

Abstract
Here, we report a Chinese case of Creutzfeldt–Jakob disease (CJD) with a rare mutation in the prion protein gene (PRNP) leading to an exchange of amino acid from valine (Val) to isoleucine (I) at codon 203 (V203I). The 80-y-old male presented with sudden memory loss, rapid loss of vocabulary, inattention and slow responses, accompanied by dizziness, blurred vision and ataxia. Two weeks after admission, he exhibited tremor, myoclonus and bilateral Babinski signs. At the end of the clinical course, he developed severe akinetic mutism. The cerebrospinal fluid (CSF) was positive for 14-3-3 protein. Increased bilateral signal intensity in the frontal and parietal lobes was seen on diffusion-weighted imaging (DWI); periodic activity was recorded on an electroencephalogram (EEG). There was no family history of similar symptoms. The total clinical course was approximately two months.
Keywords: 14-3-3 protein, PRNP, Creutzfeldt–Jakob disease, V203I, mutation
Introduction
Human prion diseases, including Creutzfeldt–Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), fatal familial insomnia (FFI) and Kuru, are fatal neurodegenerative disorders characterized by the accumulation of an abnormal isoform (PrPSc) of the host encoded cellular prion protein (PrPC) in the brain.1 Brain extracts which transmit experimentally the disease to recipient animals contain abnormal PK-resistant PrP. Of these, CJD is the most frequent, with an estimated case incidence of one person per million per year. About 85% of all CJD cases are sporadic, 10–15% are inherited and less than 1% are infectious.2 The PRNP gene is located on chromosome 20p12. It encodes the PrP protein, which undergoes a conformational conversion to an abnormal PrPSc form believed to be responsible for the development of the disease. More than 35 insertion and missense mutations and one nonsense mutation, in the open reading frame (ORF) of the PRNP gene are known. All follow an autosomal dominant inheritance pattern. Worldwide, the most common mutations are E200K in genetic CJD (gCJD), D178N coupled with methionine at codon 129 in a mutated FFI allele, and P102L in GSS.
Since CJD surveillance began in China in 2006, more than 30 different gCJD cases have been identified. The most frequent were D178N FFI3 and T188K gCJD.4 Here, we report the first Chinese CJD case with a V203I mutation homozygous for methionine at codon 129 and for glutamic acid at codon 219.
Case Presentation
An 80-y-old male was referred to the Department of Neurology, Qianfoshan Hospital, Shandong Province, China in 2011 and reported to the China National Surveillance Network for CJD (CNSNC) as a CJD suspected case based on the diagnostic criteria recommended by World Health Organization (WHO). The clinical data were collected by hospital neurologists; epidemiologic information was collected by the staff of the local Centers for Disease Control (CDC). Blood and cerebrospinal fluid (CSF) were collected for CSF 14-3-3 protein assay and PRNP sequencing at the CNSNC.
The patient was admitted to hospital about one week after exhibiting a sudden loss of memory accompanied by dizziness. He had a 30 y history of chronic gastritis, a 20 y history of coronary disease, 10 y history of encephalatrophy and a 10 y history of hypertension with the highest recorded blood pressure of 170/90 mmHg. At admission, he presented with memory loss, loss of vocabulary, inattention and slow responses, accompanied by dizziness, blurred vision and ataxia. He could walk without assistance. About one week later, his symptoms had progressed markedly and he could not do arithmetic calculations beyond Mini Mental State Examination (MMSE) grade 19. He experienced obvious myoclonic jerks and was not able to walk by himself. Two weeks after admission, tremor and myoclonus occurred frequently, he stayed in bed for most of the day, and his memory loss had worsened. Bilateral Babinski signs were easily released. At the end of the clinical course, he developed severe akinetic mutism. During hospitalization, the patient received symptomatic therapy (e.g., for hypertension, hyperlipidemia and neurotrophic therapy) and supportive care.
Brain MRI performed 10 d after the onset of clinical symptoms showed an ischemic infarct in the left centrum semiovale and bilateral frontal lobes. Diffusion-weighted imaging (DWI) showed increased bilateral signal intensity in the frontal and parietal lobes. CSF was obtained 20 d after the onset of clinical symptoms by routine lumbar puncture. The CSF was clear, the pressure was 100 mm H2O, albumin was slightly elevated at 479 mg/L (normal range is 0–350 mg/L). The CSF White Blood Cell (WBC) count was 10 × 106/L and the Red Blood Cell (RBC) count was 2 × 106/L. Western blotting identified a large, 30 kDa 14-3-3 protein specific signal in the CSF (Fig. 1). For western blotting, a 20 μL CSF sample was separated on a 12% SDS-PAGE gel and electronically transferred onto a nitrocellulose membrane. Blots were incubated with a 14-3-3 polyclonal antibody (1:1000 dilution, Santa Cruz Biological) and then incubated with a Horseradish Peroxidase conjugated goat anti-Rabbit IgG (1:5000 dilution, Santa Cruz Biological). Immunoreactive bands were assayed by an electrochemoluminescence (ECL) method (Amersham Life Sciences). Periodic sharp-waves (PSWs) were observed on an electroencephalogram (EEG) at 25 d after onset. His general condition deteriorated progressively and he died in the hospital two months after onset. Interviews with family members did not reveal anyone with a history of similar symptoms. His father died of cardiac failure and his mother died of a hemorrhagic stroke. His children are currently healthy. No brain autopsy was performed; family members refused further genetic testing.
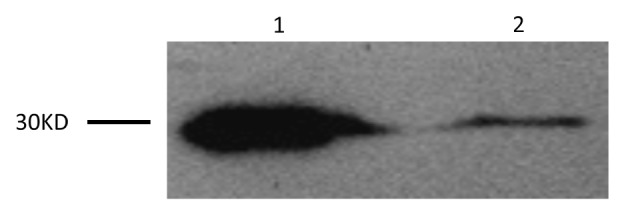
Figure 1. Western blot analysis for protein 14-3-3 in the patient’s CSF is shown in Lane 1. Lane 2 is the positive control.
Genomic DNA was extracted from peripheral blood leukocytes using a commercial kit (QIAGEN, Germany). One hundred nanograms of the extracted DNA were amplified by Polymerase Chain Reaction (PCR) using specific PRNP primers (forward primer: 5′-GGCAAACCTTGGATGCTGG-3′ and reverse primer: 5′-CCCACTATCAGGAAGATGAGG-3′). Direct sequencing of the PCR product showed a missense mutation at codon 203 of the PRNP gene (A to G), leading to a substitution of valine (Val) by isoleucine (Ile) in the PrP protein (Fig. 2). No additional nucleotide exchanges were found in other regions of the PRNP sequence. The patient’s PRNP DNA also contained a methionine homozygosity at codon 129 (M129M) and a glutamic acid homozygosity at codon 219 (E219E) (Fig. 3).
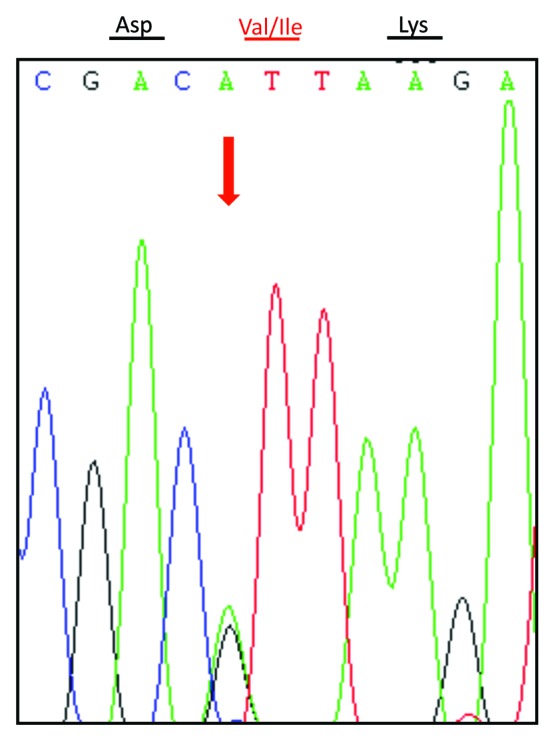
Figure 2. Graphical representation of the PRNP sequence analysis showing a G to A heterozygous transition at codon 203 in one PRNP allele, leading to substitution of valine (V) by isoleucine (I). The arrow above the curve indicates the position where both V and I are present.

Figure 3. Graphical representation of the Met/Met homozygous polymorphism at codon 129 (A) and Glu/Glu homozygosity at codon 219 (B) of PRNP in this patient.
Discussion
With direct sequencing, we characterized a V203I mutation in the coding region of the PRNP gene from a suspected CJD case. This is the first report of a V203I gCJD case in China; and to our knowledge, only two other gCJD cases associated with a V203I mutation have been reported worldwide. The first, a 69-y-old Italian male homozygous (M/M) at codon 129,5 was reported in 2000, and the second was a 66-y old Korean female heterozygous (M/V) at codon 129 was reported in 2010.6
The clinical characteristics of this V203I gCJD case were similar to sporadic CJD (sCJD); with sudden occurrence and rapid, progressive memory loss accompanied by obvious ataxia, myoclonic jerks and tremor. Progressive cognitive dysfunction, gait disturbance, myoclonus, tremor and rigidity were also described in the two other V203I gCJD cases.5,6 The disease progressed rapidly in all three V203I gCJD cases, approximately 2 mo after onset for the Korean and Chinese patients and 25 d for the Italian patient. The course in all 3 patients was significantly shorter than for the sCJD cases described in the literature.7 At 80 y of age, the Chinese patient in this report was much older than the other two, who were both 60 y of age at onset (Table 1). None of the three patients had a family history, indicating that disease occurred sporadically.
Table 1. Clinical, electroencephalographic, CSF protein 14-3-3 and genetic features of three patients with the PRNP V203I mutation.
| Author, year, country | Age at onset (years), gender | Initial clinical manifestation | Clinical symptoms | PSWCs in EEG | 14-3-3 in CSF |
MRI data | PrPSc in brain | Codon 129 | Genealogical information | Clinical course (months) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peoc'h et al. 2000, Italy |
69, Male |
Monocular diplopia, dizziness |
Hallucination, tremor, cerebellar gait, coordination deficit, multidirectional nystagmus, myoclonus |
+ |
+ |
Ø |
+ |
M/M |
Father died at age 60 from cardiac failure; mother died at age 62 from traffic accident |
1 |
||
| Jeong et al. 2010, Korea |
66, Female |
Gait disturbance, progressive cognitive dysfunction |
Tremor, mild dysphonation, cogwheel rigidity, myoclonus, dysphonation, dysphagia, quadriparesis, dyspnoea, stuporous state |
Ø |
+ |
Gyriform hyperintensity in the cerebral cortex on diffusion-weighted images and T2-weighted images |
+ |
M/V |
No information available for her parents |
2 |
||
| Shi et al. this report, China | 80, Male | Memory loss, inattention, slow responses | Dizziness, blurred vision, ataxia, tremor, myoclonus, bilateral Babinski signs, akinetic mutism | + | + | Increased signal intensity in bilateral frontal and parietal lobes | Ø | M/M | Father died from cardiac failure, mother died from stroke; ages at death not known | 2 | ||
Key: CSF, cerebrospinal fluid; PSWC, periodic sharp wave complex; MRI, magnetic resonance image; +, positive; -, negative; Ø, not available.
In addition to the characteristics described above, the three V203I gCJD patients had some similar clinical and laboratory features. All were positive for 14-3-3 protein in the CSF. The Italian and Chinese patients had periodic activities typical of sCJD on the EEG.8 The MRI results in the Korean case showed gyriform hyperintensity in the cerebral cortex in T2-weighted and diffusion-weighted images.6 Increased bilateral signal intensity was also observed in the frontal and parietal lobes with DWI in the Chinese case. For the MRI of ischemic infarct, the centrum semiovale is the common affected position and the shape is usually plaque or roundness. At the early stage of the disease, it may show high signal in DWI at the nidus. Along with the development of the disease, the signal will reduce or disappear. For the MRI of CJD, the high signal of DWI always appears in the cortex along with the gyrus. As the progression of CJD, the signal may aggravate or distribute to more extensive areas. In the Korean case, postmortem neuropathological data revealed type-1 accumulation of PrPSc, astrogliosis, vacuolization and neuronal loss. Brain extracts from the Italian case revealed an abnormal PK-resistant form of PrP. Thus, the neuropathological changes of these V203I gCJD cases were similar those seen in sporadic CJD.
Polymorphism at codon 129 of PRNP has been related to the onset age and survival times of several subtypes of human prion diseases.9 One of the three V203I mutation cases was heterozygous (M/V) and two were homozygous (M/M) at codon 129. The data are limited, but suggest that polymorphism at codon 129 does not have a strong effect on the pathogenesis of V203I gCJD, independent of the onset age or survival time. Whether the homozygous Val/Val at codon 129 contributed to the pathogenic effect of the V203I mutation remains unknown. Nuclear magnetic resonance (NMR) shows that the V203I mutation is located within the third α helix of the PrP protein. The amino acid residue of codon 203 in the PrP proteins is valine in primates, mice and rats and isoleucine in hamsters, sheep and cattle. It is located in the hydrophobic core of PrP.10 More than 10 missense mutations have been reported in the segment between amino acids 196 and 217 in PRNP. It is presumed that the mutations in that region result in reduction of the stability of PrP protein. Nevertheless, the lack of a family history in all three V203I gCJD cases indicates that the V203I mutation might have a low pathogenicity.
Acknowledgments
We thank the staff from Qianfoshan Hospital for supplying patient clinical information. This work was supported by the China Mega-Project for Infectious Disease (2011ZX10004–101, 2012ZX10004215), the Young Scholar Scientific Research Foundation of China CDC (2012A102) and SKLID Development Grant (2012SKLID102, 2011SKLID211)
Disclosure of Potential Conflicts of Interest
The authors report no conflicts of interest
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/24674
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sikorska B, Knight R, Ironside JW, Liberski PP. Creutzfeldt-Jakob disease. Adv Exp Med Biol. 2012;724:76–90. doi: 10.1007/978-1-4614-0653-2_6. [DOI] [PubMed] [Google Scholar]
- 3.Shi Q, Chen C, Gao C, Tian C, Zhou W, Zhang B, et al. Clinical and familial characteristics of ten chinese patients with fatal family insomnia. Biomed Environ Sci. 2012;25:471–5. doi: 10.3967/0895-3988.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Chen C, Shi Q, Zhou W, Zhang XC, Dong JH, Hu XQ, et al. Clinical and familial characteristics of eight Chinese patients with T188K genetic Creutzfeldt-Jakob disease. Infect Genet Evol. 2013;14:120–4. doi: 10.1016/j.meegid.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 5.Peoc’h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, et al. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000;15:482. doi: 10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 6.Jeong BH, Jeon YC, Lee YJ, Cho HJ, Park SJ, Chung DI, et al. Creutzfeldt-Jakob disease with the V203I mutation and M129V polymorphism of the prion protein gene (PRNP) and a 17 kDa prion protein fragment. Neuropathol Appl Neurobiol. 2010;36:558–63. doi: 10.1111/j.1365-2990.2010.01094.x. [DOI] [PubMed] [Google Scholar]
- 7.Das K, Davis R, Dutoit B, Parsons B. Sporadic Creutzfeldt-Jakob disease: a description of two cases. Int Psychogeriatr. 2012;24:1183–5. doi: 10.1017/S1041610212000373. [DOI] [PubMed] [Google Scholar]
- 8.Bouwman NA, Verhagen WI, Meulstee J. EEG abnormalities in poikilothermia suggesting Creutzfeldt-Jakob disease. Clin EEG Neurosci. 2009;40:196–9. doi: 10.1177/155005940904000313. [DOI] [PubMed] [Google Scholar]
- 9.Webb TE, Whittaker J, Collinge J, Mead S. Age of onset and death in inherited prion disease are heritable. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:496–501. doi: 10.1002/ajmg.b.30844. [DOI] [PubMed] [Google Scholar]
- 10.Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A. 1998;95:11667–72. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]