North America and between Europe and North America is clear (Goss et al. 2009b, 2011). At a more local scale, the presence of genetically identical forest populations at great distances, a scenario that can only be justified by human-mediated transport of the pathogen, has also been explained as the result of the concurrent movement of infected ornamental plants in different locations (Mascheretti et al. 2008, 2009). Despite the contributions of these previous studies to our understanding of the mode of spread and of the epidemic history of P. ramorum in California wild lands, many important aspects remain unresolved. First, the original coalescent inferences of migration routes within California were tentative and often inconclusive because not all infested localities were sampled, and all genetically indistinguishable populations were pooled into “meta-populations” regardless of location (Mascheretti et al. 2009). Second, no robust method existed for placing infestations caused by singleton genotypes within the genetic reconstruction of the epidemic. Because new wild infestations of most diseases are often characterized by the presence of a single genotype, this was a significant shortcoming. Third, while 3 genotypes were suggested as being the original founders of all forest infestations in California, that inference was based only on their relative dominance and positioning within a minimum-spanning network (Mascheretti et al. 2008, 2009). Previous analyses did not include historical isolates (i.e. collected before 2005) to corroborate such hypothesis, thus ignoring the possibility that current genotypes may be the result of rapid adaptation and may not represent the original founder individuals.
Here, to reconstruct the history and the routes of the invasion, we use a greatly expanded data set, encompassing samples from all known significant SOD locations, and for which age of the infestation is known. Additionally, genetically similar populations from spatially heterogeneous locations are not collapsed, but are treated as separate units, and two additional microsatellite loci are used to increase the power of the analysis. We extend our approaches to include the positioning of individual genotypes into a robust backbone population tree, and, finally, we objectively integrate historical data (e.g. infestation age) without assumptions of directionality and without subjectively positioning any population as basal or derived, into genealogical coalescence-based migration model choice. To shed light on the microevolutionary history of the pathogen in its new California range, we use historical isolates collected in the first 5 years following the discovery of P. ramorum in California to corroborate the likely number of founder individuals.
Specifically, our aims were: (1) to reconstruct the migration history of the pathogen with high resolution; (2) to identify through network analyses the multilocus genotypes (MGs) that most closely resemble those that were first introduced into California, using historical samples collected around the time when P. ramorum was first identified in Californian forests; (3) to reconstruct the evolution of MGs throughout the time-course of the epidemic and infer the likely sources of newly discovered infestations, even when only a single isolate per new infestation may be available. These aims are relevant to studies of all invasive organisms.
Materials and methods
Phytopthora ramorum sampling and microsatellite genotyping
A total of 813 isolates of P. ramorum were collected, mostly from bay laurel and occasionally from coast live oak and tanoak in 60 forests stands across 13 California counties (Alameda, San Mateo, Sonoma, Napa, Santa Cruz, Monterey, Humboldt, Marin, Santa Clara, Solano, Mendocino, Contra Costa and San Francisco) between 2002 and 2010 (Fig. 1). Additionally, the analysis included 14 samples collected from 12 nurseries in 12 US States in 2004, and 5 samples collected from a California nursery located near one of the two sites that were identified as the source of the entire California epidemic in Mascheretti et al. (2008). For some localities, only a single sample was available. Figure 1 and Supplemental Table 1 show the localities where the samples were collected. P. ramorum was cultured and isolated from the symptomatic portion of leaves and DNA extracted as previously described (Mascheretti et al. 2008).
The following microsatellite loci were genotyped: PrMS39a, PrMS39b, PrMS43a, PrMS43b, PrMS45 (Prospero et al. 2007), locus 18, locus 64 (Ivors et al. 2006) and loci ILVO145PrMS145 (a, c) (Vercauteren et al. 2010). For the full genotyping methodology see the Supplementary data.
Statistical analyses I: populations
Iterative collapsing of genetically indistinguishable populations
In order to avoid making population genetic inferences among populations which were not statistically significantly differentiated, populations were recursively clustered based upon minimum Φ ST, an analogue of Wright’s F ST that takes the evolutionary distance between individual haplotypes or alleles into account (Excoffier et al. 1992). The genetic differentiation (Φ ST) among all 62 pairs of P. ramorum forest and nursery populations was evaluated as the between population proportion of molecular variance from amova (Excoffier et al. 1992), implemented by arlequin v.3.5 (Excoffier et al. 2005). The Bruvo distance (Bruvo et al. 2004) among each pair of unique MGs was used as the evolutionary distance and given to arlequin as an external file, and statistical significance evaluated by permuting individuals across locations 10,000 times. Populations were recursively clustered by pooling the pair of populations or clusters that yielded the minimum non-significant Φ ST at each round until no further insignificant clustering (i.e. minimum Φ ST P > 0.05) was possible (Roewer et al. 2005; Mascheretti et al. 2008, 2009). All populations with fewer than five isolates were excluded from this analysis to minimize the influence of stochastic sampling error on the greedy clustering algorithm (singleton samples were analyzed separately: see Materials and Methods: Statistical Analysis—Singletons; below). The algorithm was supervised by applying it only to populations from within the same county. Following the county-based clustering, the algorithm was continued to completion, permitting collapses among counties. Additionally, we re-ran the algorithm without supervision. The results were evaluated based upon knowledge from previous analyses of P. ramorum populations (Mascheretti et al. 2008, 2009) and by subjecting the final set of populations from each run (by county, overall, and unsupervised) to a traditional hierarchical amova—evaluating the Φ CT value, which provides an unbiased way to judge population groupings i.e. by maximizing the ‘among group’ variation (whilst minimizing the ‘within group variation’) (Dupanloup et al. 2002). Examination of Φ CT the values indicated that the supervised clustering (by county Φ CT = 0.3083, P < 0.001) was superior to the unsupervised algorithm (unsupervised Φ CT = 0.2879, P < 0.001), but that allowing collapsing among counties offered little further improvement (overall Φ CT = 0.3099, P < 0.001). Since collapses among counties may reflect recent shared source populations rather than direct migration between these populations, the ‘by county’ set of populations was conservatively chosen as the basis for subsequent analyses.
Evaluation of the genetic relationships among populations
The genetic relationships among the final set of populations (post-clustering) were visualized by estimating the matrix of average pair-wise Bruvo genetic distances (Bruvo et al. 2004) among all populations with 5 or more samples and using these to construct the shortest (minimum evolution) tree using the neighbor-joining (NJ) clustering method with branch-swapping as implemented by FastME v.2.07 (Desper and Gascuel 2002). Bootstrap values (1000) were generated similarly and summarized using consense [phylip package v.3.6 (Felsenstein 2005)]. Populations with two-to-four samples were then added to this tree by supplying paup* v.4.0b10 (Swofford 1999) with the matrix of pair-wise distances for all populations and estimating an NJ tree using the FastME tree (without branch-lengths) as a backbone constraint.
Bayesian inference of minimal infection routes
We have previously (Mascheretti et al. 2009) attempted to infer infection routes by estimating all bidirectional migration rates M among populations using migrate-n (Beerli and Felsenstein 2001; Beerli 2006). Migrate-n uses coalescent simulations of genetic data to infer effective population sizes and past migration rates among n populations. It assumes that the migration matrix model is asymmetric and that population sizes need not be equal, it is therefore a very flexible and powerful tool. However, our previous use of this approach suffered from three significant limitations. First, the number of parameters to be estimated increases dramatically with the number of populations: limiting accuracy and increasing computational complexity. Second, samples were probably over-collapsed such that some populations with quite different histories were merged, potentially confounding the analysis. Third, it is very unlikely that most P. ramorum populations approach genetic equilibrium, violating the assumptions of migrate-n and rendering estimates of M unreliable. Here, rather than estimate M, we used the output ln marginal likelihoods (ln(ml)) to choose among different migration models (Beerli and Palczewski 2010) and reconstruct the minimal, most probable, set of unidirectional migration routes among the new and highly-resolved population dataset. Twenty-nine populations (n ≥ 5) yield a computationally intractable number of possible models and we therefore took a stepwise approach to make the analysis computationally tractable as described in Croucher et al. (2012) (Details in supplemental data). The number of possible models was limited by incorporating “epidemiological” data (known age of infestation—6 categories: 20+ years; 15–20 years; 10–15 years; <10 years; <8 years; <5 years)—disallowing models that included migration from a younger to an older population.
Statistical analyses II: genetic clustering and placement of singletons and ‘historical’ samples
Relationships of single isolate samples to the other populations were evaluated by re-running the NJ tree with the singletons (using the backbone tree) in paup* v.4.0b10 (Swofford 1999).
All individuals and populations were also subject to genetic clustering analysis using the non-admixing model in structure (Pritchard et al. 2000). Structure is a Bayesian model-based clustering procedure that assigns individuals to a predefined number of genetic clusters k in such a way as to minimize Hardy–Weinberg disequilibrium and linkage disequilibrium. In a fully clonal organism, such P. ramorum, the genotypes at the loci within an individual will not be independent, violating the assumptions of structure. Since there is only a single coalescent tree for a clonal organism, any clusters returned by structure will simply reflect the major clades of that tree and will therefore describe the structure of the variation present in the species rather than defining a population structure. Additionally, Structure has previously been applied to clonally reproducing organisms including P. ramorum (Goss et al. 2009b). Our aim was to ascertain whether there was any clear sub-structure in the historical patterns of clonal variation present in California, in particular in terms of whether the outbreak may have resulted from one or more introduced MGs. The posterior probability of membership in each of the identified genetic clusters was examined for all populations, singletons, and ‘historical’ isolates. Full details of the structure analysis are given in the supplementary information.
A sub-structure consisting of four genetic clusters or coalescent clades (k = 4) was inferred from the structure analysis (see Results). The percentage posterior probability of membership in each of the four genetic clusters was examined. In order to visualize overall population membership in each of the genetic clusters, the genetic overlap (Ayala 1983) between each pair of populations (A, B) was defined based upon the posterior probability of cluster membership i.e. min(Pr(Cluster1A),Pr(Cluster1B)) + min(Pr(Cluster2A),Pr(Cluster2B)) + min(Pr(Cluster3A),Pr(Cluster3B)) + min(Pr(Cluster4A),Pr(Cluster4B)). This estimate was converted to a Genetic Cluster Dissimilarity (GCD) by subtracting the value from 1, and the matrix of pair-wise estimates clustered (NJ) using FastME v.2.07 (Desper and Gascuel 2002) (Fig. 2b).
Fig. 2.
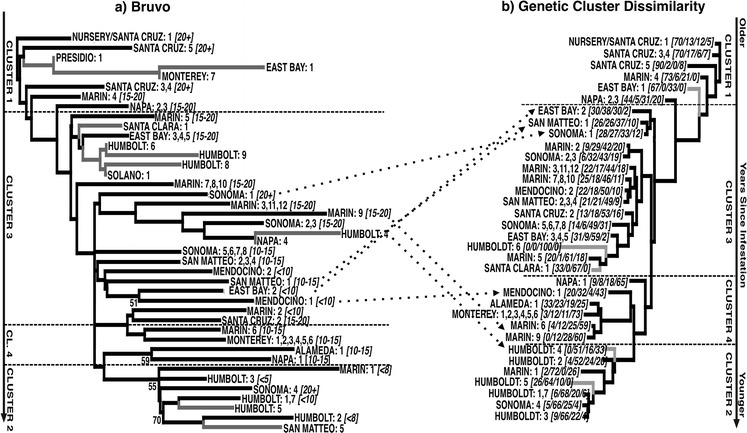
Neighbor-joining representations of the genetic relationships among P. ramorum populations. a NJ-tree based upon the Bruvo genetic distance: backbone-tree generated using populations with sample sizes ≥5, with populations of sample sizes 1–5 (grey branches) appended in PAUP* in a second step. Numbers in parentheses following the population names are years since infestation. b All populations with n > 1 clustered (NJ) according to Genetic Cluster Dissimilarity (1 genetic overlap) as derived from the structure defined genetic clusters. The numbers in parenthesis indicate the percent posterior probability of membership in each of the genetic clusters (1, 2, 3, 4) identified by structure. The vertical arrows to either side of the figures indicate the time-course from older to younger infestations. The horizontal dashed lines mark the boundaries of the genetic clusters as defined by NJ analysis (clustering) of the Genetic Cluster Dissimilarity index (the location of these lines is approximate in Fig. 2a). Populations moving markedly between representations in a, b are indicated by dotted arrows
The mutational steps and likely evolutionary relationships between individual MGs were examined by generating a minimum spanning network (MSN) using splitstree 4 (Huson and Bryant 2006) from the Bruvo (Bruvo et al. 2004) genotypic distance matrix. Microsatellite ILVO145PrMS145 (a, c) was excluded to avoid ambiguity since this locus was not able to be genotyped for all individuals. In total, 63 MGs were analyzed (non-historical singleton MGs were excluded from the network for simplicity—see supplemental information) and the MGs of the historical isolates were then located on the network.
Results
Genetic variability
With the exception of one isolate (MG117), microsatellite PrMS39a was invariant. All the other loci exhibited between 4 (locus 64) and 12 (locus 43b) different alleles. Since ILVO145PrMS145 a and c were unable to be amplified for some samples, MGs were defined and numbered on the basis of the other markers (Mascheretti et al. 2009), with sub-genotypes (a, b, c etc.) designated according to the alleles present at the ILVO145PrMS145 a and c loci (see Supplemental Table 1). Including ILVO145PrMS145 (a, c), 224 MGs were defined (117 excluding these markers). More than half of the MGs (139 (62.1 %); 66 (57.4 %) were singletons. The most frequent genotypes were MG38 (n = 95; 11.4 %); MG42 (n = 113; 13.5 %); MG46 (n = 122; 14.6 %); corresponding to the three frequent and possible founder MGs (#13, #14, and #15; respectively) originally identified in Mascheretti et al. (2008).
Population structure
Iterative within-county collapsing reduced the initial set of 43 populations (n > 5) to 29 (see Fig. 1 and Supplementary Table 1). AMOVA results are given in Table 1. Although the proportions of genetic variation within individual populations (68 %) and among groups (31 %) were high, variation among populations within groups (the final populations) was extremely low (1 %), indicating that this sample-grouping scheme is reasonable. The NJ tree, rooted through the Nursery-Santa Cruz 1 sample is shown in Fig. 2a. The time since infestation (20+ years; 15–20 years; 10–15 years; <10 years; <8 years; <5 years) is indicated. The tree accurately reflects the age of each infestation, with the oldest infestations (except Sonoma 4 and Sonoma 1) all close to the root and the youngest infestations furthest from the root. Although bootstrap values were low (expected, given the few loci employed), the remarkable congruence between age and topology indicates that the tree accurately reflects the genetic relationships among populations and that these genetic relationships reflect the progression of the infestation from older to younger populations.
Table 1.
Analysis of molecular variance results for the 29 populations defined by “within-county” collapsing
| Group | df | SS | Variance components | % Variation |
|---|---|---|---|---|
| Among groups | 28 | 17.975 | 0.0214 | 30.83 |
| Among populations within groups | 15 | 0.871 | 0.0472 | 1.17 |
| Within populations | 767 | 36.236 | 0.0695 | 68.00 |
| SUM | 810 | 55.081 | ||
| Φ SC | 0.0170NS | |||
| Φ ST | 0.3200*** | |||
| Φ CT | 0.3083*** |
NS = P > 0.05
*** P < 0.0001
Structure (Pritchard et al. 2000) was used to analyze all populations in terms of the coalescent structure of the genetic variation (not to assign samples to genetic populations per se). The ln probability of the data, for each replicate at each value of k, was summarized using structure harvester (Earl and vonHoldt 2011). Ln Pr(X|k) increased with each value of k and did not have a clear maximum. Although typical of many datasets, this pattern is precisely that which would be expected from a process of continual population splitting from a single initial source. The method of Evanno et al. (2005) indicated an optimal ΔK at k = 2. Since this method cannot evaluate k = 1, it is therefore probable that the ‘true’ k = 1—again indicating that the entire epidemic likely results from single introduced MG. A secondary maximum ΔK was observed at k = 4, and this value was therefore examined as an indicator of possible sub-structure in the data. It is important to remember that this does not imply that the P. ramorum epidemic in California consists of four genetic populations, but simply that there are four marked divisions or clades within the population branching process that led to the current distribution. (Determination of k and bar-plots for k = 4 are given in supplemental Figs. 1 and 2).
The NJ representation of the relative membership of populations within the coalescent clusters based upon GCD (see methods) in Fig. 2b, maps closely to the Bruvo NJ tree in Fig. 2a (the approximate boundaries of the clusters are indicated by broken lines on both figures). Cluster 1 represents the oldest infestations, cluster 3 the next oldest and the majority of populations, then cluster 4, and finally cluster 2 maps to the youngest populations. Although the genetic clusters follow an age progression, as may be expected given that the clusters represent clades of the coalescent history, it is important to note that the clusters may sometimes contain populations having more than one source and that the depiction in Fig. 2 does not therefore represent an accurate population history, which may be more accurately reconstructed in the coalescence network (see below). As such, a few populations moved notably between these two depictions of genetic relatedness (arrows between Fig. 2a, b). In general these were populations with small sample sizes and were more logically grouped based upon GCD (Fig. 2b), e.g. Humboldt 4 (n = 3) groups with the other young Humboldt populations in Cluster 2; East Bay 2 (n = 6) moves to cluster 1 in which East Bay 1 also occurs; SO1 (n = 5) joins other old populations in cluster 1. Despite these apparent (albeit informative) unstable placements, the overall congruence of the structure genetic clusters and the NJ-topology indicates the robustness of the NJ framework and its ability to place small population samples accurately.
Evolution of multilocus genotypes
As in previous analyses (Mascheretti et al. 2008, 2009), the MSN (not shown; see supplementary information) revealed a few centrally located and frequent MGs with less abundant MGs located at the edges of the network. This pattern is consistent with one or a few closely related initial clonal founder individuals giving rise to occasional new genotypes by mutation over time. Although we have previously suggested that the most abundant genotypes represent the founders of the California epidemic (Mascheretti et al. 2008, 2009), until now we have not been able to provide additional support in favor of this hypothesis.
The structure analysis identified four genetic clusters and each individual MG was therefore classified according to the genetic cluster with which it showed greatest affinity based upon the posterior probability of cluster membership output by structure. Results for all MGs are given in Supplemental Table 2. The four most common MGs each fall centrally into one of these clusters and alleles at two SSR loci (MS43a and b) within each of these MGs appear to define the clusters. MG46/96 (MG96 was merged with MG46 because of missing/null alleles at MS18) (n = 123) is at the heart of cluster 1, MG42 (n = 134) at the heart of cluster 3, and MG38 (n = 95) at the heart of cluster 4. These MGs are identical except at locus MS43b where MG46/96 carries genotype 93/93 (cluster 1), MG42 has genotype 92/92 (cluster 3), and MG38 has genotype 91/91 (cluster 4). The fact that cluster 1 is basal in the NJ-tree (Fig. 2b) therefore suggests that MG46/96 could represent the original founding genotype in the Californian P. ramorum infestation. MGs 42, 46/96 and 38 correspond precisely to the three MGs originally suggested as being possibly ancestral by (Mascheretti et al. 2008). Cluster 2 is defined by MG24 (n = 60), which like MG42 has genotype 92/92 at MS43b, but differs from MG42 by having genotype 69/69 at MS43a (instead of 70/70 for MG42). The one-step differences between these MGs, together with their commonness, supports the notion that these genotypes are early founders of the SOD epidemic and form the base of the four clades corresponding to the genetic clusters identified by structure. The putative evolutionary relationship between these four MGs is depicted as a simple mutational network in Fig. 3. According to the NJ analyses (Fig. 2), cluster 2 is the youngest and, examination of the MSN (supplemental), suggests that it may have at least two origins as indicated by the fact that some MGs are not directly connected to others in the network. This is supported by the values of clonal genotype diversity (R) and Nei’s genetic diversity (D) (Fig. 3), with R for cluster 2 being twice that of cluster 3, and D also being markedly higher. Cluster 4 also showed a much less marked increase in these values.
Fig. 3.
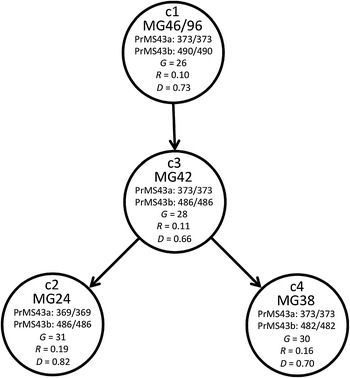
Minimal representation of mutational shifts between the four common and putatively founding multi-locus genotypes (MGs) (MG46/96, MG42, MG24, MG38) that define each of the four genetic clusters or “clades” (c1–c4). The key changes (bp) at the two microsatellite loci PrMS43a and PrMS43b are indicated. G Number of MGs in cluster, R clonal genotype diversity, D genetic diversity (1−Σg 2i), g is the frequency of MG i. (MG96 was merged with MG46 as they are identical except for missing/null alleles at MS18)
The inclusion of 59 historical isolates collected between 2001 and 2004 allows for the testing of the hypothesis that the most common genotypes are also the oldest ones, and that new genotypes have evolved by local mutation. The MGs on the MSN were therefore annotated as “historical” if a historical isolate representing that MG was present in the data. The number of individuals representing an historical MG was then calculated and plotted (Fig. 4). This plot demonstrates that the majority of individuals are represented by a few common MGs, and that the majority of individuals also represent historical MGs (n = 582; 73 %), in other words the most common MGs are also historical. As expected, MGs 42, 46/96, 38, and 24 corresponded to historical isolates. To test this further, we asked if one-step neighbors of MGs 42, 46/96, 38 and 24 were more likely to be historical than MGs that were two or more steps removed (Fig. 4, inset). The result was highly significant (χ2 = 45.96; P < < 0.0001) with one-step neighbors of the three common MGs being more than ten-times as likely to be historical than MGs two or more steps away (Odds ratio = 10.83; 95 % CI 4.82–24.33). It should also be noted that non-historical singleton MGs were excluded from the network for clarity (see supplemental information), meaning that the number of non-historical MGs that were more than two-steps away from the most common MGs is in fact larger than reported here. Thus, the results of this analysis are conservative.
Fig. 4.
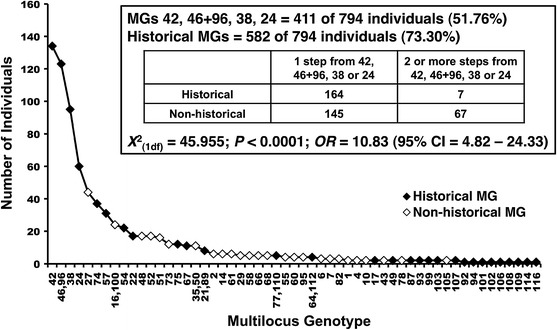
Plot showing the occurrence of 61 multi-locus genotypes (MGs) from the minimum spanning network MSN (see supplemental information), and whether they are “historical” or “non-historical”. Double labels on the x-axis (e.g. 46.96 or 16.100) indicate genotypes that had no “distance” between them because they are identical except at null alleles. Inset test indicating that common MGs are more likely to be old (“historical”), with one-step neighbors of the four most common MGs being almost 11 times more likely to be historical than MGs two-or more steps removed from the most common genotypes. Non-historical singleton MGs were excluded from this plot and the inset—including them makes one-step neighbors of the four most common MGs being almost 20 times more likely to be historical than MGs two-or more steps removed from the most common genotypes (OR = 19.55 (95 % CI = 8.84–43.26); χ2 = 86.117, P ≪ 0.0001)
Colonization history of P. ramorum in California
A coalescent approach to migration model choice (Beerli and Palczewski 2010) was adapted in order to determine the possible routes of spread of P. ramorum. Historical data (years since infestation) were incorporated into this analysis by excluding models that would imply colonization from younger to older infestations. The resulting model is displayed in Fig. 5: the model cannot place accurately all populations with statistically significant confidence, however the source of 23 out of 29 (79 %) populations is supported in over 50 % of the replications, while the source of the remaining 21 % is open to interpretation.
Fig. 5.
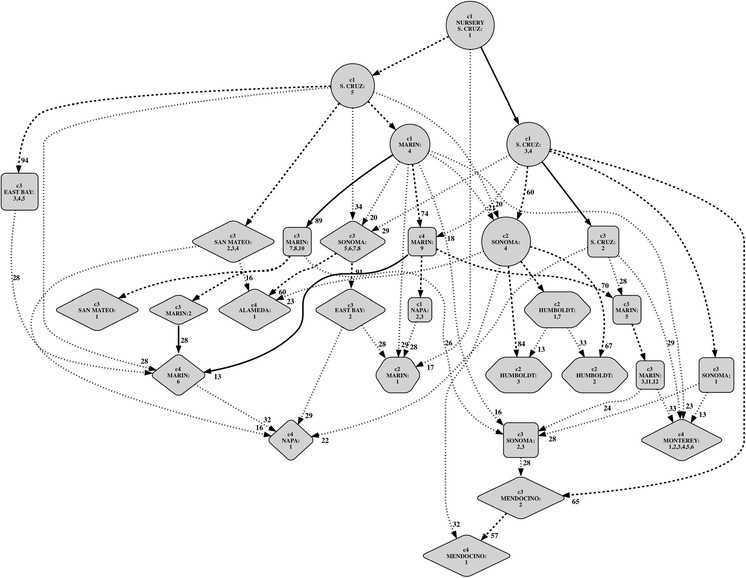
Migrate-n model-choice based reconstruction of the possible colonization routes of P. ramorum during the Californian SOD epidemic. Population names are indicated at nodes along with genetic cluster (“clade”) membership. Nodes shapes correspond approximately to time since infestation: circle 20+ years; square 15–20 years; diamond 10–15 years; hexagon ≪10 years. Solid arrows connecting nodes indicate populations <10 km apart (i.e. natural spread likely), otherwise bold-dashed arrows indicate connections supported more than 50 % of the time; dotted arrows were poorly supported. Numbers next to arrows indicate the proportion of times a migration route was supported (100 % if not indicated)
Importantly, the Nursery—Santa Cruz 1 population emerged naturally as the source population in the initial pair-wise analyses and prior to incorporating the historical data, thus corroborating independently that the SOD epidemic in California started in the Santa Cruz area and originally came from nursery infestations. Secondary sources or stepping stones for the pathogen appear to be all located in areas with significant forest cover and are all characterized by cooler climate (several sites in Marin and Sonoma counties), while old infestation in more urban areas characterized by warmer climate (e.g. in the San Francisco East Bay and in the Napa Valley) were rather inconsequential for the further spread of the pathogen (Fig. 1). The analysis also revealed that in several cases, multiple infestation routes involving different sources have infested the same counties, proving that many counties were each infested more than once during the brief history of the disease. The vast majority of linkages between sites uncovered by the coalescence analysis are at distances of tens of km, the implications of this finding are discussed below.
Discussion
Employing 832 isolates from 60 forest localities, we have revealed a far greater degree of spatial-genetic structuring in P. ramorum than previously indicated (Mascheretti et al. 2009). This increased resolution stems from: (1) the inclusion of two additional loci; (2) the inclusion of all the data [previously studies employed haplotypes, because the largely invariant second allele was removed from the clonal genotypes (Mascheretti et al. 2008, 2009)]; (3) the use of the (Bruvo et al. 2004) evolutionary genetic distance as the basis of the AMOVA and Φ ST calculations. The latter, although originally designed for polyploid data, is advantageous here for two main reasons: (a) Unlike the ‘R ST-like’ method built into Arlequin, the measure can be calculated for partial microsatellite repeats, avoiding the exclusion of such data; (b) the method accounts for the decreasing probability of a mutational change with increasing numbers of repeats (rather than increasing as the square of the repeat difference as in R ST)—minimizing the influence on genetic distance of rarer MGs with multi-step repeat differences (Bruvo et al. 2004).
Although we cannot know the rate at which new genotypes are generated and become extinct through mitotic or somatic recombination events in the absence of meiosis, it is reasonable to assume that the rate of generation of new SSR alleles may be lower than that of 10−4 suggested for shifts in number of repeats at SSR loci during meiosis (Estoup et al. 2002) and approaching ca. 10−9, the general neutral mutation rate reported across all kingdoms (Kasuga et al. 2002). Thus, the generation of more than 200 genotypes from 1 to 4 original founders over approximately 25 years implies populations of extremely large sizes: this is not surprising given the extent of establishment of the pathogen in California and the large numbers of mitospores it produces on the leaves of infectious hosts (Reviewed in Garbelotto and Hayden 2012). While it is true that the number of polymorphic repeats in SSR loci of individuals from California forests appears to reflect a stepwise mutation process, and only includes incremental or small jumps in the number of repeats as confirmed by published sequencing results (Ivors et al. 2006, Prospero et al. 2004, 2007, 2009; Vercauteren et al. 2010), the pattern of homozygosity at many of the loci is surprising. Nonetheless, similar patterns of “loss of heterozygosity” (LOH) appear to be a common feature of the genetics of Phytopthoras and LOH as a result of very frequent mitotic crossing has been demonstrated to be the norm in clonal isolates of P. capsici (Lamour et al. 2012) and P. cinnamoni (Dobrowolski et al. 2003). Indeed, it has been suggested that clonality may be advantageous to invasive organisms and that mitotic recombination may act to purge clonal organisms of recessive deleterious mutations (as meiosis can for sexual organisms in stable environments) (Shaw 1983; Dobrowolski et al. 2003). In any case, it is unlikely that binding site mutations have resulted in consistent hemizygous PCR amplification at so many loci and across numerous studies.
Although we have previously presented results of AMOVAs (Mascheretti et al. 2008, 2009), this study hierarchically merged all populations with insignificant pairwise Φ ST values only if within the same county, resulting in a low 1 % genetic variance within metapopulations. This value represents a significant one-order of magnitude reduction from results of previous analyses (Mascheretti et al. 2009) and indicates metapopulations are better defined here than in previous studies. The collapsing of populations into metapopulations based on Φ ST without any geographic supervision as done in previous studies (Mascheretti et al. 2008, 2009) had resulted in the merging of populations with different histories, and thus had reduced our ability to reconstruct the invasion routes followed by the pathogen.
Genetic relationships among populations were examined by drawing NJ-trees. To ensure the robustness of this analysis we used a two-tiered approach. First, the shortest possible NJ-tree with populations of sample size ≥5 was constructed. Second, this was used as a backbone constraint to which the remaining populations were appended. This yielded a depiction of the genetic relationships among the populations (Fig. 2a) mirroring the known age of the infestations, with the oldest populations near the root (Nursery-Santa Cruz 1) and the youngest infestations furthest from the root. It is important to remember that the NJ-tree does not represent a phylogeny, branches do not represent splitting events, but merely genetic distances. The level of structure present in the NJ-tree reflects the evolutionary process by which the MGs have evolved. Many infestations probably experience a strong founder effect, with only one or a few MGs being introduced and very little subsequent gene flow into or out of the populations. Although the MGs within each population subsequently diversify by mutation and somatic recombination, most new MGs remain rare (Mascheretti et al. 2009), and many are eliminated seasonally as populations crash during the drier summer months (Prospero et al. 2007). This, combined with the fact that P. ramorum currently reproduces in an exclusively clonal manner (Ivors et al. 2006; Goss et al. 2009a), means that the historical signal of colonization remains intact and the NJ-tree not only reflects the genetic similarity among the populations, but also the relative timing of the colonization events. In this study, it was possible to define four very closely related core genotypes that have generated four genetic clusters: MG46/96: cluster 1; MG42: cluster 3; MG38: cluster 4; and MG24: cluster 2. These clusters map to the known age since infestation and they likely represent clades of the branching process that created the current pattern of genetic variation. Indeed, when the relationships among the populations were plotted as an NJ-tree using a distance measure (GCD) based-upon cluster membership, the tree even more closely mirrored infestation age than the Bruvo-based tree (Fig. 2b). Hence, we can deduce that MG46/96 represents the most ancestral of all these MGs. We conclude that despite limited information content of the markers used, the biology of P. ramorum allows for a solid reconstruction of the actual order of events during the epidemic.
MGs 46/96, 42 and 38 are identical to those originally suggested as the original MGs introduced into Californian forests (Mascheretti et al. 2008). This is further supported by the analysis of historical isolates that were collected in the early days (i.e. between 2001 and 2004) of the research on SOD. These historical isolates include many of the most numerous MGs, with the four common MGs and their immediate one-step neighbors in the MSN accounting for 99 % of individuals within historical MGs and only for 68 % of those within non-historical MGs. Hence, the most common genotypes are the oldest and consequently MG 46/96, and subsequently MGs 42, 38 and 24, may indeed represent the original progenitors of the SOD epidemic. Interestingly, each area is dominated by a single cluster, suggesting founder individuals may maintain an advantage over other isolates, whether subsequently introduced or locally generated.
The above results suggest that it might be possible to reconstruct the route of the epidemic in detail. We attempted to do this using Migrate-n to choose among migration models on the basis of their ln marginal likelihood, incorporating the known time since infestation into the model. The analysis of genetic data in concert with epidemiological data is something that has rarely been done in disease ecology (Milgroom and Peever 2003). We assumed that each population has only a single source population and this may be mostly be true given the limited natural dispersal abilities of P. ramorum (Mascheretti et al. 2008, 2009). However, especially for some of the oldest infestations occurring before the SOD epidemic was recognized, multiple transmissions among sites may have occurred. Additionally, the reconstruction must be interpreted cautiously because it is subject to stochastic sampling errors, and because the algorithm may be mislead by populations that are by chance genetically similar (because they share founders with similar genotypes) but actually derive not from each other but from parental or grand-parental populations. Finally, populations that appear to derive from the same parent may actually derive from one another. Nonetheless, this reconstruction not only corroborates many expectations and observations from earlier migration-based analyses, but also provides some remarkable new insights.
Both NJ and coalescent analyses indicate that multiple infestations, linked to different sources, occurred in different locations within several counties. Only noted exceptions were in Santa Cruz County, now confirmed as the primary site of the original release of the pathogen from nurseries into the wild, and in the geographically isolated Big Sur region and in Humboldt County.
It has been previously suggested (Mascheretti et al. 2009; Prospero et al. 2009) that nurseries are not currently contributing to wild infestations because of significant F ST or Φ ST values observed between nursery and wild populations. This is reasonable due to the current enormous size of wild populations compared to nursery populations. However, our ability to place individual genotypes on the robust NJ tree (Fig. 2a) represents a major advancement in our ability to determine the source of new small infestations. This approach indicated that several minor infestations have been caused by isolates in cluster 1 and are likely to have originated, perhaps as recently as 2005–2006 in the case of the San Francisco Presidio, from infected ornamental plants. It should be noted that in general these infestations are minor because they occurred in areas that are not particularly suitable to outbreaks due to the lack of infectious hosts (Prunedale, Monterey County), because in an urban environment (Presidio of San Francisco), or because in areas with highly fragmented habitat characterized by warmer climate (East San Francisco Bay). However, the overall limited representation of these nursery-linked introductions may mask a constant flux of genotypes from ornamental plantings to wild-lands.
One further question that arises from the results presented in this study is whether clusters 2, 3 and 4 may be better adapted to forest environments than cluster 1 isolates which are strictly associated to nurseries, to the few minor introductions mentioned above, and to those Santa Cruz forests adjacent to the nursery identified as one of the original sources of the California infestation in Mascheretti et al. (2008). Although this conclusion requires further supporting evidence, the limited dispersal of the basal cluster 1 relative to the other clusters is suggestive of adaptation in the other three derived clusters. Notwithstanding a possible reduced fitness of nursery genotypes (e.g. cluster 1 of the NA1 lineage) in forests, California nurseries are known to harbor three genetically and phenotypically distinct lineages of the pathogen (Brasier 2003; Elliot et al. 2009; Ivors et al. 2006), and a low level of migration between ornamental plantings and wild-lands could lead to the introduction of the NA2 and EU1 lineages to California forests, where currently only the NA1 lineage is present. The introduction of the EU1 lineage (A1 mating type—NA1 and NA2 have the A2 mating type, reviewed in Grünwald et al. 2012) could permit sexual reproduction and the production of hardy sexual reproductive structures (oospores) that may increase the survival of the pathogen, and allow faster adaptation of the pathogen to new habitats and hosts.
One important additional finding of this study is that the significant outbreak recently discovered in Northern Humboldt County (Humboldt 3) and equidistant to the closest infestations both North (Oregon) and South (Southern Humboldt, Humboldt 7–9), was linked both by NJ and coalescent analysis to the same source as the Southern Humboldt infestation (approximately 100 km to the South). The infestation was originally discovered by baiting the pathogen 20 miles downstream (Humboldt 5) in Redwood Creek, a waterway that crosses the aerial infestation. Genotyping showed the genotypes in the stream were the same found in the terrestrial infestation 20 miles upstream, and that enough time had lapsed since the introduction of the pathogen for several genotypes to have been generated locally in such site (Supplemental Table S1). These findings underlined the importance of genetic data to: (a) link baiting results from waterways with a putative terrestrial source; (b) date an infestation, highlighting that even if recently discovered, some infestations may be older and simply unnoticed in spite of significant surveying efforts; (c) determine that the likely source of this recently discovered outbreak was neither current nursery genotypes, nor the neighboring Oregon infestation, but the infestation appeared rather to have been started from the same source that started the Southern Humboldt infestation. Given the long distance between Sonoma and Humboldt, the most likely pathway had to be that of the movement of infected ornamental plants, a pathway that has been shown (Goss et al. 2011, b; Mascheretti et al. 2008, 2009; this study) to be effective at the long distance movement of P. ramorum. Although possibly coincidental, the source of the Humboldt infestation (Sonoma 4) includes not only a large wild infestation, but also a nursery that although not sampled is adjacent (<3 km) to some of our sampling transects.
The coalescent analysis in this study clearly identifies several large wild infestations in cool areas of Santa Cruz, Marin, and Sonoma counties as stepping stones to further introductions, providing the first solid evidence that large pathogen population size may be the most important factor leading to long distance spread of the disease. Previously, only genetically indistinguishable populations at a significant distance could be taken as evidence of human- mediated transport, the coalescence network produced in this study (Fig. 5) shows a large number of linkages (18 of 22 well supported linkages represented by dashed lines) to be between sites at distances much larger than that observed or inferred as the maximum distance at which the pathogen may naturally spread (Hansen et al. 2008; Mascheretti et al. 2008, 2009). Thus, humans played a major role in determining the current distribution of the pathogen. Overall, the coalescence network provides solid evidence that the current distribution of the pathogen in California is explained by multiple independent introductions of P. ramorum and not by a gradual spread from one or two introduction points.
The entire coalescent network appears to have been originally generated by nursery genotypes, thus suggesting a significant number of introductions is likely to have been caused by the movement of infected ornamental plants both at short and long distances. It appears that all genotypes belonging to cluster 1 have a direct relationship with the ornamental trade, thus all infestations with cluster 1 genotypes (8 of 18 supported linkages between populations too distant for natural spread shown as dashed lines originating from cluster 1 populations in Fig. 5), or those immediately derived from cluster 1 (7 additional linkages in Fig. 5) in the coalescent analysis, can be reputed to be related to the movement of infected ornamental plants. Thus, in total, the pathogen appears to have been introduced 8–14 times in different California locations through the movement of infected ornamental plants. Several introductions may explain both the high success of establishment of the pathogen and the presence of four distinct clusters of populations.
In contrast to nursery isolates and large wild infestations, old infestations that did not develop early on into outbreaks such as San Mateo 1 and East Bay 1 appear to have had a limited role in the further dispersal of the pathogen. Thus, limiting the population size in individual areas, in addition to blocking pathways of spread, may be of paramount importance in slowing the spread of a disease such as SOD. This observation may be important, since favorable weather conditions in 2010–2012 (www.sodmap.org) have resulted in a clear local expansion of the disease in many locations, potentially generating new sources for future long-distance dispersal.
Both the NJ and coalescent analyses indicate that the pathogen has often spread to adjacent areas from many of these initial discrete introductions, most likely through natural movement of its airborne propagules. In particular, the coalescent analysis performed in this study allows the reconstruction of spread pathways within Marin and Santa Cruz counties (solid lines in Fig. 5). Although the coalescent analyses ignored relationships among genetically undistinguishable populations if within the same county, age of the infestation can be used to determine spread pathways among these. For instance, in the Big Sur region the oldest infestations are centrally located (MO1, MO6) with younger infestations present both North (MO5, MO3) and South (MO4, MO2) of what is obviously the first local infestation for the entire region, responsible for the spread of the pathogen both North- and South-ward.
In this study, we have reconstructed the evolutionary epidemiological history of the SOD epidemic in California in detail. This has been possible because of the almost complete sampling coverage of infestations in California, and because of the limited natural dispersal and of the exclusively clonal reproductive biology of P. ramorum. With the current study, we are now able to demonstrate that 1–4 genotypes (most likely 1, rapidly diverged into 4) were originally introduced in California, that most counties were infested more than once by different founders, and that, while new recent outbreaks are linked mostly to pathogen movement (both natural and human-mediated) from large wild infestations, in some cases the source can still be identified in infected ornamental plants. Based on the different incidence of the four clusters, we can also suggest that some genotypes may be more adapted to cause forest outbreaks than others. Further, the identification of several old infestations in the East Bay, Mendocino, and Napa counties that did not develop into epidemic outbreaks like other equally-aged or even younger infestations (Big Sur, Southern Humboldt, West Marin) suggests the presence of strong and different local ecological constraints to disease progression.
Finally, the coalescent network here generated shows probable historical linkages between infestations, thus facilitating the modeling of both future expansions and of how the epidemic might have been affected by attempts to eradicate the pathogen at different temporal stages. Such analyses are urgently needed to aid policy makers decide on whether, and at what stage, strong regulations attempting eradication may be beneficial after the introduction of an exotic pathogen.
Good reconstructions of invasion patterns for other forest diseases have been generated (reviewed in Garbelotto 2008; see also Dutech et al. 2012), but this is the first detailed reconstruction of an emergent exotic forest diseases in a time-frame of <20 years. We believe that the approaches used here may not only be computationally less intensive but may also be applicable to other introduced organisms with reproductive and dispersal strategies comparable to P. ramorum. Although we are aware that the intensive sampling described here may not always be feasible, we would like to note that including field epidemiological information to guide the computational algorithms used to infer relationships among populations was key to the success of the analysis and should become a common practice when attempting to reconstruct the invasion history of an exotic organism.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was funded by the Ecology of Infectious Diseases grant EF-062654, provided jointly by the NSF and NIH and by the Gordon and Betty Moore Foundation. PC was funded by NSF (DEB 0919215) Collection of samples predating the NSF-NIH grant was made possible through funding from USDA-APHIS and by the USFS, State and Private Forestry. Clay de Long is the author of Fig. 1.
Footnotes
Peter J. P. Croucher and Silvia Mascheretti contributed equally to this paper.
References
- Ayala FJ. Enzymes as taxonomic characters. In: Oxford GS, Rollinson D, editors. Protein polymorphism: adaptive and taxonomic significance. Systematics Association Special Volume No 24. London: Academic Press; 1983. pp. 3–26. [Google Scholar]
- Beerli P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 2006;22:341–345. doi: 10.1093/bioinformatics/bti803. [DOI] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J. Maximum-likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA. 2001;98:4563–4568. doi: 10.1073/pnas.081068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli P, Palczewski M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics. 2010;185:313–326. doi: 10.1534/genetics.109.112532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracho MA, Gosalbes MJ, Blasco D, Moya A, Gonzalez-Candelas F. Molecular epidemiology of a hepatitis C virus outbreak in a hemodialysis unit. J Clin Microbiol. 2005;43:2750–2755. doi: 10.1128/JCM.43.6.2750-2755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier CM. Sudden oak death: Phytopthora ramorum exhibits transatlantic differences. Mycol Res. 2003;107:257–259. doi: 10.1017/S0953756203217664. [DOI] [PubMed] [Google Scholar]
- Bruvo R, Michiels NK, D’Souza TG, Schulenburg H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol. 2004;13:2101–2106. doi: 10.1111/j.1365-294X.2004.02209.x. [DOI] [PubMed] [Google Scholar]
- Cottam EM, Thébaud G, Wadsworth J, et al. Integrating genetic and epidemiological data to determine transmission pathways of foot-and-mouth disease virus. Proc R Soc Lond B Biol. 2008;275:887–889. doi: 10.1098/rspb.2007.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher PJP, Oxford GS, Lam A, Mody N, Gillespie RG (2012) Colonization history and population genetics of the color-polymorphic Hawaiian happy-face spider Theridion grallator (Araneae, Theridiidae). Evolution. doi:10.1111/j.1558-5646.2012.01653.x [DOI] [PubMed]
- Cummings CA, Relman DA. Microbial Forensics—“cross-examining pathogens”. Science. 2002;296:1976–1979. doi: 10.1126/science.1073125. [DOI] [PubMed] [Google Scholar]
- Davidson JM, Warres S, Garbelotto M, Hansen EM, Rizzo DM. Sudden oak death and associated disaeses caused by Phytophthora ramorum. Plant Health Progress. 2003 [Google Scholar]
- Davidson JM, Wickland AC, Patterson HA, Falk KR, Rizzo DM. Transmission of Phytophthora ramorum in mixed evergreen forest in California. Phytopathology. 2005;95:587–596. doi: 10.1094/PHYTO-95-0587. [DOI] [PubMed] [Google Scholar]
- Desper R, Gascuel O. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J Comput Biol. 2002;9:687–705. doi: 10.1089/106652702761034136. [DOI] [PubMed] [Google Scholar]
- Dobrowolski MP, Tommerup IC, Sheare BL, O’Brien PA. Three clonal lineages of Phytopthora cinnamomi in Australia revealed by microsatellites. Phytpathology. 2003;93:695–704. doi: 10.1094/PHYTO.2003.93.6.695. [DOI] [PubMed] [Google Scholar]
- Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11:2571–2581. doi: 10.1046/j.1365-294X.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- Dutech C, Barres B, Bridier J, Robin C, Milgroom MG, Ravigne V (2012) The chestnut blight fungus world tour: successive introduction events from diverse origins in an invasive plant fungal pathogen. Mol Ecol. doi:10.1111/j.1365-294X.2012.05575.x [DOI] [PubMed]
- Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2011 [Google Scholar]
- Elliot M, Sumampong G, Varga A, et al. PCR-RFLP markers identify three lineages of the North American and European populations of Phytopthora ramorum. For Pathol. 2009;39:266–278. doi: 10.1111/j.1439-0329.2008.00586.x. [DOI] [Google Scholar]
- Engelbrecht CJB, Harringotn TC, Steimel J, Capretti P. Genetic variation in eastern North American and putatively introduced populations of Ceratocystis fimbriata f. platani. Mol Ecol. 2004;13:2995–3005. doi: 10.1111/j.1365-294X.2004.02312.x. [DOI] [PubMed] [Google Scholar]
- Estoup A, Jarne P, Cornuet J-M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol. 2002;11:1591–1604. doi: 10.1046/j.1365-294X.2002.01576.x. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse P, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S (2005) Arlequin (Version 3.01): an integrated software package for population genetics data analysis. Computation and Molecular Population Genetics Lab (GMPG), Institute of Zoology University of Berne, Berne, Switzerland
- Felsenstein J (2005) PHYLIP (phylogeny inference package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
- Fitzpatrick BM, Fordyce JA, Niemiller ML, Reynolds RG. What can DNA tell us about biological invasions? Biol Invasions. 2012;14:245–253. doi: 10.1007/s10530-011-0064-1. [DOI] [Google Scholar]
- Fletcher J, Bender C, Budowle B, et al. Plant pathogen forensics: capabilities, needs, and recommendations. Microbiol Mol Biol R. 2006;70:450–471. doi: 10.1128/MMBR.00022-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbelotto M. Molecular analyses to study invasions by forest pathogens: examples from Mediterranean ecosystems. Phytopathol Mediterr. 2008;47:183–203. [Google Scholar]
- Garbelotto M, Hayden K. Sudden oak death: interactions of the exotic oomycete Phytophthora ramorum with naïve North American hosts. Eukaryot Cell. 2012;11(11):1313–1323. doi: 10.1128/EC.00195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbelotto M, Svihra P, Rizzo DM. Sudden oak death syndrome fells 3 oak species. Calif Agric. 2001;55:9–19. doi: 10.3733/ca.v055n01p9. [DOI] [Google Scholar]
- Gonthier P, Garbelotto M. Amplified fragment length polymorphism and sequence analyses reveal massive gene introgression from the European fungal pathogen Heterobasidion annosum into its introduced congener H. irregulare. Mol Ecol. 2011;20:2756–2770. doi: 10.1111/j.1365-294X.2011.05121.x. [DOI] [PubMed] [Google Scholar]
- Goss EM, Carbone I, Grunwald NJ. Ancient isolation and independent evolution of the three clonal lineages of the exotic sudden oak death pathogen Phytophthora ramorum. Mol Ecol. 2009;18:1161–1174. doi: 10.1111/j.1365-294X.2009.04089.x. [DOI] [PubMed] [Google Scholar]
- Goss EM, Larsen MM, Chastagner GA, Givens DR, Grünwald NJ. Population genetic analysis infers migration pathways of Phytopthora ramorum in US Nurseries. PLoS Pathog. 2009;5:e1000583. doi: 10.1371/journal.ppat.1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss EM, Larsen MM, Vercauteren A, et al. Phytopthora ramorum in Canada: evidence for migration with North America and from Europe. Phytopathology. 2011;101:166–171. doi: 10.1094/PHYTO-05-10-0133. [DOI] [PubMed] [Google Scholar]
- Grünwald NJ, Goss EM. Evolution and population genetics of exotic and re-emerging pathogens: novel tools and approaches. Annu Rev Phytopathol. 2011;49:249–267. doi: 10.1146/annurev-phyto-072910-095246. [DOI] [PubMed] [Google Scholar]
- Grünwald NJ, Goss EM, Larsen MM, et al. First report of the European lineage of Phytophthora ramorum on Viburnum and Osmanthus spp. in a California nursery. Plant Dis. 2008;92:314. doi: 10.1094/PDIS-92-2-0314B. [DOI] [PubMed] [Google Scholar]
- Grünwald NJ, Garbelotto M, Goss E, Heungens K, Prospero S. Emergence of the sudden oak death pathogen Phytophthora ramorum. Trends Microbiol. 2012;20:131–138. doi: 10.1016/j.tim.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Hansen EM, Reeser PW, Sutton W, Winton L, Osterbauer N. First report of A1 mating type of Phytophthora ramorum in North America. Plant Dis. 2003;87:1267. doi: 10.1094/PDIS.2003.87.10.1267A. [DOI] [PubMed] [Google Scholar]
- Hansen E, Kanaskie A, Prospero S, McWilliams M, Goheen E, Osterbauer N, Reeser P, Sutton W. Epidemiology of Phytophthora ramorum in Oregon tanoak forests. Can J For Res. 2008;38:1133–1143. doi: 10.1139/X07-217. [DOI] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Ivors KL, Hayden KJ, Bonants PJ, Rizzo DM, Garbelotto M. AFLP and phylogenetic analyses of North American and European populations of Phytophthora ramorum. Mycol Res. 2004;108:378–392. doi: 10.1017/S0953756204009827. [DOI] [PubMed] [Google Scholar]
- Ivors K, Garbelotto M, Vries ID, et al. Microsatellite markers identify three lineages of Phytophthora ramorum in US nurseries, yet single lineages in US forest and European nursery populations. Mol Ecol. 2006;15:1493–1505. doi: 10.1111/j.1365-294X.2006.02864.x. [DOI] [PubMed] [Google Scholar]
- Kasuga T, White TJ, Taylor J. Estimation of nucleotide substitution rates in eurotiomycete fungi. Mol Biol Evol. 2002;19:2318–2324. doi: 10.1093/oxfordjournals.molbev.a004056. [DOI] [PubMed] [Google Scholar]
- Lamour KH, Mudge J, Gobena D, et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytopthora capsici. MPMI. 2012;25:1350–1360. doi: 10.1094/MPMI-02-12-0028-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner T, Albert J. The molecular clock of HIV-1 unveiled through analysis of know transmission history. Proc Natl Acad Sci USA. 1999;96:10752–10757. doi: 10.1073/pnas.96.19.10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lim SL, Ruan Y, et al. SARS transmission pattern in Singapore reassessed by viral sequence variation analysis. PLoS Med. 2005;2:e43. doi: 10.1371/journal.pmed.0020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascheretti S, Croucher PJ, Vettraino A, Prospero S, Garbelotto M. Reconstruction of the sudden oak death epidemic in California through microsatellite analysis of the pathogen Phytophthora ramorum. Mol Ecol. 2008;17:2755–2768. doi: 10.1111/j.1365-294X.2008.03773.x. [DOI] [PubMed] [Google Scholar]
- Mascheretti S, Croucher PJ, Kozanitas M, Baker L, Garbelotto M. Genetic epidemiology of the sudden oak death pathogen Phytophthora ramorum in California. Mol Ecol. 2009;18:4577–4590. doi: 10.1111/j.1365-294X.2009.04379.x. [DOI] [PubMed] [Google Scholar]
- Milgroom MG, Peever TL. Population biology of plant pathogens. Plant Dis. 2003;87:608–617. doi: 10.1094/PDIS.2003.87.6.608. [DOI] [PubMed] [Google Scholar]
- Paoletti M, Buck KW, Brasier CM. Selective acquisition of novel mating type and vegetative incompatibility genes via interspecies gene transfer in the globally invading eukaryote Ophiostoma novo-ulmi. Mol Ecol. 2006;15:249–262. doi: 10.1111/j.1365-294X.2005.02728.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prospero S, Black JA, Winton LM. Isolation and characterization of microsatellite markers in Phytophthora ramorum, the causal agent of sudden oak death. Mol Ecol Notes. 2004;4:672–674. doi: 10.1111/j.1471-8286.2004.00778.x. [DOI] [Google Scholar]
- Prospero S, Hansen EM, Grünwald NJ, Winton LM. Population dynamics of the sudden oak death pathogen Phytophthora ramorum in Oregon from 2001 to 2004. Mol Ecol. 2007;16:2958–2973. doi: 10.1111/j.1365-294X.2007.03343.x. [DOI] [PubMed] [Google Scholar]
- Prospero S, Grünwald NJ, Winton LM, Hansen EM. Migration patterns of the emerging plant pathogen Pythopthora ramorum on the west coast of the United States of America. Phytopathology. 2009;99:739–749. doi: 10.1094/PHYTO-99-6-0739. [DOI] [PubMed] [Google Scholar]
- Rizzo DM, Garbelotto M. Sudden oak death: endangering California and Oregon forest ecosystems. Front Ecol Environ. 2003;1:197–204. doi: 10.1890/1540-9295(2003)001[0197:SODECA]2.0.CO;2. [DOI] [Google Scholar]
- Rizzo DM, Garbelotto M, Davidson JM, Slaughter GW, Koike ST. Phytophthora ramorum as the cause of extensive mortality of Quercus spp. and Lithocarpus densiflora in California. Plant Dis. 2002;86:205–214. doi: 10.1094/PDIS.2002.86.3.205. [DOI] [PubMed] [Google Scholar]
- Rizzo DM, Garbelotto M, Hansen EM. Phytophthora ramorum: integrative research and management of an emerging pathogen in California and Oregon forests. Annu Rev Phytopathol. 2005;43:309–335. doi: 10.1146/annurev.phyto.42.040803.140418. [DOI] [PubMed] [Google Scholar]
- Roewer L, Croucher PJ, Willuweit S, et al. Signature of recent historical events in the European Y-chromosomal STR haplotype distribution. Hum Genet. 2005;116:279–291. doi: 10.1007/s00439-004-1201-z. [DOI] [PubMed] [Google Scholar]
- Shaw DS. The cytogenetics and genetics of Phytopthora. In: Erwin DS, Bartnicki-Garcia S, Tsao PS, editors. Phytopthora: its biology, taxonomy, ecology and pathology. St. Paul, MN: The American Phytopathological Society; 1983. pp. 81–94. [Google Scholar]
- Swofford DL. PAUP*: phylogenetic analysis using parsimony. Sunderland, MA: Sinauer Associates; 1999. [Google Scholar]
- Van Poucke K, Heungens K, Franceschini S, Webber J, Brasier C (2012) Phytophthora ramorum in Europe: a research update. In: Frankel SJ, Palmieri KM, Alexander J (eds) Proceedings of the fifth sudden oak death science symposium, 19–22 June 2012, Petaluma, CA. USDA Forest Service, Albany, CA (in press)
- Vercauteren A, Van Bockstaele E, De Dobbelaere I, et al. Clonal expansion of the Belgian Phytophthora ramorum populations based on new microsatellite markers. Mol Ecol. 2010;19:92–107. doi: 10.1111/j.1365-294X.2009.04443.x. [DOI] [PubMed] [Google Scholar]
- Walsh PD, Biek R, Real LA. Wave-like spread of Ebola Ziare. PLoS Biol. 2005;3:e371. doi: 10.1371/journal.pbio.0030371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werres S, Marwitz R, In’t Veld WA, et al. Phytophthora ramorum p. nov., a new pathogen on Rhododendron and Viburnum. Mycol Res. 2001;105:1155–1165. doi: 10.1016/S0953-7562(08)61986-3. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.