

Alkali metal zincates are among an increasing number of mixed-metal organoreagents that are attracting widespread attention because of their ability to exhibit synergic reactivity. Such special behaviour can be defined as reactions arising from the cooperative effects of the two distinct metals, the hard alkali metal and soft zinc, within the multicomponent zincate that cannot be reproduced by either the single alkali metal or zinc component on its own. This synergism has been particularly prominent in metallation (metal-hydrogen exchange) applications.1 Alkylzinc (R2Zn) or amidozinc [RZn(NR′2)/Zn(NR′2)2] reagents are generally notoriously poor kinetic bases incapable of directly metallating (zincating) aromatic substrates to any synthetically useful extent, but combined with an alkali metal compound2 or related component3 they can transform into highly reactive “zincators”. Kondo and Uchiyama’s “LiZn(TMP)tBu2” (TMP = 2,2,6,6-tetramethylpiperidide), Mongin’s “LiZn(TMP)3”,[2a–j] and our own [(TMEDA)Na(TMP)(tBu)Zn(tBu)][2p–t] (TMEDA = N,N,N′,N′-tetramethylethylenediamine) belong in this category and although fundamental differences exist between these powerful amide-based zincators, their Zn-H exchange reactions can be grouped together as alkali-metal-mediated zincations “AMMZn’s”.1 Exemplified by TMP, the amide components involved in AMMZn chemistry are invariably monoanionic ligands derived from secondary amines containing one acidic N–H bond. Secondary diamines containing two acidic N–H bonds and therefore having potentially at least two metallation sites would offer an interesting contrast but until this work they have not been investigated in this context. Reported herein, our first venture in introducing diamines to this chemistry by reacting a mixture of nBuLi and tBu2Zn (or Me2Zn) with N,N′-diisopropylethylenediamine, iPrN(H)CH2CH2N(H)iPr, has uncovered a surprising new synergic chemistry, which leads ultimately to the transformation of the saturated ethylenediamine to a dianionic unsaturated diazaethene.
To gauge whether a bimetallic mixture exhibits synergic activity, its separated monometallic components should be reacted with the substrate in control reactions. As expected, we found that tBu2Zn is too weak a base to deprotonate N,N′-diisopropylethylenediamine, but instead forms the simple Lewis acid-Lewis base adduct [tBu2Zn· {iPrN(H)CH2CH2N(H)iPr}] (1; Scheme 1: see the Supporting Information for full details). While we easily generated a crystalline product from a 1:1 reaction of the more powerful base nBuLi and the diamine, and characterised it spectroscopically as [{LiN(iPr)CH2CH2N(H)iPr}], we deemed it unnecessary to determine its crystal structure as Gardiner and Raston had previously reported4 that the same reaction with the tert-butyl homologue tBuN(H)CH2CH2N(H)tBu produced lithiation of one N–H unit in [cis-{Li[μ-N(tBu)CH2CH2N(H)tBu]}2] (2), which is dimeric with a 5,4,5-fused ring system having a (LiN)2 core (Scheme 1). From the precedents of these homometallic zinc and lithium control reactions, in the absence of any co-operative interactions between the distinct metals one might anticipate that reaction of a 1:1 mixture of tBu2Zn and nBuLi with N,N′-diisopropylethylenediamine in the presence of TMEDA (one molar equivalent) would produce a co-complex of composition [tBu2Zn{iPrN(Li⋅TMEDA)CH2CH2N(H)iPr}] (3). Kinetically it does and we obtained the crystal structure of 3 (Figure 1). However, thermodynamically, this reaction mixture in hexane solution surprisingly affords the crystalline product [(TMEDA)Li(iPrNCHCHNiPr)Zn(tBu)] (4). As depicted in Scheme 1, formation of this lithium 1,3-diaza-2-zincacyclopentene formally requires the loss of two protons and hydrogen gas to transform the neutral saturated ethylenediamine to a dianionic unsaturated variant. To check whether this transformation was the result of employing two or more equivalents of the metal reagent and/or adding TMEDA, a 3:1:0 and 3:1:3 mixture of nBuLi, diamine, and TMEDA, in the absence of tBu2Zn, was evaluated but NMR spectroscopic studies confirmed only synthetically insignificant trace amounts of a diazaethene product with the major product being an ethylenediamine complex in which the -NCH2CH2N- bridge is retained.5 On this evidence, the double (sp3) C-H bond activation and concomitant C=C formation involved in the making of the metallocycloalkene 4 can be attributed at least in part to a special bimetallic synergic effect under the particular conditions studied, though other factors such as changing concentration may also be important as C=C bond formation has been observed to occur in concentrated monometallic systems (see below).
Scheme 1.
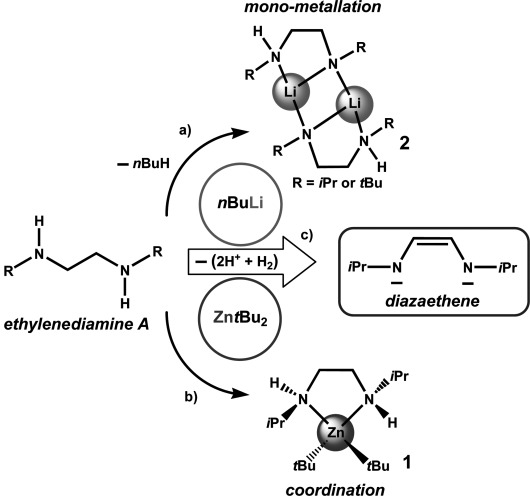
Comparison of non-cooperative reactions [nBuLi (a) or tBu2Zn (b)] and cooperative reactions [nBuLi and tBu2Zn (c)] of metal alkyls with an ethylenediamine A. Reaction conditions: a) hexane at 25 °C, 1 h; b) hexane at 25 °C, 24 h and at 69 °C, 10 min; and c) hexane at 69 °C, 2 h.
Figure 1.
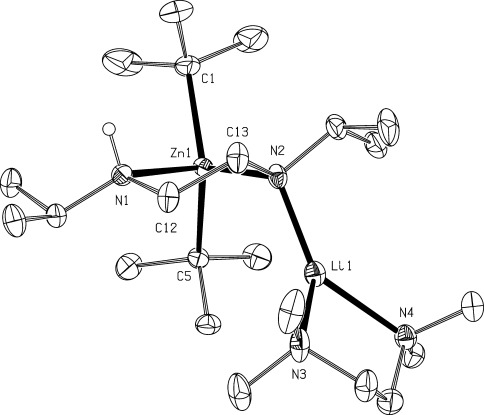
Molecular structure of 3 with hydrogen atoms (except N-H) omitted for clarity. Selected bond lengths [Å] and angles [o]: Zn1-C1 2.073(2), Zn1-C5 2.068(2), Zn1-N1 2.326(1), Zn1-N2 2.127(1), Li1-N2 1.993(3), Li1-N3 2.143(3), Li1-N4 2.173(3), N1-C12 1.470(2), N2-C13 1.461(2), C12-C13 1.517(2); C5-Zn1-C1 125.65(7), C5-Zn1-N2 111.49(6), C5-Zn1-N1 108.98(5), C5-Zn1-N2 111.49(56), C1-Zn1-N1 102.67(6), N2-Zn1-N1 83.10(5), N2-Li1-N3 127.12(15), N2-Li1-N4 141.1(2), N3-Li1-N4 87.1(1).
Determined by X-ray crystallography, the molecular structures of 3 (Figure 1) and 4 can be classed as contact ion pair zincates comprising a TMEDA-chelated lithium cation and an alkyl(diamido)zinc anion. Within 3 distorted tetrahedral Zn1 completes a highly puckered C2N2Zn metallacyclic ring (see, for instance, the torsion angle Zn1-N2-C13-C12 = −48.82(17)o) with an N-Zn-N bite angle of 83.10(5)° with exo tBu substituents on Zn and iPr/H and iPr/Li substituents on N1 and N2, respectively. The iPr groups both occupy equatorial sites and lie anti to each other across the five-atom ring which exhibits an ethylene C12-C13 bond length of 1.517(2) Å consistent with a single bond. Trigonal-planar Li1 has an N3 coordination comprising one diamine atom and two N atoms of TMEDA with the formal anionic Li-N2 bond (1.993(3) Å) being shorter than the latter dative Li-N bonds (mean length, 2.158 Å). Though the connectivity within 4 can be crystallographically determined, generic twinning of the samples adversely effected all modelling attempts and rules out discussion of its bonding dimensions. However, using an identical procedure to that for 4, we synthesised and crystallographically characterised the isostructural methyl homologue [(TMEDA)Li(iPrNCHCHNiPr)Zn(Me)] (5), the crystal data for which are more accurate allowing such a discussion. In the molecular structure of 5 (Figure 2) the anionic moiety shows N,N′-chelation by the N-C=C-N unit to zinc (bite angle, 83.98(9)°) to build a five-atom metallacycle with exo iPr and Me substituents on N and Zn atoms, respectively. Distorted trigonal planar zinc deviates modestly from the C2N2 plane, bonding symmetrically to the N atoms (lengths, 1.966(2) and 1.964(2) Å; Zn 0.459(3)Å out of plane). In a typical range for sp2–sp2 C=C bonds, the C5-C6 bond length is 1.349(3) Å. The Li+ ion of the cationic moiety π bonds (η4-) asymmetrically to the N-C=C-N unit, closer to one end (Li1-N1, 2.165(4), Li1-C5, 2.235(4) Å) than the other (Li1-N2, 2.301(4); Li1-C6, 2.286(4) Å). This asymmetry continues with Li anti to Zn, with respect to the C2N2 plane, and separated from it by 2.688(4) Å. Though new for Li:Zn combinations, the alkali metal face-capped 1,3-diaza-2-metallocyclopentene motif of 4 and 5 is known for other combinations including for example Na:Zn,6 K:Zn,7 K:Mg,8 Li:Ga,9 or K:Ga.10 Most of these structural precedents originate from elemental alkali metal reduction of 1,4-diaza-1,3-butadienes or related unsaturated molecules which sets them synthetically apart from 4 and 5 as to the best of our knowledge these two Li:Zn examples represent the first to be synthesised from a completely non-activated aliphatic diamine.
Figure 2.
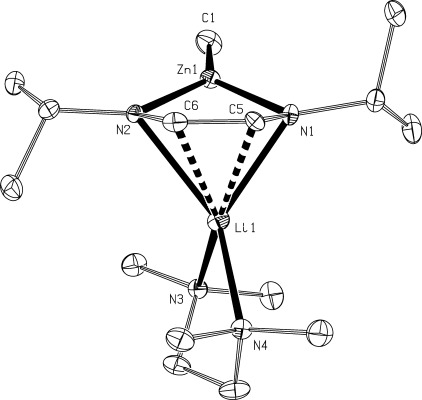
Molecular structure of 5 with hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [o]: Zn1-C1 1.956(3), Zn1-N1 1.966(2), Zn1-N2 1.964(2), Zn1-Li1 2.688(4), Li1-N1 2.165(4), Li1-N2 2.301(4), Li1-N3 2.145(4), Li1-N4 2.106(4), Li1-C5 2.235(4), Li1-C6 2.286(4), N1-C5 1.398(3), N2-C6 1.402(3), C5-C6 1.349(3), N1-C5 1.398(3); C1-Zn1-N1 136.6(1), C1-Zn1-N2 138.69(9), N2-Zn1-N1 83.98(9), N2-Li1-N3 112.5(2), N4-Li1-N3 86.51(16), N4-Li1-N1 126.8(2), N3-Li1-N1 124.6(2), N4-Li1-N2 139.9(2), N1-Li1-N2 72.1(1).
The closest synthetic analogy to the reaction producing 4 and 5 is Veith’s report[11a] of the 1,3-diaza-2-silacyclopentene 6 a as it involves dilithiation of tBuN(H)CH2CH2N(H)tBu (Scheme 2). Distinct from our method, the anticipated dianionic diamide was not isolated but trapped in situ with a dichlorosilane to generate the neutral, as opposed to our anionic, heterocyclopentene. Oddly, 6 a formed only in highly concentrated solutions,[11a] whereas dilute solutions,[11b] more akin to that employed in our study gave an alternative heterocyclopentane product 6 b. Veith et al. conceded that the reason for the double hydrogen abstraction from the ethylene backbone in forming 6 a was unclear. In theory 4 and 5 having potentially labile metal centres primed for participation in salt metathesis reactions could be regarded as intermediates in conversions of this type.
Scheme 2.
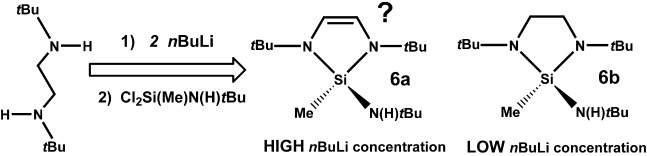
Veith’s reaction of tBuN(H)CH2CH2N(H)tBu with nBuLi and further trapping with a dichlorosilane.
A repeat reaction of tBu2Zn, nBuLi and the diamine in the additional presence of the bulky ketone (tBu)2C=O (Scheme 3) may have provided an initial clue to the mechanism behind the formation of 4. An NMR analysis of the crude reaction revealed a complicated mixture of products,5 among which, significantly, 4 and the lithium alkoxide [{tBu2C(H)OLi}4] (7), were clearly identified. The existence of 7, the result of a hydride addition to the electrophilic C=O of the ketone, hints at the possible participation of an intermediate hydride species in the N-CH2-CH2-N to N-CH=CH-N transformation. While the source of the hydride cannot be ascertained with absolute certainty at this juncture, we could straightforwardly rule out that it was coming from either nBuLi or tBu2Zn,12 suggesting that it originates from the ethylene backbone of the diamine. With monoamine dibenzylamine [(PhCH2)2NH], it has been postulated13 that metallation of N-H, followed by β-hydride elimination of “M+H−” and its subsequent metallation of the remaining NCH2 unit (accompanied by H2 evolution) accounts for the formation of the 2-aza-allyl anion [{PhC(H)=N-C(H)Ph}−]. Similar metallation/β-hydride elimination/re-metallation sequences could be operating in the reaction yielding 4. For completeness, isolated 7 was also crystallographically characterised. Its molecular structure (see the Supporting Information) is tetrameric with a heterocubane [(μ3-OR)4Li4] core of a type well documented for lithium alkoxides.14 These precedents commonly possess additional donor ligands at the Li corners, so 7 belongs to a rarer type15 wherein the large bulk of the R group promotes steric saturation of Li at the expense of electronic saturation.
Scheme 3.
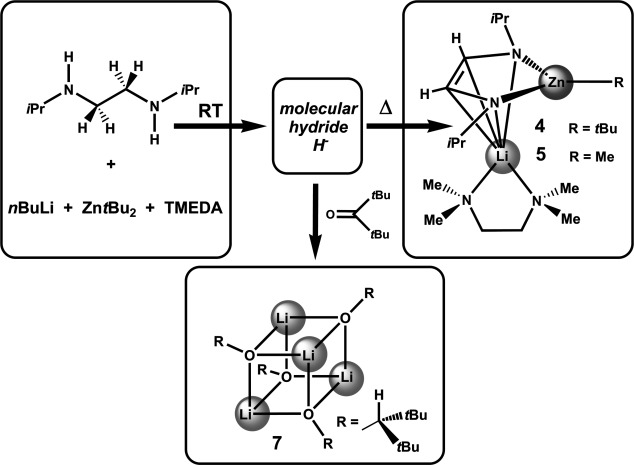
Reaction of iPrN(H)CH2CH2N(H)iPr with a nBuLi/R2Zn/TMEDA synergic mixture to form 4 or 5 via a putative intermediate hydride which can also be trapped with a ketone to generate a lithium alkoxide.
Noting that the N-CH2-CH2-N to N-CH=CH-N transformation outlined here for certain will not involve redox processes like that reported by Brookhart et al.16 for late transition metal catalysed intramolecular dehydrogenations, in future work we plan to carry out a comprehensive computational study to elucidate the mechanism of this transformation with emphasis on the precise role of the lithium–zinc co-operativity.
Experimental Section
General methods: All reactions and manipulations were carried out in an atmosphere of dry pure argon gas using standard Schlenk and glovebox techniques. n-Hexane was distilled from sodium benzophenone. NMR spectra were recorded on a Bruker AVANCE 400 NMR spectrometer, operating at 400.13 MHz for 1H, 155.50 MHz for 7Li and 100.62 MHz for 13C. Data for X-ray crystal structure determination were obtained with a Oxford Diffraction Gemini diffractometer using MoKα (λ = 0.71073 Å; compounds 5 and 7) and CuKα (λ = 1.54180 Å; compounds 1 and 3) graphite-monochromated radiations. Satisfactory elemental analyses of the compounds could not be obtained due to their high air- and moisture-sensitive nature.
Synthesis of 1: A Schlenk tube was charged with of ZntBu2 (4 mmol, 0.72 g), which was dissolved in hexane (20 mL) and one equivalent of iPr(H)NCH2CH2CH2N(H)iPr (4 mmol, 0.72 mL) was added by using a syringe. The resultant colourless solution was allowed to stir overnight at room temperature and heated at reflux temperature for 10 min. To aid crystallisation the solution was concentrated under reduced pressure to a final volume of 2–3 mL and, after standing overnight at −27 °C, colourless crystals of 1 (suitable for X-ray crystallographic analysis) were obtained (0.20 g, 15 %). The low crystalline yield obtained for 1 is just a reflection of its high solubility, being the overall reaction yield almost quantitative as determined by NMR spectroscopic analyses of both 1 and reaction filtrates. 1H NMR (400.13 MHz, C6D6, 293 K): δ=2.62 (m, 2 H, CH, iPr), 2.02 (m, 4 H, CH2), 1.34 (s, 18 H, CH3, tBu), 0.90 (d, J=5.2 Hz, 12 H, CH3, iPr), 0.85 ppm (s, br, 2 H, NH); 13C{1H} NMR (100.62 MHz, C6D6, 293 K): δ=49.5 (CH, iPr), 47.2 (CH2), 35.8 (CH3, tBu), 23.1 (CH3, iPr), 19.9 ppm (C(CH3), iPr).
Crystallisation of 3: nBuLi (1.25 mL, 2 mmol) was added dropwise to a solution of DPEDA(H2) (0.36 mL, 2 mmol) in hexane (10 mL) at 0 °C. This temperature was maintained as TMEDA (0.3 mL, 2 mmol) and a solution of tBu2Zn (0.36 g, 2 mmol) in hexane (10 mL) were added giving a pale yellow solution with some white solid. This solution was stored immediately at −27 °C giving a crop of colourless crystals suitable for X-ray crystallographic analysis corresponding to complex 3. Attempts to characterise the kinetic product 3 by NMR spectroscopy were unsuccessful due to its high thermal instability.
Synthesis of 4: nBuLi (1.25 mL, 2 mmol) was added dropwise to a solution of DPEDA(H2) (0.36 mL, 2 mmol) in hexane (10 mL) at 0 °C. This temperature was maintained as TMEDA (0.3 mL, 2 mmol) and a solution of tBu2Zn (0.36 g, 2 mmol) in hexane (10 mL) were added giving a pale yellow solution with some white solid. This solution was refluxed for 2 h producing a bright orange solution. Storing the solution at −70 °C gave a crop of yellow crystals of 4 which were isolated in a 38.9 % (0.30 g) crystalline yield. 1H NMR (400.13 MHz, C6D6, 300 K): δ(ppm)=5.83 (s, 2 H, CH, CH=CH), 3.48 (m, 2 H, CH, iPr), 1.81 (s, 12 H, CH3, TMEDA), 1.60 (s, 9 H, CH3, tBu), 1.58 (s, 4 H, CH2, TMEDA), 1.40 (d, J=6.3 Hz, 6 H, CH3, iPr), 1.32 ppm (d, J=6.3 Hz, 6 H, CH3, iPr); 13C{1H} NMR (100.62 MHz, C6D6, 300 K): δ(ppm)=116.3 (CH, CH=CH), 56.0 (CH2, TMEDA), 52.7 (CH, iPr), 45.6 (CH3, TMEDA), 35.4 (CH3, tBu), 28.6 (CH3, iPr), 28.0 (CH3, iPr), 20.6 ppm (C(CH3), tBu); 7Li (155.50 MHz, C6D6, 300 K): δ(ppm)= −2.40 ppm.
Acknowledgments
We gratefully acknowledge the generous support of the UK EPSRC (grant award no. EP/F063733/1) and the Royal Society/Wolfson Foundation (research merit award to R.E.M). We also thank our colleague Dr Eva Hevia for many enlightening discussions. This research was also sponsored by a Marie Curie Intra European Fellowship within the 7th European Community Framework Programme (for P.G.-A.).
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- 1a).Mulvey RE. Organometallics. 2006;25:1060. [Google Scholar]
- b).Mulvey RE, Mongin F, Uchiyama M, Kondo Y. Angew. Chem. 2007;119:3876. doi: 10.1002/anie.200604369. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:3802. [Google Scholar]
- c).Mulvey RE. Acc. Chem. Res. 2009;42:743. doi: 10.1021/ar800254y. For recent reviews on alkali metal zincates and their applications in metallation applications, see: [DOI] [PubMed] [Google Scholar]
- 2a).L’Helgoual’ch J-M, Bentabed-Ababsa G, Chevallier F, Yonehara M, Uchiyama M, Derdour A, Mongin F. 2008. Chem. Commun. 5375. [DOI] [PubMed]
- b).Dayaker G, Sreeshailam A, Chevallier F, Roisnel T, Krishna PR, Mongin F. Chem. Commun. 2010;46:2862. doi: 10.1039/b924939g. [DOI] [PubMed] [Google Scholar]
- c).Snégaroff K, L’Helgoual’ch J-M, Bentabed-Ababsa G, Nguyen TT, Chevallier F, Yonehara M, Uchiyama M, Derdour A, Mongin F. Chem. Eur. J. 2009;15:10280. doi: 10.1002/chem.200901432. [DOI] [PubMed] [Google Scholar]
- d).Seggio A, Priem G, Chevallier F, Mongin F. 2009. Synthesis 3617.
- e).Seggio A, Lannou M-I, Chevallier F, Nobuto D, Uchiyama M, Golhen S, Roisnel T, Mongin F. Chem. Eur. J. 2007;13:9982. doi: 10.1002/chem.200700608. [DOI] [PubMed] [Google Scholar]
- f).Seggio A, Chevallier F, Vaultier M, Mongin F. J. Org. Chem. 2007;72:6602. doi: 10.1021/jo0708341. [DOI] [PubMed] [Google Scholar]
- g).L’Helgoual’ch J-M, Seggio A, Chevallier F, Yonehara M, Jeanneau E, Uchiyama M, Mongin F. J. Org. Chem. 2008;73:177. doi: 10.1021/jo7020345. [DOI] [PubMed] [Google Scholar]
- h).Uchiyama M, Matsumoto Y, Usui S, Hashimoto Y, Morokuma K. Angew. Chem. 2007;119:944. doi: 10.1002/anie.200602664. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:926. [Google Scholar]
- i).Uchiyama M, Kobayashi Y, Furuyama T, Nakamura S, Kajihara Y, Miyoshi T, Sakamoto T, Kondo Y, Morokuma K. J. Am. Chem. Soc. 2008;130:472. doi: 10.1021/ja071268u. [DOI] [PubMed] [Google Scholar]
- j).Kondo Y, Morey JV, Morgan JC, Naka H, Nobuto D, Raithby PR, Uchiyama M, Wheatley AEH. J. Am. Chem. Soc. 2007;129:12734. doi: 10.1021/ja072118m. [DOI] [PubMed] [Google Scholar]
- k).García F, McPartlin M, Morey JV, Nobuto D, Kondo Y, Naka H, Uchiyama M, Wheatley AEH. 2008. Eur. J. Org. Chem. 644.
- l).Clegg W, Graham DV, Herd E, Hevia E, Kennedy AR, McCall MD, Russo L. Inorg. Chem. 2009;48:5320. doi: 10.1021/ic900313b. [DOI] [PubMed] [Google Scholar]
- m).Hevia E, Kennedy AR, Klett J, McCall MD. 2009. Chem. Commun. 3240.
- n).Armstrong DR, Herd E, Graham DV, Hevia E, Kennedy AR, Clegg W, Russo L. 2008. Dalton Trans. 1323. [DOI] [PubMed]
- o).Armstrong DR, Drummond AM, Balloch L, Graham DV, Hevia E, Kennedy AR. Organometallics. 2008;27:5860. [Google Scholar]
- p).Clegg W, Dale SH, Hevia E, Hogg LM, Honeyman GW, Mulvey RE, O’Hara CT. Angew. Chem. 2006;118:6698. doi: 10.1002/anie.200602288. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:6548. [Google Scholar]
- q).Armstrong DR, Garcia-Alvarez J, Graham DV, Honeyman GW, Hevia E, Kennedy AR, Mulvey RE. Chem. Eur. J. 2009;15:3800. doi: 10.1002/chem.200801928. [DOI] [PubMed] [Google Scholar]
- r).Armstrong DR, Clegg W, Dale SH, Hevia E, Hogg LM, Honeyman GW, Mulvey RE. Angew. Chem. 2006;118:3859. doi: 10.1002/anie.200600720. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:3775. [Google Scholar]
- s).Armstrong DR, Balloch L, Clegg W, Dale SH, Garcia-Alvarez P, Hevia E, Hogg LM, Kennedy AR, Mulvey RE, O’Hara CT. Angew. Chem. 2009;121:8831. doi: 10.1002/anie.200904506. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:8675. [Google Scholar]
- t).Andrikopoulos PC, Armstrong DR, Barley HRL, Clegg W, Dale SH, Hevia E, Honeyman GW, Kennedy AR, Mulvey RE. J. Am. Chem. Soc. 2005;127:6184. doi: 10.1021/ja050860l. [DOI] [PubMed] [Google Scholar]
- 3a).Wunderlich S, Knochel P. 2008. Chem. Commun. 6387.
- b).Mosrin M, Monzon G, Bresser T, Knochel P. 2009. Chem. Commun. 5615. [DOI] [PubMed]
- c).Wunderlich SH, Rohbogner CJ, Unsinn A, Knochel P. Org. Process Res. Dev. 2010;14:339. [Google Scholar]
- d).Wunderlich S, Knochel P. Org. Lett. 2008;10:4705. doi: 10.1021/ol802118e. [DOI] [PubMed] [Google Scholar]
- e).Mosrin M, Knochel P. Chem. Eur. J. 2009;15:1468. doi: 10.1002/chem.200801831. [DOI] [PubMed] [Google Scholar]
- f).Mosrin M, Knochel P. Org. Lett. 2009;11:1837. doi: 10.1021/ol900342a. [DOI] [PubMed] [Google Scholar]
- g).Dong Z, Clososki GC, Wunderlich SH, Unsinn A, Li J, Knochel P. Chem. Eur. J. 2009;15:457. doi: 10.1002/chem.200801558. [DOI] [PubMed] [Google Scholar]
- h).Hevia E, Chua JZ, Garcia-Alvarez P, Kennedy AR, McCall MD. Proc. Natl. Acad. Sci. USA. 2010;107:5294. doi: 10.1073/pnas.0913307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardiner M G, Raston C L. Inorg. Chem. 1996;35:4047. doi: 10.1021/ic951251p. [DOI] [PubMed] [Google Scholar]
- 5. See the Supporting Information for full experimental and crystallographic details.
- 6.Yang X-J, Yu J, Liu Y, Xie Y, Schaefer H F, Liang Y, Wu B. 2007. Chem. Commun. 2363.
- 7.Rijnberg E, Boersma J, Jastrzebski J T B H, Lakin M T, Spek A L, Van Koten G. 1995. Chem. Commun. 1839.
- 8.Liu Y, Li S, Yang X-J, Yang P, Wu B. J. Am. Chem. Soc. 2009;131:4210. doi: 10.1021/ja900568c. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt E S, Mitzel N W, Schmidbaur H. Z. Naturforsch. B. 2001;56:937. [Google Scholar]
- 10.Aldridge S, Baker R J, Coombs N D, Jones C, Rose R P, Rossin A, Willock D J. 2006. Dalton Trans. 3313. [DOI] [PubMed]
- 11a).Veith M, Schillo B, Huch V. Angew. Chem. 1999;111:131. [Google Scholar]; Angew. Chem. Int. Ed. 1999;38:182. [Google Scholar]
- b).Veith M, Rammo A. Z. Anorg. Allg. Chem. 1997;623:861. [Google Scholar]
- 12. Note that in the synthesis of 4 the order of addition of the metal reagents to the diamine is not crucial, as tBu2Zn or nBuLi can be introduced first. In the latter case all the nBuLi is consumed in forming a lithium amide before the zinc reagent is applied. This rules out the possibility that in the ketone reaction the hydride comes from the lithium reagent, a point confirmed by a control reaction between nBuLi and the ketone which did not produce a reduction product. The other possibility that H− was coming from tBu groups on zinc was also ruled out as the reaction proved repeatable with Me2Zn forming 5.
- 13a).Andrews PC, Armstrong DR, Baker DR, Mulvey RE, Clegg W, Horsburgh L, O’Neil PA, Reed D. Organometallics. 1995;14:427. [Google Scholar]
- b).Andrews PC, Duggan PJ, Fallon GD, McCarthy TD, Peatt AC. 2000. J. Chem. Soc. Dalton Trans. 2505.
- c).Andrews PC, Duggan PJ, Maguire M, Nichols PJ. 2001. Chem. Commun. 53.
- 14a).Boyle TJ, Pedrotty DM, Alam TM, Vick SC, Rodriguez MA. Inorg. Chem. 2000;39:5133. doi: 10.1021/ic000432a. [DOI] [PubMed] [Google Scholar]
- b).Boyle TJ, Alam TM, Peters KP, Rodriguez MA. Inorg. Chem. 2001;40:6281. doi: 10.1021/ic0106569. See, for example. [DOI] [PubMed] [Google Scholar]
- 15a).Piarulli U, Williams DN, Floriani C, Gervasio G, Viterbo D. 1994. Chem. Commun. 1409.
- b).Reisinger A, Trapp N, Krossing I. Organometallics. 2007;26:2096. See, for example. [Google Scholar]
- 16a).Diaz-Requejo M M, Disalvo D, Brookhart M. J. Am. Chem. Soc. 2003;125:2038. doi: 10.1021/ja029393n. [DOI] [PubMed] [Google Scholar]
- b).Bolig AD, Brookhart M. J. Am. Chem. Soc. 2007;129:14544. doi: 10.1021/ja075694r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.