

Abstract
Seven proteins in the human blood clotting cascade bind, via their GLA (γ-carboxyglutamate-rich) domains, to membranes containing exposed phosphatidylserine (PS), although with membrane binding affinities that vary by three orders of magnitude. Here we employed Nanodiscs of defined phospholipid composition to quantify the phospholipid binding specificities of these seven clotting proteins. All bound preferentially to nanobilayers in which PS headgroups contained L-serine versus D-serine. Surprisingly, however, nanobilayers containing phosphatidic acid (PA) bound substantially more of two of these proteins—factor VIIa and activated protein C—than did equivalent bilayers containing PS. Consistent with this finding, liposomes containing PA supported higher proteolytic activity by factor VIIa and activated protein C toward their natural substrates (factors X and Va, respectively) than did PS-containing liposomes. Moreover, treating activated human platelets with phospholipase D enhanced the rates of factor X activation by factor VIIa in the presence of soluble tissue factor. We hypothesize that factor VII and protein C bind preferentially to the monoester phosphate of PA because of its accessibility and higher negative charge compared to the diester phosphates of most other phospholipids. We further found that phosphatidylinositol 4-phosphate, which contains a monoester phosphate attached to its myo-inositol headgroup, also supported enhanced enzymatic activity of factor VIIa and activated protein C. We conclude that factor VII and protein C bind preferentially to monoester phosphates, which may have implications for the function of these proteases in vivo.
Keywords: Membranes, Phospholipids, Blood clotting, Phosphatidylinositol phosphates
It has long been known that changes in the phospholipid composition of cell membranes profoundly regulate blood clotting.1 This is because many of the proteins of the plasma clotting cascade, both procoagulant and anticoagulant, bind reversibly to membrane bilayers containing exposed anionic phospholipids, with phosphatidylserine (PS) being reported in a wealth of studies to have the greatest ability to accelerate blood coagulation. In this paper we provide evidence that, surprisingly, phosphatidic acid (PA) enhances the activity of certain membrane-bound blood clotting reactions more effectively than does PS.
The most common membrane-binding domains of proteins in the mammalian blood clotting cascade are γ-carboxyglutamate-rich (GLA) domains, located at the N-terminus of four procoagulant proteins—prothrombin (PT) and factors VII (fVII), IX (fIX) and X (fX)—and three anticoagulant proteins—protein C (PrC), protein S (PrS) and protein Z (PrZ). Although these seven GLA domains are very similar in sequence and structure, their membrane binding affinities vary by about three orders of magnitude.2,3 For example, fVIIa and activated PrC (APC) bind weakly to PS-containing membranes, while fX binds with relatively high affinity. To our knowledge, however, no study has directly compared the membrane binding properties of all seven human GLA domain-containing blood clotting proteins under the same conditions. It has previously been shown that several of the plasma clotting proteins, including components of the complexes of fXa with factor Va (fVa), fIXa with factor VIIIa (fVIIIa), and fVIIa with tissue factor (TF), bind much more weakly to membranes if PS has D-serine in its headgroup (D-PS) versus the naturally-occurring L-serine (L-PS),1,4 but to our knowledge the PS headgroup stereospecificity of GLA domain binding has not been evaluated for all seven of the human GLA domain-containing clotting proteins.
Accordingly, in this study we used a surface plasmon resonance (SPR)-based approach to quantify the binding of the seven human GLA domain-containing clotting proteins to supported nanoscale phospholipid bilayers (Nanodiscs)5 containing 50% L-PS, 50% D-PS (with the balance being 50% phosphatidylcholine, PC). As a negative control, we also used Nanodiscs with 50% PA, which has the simplest structure of any phospholipid. We now report that, while all seven proteins bound more extensively to nanobilayers containing L-PS versus D-PS, two of them—fVIIa and APC—bound more extensively to nanobilayers containing PA rather than PS. Furthermore, the proteolytic activities of fVIIa and APC were enhanced when liposomes contained PA rather than PS, although mixtures of PS and PA gave the highest activities. Finally, we report that phosphatidylinositol 4-phosphate (PI4P) also supported enhanced enzymatic activity of fVIIa and APC.
EXPERIMENTAL PROCEDURES
Materials
Materials were from the following sources: phospholipase D (Streptomyces sp.), Biomol International (now Enzo Life Sciences, Ann Arbor, MI); Tyrode’s salts, TRAP (thrombin receptor activating peptide), A23187 and D-serine, Sigma-Aldrich (St. Louis, MO); human fVa, APC, PrZ, PrS and DAPA, Haematologic Technologies (Essex Junction, VT); bPT and human fX, fIX, and PT, Enzyme Research Laboratories (South Bend, IN); NTA Biacore sensor chips, GE Healthcare (Piscataway, NJ); and RGDS (H-Arg-Gly-Asp-Ser-OH), Bachem (Torrance, CA). Recombinant human fVIIa L0678 was produced in the milk of transgenic goats and kindly provided by rEVO Biologics (Framingham, MA). Recombinant human fVIIa was also obtained from American Diagnostica (now Sekisui Diagnostics, Stamford, CT). Comparisons (data not shown) of fVIIa binding to PS- and PA-containing Nanodiscs, and of rates of fVIIa-mediated fX activation on liposomes of varying phospholipid composition, indicated that the two recombinant fVIIa preparations were equivalent to each other and to plasma-derived fVIIa (from Haematologic Technologies). The data reported in this study were obtained using fVIIa L0678.
Phospholipids were from Avanti Polar lipids (Alabaster, AL) and contained 1-palmitoyl-2-oleoyl acyl chains, except for PE which had 1,2-dioleoyl acyl chains, and PI4P which was from porcine brain. D-PS was synthesized from PC and D-serine as described.6 Liposomes were prepared using Bio-Beads® SM-2 (Bio-Rad; Hercules, CA) and 20 mM sodium deoxycholate as described.7 Recombinant membTF,8 sTF9,10 and membrane scaffold protein11 were expressed in E. coli and purified as described.
SPR Analyses of Protein Binding to Nanoscale Bilayers
Nanodiscs of desired phospholipid composition were prepared and used to quantify protein binding via SPR using a Biacore 3000 instrument (GE Healthcare) as previously described,5 except that 0.2% bovine serum albumin was included in the buffers in which fVIIa binding was quantified. Briefly, the maximal, steady state RU values were plotted vs. protein concentration, to which the single-site ligand binding equation was fitted to yield Kd values and maximal binding stoichiometries. Representative SPR profiles for fX binding to Nanodiscs with 50% L-PS/50% PC are given in Figure S1 (Supporting Information).
Rates of FX Activation and FVa Inactivation
Initial rates of fX activation by fVIIa or sTF:fVIIa in the presence of liposomes were quantified at room temperature as described,5,12 typically using 100 nM fX and either 1 to 10 nM fVIIa without sTF, or 10 to 100 pM fVIIa with 20 nM sTF. Initial rates of fX activation by TF:fVIIa assembled on TF-liposomes were quantified as described previously,5,12 typically using 100 nM fX, 1 to 5 pM fVIIa, and 500 pM TF. Initial rates of fVa inactivation by APC were quantified at room temperature in a two-stage assay as described13 with minor modifications. In the first stage, 50 pM APC, 3 nM fVa and 20 μM phospholipids were incubated for 0–30 min.
Phospholipase D (PLD) Treatment of Activated Human Platelets
Citrated blood was collected via atraumatic venipuncture from healthy human volunteers, and platelets were purified from platelet-rich plasma by gel filtration on Sepharose 2B14 using calcium-free Tyrode’s buffer containing 1% bovine serum albumin. Platelets were resuspended at 2.05 × 108 platelets/ml in Tyrode’s buffer containing 300 μg/mL RGDS peptide to prevent aggregation, then activated at 37°C with agitation for 10 min with 10 μM TRAP followed by an additional 10 min with 20 μM A23187. Activated platelets were then incubated with varying concentrations of PLD for 1 hr at 37°C, after which they were diluted to 1.02 × 106 platelets/ml and used in place of liposomes to quantify initial rates of fX activation by sTF:fVIIa as described above.
RESULTS
Phospholipid Specificity in Binding of Clotting Proteins to Membranes
To evaluate the phospholipid specificity of membrane binding for all seven of the known GLA domain-containing clotting proteins, we quantified their binding to nanoscale phospholipid bilayers immobilized on Biacore chip surfaces, using Nanodiscs containing 50% L-PS or D-PS (balance = 50% PC). As a general control for anionic phospholipids, we also used Nanodiscs containing 50% PA (balance = 50% PC). In addition to examining the seven human clotting proteins with GLA domains, we also examined bovine PT (bPT; abbreviations without a preceding “b” all refer to human clotting proteins), whose PS-binding properties have been studied extensively in the past. The binding results presented in Figure 1 show that all these proteins bound preferentially to bilayers containing L-PS compared to D-PS; thus, their membrane binding is stereospecific for L-serine in the PS headgroup.
Figure 1.
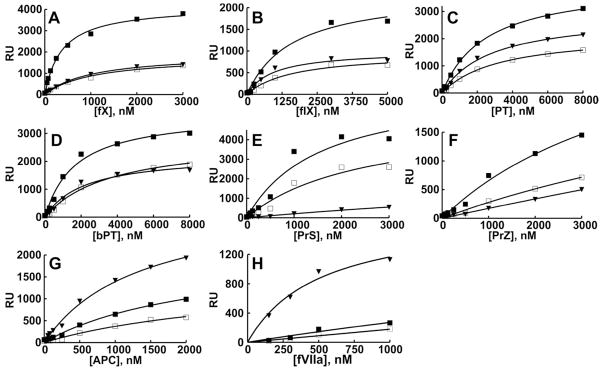
Phospholipid specificity of the binding of GLA domain-containing clotting proteins to membranes. (A–H) Nanodiscs containing either 50% L-PS (■), 50% D-PS (□), or 50% PA (▼) (balance = 50% PC) were immobilized on Biacore sensorchips, after which maximal steady-state levels of protein binding were quantified (RU values). The single-site ligand binding equation was fitted to the data, from which the binding parameters in Table 1 were derived. Shown are representative binding isotherms for each protein, from 3 independent experiments each.
While most of the proteins exhibited saturable binding to 50% L-PS nanobilayers, fVIIa gave no evidence of saturation even at 1 μM fVIIa (Figure 1H), consistent with previous estimates of its Kd ranging from 4 to 17 μM for binding to PS/PC liposomes.2,15,16 The binding isotherms for both PrZ and PrS were sigmoidal (possibly due to protein aggregation). Therefore, although reliable binding parameters for fVIIa, PrS and PrZ could not be calculated using 50% L-PS nanobilayers, qualitative differences in the phospholipid binding specificities to L-PS versus D-PS or PA can be discerned from inspection of the binding curves for these proteins.
Table 1 summarizes the membrane binding parameters for fX, fIX, PT, bPT, and APC. The Kd values for binding of these proteins to 50% L-PS nanobilayers ranged from 0.38 μM (fX) to about 3 μM (APC and PT). The binding stoichiometries for fX, fIX, PT and bPT, expressed as the number of protein molecules bound at saturation per leaflet on 50% L-PS nanobilayers, were similar to each other, ranging from 5.0 to 6.6 protein molecules/leaflet (Table 1). We previously demonstrated that the type of Nanodiscs used in this study contain 67 ± 1 total phospholipid molecules per leaflet.5 Therefore, for these four proteins, we calculate one protein binding site per 5 to 7 L-PS molecules, which is in reasonably good agreement with our previous calculation of one fX binding site for every 7.9 L-PS molecules.5 On the other hand, the maximal binding stoichiometry for APC on 50% L-PS nanobilayers was only 3.3 protein molecules/leaflet, corresponding to one protein binding site per 10.2 L-PS molecules. Although fX’s binding affinity for D-PS nanobilayers was almost three fold weaker than its affinity for L-PS bilayers, the binding affinities to D-PS versus L-PS bilayers for fIX, PT, bPT and APC were similar. However, fX, fIX, PT, bPT, and APC all exhibited higher binding stoichiometries to L-PS versus D-PS nanobilayers (Table 1), which is also readily apparent from inspection of their binding isotherms (Figure 1).
Table 1.
Binding parameters for clotting proteins on Nanoscale bilayers of varying anionic phospholipid composition
| phospholipid compositiona | fX | fIX | PT | bPT | APC | fVIIa |
|---|---|---|---|---|---|---|
|
| ||||||
| Kd (μM)b | ||||||
| 50% L-PS | 0.38 ± 0.005 | 1.5 ± 0.1 | 3.1 ± 0.3 | 2.0 ± 0.2 | 3.0 ± 0.4 | ndc |
| 50% D-PS | 1.07 ± 0.06 | 1.2 ± 0.2 | 4.2 ± 0.6 | 3.2 ± 0.3 | 3.6 ± 0.3 | nd |
| 50% PA | 0.85 ± 0.18 | 1.08 ± 0.1 | 3.3 ± 0.4 | 2.4 ± 0.1 | 2.1 ± 0.4 | 1.7 ± 0.1 |
|
| ||||||
| binding stoichiometry (protein molecules bound/leaflet)b
| ||||||
| 50% L-PS | 5.6 ±0.4 | 6.6 ± 0.2 | 5.0 ± 0.1 | 6.2 ± 0.2 | 3.3 ± 0.1 | nd |
| 50% D-PS | 2.9 ± 0.2 | 2.9 ± 0.4 | 3.0 ± 0.2 | 4.8 ± 0.1 | 2.5 ± 0.2 | nd |
| 50% PA | 2.8 ± 0.04 | 3.3 ± 0.5 | 3.3 ± 0.2 | 4.4 ± 0.3 | 6.8 ± 1.0 | 4.7 ± 1.5 |
Nanodiscs contained the indicated anionic phospholipid (balance = 50% PC).
Values (mean ± standard error; n = 3) are derived from fitting the one-site ligand binding equation to binding isotherms such as those depicted in Figure 1.
nd, not determined.
As an additional control for phospholipid headgroup specificity, we quantified the binding of all eight proteins to nanobilayers containing 50% PA, the simplest phospholipid. As expected, six of them (fIX, fX, PT, bPT, PrS and PrZ) showed less binding to PA-containing bilayers relative to L-PS-containing bilayers, with PrS showing the greatest preference for L-PS over PA (Figure 1A–F). Very unexpectedly, however, APC and fVIIa bound considerably better to nanobilayers containing PA than L-PS (Figure 1G,H). Thus, APC bound to PA with a lower Kd, and with a greater than twofold higher binding stoichiometry, compared to its binding to L-PS (Table 1). This corresponds to about one APC binding site per 4.9 PA molecules, compared to one APC binding site per 10.2 L-PS molecules. The effect was even more striking for fVIIa, which bound to PA with a Kd of 1.7 μM and a binding stoichiometry of 4.7 protein molecules per leaflet (i.e., one fVIIa binding site per 7.1 PA molecules). In contrast, the binding of fVIIa to L-PS showed no evidence of saturation over the concentration range tested. APC and fVIIa therefore recognize PA preferentially over L-PS or D-PS when binding to membranes.
Enhancement of FVIIa Enzymatic Activity by PA
It is well known that reactions of the blood clotting cascade involving GLA domain-containing proteins are greatly accelerated in the presence of membranes containing anionic phospholipids, with PS exhibiting the highest activity by far.1 (For the remainder of this report, we refer to L-PS simply as “PS.”) The surprising ability of fVIIa to bind to PA-containing nanobilayers prompted us to examine how liposomes with PA modulate the rate of fX activation by fVIIa, with or without its protein cofactor, TF. In particular, we used either the isolated ectodomain of TF (sTF) or fully membrane-anchored TF (membTF).
We measured rates of fX activation by sTF:fVIIa in the presence of 50 μM liposomes with varying proportions of PA and PS (balance = PC), and normalized the rates to that observed with 30% PS/70% PC. As expected, binary mixtures of PS and PC (open squares in Figure 2A) showed increasing rates of fX activation as the PS content increased. In parallel experiments, we varied the proportions of PA and PS such that the total PA + PS was kept at 30% and the balance was 70% PC (open circles in Figure 2A). Liposomes containing 30% PA/70% PC (y-intercept) supported a rate of fX activation that was 6.8-fold higher than liposomes with 30% PS/70% PC. The maximal rate was observed with liposomes containing 5% PS/25% PA/70% PC (17.5-fold higher than with 30% PS/70% PC). When mixtures of PS + the anionic phospholipid, phosphatidylglycerol (PG), were also examined (total PS + PG was kept at 30%; balance = PC), the rates of fX activation showed insignificant enhancement by PG. This finding indicates that the rate enhancement of sTF:fVIIa by PA is not replicated by another anionic lipid.
Figure 2.

Enhancement of fVIIa enzymatic activity by PA. (A) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing either 0–30% PS (□), mixtures of PS and PG such that PS + PG = 30% (▼), or mixtures of PS and PA such that PS + PA = 30% (○). Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 30% PS/70% PC liposomes (mean, 0.8 ± 0.01 min−1). (B) Rates of fX activation by fVIIa (no sTF) in the presence of liposomes (balance = PC) containing either 0–30% PS (□), or mixtures of PS and PA such that PS + PA = 30% (○). Rates were normalized in each experiment to that of fX activation by fVIIa on 30% PS/70% PC liposomes (mean, 0.5×10−3 ± 0.2×10−3 min−1). (C) Rates of fX activation by fVIIa in the presence of membTF-liposomes (balance = PC) containing either 0–30% PS (□), mixtures of PS and PE such that PS + PE = 30% (■), or mixtures of PS and PA such that PS + PA = 30% (○). Rates were normalized in each experiment to that of fX activation by TF-fVIIa on 30% PS/70% PC liposomes (mean, 401.4 ± 38.8 min−1). (D) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing 30% PE plus 0–30% PS (■), or 30% PE plus mixtures of PS and PA such that PS + PA = 30% (●). Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 30% PS/70% PC liposomes (mean, 0.8 ± 0.01 min−1). (E) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing a mixture of 5% PS, 30% PE, and 0–5% PA. The horizontal dotted line is the value for liposomes with 10% PS, 30% PE, 0% PA. Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 5% PS/30% PE/65% PC liposomes (mean, 0.70 ± 0.03 min−1). (F) Rates of fX activation by sTF:fVIIa in the presence of activated platelets that had been pretreated with the indicated PLD concentrations. Rates were normalized in each experiment to the rate with activated platelets not treated with PLD (mean, 1.6×10−2 ± 0.3×10−2 min−1). Data in all panels are mean ± standard error (n = 3 to 7).
We also measured rates of fX activation by fVIIa alone, which showed a very similar ability of PA to enhance fX activation (Figure 2B), although higher fVIIa concentrations were required in the absence of its protein cofactor. As with sTF:fVIIa, the optimal liposome composition was 5% PS/25% PA/70% PC and yielded a similar increase in activity (23.1-fold) over that observed with 30% PS/70% PC. This optimal phospholipid composition is likely a compromise between preferential binding of fVIIa to PA (with or without sTF) and preferential binding of fX to PS.
In contrast to experiments with sTF, when membTF is employed as the cofactor for fVIIa, almost all the binding energy holding the two proteins together on the membrane surface comes from protein-protein interactions; furthermore, the GLA domain of fVIIa is not required for this protein to bind productively to membTF.17 Consistent with this notion, liposomes containing combinations of PA and PS (totaling 30%) supported rates of fX activation by membTF:fVIIa that were never significantly higher than the rate observed with 30% PS/70% PC liposomes (Figure 2C). This argues that the contribution of PA to enhancing the rate of fX activation by fVIIa or sTF:fVIIa results directly from its superior ability over PS to recruit fVIIa to the membrane surface, rather than effects of fX or TF.
Synergy Between PA and Phosphatidylethanolamine (PE) to Enhance sTF:fVIIa Activity
We previously showed that PE synergizes strongly with PS to promote fX activation by the membTF:fVIIa complex, even though PE supports very little activity without PS.4,10 Furthermore, we demonstrated that most glycerophospholipids other than PC also synergize with PS to promote fX activation by membTF:fVIIa, in a mechanism we termed the ABC (Anything But Choline) hypothesis.4 In fact, Figure 2C demonstrates typical synergy between either PA or PE with PS to enhance fX activation by membTF:fVIIa. We were curious, however, whether PE could synergize with PA (with or without PS) to enhance fX activation by sTF:fVIIa. Figure 2D (solid symbols) shows that PE increased all the observed rates of fX activation by sTF:fVIIa, with or without PA, with maximal activity supported by liposomes containing 2% PS/28% PA/30% PE/40% PC. This finding argues strongly that the ability of PA to enhance fX activation by sTF:fVIIa is not just due to the previously-documented “synergy” that can occur between PS and most other non-PC phospholipids to enhance fX activation by membTF:fVIIa (which appears largely to be due to enhanced recruitment of fX to the membrane surface4).
In cell membranes, PA is a minor phospholipid, typically constituting less than 4% of total cellular lipid.18 We therefore explored whether (patho)physiologically relevant levels of PA could enhance fVIIa enzyme activity. Accordingly, we examined rates of fX activation using liposomes containing 0–5% PA, along with 5% PS and 30% PE (balance = PC), which should better reflect the composition of plasma membranes. The results in Figure 2E, normalized to the rate observed with 5% PS/30% PE/65% PC, show that even low levels of PA measurably enhanced fX activation by sTF:fVIIa. Thus, liposomes with 1.5% PA approximately doubled the rate of fX activation compared to liposomes without PA, while liposomes with 5% PA exhibited a 7.5-fold increase in activity.
Treating Platelets with Phospholipase D (PLD) to Enhance sTF:fVIIa Activity
PLD is a highly regulated enzyme in mammalian cells and also expressed by certain pathogenic microorganisms.18 It generates PA by removing phospholipid headgroups. We postulated that treating activated platelets with PLD would expose additional PA on the platelet surface, which in turn should enhance the rate of fX activation by sTF:fVIIa. Indeed, PLD treatment of activated human platelets enhanced their ability to support sTF:fVIIa-mediated fX activation about sixfold (Figure 2F).
Enhancement of sTF:fVIIa Enzyme Activity and FVIIa Membrane Binding by PI4P
We hypothesized that it was the presence of a monoester phosphate in PA that was responsible for its ability to enhance the binding of fVIIa and APC to membranes. Notably, phosphorylated derivatives of phosphatidylinositol (PI) contain one or more monoester phosphates on the myo-inositol moiety, leading us to predict that PI phosphates (PIPs) may also support enhanced fVIIa enzymatic activity. We screened PI singly phosphorylated at the 3, 4, or 5 position of myo-inositol, as well as doubly and triply phosphorylated PI, and found that liposomes with PI4P supported the highest rates of fX activation by sTF:fVIIa (Figure 3). Thus, PI4P significantly enhanced the activity of sTF:fVIIa, as exemplified by normalized rates of fX activation in the presence of liposomes with varying proportions of PI4P and PS (such that PI4P + PS = 30%; balance = 70% PC; Figure 4A). The results were qualitatively similar to those observed with PA (cf. Figure 2A), although maximal enzymatic activity was observed with 20% PS/10% PI4P/70%PC compared to 5% PS/25% PA/70% PC, and the magnitude of the maximal enhancement of sTF:fVIIa enzyme activity was also lower (3.72-fold enhancement with PI4P compared to 17.5-fold with PA).
Figure 3.
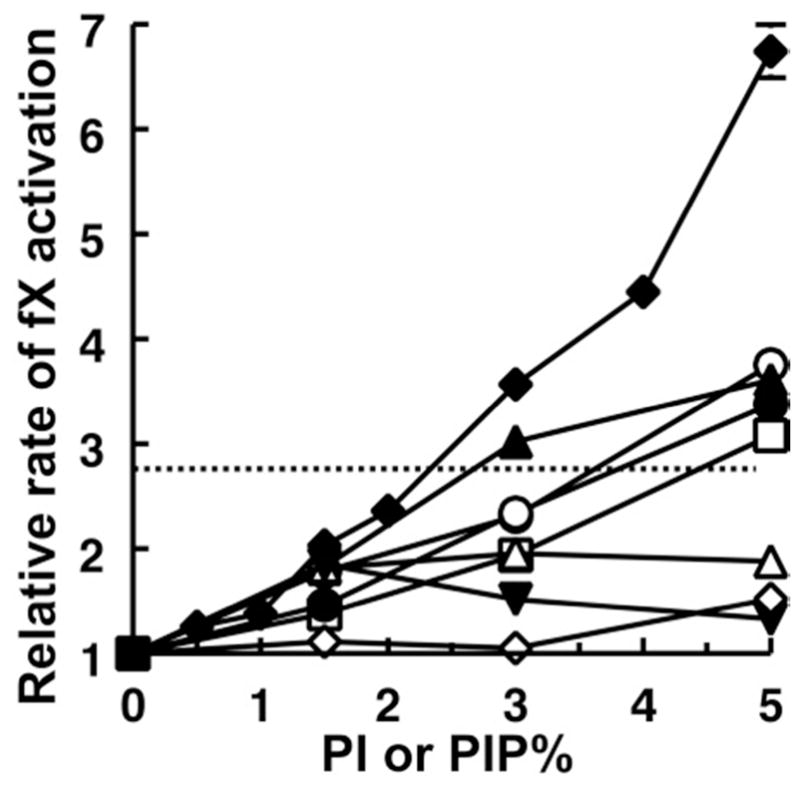
Enhancement of fVIIa enzymatic activity by PI and PIPs. Plotted are normalized rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing 5% PS, 30% PE, and 0–5% of the following lipids: PI (□), PI3P (○), PI5P (●), PI4P (◆), PI(3,4)P2 (△), PI(3,5)P2 (▲), PI(4,5)P2 (▼), or PI(3,4,5)P3 (◇). The horizontal dotted line is the value for liposomes with 10% PS, 30% PE, 70% PC (i.e., no PI or PIPs). Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 5% PS/30% PE/65% PC liposomes (mean, 0.70 ± 0.03 min−1). Data are mean ± standard error (n = 3).
Figure 4.

Enhancement of fVIIa enzymatic activity and membrane binding by PI4P. (A) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing either 0–30% PS (□), or mixtures of PS and PI4P such that PS + PI4P = 30% (◇). Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 30% PS/70% PC liposomes (mean, 0.8 ± 0.01 min−1). (B) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing 30% PE plus either 0–30% PS (■), or mixtures of PS and PI4P such that PS + PI4P = 30% (◆). Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 30% PS/70% PC liposomes (mean, 0.8 ± 0.01 min−1). (C) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing a mixture of 5% PS, 30% PE, and 0–5% PI4P. The horizontal dotted line is the value for liposomes with 10% PS, 30% PE, 0% PI4P. Rates were normalized in each experiment to that of fX activation by sTF:fVIIa on 5% PS/30% PE/65% PC liposomes (mean, 0.70 ± 0.03 min−1). (D) Rates of fX activation by sTF:fVIIa in the presence of liposomes (balance = PC) containing mixtures of PA and PI4P such that PA + PI4P = 30%. Rates were normalized in each experiment to that of fX activation on 30% PI4P/70% PC liposomes (mean, 0.31 ± 0.01 min−1). (E) Binding of fVIIa to Nanodiscs containing either 20% L-PS (■), 20% PA (▼), or 20% PI4P (●) (balance = 80% PC). Binding data were acquired and analyzed as in Figure 1. Data in panels A–D are mean ± standard error (n = 3 to 6).
Incorporating 30% PE into liposomes with mixtures of PI4P and PS further enhanced sTF:fVIIa enzymatic activity (Figure 4B), similar to what was observed for PA. When liposomes contained 5% PS and 30% PE, the rate of fX activation by sTF:fVIIa was enhanced by incorporating up to 5% PI4P, and was approximately doubled with 1.5% PI4P (Figure 4C), which is also very similar to the effect of PA (cf. Figure 2E).
If PA and PI4P work by the same mechanism to increase the rate of fX activation by sTF:fVIIa, then their effects should be additive, not synergistic. Indeed, when rates of fX activation by sTF:fVIIa were measured on liposomes with varying amounts of PA and PI4P (such that PA + PI4P was a constant 30%; balance = 70% PC), we found a linear relationship between PA or PI4P content and enzyme activity (Figure 4D).
We tested whether PI4P could support enhanced fVIIa binding using the same SPR-based approach as above, but with Nanodiscs containing 20% PI4P, PA or PS (Figure 4E). (We used a lower content of anionic lipid in these experiments, owing to the higher negative charge on PI4P.) We found that fVIIa bound similarly to PA and PI4P, with the following binding parameters: For PA, the Kd was 0.63 ± 0.11 μM and the stoichiometry was 4.04 ± 0.69 fVIIa molecules per leaflet. For PI4P, the Kd was 1.05 ± 0.2 μM and the stoichiometry was 5.6 ± 0.8 fVIIa molecules per leaflet.
Enhancement of APC Enzymatic Activity by PA and PI4P
We tested the ability of PA- and PI4P-containing liposomes to accelerate the rate of proteolytic inactivation of factor Va (fVa) by APC. Liposomes had either binary mixtures of PS and PC (0–30% PS; balance = PC), or mixtures of PS plus either PA or PI4P in which the total amount of anionic lipid equaled 30% and the balance was 70% PC. Both PA and PI4P enhanced the rate of fVa inactivation by APC; maximal rates were observed with PA-liposomes containing 5% PS/25% PA/70% PC, while PI4P-liposomes showed a broader optimum (5–20% PS/10–25% PI4P/70% PC (Figure 5). The rates were increased 5-fold and 2.5-fold by PA and PI4P respectively, compared to 30% PS liposomes. Thus, like fVIIa, APC binds more extensively to bilayers containing PA than PS, and its enzymatic activity is enhanced when either PA or PI4P are included in liposomes that also contain PS.
Figure 5.
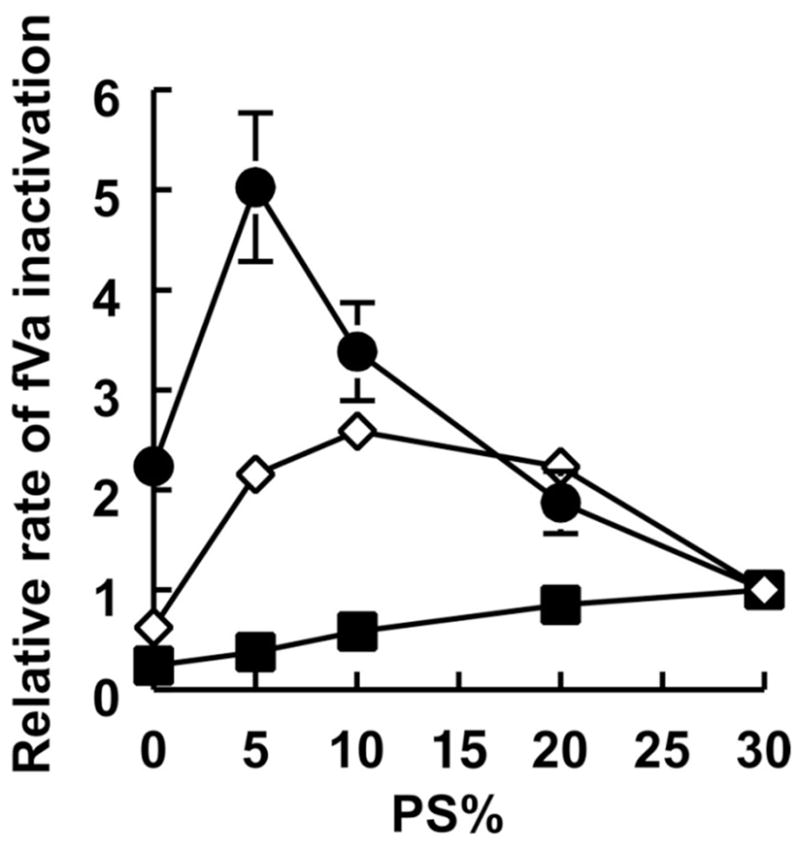
Enhancement of APC enzymatic activity by PA and PI4P. Rates of fVa inactivation by APC were quantified in the presence of liposomes (balance = PC) containing either 0–30% PS (■), mixtures of PS and PA such that PS + PA = 30% (●), or mixtures of PS and PI4P such that PS + PI4P = 30% (◇). Rates were normalized to 30% PS/70% PC. Data are mean ± standard error (n = 3).
DISCUSSION
For years it has been known that clotting proteins with GLA domains bind to membrane bilayers containing exposed PS.1,18 Of these seven proteins in the human coagulation system, APC and fVIIa have the weakest membrane affinities.2,3 In particular, fVIIa binds to PS-containing bilayers with a Kd of about 17 μM, which is 1700 times its plasma concentration.19 In this study we report the surprising finding that human APC and fVIIa are actually PA-binding proteins, in that they show substantial preference for interacting with PA over PS. This was especially prominent for fVIIa.
The reactions catalyzed by fVIIa and APC require both enzyme and substrate to bind to membranes for full activity. Accordingly, we found that the increased binding of fVIIa and APC to PA over PS translated into substantially increased enzymatic activities for these proteases, which again was most striking for fVIIa. Thus, incorporating PA into PS/PC liposomes enhanced fVIIa enzymatic activity either 23-fold (for fVIIa alone) or 18-fold (for sTF:fVIIa). By comparison, APC activity was enhanced about 5-fold when PS/PC liposomes also included PA.
PA contains a monoester phosphate, while other phospholipids contain a diester phosphate to which an alcohol (the headgroup) is esterified. We therefore hypothesized that it was the presence of the monoester phosphate on PA that was important for enhanced binding of fVIIa and APC. To test this hypothesis, we examined PIPs, which also contain one or more phosphomonoesters in their headgroups. PI4P enhanced the membrane binding of fVIIa, and also the enzymatic activities of both fVIIa and APC.
The increased binding of fVIIa and APC to PA and PI4P could be due to less steric hindrance in binding to monoester phosphates relative to diester phosphates, and/or the unique ionization properties of such monoester phosphates. Thus, PA has two ionizable groups, with pKa values of 3.0 and 6.9–7.9, respectively.20 The second pKa is near physiologic pH; therefore, PA in cell membranes can carry a charge from −1 to −2, depending on local pH and the presence of divalent metal ions or hydrogen bonding partners.21 Thus, the special ionization nature of the phosphomonoester group of PA and PI4P might be the reason that fVIIa and APC bound preferentially to these phospholipids over other anionic lipids like PS or PG, which have a net −1 charge. Interestingly, the phosphomonoester in PI4P has the lowest pKa of any PIP,22 so the fact that this was the most active of all the PIPs tested is consistent with the requirement that the phosphate group have a net charge that is more negative than −1.
The effects of PA and PI4P on these proteases were distinct from the previously reported “synergy” between PE and PS (discussed in Tavoosi et al.4), because, unlike PE, we found that PA dramatically increased the enzymatic activity of fVIIa and APC rather than just shifting the PS-dependence. In addition, PE still synergized with mixtures of PA and PS, which is further evidence that PE acts by a mechanism distinct from that of PA. The most straightforward explanation is that PA and PI4P function to recruit the enzyme (fVIIa or APC) to the membrane surface, while PS or PS/PE mixtures function to recruit the substrate (fX or fVa, respectively) to the membrane. For this reason, maximal fVIIa and APC proteolytic activities required both PA and PS in liposomes, which likely represents a compromise in phospholipid composition to enhance membrane binding of both enzyme and substrate.
PA makes up roughly 1–2% of in the phospholipid in cell membranes23 and has diverse cellular roles, including serving as the intermediate for phospholipid synthesis, participating in intracellular signaling pathways, and altering membrane dynamics. PA levels can be rapidly up-regulated, being produced via phosphorylation of diacylglycerol, acylation of lysophosphatidic acid, or PLD-mediated hydrolysis of lipids such as PC.24 Like PS and PE, PA is generally localized to the cytoplasmic face of the plasma membrane, although roles for extracellular PA have been reported.25,26 There are at least four possible mechanisms for exposure of PA to plasma: One, asymmetry of the plasma membrane is not absolute, with about 20% of PA residing on the outer leaflet.27,28 Two, the blood clotting system is most active at locations of tissue damage and trauma; thus, lysed or damaged cells will expose all their intracellular phospholipids, including PA and PIPs. Three, phospholipid scramblase, which is responsible for externalizing PS and PE in apoptosis and platelet activation, is thought to scramble the bilayer orientation of all phospholipids, including PA.29 And four, a number of infectious microorganisms secrete PLD,30–32 which can generate cell-surface PA. The first three of these mechanisms apply equally well to PS, whose exposure to plasma is also required for efficient triggering of blood clotting reactions.1
PA did not enhance the enzymatic activity of fVIIa bound to membrane-anchored TF. This result is consistent with our earlier finding that protein-protein interactions outside fVIIa’s GLA domain provide most of the binding energy holding the membTF:fVIIa complex together.17 Thus, PA would not be expected to contribute substantially to recruiting fVIIa to membTF. Interestingly, recombinant versions of fVIIa and APC have been used as drugs to treat bleeding and sepsis, respectively.33,34 At the high concentrations administered to control bleeding, recombinant fVIIa is thought to act in a largely TF-independent manner by binding to exposed PS on activated platelets and/or damaged cells.35 In fact, because fVIIa and APC bind so weakly to membranes, some investigators have mutated residues in their GLA domains to enhance their affinities for PS/PC bilayers, in hopes of creating drugs with increased efficacy.15,36 We propose that the binding of recombinant fVIIa to exposed PA may also play a role in its effectiveness in controlling bleeding in vivo.
Supplementary Material
Acknowledgments
Funding
This study was supported by US NIH grant R01 HL103999 from the National Heart, Lung and Blood Institute.
We thank Dr. Stephen Sligar for providing membrane scaffold protein and advice on Nanodisc technology.
Abbreviations
- D-PS
PS with D-serine
- fV
factor V
- fVII
factor VII
- fVIII
factor VIII
- fIX
factor IX
- fX
factor X
- GLA domain
γ-carboxyglutamate-rich domain
- L-PS
PS with L-serine
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI4P
phosphatidylinositol 4-phosphate
- PS
phosphatidylserine
- PLD
phospholipase D
- PrC
protein C
- PrS
protein S
- PrZ
protein Z
- PT
prothrombin
- RGDS
H-Arg-Gly-Asp-Ser-OH
- SPR
surface plasmon resonance
- TF
tissue factor
- TRAP
thrombin receptor activating peptide
Footnotes
The authors declare no competing financial interests.
One supplemental figure (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zwaal RFA, Comfurius P, Bevers EM. Lipid-protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376:433–453. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 2.McDonald JF, Shah AM, Schwalbe RA, Kisiel W, Dahlback B, Nelsestuen GL. Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry. 1997;36:5120–5127. doi: 10.1021/bi9626160. [DOI] [PubMed] [Google Scholar]
- 3.Nelsestuen GL, Kisiel W, Di Scipio RG. Interaction of vitamin K dependent proteins with membranes. Biochemistry. 1978;17:2134–2138. doi: 10.1021/bi00604a017. [DOI] [PubMed] [Google Scholar]
- 4.Tavoosi N, Davis-Harrison RL, Pogorelov TV, Ohkubo YZ, Arcario MJ, Clay MC, Rienstra CM, Tajkhorshid E, Morrissey JH. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286:23247–23253. doi: 10.1074/jbc.M111.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007;282:6556–6563. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 6.Boettcher JM, Davis-Harrison RL, Clay MC, Nieuwkoop AJ, Ohkubo YZ, Tajkhorshid E, Morrissey JH, Rienstra CM. Atomic view of calcium-induced clustering of phosphatidylserine in mixed lipid bilayers. Biochemistry. 2011;50:2264–2273. doi: 10.1021/bi1013694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith SA, Morrissey JH. Rapid and efficient incorporation of tissue factor into liposomes. J Thromb Haemost. 2004;2:1155–1162. doi: 10.1111/j.1538-7836.2004.00772.x. [DOI] [PubMed] [Google Scholar]
- 8.Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987;50:129–135. doi: 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 9.Rezaie AR, Fiore MM, Neuenschwander PF, Esmon CT, Morrissey JH. Expression and purification of a soluble tissue factor fusion protein with an epitope for an unusual calcium-dependent antibody. Protein Expression Purif. 1992;3:453–460. doi: 10.1016/1046-5928(92)90062-2. [DOI] [PubMed] [Google Scholar]
- 10.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34:13988–13993. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 11.Bayburt TH, Grinkova YV, Sligar SG. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002;2:835–856. [Google Scholar]
- 12.Waters EK, Morrissey JH. Restoring full biological activity to the isolated ectodomain of an integral membrane protein. Biochemistry. 2006;45:3769–3774. doi: 10.1021/bi052600m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–819. [PubMed] [Google Scholar]
- 14.Tangen O, Berman HJ, Marfey P. Gel filtration. A new technique for separation of blood platelets from plasma. Thromb Diath Haemorrh. 1971;25:268–278. [PubMed] [Google Scholar]
- 15.Shah AM, Kisiel W, Foster DC, Nelsestuen GL. Manipulation of the membrane binding site of vitamin K-dependent proteins: Enhanced biological function of human factor VII. Proc Natl Acad Sci U S A. 1998;95:4229–4234. doi: 10.1073/pnas.95.8.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harvey SB, Stone MD, Martinez MB, Nelsestuen GL. Mutagenesis of the gamma-carboxyglutamic acid domain of human factor VII to generate maximum enhancement of the membrane contact site. J Biol Chem. 2003;278:8363–8369. doi: 10.1074/jbc.M211629200. [DOI] [PubMed] [Google Scholar]
- 17.Neuenschwander PF, Morrissey JH. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J Biol Chem. 1994;269:8007–8013. [PubMed] [Google Scholar]
- 18.Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta Rev Biomembranes. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Fair DS. Quantitation of factor VII in the plasma of normal and warfarin-treated individuals by radioimmunoassay. Blood. 1983;62:784–791. [PubMed] [Google Scholar]
- 20.Kooijman EE, Carter KM, van Laar EG, Chupin V, Burger KN, de Kruijff B. What makes the bioactive lipids phosphatidic acid and lysophosphatidic acid so special? Biochemistry. 2005;44:17007–17015. doi: 10.1021/bi0518794. [DOI] [PubMed] [Google Scholar]
- 21.Kooijman EE, Burger KN. Biophysics and function of phosphatidic acid: a molecular perspective. Biochim Biophys Acta. 2009;1791:881–888. doi: 10.1016/j.bbalip.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Shin JJ, Loewen CJ. Putting the pH into phosphatidic acid signaling. BMC Biol. 2011;9:85. doi: 10.1186/1741-7007-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vance JE, Steenbergen R. Metabolism and functions of phosphatidylserine. Prog Lipid Res. 2005;44:207–234. doi: 10.1016/j.plipres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 24.English D, Cui Y, Siddiqui RA. Messenger functions of phosphatidic acid. Chem Phys Lipids. 1996;80:117–132. doi: 10.1016/0009-3084(96)02549-2. [DOI] [PubMed] [Google Scholar]
- 25.English D. Phosphatidic acid: a lipid messenger involved in intracellular and extracellular signalling. Cell Signal. 1996;8:341–347. doi: 10.1016/0898-6568(95)00076-3. [DOI] [PubMed] [Google Scholar]
- 26.Krachler AM, Ham H, Orth K. Outer membrane adhesion factor multivalent adhesion molecule 7 initiates host cell binding during infection by gram-negative pathogens. Proc Natl Acad Sci U S A. 2011;108:11614–11619. doi: 10.1073/pnas.1102360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinn PJ. Plasma membrane phospholipid asymmetry. Subcell Biochem. 2002;36:39–60. doi: 10.1007/0-306-47931-1_3. [DOI] [PubMed] [Google Scholar]
- 28.Gascard P, Tran D, Sauvage M, Sulpice JC, Fukami K, Takenawa T, Claret M, Giraud F. Asymmetric distribution of phosphoinositides and phosphatidic acid in the human erythrocyte membrane. Biochim Biophys Acta. 1991;1069:27–36. doi: 10.1016/0005-2736(91)90100-m. [DOI] [PubMed] [Google Scholar]
- 29.Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res. 2003;44:233–242. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Edwards JL, Entz DD, Apicella MA. Gonococcal phospholipase D modulates the expression and function of complement receptor 3 in primary cervical epithelial cells. Infect Immun. 2003;71:6381–6391. doi: 10.1128/IAI.71.11.6381-6391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKean SC, Davies JK, Moore RJ. Expression of phospholipase D, the major virulence factor of Corynebacterium pseudotuberculosis, is regulated by multiple environmental factors and plays a role in macrophage death. Microbiology. 2007;153:2203–2211. doi: 10.1099/mic.0.2007/005926-0. [DOI] [PubMed] [Google Scholar]
- 32.Lucas EA, Billington SJ, Carlson P, McGee DJ, Jost BH. Phospholipase D promotes Arcanobacterium haemolyticum adhesion via lipid raft remodeling and host cell death following bacterial invasion. BMC Microbiol. 2010;10:270. doi: 10.1186/1471-2180-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedner U, Lee CA. First 20 years with recombinant FVIIa (NovoSeven) Haemophilia. 2011;17:e172–182. doi: 10.1111/j.1365-2516.2010.02352.x. [DOI] [PubMed] [Google Scholar]
- 34.Marti-Carvajal AJ, Sola I, Gluud C, Lathyris D, Cardona AF. Human recombinant protein C for severe sepsis and septic shock in adult and paediatric patients. Cochrane Database Syst Rev. 2012;12:CD004388. doi: 10.1002/14651858.CD004388.pub6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hedner U. Mechanism of action, development and clinical experience of recombinant FVIIa. J Biotechnol. 2006;124:747–757. doi: 10.1016/j.jbiotec.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 36.Nelsestuen GL. Enhancement of vitamin-K-dependent protein function by modification of the γ-carboxyglutamic acid domain: Studies of protein C and factor VII. Trends Cardiovasc Med. 1999;9:162–167. doi: 10.1016/s1050-1738(99)00024-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.