

Abstract
Background/Aims
Helicobacter pylori infection induces cyclooxygenase-2 (COX-2) and epidermal growth factor receptor (EGFR) overexpression, and these factors may engage in cross-talk. The aim of the present study was to evaluate the effect of H. pylori on EGFR signaling pathways and to determine whether celecoxib has an inhibitory effect on this pathway.
Methods
The AGS cell line was cocultured with H. pylori G27 and the isogenic cagE- mutant. The expression of COX-2, EGFR, heparin binding-epidermal growth factor (HB-EGF), and transforming growth factor-β (TGF-β) was measured by real time-polymerase chain reaction (RT-PCR). Next, Western blot analyses of COX-2, EGFR, total Akt, phosphorylated Akt (pAkt), and phosphorylated glycogen synthase kinase-3β (pGSK3β) were performed after incubating H. pylori-treated AGS cells for 24 hours with various concentrations of celecoxib (0, 10, 20, and 30 µmol/L).
Results
H. pylori infection upregulated the mRNA levels of COX-2, EGFR, HB-EGF, and TGF-β, as detected by RT-PCR. However, AGS cells treated with cagE- mutants, which have a defective type IV secretion system, did not exhibit EGFR upregulation. Celecoxib had inhibitory effects on the H. pylori-induced overexpression of COX-2 (p=0.015), EGFR (p=0.025), pAkt (p=0.025), and pGSK3β (p=0.029) by Western blot analysis.
Conclusions
H. pylori with an intact type IV secretion system activated the COX-2 and EGFR-Akt pathways in the AGS cell line. As celecoxib exhibited inhibitory effects on the EGFR signaling pathway, the cross-talk of COX-2 and EGFR likely mediates H. pylori-induced gastric cancer.
Keywords: Gastric carcinoma; Helicobacter pylori; Cyclooxygenase 2; Receptor, epidermal growth factor; Celecoxib
INTRODUCTION
Helicobacter pylori infection is a major risk factor of gastric cancer.1,2 However, only small portion of infected hosts develop gastric cancer, clinical outcome of the infection is determined by interaction of host genetics, environmental factors and the virulence of the bacteria such as vacuolating cytotoxin (vacA) and cag pathogenicity island (cag PAI).3 VacA protein induces vacuolization in epithelial cells and cag PAI encodes CagA and other proteins forming type IV secretion apparatus, through which translocation of CagA occurs.4,5
Cyclooxygenase (COX), an enzyme that catalyzes the conversion of arachidonic acids to prostaglandins, plays an important role in physiological and pathological pathways.6 COX-2 is inducible form and expressed in response to inflammation and carcinogenesis. Several epidemiological and clinical studies have shown the relationship of COX-2 expression to gastric cancer progression.7,8 In addition, it is known that H. pylori infection causes COX-2 overexpression in the early step of gastric carcinogenesis.9,10 Therefore, there have been efforts to target COX-2 to prevent the development of gastric cancer. Nonsteroidal anti-inflammatory drugs, especially, selective COX-2 inhibitors, such as celecoxib, have been suggested to reduce the risk of gastric cancer in vivo and in vitro.11-13 The anticancer effect of celecoxib is mediated by COX-2 independent pathways as well as by COX-2 dependent pathway.14-17 Our previous study showed that one of the anticancer mechanisms of celecoxib is downregulation of Akt signaling in gastric cancer cell lines.16
Epidermal growth factor receptor (EGFR) is a member of the ErbB family and plays important roles in cell survival, proliferation, differentiation, and migration. EGFR overexpression is frequently detected in human gastric cancers.18 It has been proposed that H. pylori VacA up-regulates EGFR and the downstream targets including Akt signaling pathway,19-21 which is also proved to be inhibited by selective COX-2 inhibitor. However, the relationship of COX-2 and EGFR pathway has not been clarified in the gastric cancer cells.
From this background, the aim of this study was to evaluate the effect on expression of COX-2, EGFR and the downstream targets after H. pylori infection in the gastric cancer cell line. In addition, we investigated whether celecoxib, COX-2 selective antagonist, has inhibitory effect on H. pylori-induced EGFR signal conduction pathway.
MATERIALS AND METHODS
1. Materials and reagents
Purified celecoxib was provided by Pfizer Pharmaceuticals Korea (Seoul, Korea) and was dissolved in 100% dimethyl sulfoxide (DMSO); final DMSO concentrations in all cultures were below 0.1%.
2. Cell culture and H. pylori strain
G27 strain (cagA+, vacA+ [s1, m1]) wild type and cagE- isogeneic mutant of the strain, obtained from Professor S. Falkow (Stanford University, Stanford, CA, USA), G69a strain (cagA+, vacA+), expressing green fluorescence protein (a gift from Dr. Reiner Haas, Munich, Germany) and HP 99 strain (cagA+, vacA+), were used in this experiment.22 Bacteria were cultured under microaerobic conditions (5% O2, 10% CO2, and 85% N2) at 37℃ on chocolate agar plates for 24 to 36 hours. Bacteria were harvested and resuspended in RPMI-1640 (Gibco, GrandIsland, NY, USA). Then, those were supplemented with 10% fetal bovine serum, and 100 U/mL penicillin and 100 mg/mL streptomycin for coculture with the AGS cells (ATCC CRL 1739; obtained from American type culture collection, Bethesda, MD, USA) at a multiplicity of infection of 100:1.
After 6 hours for real time-polymerase chain reaction (RT-PCR) and 24 hours for Western blotting, the cells were rinsed with phosphate-buffered saline (PBS; pH 7.4, 37℃) and various concentrations (0, 10, 20, and 30 µmol/L) of celecoxib were added and incubated for another 6 or 24 hours with serum starvation.
3. RT-PCR
To extract total RNAs from AGS cell lines, we used TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) as recommended by the manufacturer and the collected RNA was purified using RNeasy mini kits (Qiagen, Valencia, CA, USA). RNA samples were diluted to a final concentration of 500 ng/µL in RNase-free water and stored at 80℃, until use. RT-PCR reaction was performed on the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) in 20L SYBR Premix Ex Taq™ (Takara Bio, Shiga, Japan) using 200 ng of cDNA. The thermal cycler conditions were 10 seconds hold at 95℃, followed by 40 to 45 cycles of 5 seconds at 95℃, and 33 seconds at 60℃. The following primers were used: COX-2 forward, TTCAAATGAGATTGTGGGAAAATTGCT; COX-2 reverse, AGATCATCTCTGCCTGAGTATCTT; heparin binding-epidermal growth factor (HB-EGF) forward, CTCTTTCTGGCTGCAGTTCTC; HB-EGF reverse, AGCTGGTCCGTGGATACAGT; EGFR forward, CTATGAGATGGAGGAAGACG; EGFR reverse, CAGAGGAGGAGTATGTGTGA; TGF-β forward, GTATGGGGTCGCAGGGTGTT; TGF-β reverse, CAGATGCGCTGTGGCTTTGC; Homo sapiens actin, β forward, TTCGAGCAAGAGATGGCCAC; Homo sapiens actin, β reverse, CGGATGTCCACGTCACACTT. All equipment and reagents were purchased from Applied Biosystems and used according to their recommended protocols.
4. Western blotting
In Western blotting, H. pylori G69a strain was mainly used and G27 strain (wild type and cagE- isogenic mutant) was used additionally to evaluate protein expression of EGFR. After washing twice with PBS (pH 7.4, 37℃), the AGS cells were treated with cell lyses buffer (Sigma Chemical Co., St. Louis, MO, USA), and the protein concentration was measured with the BCA TM protein assay kit (Pierce, Rockford, IL, USA). Cell extracts (20 µg protein) were subjected to 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis and the separated proteins were transferred to polyvinylidene difluoride membranes. After blocking the nonspecific binding sites with nonfat dry milk, the membranes were incubated with anti-COX-2, anti-EGFR, anti-phospho-Ser473-Akt, anti-Akt, antiphosphorylated glycogen synthase kinase-3β, and antiactin. The COX-2 membrane incubation with COX-2 antibody (goat polyclonal immunoglobulin G antibody, 1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was carried out at 4℃ overnight, then the blots were incubated with secondary antibody (donkey antigoat antibody, 1:1,000). Western blots were analyzed and quantified using the Luminescent image analyzer LAS 1000-plus (Fuji Photo Film, Tokyo, Japan) and the Image Gauge version 3.12 (Fuji Photo Film).
5. Statistical analyses
SPSS version 18.0 for Windows (IBM Co., Armonk, NY, USA) was used for the statistical analysis. The level of mRNA was expressed as fold changes (mean±SEM) relative to the control groups. The level of protein was expressed as pg/mL (mean±SEM). Mann-Whitney U test was used for the comparison between two groups. Statistical significance was set at p<0.05.
RESULTS
1. H. pylori stimulates COX-2 and EGFR expression
After 6-hour incubation with G27 wild type, the expression of COX-2 (n=8), HB-EGF (n=9), EGFR (n=11), and TGF-β (n=7) were increased in AGS cell lines (Table 1, Fig. 1). Similar patterns of mRNA expression were observed using different strains of H. pylori, such as G69a and HP99 (data not shown). However, AGS cells treated with cagE- mutant of G27 did not show any significant difference compared to control (Table 1, Fig. 1). In addition, in comparison to AGS cells treated with G27 wild type, those treated with cagE- mutant showed significant lower level of mRNA expression in HB-EGF and EGFR (Fig. 1).
Table 1.
Real-Time Polymerase Chain Reaction Analysis of COX-2, EGFR, HB-EGF, and TGF-β in AGS Cells Treated with Wild-Type Helicobacter pylori and Its Isogenic cagE- Mutant
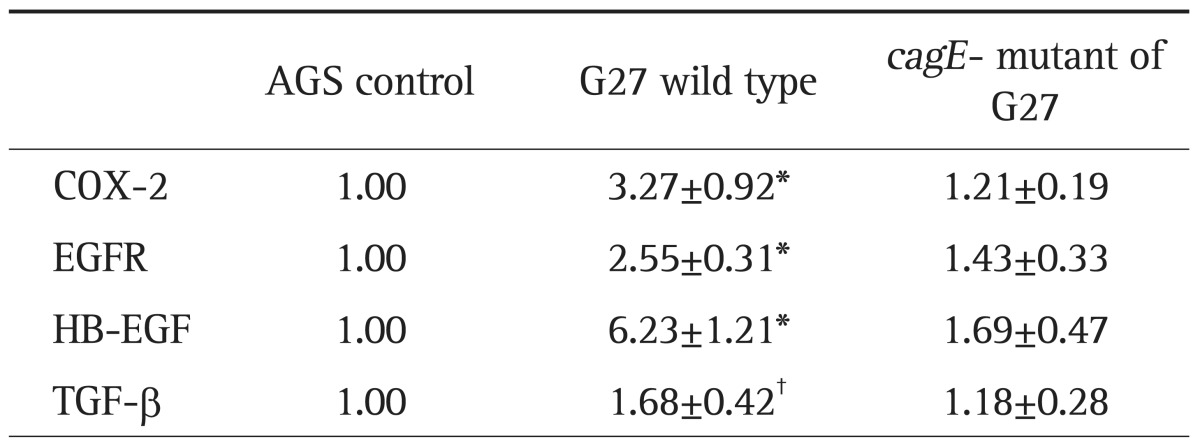
Data are presented as fold increases (mean±SEM) in mRNA.
COX-2, cyclooxygenase-2; EGFR, epidermal growth factor receptor; HB-EGF, heparin binding-epidermal growth factor; TGF-β, transforming growth factor-β.
*p<0.005; †p<0.05 compared to AGS cells.
Fig. 1.

Real-time polymerase chain reaction results for cyclooxygenase-2 (COX-2), epidermal growth factor receptor (EGFR), heparin binding-epidermal growth factor (HB-EGF), and transforming growth factor-β (TGF-β) in the AGS cell line after treatment with Helicobacter pylori. The incubation of AGS cells treated with wild type G27 or the cagE- isogenic mutant resulted in different mRNA expression. (A) COX-2, (B) EGFR, (C) HB-EGF, and (D) TGF-β. Overexpression of COX-2, EGFR, HB-EGF, and TGF-β was observed in AGS cells treated with the wild type H. pylori strain G27. There was no significant difference between the levels of mRNA in the cells treated with the cagE- isogenic mutant and in the AGS control. Compared with AGS cells treated with wild type G27, the expression of HB-EGF (p=0.001) and EGFR (p=0.010) was significantly lower in cells treated with the cagE- isogenic mutant.
HP, Helicobacter pylori.
*p<0.05 compared with AGS cells; †p<0.05 compared with wild type G27.
2. Western blotting for COX-2, EGFR, Akt, GSK3β with or without celecoxib treatment
To assess whether H. pylori-induced protein expressions of COX-2 and EGFR-Akt pathway are suppressed by celecoxib, AGS cell lines were incubated with H. pylori strain G69a in the presence or absence of celecoxib. The effects of 0, 10, 20, and 30 µmol/L of celecoxib treatment in AGS cells on the expression of COX-2, EGFR, Akt, and pGSK3β were evaluated by Western blot. The 10, 20, and 30 µmol/L concentrations of celecoxib showed significant inhibitory effects on the expression of COX-2 at 24 hours of incubation in the AGS cell lines (n=7) (Table 2, Fig. 2A). The 20 µmol/L concentration of celecoxib showed significant inhibitory effects on the overexpression of COX-2 by H. pylori infection (Table 2, Fig. 2A). For EGFR, H. pylori infection induced EGFR overexpression with G69a strain (Fig. 2B) and with G27 wild type (data not shown). However, overexpression of EGFR was not observed in AGS cell lines treated with cagE- mutant of G27 strain (data not shown). The 20 and 30 µmol/L concentrations of celecoxib showed significant inhibitory effects on the expression of EGFR in H. pylori treated AGS cell lines with G69a strain (n=12) (Table 2, Fig. 2B).
Table 2.
Effects of Celecoxib on the Protein Expression of COX-2, EGFR, tAkt, pAkt, and pGSK3β in an AGS Control and in Helicobacter pylori (HP)-Infected AGS Cells
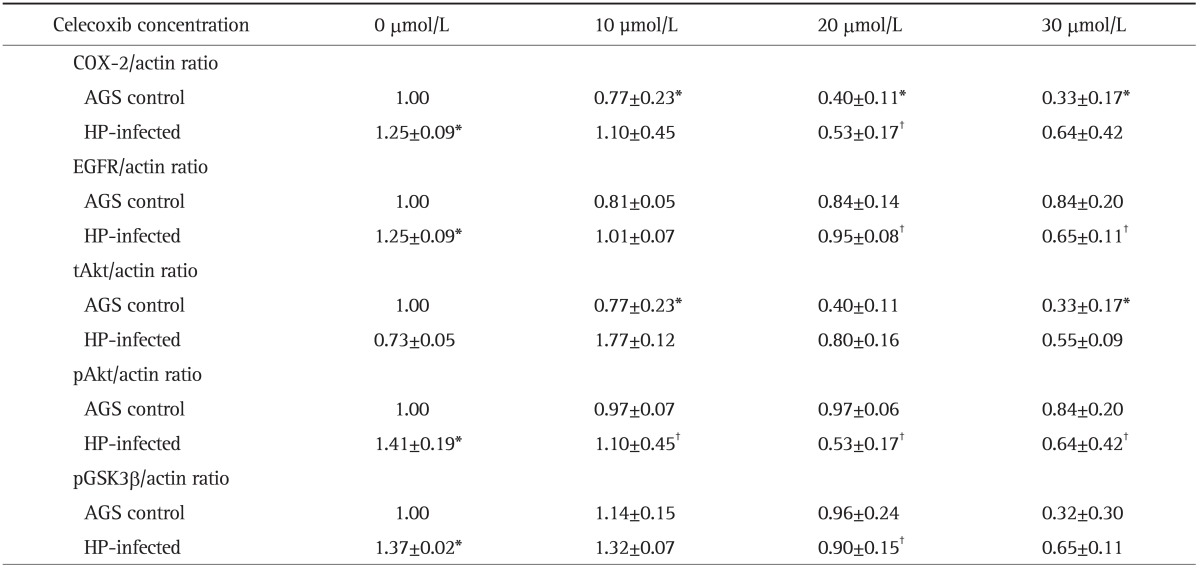
Data are presented as mean±SEM.
COX-2, cyclooxygenase-2; EGFR, epidermal growth factor receptor; tAkt, total Akt; pAkt, phosphorylated Akt; pGSK3β, phosphorylated glycogen synthase kinase-3β; HP, Helicobacter pylori.
*p<0.05 compared with AGS control; †p<0.05 compared with HP-infected cells without celecoxib treatment.
Fig. 2.

Western blotting for (A) cyclooxygenase-2 (COX-2), (B) epidermal growth factor receptor (EGFR), (C) total Akt (tAkt), (D) phosphorylated Akt (pAKt), and (E) phosphorylated glycogen synthase kinase-3β (pGSK3β) in Helicobacter pylori-infected AGS cells. The incubation of AGS cells infected by the H. pylori G69a strain and an AGS cell control with various concentrations (0, 10, 20, and 30 µmol/L) of celecoxib for 24 hours resulted in different protein expression. (A) AGS cells treated with the H. pylori G69a strain exhibited COX-2 overexpression (p=0.001). The 10, 20, and 30 µmol/L concentrations of celecoxib had inhibitory effects on the protein expression of COX-2 in the AGS control (p=0.026. p=0.001, and p=0.017, respectively). The 20 µmol/L concentration of celecoxib had inhibitory effects on the protein expression of COX-2 in H. pylori-treated AGS cells (p=0.015). (B) AGS cells treated with the H. pylori G69a strain exhibited significant EGFR overexpression (p<0.001). The 20 and 30 µmol/L concentrations of celecoxib had inhibitory effects on the protein expression of EGFR in H. pylori-treated AGS cells (p=0.025 and p=0.004, respectively). (C, D) AGS cells treated with the H. pylori G69a strain exhibited overexpression of pAkt (p<0.001) but not tAkt. The 30 µmol/L concentration of celecoxib had inhibitory effects on the expression of tAkt in the AGS control (p=0.020). There was a significant increase in pAkt expression after H. pylori infection (p=0.001). The 10, 20, and 30 µmol/L concentrations of celecoxib inhibited the overexpression of pAkt after 24 hours of incubation with the AGS control (p=0.026, p=0.001, and p=0.017, respectively). The 20 µmol/L concentration of celecoxib inhibited overexpression of pAkt after 24 hours of incubation with AGS cells treated with the G69a strain (p=0.015). (E) AGS cells treated with the H. pylori G69a strain showed significant overexpression of phosphorylated glycogen synthase kinase-3β (pGSK3β) (p=0029). The 20 µmol/L concentration of celecoxib had a significant inhibitory effect on the expression of pGSK3β in AGS cells treated with the G69a strain (p=0.029).
*p<0.05 compared with the AGS control; †p<0.05 compared with H. pylori-infected cells without celecoxib treatment.
In addition, we measured total Akt (tAkt), phosphorylated Akt (pAkt), and pGSK3β as downstream targets of EGFR-Akt signaling. AGS cell lines treated with H. pylori G69a strain showed significant overexpression of pAkt (p<0.001, n=7) (Fig. 2D), not tAkt (n=8) (Fig. 2C). Celecoxib showed significant inhibitory effects on the expression of pAkt in AGS control and H. pylori infected AGS cell lines (Fig. 2D). In case of pGSK3β, H. pylori induced overexpression of pGSK3β, which was inhibited by treatment of 20 µmol/L of celecoxib (p<0.029, n=4) (Fig. 2E).
DISCUSSION
Our experiments show that H. pylori infection up-regulated COX-2, TGF-β, HB-EGF, EGFR, and PI3K/Akt pathways in AGS cell lines. Importantly, celecoxib, a selective COX-2 antagonist, had inhibitory effect on EGFR and its downstream targets, which implies that H. pylori-induced EGFR activation may cross-talk with COX-2. Previously, other investigators reported H. pylori infection can initiate HB-EGF release, transactivate EGFR and subsequently activate ERK1/2 MAP kinase and PI3K/Akt signaling.19-21 Akt, a threonine protein kinase, plays a role as a regulator of cell proliferation apoptosis, glycogen metabolism, migration, and cell survival.23 It is one of major downstream target of EGFR-PI3K pathway and its complete activation need phosphorylation of regulate sites. Phosphorylated Akt targets glycogen synthase kinase 3 (GSK3), subsequently phosphorylates GSK3β and GSK3α. As overexpression of EGFR correlates with poor prognosis in several malignancies,24 inhibiting the downstream targets of EGFR activation by H. pylori is crucial. There have been reports that inhibition of Akt/PI3K enhances H. pylori-induced apoptosis.21 Our result coincides well with previous studies and supports the importance of blocking EGFR signaling pathways in carcinogenesis.19-21
Interestingly, we found out that AGS cells treated with cagE- strain did not show overexpression of EGFR compared to those treated with wild type. There have been numerous studies that H. pylori strains that possess the cag PAI induce more severe gastritis and increase the risk of peptic ulcer disease gastric adenocarcinoma.25,26 CagE, encoded by cag PAI, is one of structural protein of type IV secretion system that delivers the CagA protein into the host cell. This process is accomplished by specialized adhesin, which activates host cell integrins for subsequent delivery of CagA. Recently, there have been studies that type IV secretion system, as well as CagA, play an important role in the various signal pathways.27-29 Our study result well correspond to the previous study that cag PAI, especially cagE, is important in H. pylori-induced EGFR transactivation.19
On the other hand, previous investigators suggested that H. pylori-induced EGF activation may promote epithelial-mesenchymal transition (EMT).30 Cells undergoing EMT lose epithelial characteristics, such as, cell-to-cell adhesion and differentiation, and acquire mesenchymal properties like mobility, invasiveness and resistance to apoptosis. It is regulated by TGF-β mediated signal pathways; up-regulation of members of the EMT transcriptome, such as Snail and Slug, and down-regulation of E-cadherin.31,32 In addition, there has been evidence that COX-2 is related to EMT in colon cancer cells.33 Furthermore, there have been evidences that EGFR signaling is required to TGF-β mediated COX-2 induction in bronchial epithelial and hepatocellular carcinoma cell lines.34,35 Taken together, COX-2 might be involved in EMT as a downstream of EGFR pathway in gastric epithelial cell line and further study is undergoing.
The cross-talk between COX-2 and EGFR may be mediated by prostaglandins, especially prostaglandin E2 (PGE2). EGFR signaling activates MAP kinase activity and subsequently induces AP-1 mediated COX-2 transcription. In turn, increased COX-2 transcription enhances production of prostaglandins.34 Previously, we have demonstrated that celecoxib, not indomethacin, significantly reduced the number of viable gastric cancer cells in a dose- and time-dependent manner. Interestingly, we could find that the level of PGE2 is lower in the indomethacin-treated cells and addition of PGE2 did not make much difference in the inhibitory effect of celecoxib.16 It implies the mechanism is neither dependent on COX-2 nor PGE2 alone and might be more complex. We have limitations to conclude the direct mechanism here, as we did not measure the PGE2 levels in the presence or absence of celecoxib.
Several nonselective and selective COX-2 inhibitors have been the target of prevent cancers including gastric cancer.37,38 However, COX-2 inhibitors, such as valdecoxib and rofecoxib, were removed from the market previously due to increased risk of cardiovascular events, especially myocardial infarction.39,40 In case of celecoxib, there have been controversies in cardiovascular events. Although, CLASS study found the risk of cardiovascular events in celecoxib users,41 other studies showed that celecoxib exposure did not elevate the risk of cardiovascular events.42 Recently, a population-based intervention trial in China proved that celecoxib treatment alone could prevent progression of premalignant lesions to gastric cancer.43 Moreover, there have been various combinations of conventional chemotherapy with celecoxib have been studied,44-48 and some of them proved the effect of celecoxib cotreatment.44,45 Furthermore, there have been studies to target COX-2 and EGFR synergistically in metastatic colon cancer or recurred head and neck cancers.49,50 Given that COX-2 plays a role not only in the early stage of gastric cancer involving apoptosis, but also in the late stage of EMT, selective COX-2 inhibitor, celecoxib could be resurfaced as an anticancer agent with combination of molecular target chemotherapy.
Our study has many limitations. First, we planned to perform the same number of experiments such as six times for each issue. However, we could not avoid the variation numbers of experiments in each issue with following reasons. First, there had been insignificant change after H. pylori infection probably due to condition of H. pylori status or condition of cell line. Secondly, sometimes contamination occurred resulting in inconsistent results. In this case, we repeated the experiments again. Thirdly, we found variation of actin in the Western blotting, resulting in the difficulty of interpretation. In this case, we also repeated the experiment. Another limitation is that we did not measure the PGE2 levels with or without celecoxib. Thus, we could not conclude the direct mechanism regarding cross-talk between COX-2 and EGFR through prostaglandins, especially PGE2. We are planning to perform this experiment in the future.
In conclusion, H. pylori activated EGFR-Akt-GSK pathway in AGS cell lines, which was dependent on type IV secretion system. As celecoxib showed inhibitory effects on EGFR signal pathway, it seems that cross-talk between COX-2 and EGFR mediates the H. pylori-induced gastric cancer.
ACKNOWLEDGEMENTS
This work was supported by grant no. 02-2012-017 from the Seoul National University Bundang Hospital Research Fund.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Correa P. Helicobacter pylori and gastric cancer: state of the art. Cancer Epidemiol Biomarkers Prev. 1996;5:477–481. [PubMed] [Google Scholar]
- 2.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 3.Stoicov C, Saffari R, Cai X, Hasyagar C, Houghton J. Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: bacterial and host factors responsible for altered growth signaling. Gene. 2004;341:1–17. doi: 10.1016/j.gene.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 4.Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 5.Fischer W, Püls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams C, Shattuck-Brandt RL, DuBois RN. The role of COX-2 in intestinal cancer. Ann N Y Acad Sci. 1999;889:72–83. doi: 10.1111/j.1749-6632.1999.tb08725.x. [DOI] [PubMed] [Google Scholar]
- 7.Yamac D, Ayyildiz T, CoXMLLink_XYZkun U, et al. Cyclooxygenase-2 expression and its association with angiogenesis, Helicobacter pylori, and clinicopathologic characteristics of gastric carcinoma. Pathol Res Pract. 2008;204:527–536. doi: 10.1016/j.prp.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Tatsuguchi A, Matsui K, Shinji Y, et al. Cyclooxygenase-2 expression correlates with angiogenesis and apoptosis in gastric cancer tissue. Hum Pathol. 2004;35:488–495. doi: 10.1016/j.humpath.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Cho SO, Lim JW, Kim KH, Kim H. Involvement of Ras and AP-1 in Helicobacter pylori-induced expression of COX-2 and iNOS in gastric epithelial AGS cells. Dig Dis Sci. 2010;55:988–996. doi: 10.1007/s10620-009-0828-y. [DOI] [PubMed] [Google Scholar]
- 10.Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett. 2010;295:7–16. doi: 10.1016/j.canlet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Hu PJ, Yu J, Zeng ZR, et al. Chemoprevention of gastric cancer by celecoxib in rats. Gut. 2004;53:195–200. doi: 10.1136/gut.2003.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho SJ, Kim N, Kim JS, Jung HC, Song IS. The anti-cancer effect of COX-2 inhibitors on gastric cancer cells. Dig Dis Sci. 2007;52:1713–1721. doi: 10.1007/s10620-007-9787-3. [DOI] [PubMed] [Google Scholar]
- 13.Wang WH, Huang JQ, Zheng GF, Lam SK, Karlberg J, Wong BC. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2003;95:1784–1791. doi: 10.1093/jnci/djg106. [DOI] [PubMed] [Google Scholar]
- 14.Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Huang P, Xu X, Liu J, Guo C. Anticancer effect of celecoxib via COX-2 dependent and independent mechanisms in human gastric cancers cells. Dig Dis Sci. 2009;54:1418–1424. doi: 10.1007/s10620-008-0510-9. [DOI] [PubMed] [Google Scholar]
- 16.Kim N, Kim CH, Ahn DW, et al. Anti-gastric cancer effects of celecoxib, a selective COX-2 inhibitor, through inhibition of Akt signaling. J Gastroenterol Hepatol. 2009;24:480–487. doi: 10.1111/j.1440-1746.2008.05599.x. [DOI] [PubMed] [Google Scholar]
- 17.Caputo R, Tuccillo C, Manzo BA, et al. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res. 2003;9:2015–2021. [PubMed] [Google Scholar]
- 18.Zheng L, Wang L, Ajani J, Xie K. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004;7:61–77. doi: 10.1007/s10120-004-0277-4. [DOI] [PubMed] [Google Scholar]
- 19.Keates S, Keates AC, Nath S, Peek RM, Jr, Kelly CP. Transactivation of the epidermal growth factor receptor by cag+ Helicobacter pylori induces upregulation of the early growth response gene Egr-1 in gastric epithelial cells. Gut. 2005;54:1363–1369. doi: 10.1136/gut.2005.066977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabassam FH, Graham DY, Yamaoka Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol. 2009;11:70–82. doi: 10.1111/j.1462-5822.2008.01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan F, Cao H, Chaturvedi R, et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology. 2009;136:1297–1307. doi: 10.1053/j.gastro.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim N, Park WY, Kim JM, et al. Analysis of gene expression profile of AGS cells stimulated by Helicobacter pylori adhesion. Gut Liver. 2007;1:40–48. doi: 10.5009/gnl.2007.1.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 24.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Saadat I, Higashi H, Obuse C, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 26.Peek RM, Jr, Moss SF, Tham KT, et al. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–868. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 27.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10:1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- 28.Censini S, Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibata W, Hirata Y, Maeda S, et al. CagA protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the Mongolian gerbil model. J Pathol. 2006;210:306–314. doi: 10.1002/path.2040. [DOI] [PubMed] [Google Scholar]
- 30.Yin Y, Grabowska AM, Clarke PA, et al. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut. 2010;59:1037–1045. doi: 10.1136/gut.2009.199794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maehara Y, Kakeji Y, Kabashima A, et al. Role of transforming growth factor-beta 1 in invasion and metastasis in gastric carcinoma. J Clin Oncol. 1999;17:607–614. doi: 10.1200/JCO.1999.17.2.607. [DOI] [PubMed] [Google Scholar]
- 32.Vagenas K, Spyropoulos C, Gavala V, Tsamandas AC. TGFbeta1, TGFbeta2, and TGFbeta3 protein expression in gastric carcinomas: correlation with prognostics factors and patient survival. J Surg Res. 2007;139:182–188. doi: 10.1016/j.jss.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Jang TJ, Jeon KH, Jung KH. Cyclooxygenase-2 expression is related to the epithelial-to-mesenchymal transition in human colon cancers. Yonsei Med J. 2009;50:818–824. doi: 10.3349/ymj.2009.50.6.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu M, Yang SC, Sharma S, et al. EGFR signaling is required for TGF-beta 1 mediated COX-2 induction in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2007;37:578–588. doi: 10.1165/rcmb.2007-0100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogunwobi OO, Wang T, Zhang L, Liu C. Cyclooxygenase-2 and Akt mediate multiple growth-factor-induced epithelial-mesenchymal transition in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2012;27:566–578. doi: 10.1111/j.1440-1746.2011.06980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol. 2005;23:254–266. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 37.Futagami S, Suzuki K, Hiratsuka T, et al. Chemopreventive effect of celecoxib in gastric cancer. Inflammopharmacology. 2007;15:1–4. doi: 10.1007/s10787-006-1541-5. [DOI] [PubMed] [Google Scholar]
- 38.Yang P, Zhou Y, Chen B, et al. Aspirin use and the risk of gastric cancer: a meta-analysis. Dig Dis Sci. 2010;55:1533–1539. doi: 10.1007/s10620-009-0915-0. [DOI] [PubMed] [Google Scholar]
- 39.Juni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021–2029. doi: 10.1016/S0140-6736(04)17514-4. [DOI] [PubMed] [Google Scholar]
- 40.Kimmel SE, Berlin JA, Reilly M, et al. Patients exposed to rofecoxib and celecoxib have different odds of nonfatal myocardial infarction. Ann Intern Med. 2005;142:157–164. doi: 10.7326/0003-4819-142-3-200502010-00005. [DOI] [PubMed] [Google Scholar]
- 41.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 42.Graham DJ, Campen D, Hui R, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365:475–481. doi: 10.1016/S0140-6736(05)17864-7. [DOI] [PubMed] [Google Scholar]
- 43.Wong BC, Zhang L, Ma JL, et al. Effects of selective COX-2 inhibitor and Helicobacter pylori eradication on precancerous gastric lesions. Gut. 2012;61:812–818. doi: 10.1136/gutjnl-2011-300154. [DOI] [PubMed] [Google Scholar]
- 44.Arjona-Sánchez A, Ruiz-Rabelo J, Perea MD, et al. Effects of capecitabine and celecoxib in experimental pancreatic cancer. Pancreatology. 2010;10:641–647. doi: 10.1159/000288708. [DOI] [PubMed] [Google Scholar]
- 45.Aruajo AM, Mendez JC, Coelho AL, et al. Phase II study of celecoxib with cisplatin plus etoposide in extensive-stage small cell lung cancer. Cancer Invest. 2009;27:391–396. doi: 10.1080/07357900802232756. [DOI] [PubMed] [Google Scholar]
- 46.Csiki I, Morrow JD, Sandler A, et al. Targeting cyclooxygenase-2 in recurrent non-small cell lung cancer: a phase II trial of celecoxib and docetaxel. Clin Cancer Res. 2005;11:6634–6640. doi: 10.1158/1078-0432.CCR-05-0436. [DOI] [PubMed] [Google Scholar]
- 47.El-Rayes BF, Zalupski MM, Manza SG, et al. Phase-II study of dose attenuated schedule of irinotecan, capecitabine, and celecoxib in advanced colorectal cancer. Cancer Chemother Pharmacol. 2008;61:283–289. doi: 10.1007/s00280-007-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan CX, Loehrer P, Seitz D, et al. A phase II trial of irinotecan, 5-fluorouracil and leucovorin combined with celecoxib and glutamine as first-line therapy for advanced colorectal cancer. Oncology. 2005;69:63–70. doi: 10.1159/000087302. [DOI] [PubMed] [Google Scholar]
- 49.Chan E, Lafleur B, Rothenberg ML, et al. Dual blockade of the EGFR and COX-2 pathways: a phase II trial of cetuximab and celecoxib in patients with chemotherapy refractory metastatic colorectal cancer. Am J Clin Oncol. 2011;34:581–586. doi: 10.1097/COC.0b013e3181fe46a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kao J, Genden EM, Chen CT, et al. Phase 1 trial of concurrent erlotinib, celecoxib, and reirradiation for recurrent head and neck cancer. Cancer. 2011;117:3173–3181. doi: 10.1002/cncr.25786. [DOI] [PubMed] [Google Scholar]