

Background: SAM domain-mediated polymerization is essential for ETV6-NTRK3 (EN)-induced cellular transformation.
Results: Mutation of a salt bridge at the SAM polymer interface weakens SAM polymerization and abrogates EN transformation.
Conclusion: Intermolecular electrostatic interactions are important for SAM domain polymerization and EN transformation.
Significance: These studies provide further insights into the mechanisms by which ETV6-SAM domain mediates EN transformation.
Keywords: Cancer Biology, Cell Signaling, Ets Family Transcription Factor, Fusion Protein, Oncogene, ETV6-NTRK3, SAM Domain, Fusion Oncogene
Abstract
The ETV6-NTRK3 (EN) chimeric oncogene is expressed in diverse tumor types. EN is generated by a t(12;15) translocation, which fuses the N-terminal SAM (sterile α-motif) domain of the ETV6 (or TEL) transcription factor to the C-terminal PTK (protein-tyrosine kinase) domain of the neurotrophin-3 receptor NTRK3. SAM domain-mediated polymerization of EN leads to constitutive activation of the PTK domain and constitutive signaling of the Ras-MAPK and PI3K-Akt pathways, which are essential for EN oncogenesis. Here we show through complementary biophysical and cellular biological techniques that mutation of Lys-99, which participates in a salt bridge at the SAM polymer interface, reduces self-association of the isolated SAM domain as well as high molecular mass complex formation of EN and abrogates the transformation activity of EN. We also show that mutation of Asp-101, the intermolecular salt bridge partner of Lys-99, similarly blocks transformation of NIH3T3 cells by EN, reduces EN tyrosine phosphorylation, inhibits Akt and Mek1/2 signaling downstream of EN, and abolishes tumor formation in nude mice. In contrast, mutations of Glu-100 and Arg-103, residues in the vicinity of the interdomain Lys-99–Asp-101 salt bridge, have little or no effect on these oncogenic characteristics of EN. Our results underscore the importance of specific electrostatic interactions for SAM polymerization and EN transformation.
Introduction
ETV6 (or TEL) is an ETS family transcriptional repressor that is frequently targeted by chromosomal translocations in human cancers (1, 2). These rearrangements result in fusion genes encoding chimeric oncoproteins, in which the N-terminal SAM domain of ETV6 is linked to the C-terminal PTK3 domain of a variety of tyrosine kinases or to the DNA binding domain of other transcription factors (2). Known tyrosine kinase fusion partners of ETV6 include PDGF receptor β, ABL1/2, JAK2, NTRK3, FGFR3, and SYK (3–11). These ETV6 fusions are primarily expressed in hematopoietic malignancies. However, ETV6-NTRK3 (EN) fusion is unique in being expressed in tumors derived from several cell lineages (10). EN was initially discovered by cloning the breakpoints of the t(12;15)(p13;q25) translocation associated with congenital fibrosarcoma, a pediatric spindle cell malignancy of the soft tissues (11). Later EN expression was detected in mesoblastic nephroma (12), adult acute myeloid leukemia (13), and secretory breast carcinoma (14). More recent studies have reported EN expression in mammary analog secretory carcinoma of salivary glands (15), secretory-like skin carcinoma (16), and chronic eosinophilic leukemia (17). Similar to other ETV6-PTK fusions, SAM domain-mediated polymerization of EN is essential for constitutive activation of the PTK domain (18). This, in turn, leads to the activation of the wild type (WT) NTRK3 downstream signaling pathways, namely the PI3K-Akt and Ras-MAPK cascades, and subsequently cellular transformation (19).
The SAM domain, which is also known as the PNT domain in ETS transcription factors, has been found in several regulatory proteins and often facilitates cellular protein complex assembly by forming homotypic or heterotypic oligomers (5, 20–25). The x-ray crystallographic structure of the ETV6-SAM domain revealed an extended homopolymer in which the SAM monomers are arranged in a head-to-tail fashion (26). Two interfaces in each monomer, called the mid-loop and end-helix surfaces, form interdomain hydrophobic cores surrounded by charged residues. Due to high affinity polymerization, the SAM domain is found in the insoluble fractions when expressed in Escherichia coli. However, replacement of either of two critical hydrophobic residues to a negatively charged residue (A93D and V112E, according to the human ETV6 cDNA numbering) within the mid-loop and end-helix surfaces, respectively, renders SAM domain monomeric (26, 27). We previously showed that A93D and V112E mutants of the EN chimera are also monomeric and cannot form high molecular weight complexes in the cells (28). These mutant EN proteins fail to activate their PTK domains and downstream signaling pathways (PI3K-Akt and Ras-MAPK) and, therefore, lack transformation activity. These studies suggested that SAM-PTK fusion oncoproteins could be inactivated by blocking SAM-mediated polymer formation. However, inhibiting the polymerization of SAM-PTK fusion oncoproteins by targeting the hydrophobic interaction core may be difficult as there are no obvious small molecule binding pockets in the interaction face of SAM monomers (28). Therefore, other possible mechanisms need to be explored to identify alternative strategies for targeting SAM polymerization.
The WT ETV6 transcription factor has been reported to be sumoylated at Lys-99 within the SAM domain. Mutation of this residue resulted in decreased sumoylation and inhibited nuclear export of the transcription factor (29, 30). However, recent studies, which employed mutational or mass spectrometry-based proteomics approaches, identified Lys-11 within the unstructured N-terminal sequence preceding the SAM domain, and not Lys-99, as the major SUMO modification site in ETV6 (31, 32). This implies that Lys-99 may affect ETV6 function in a manner other than as a SUMO acceptor. Interestingly, Lys-99 lies in the interface of two SAM monomers and contributes to a salt bridge surrounding the hydrophobic interaction core of the SAM polymer (26). These findings suggest that Lys-99 may play a critical role in the self-association of SAM domain.
In this study we examined whether electrostatic interactions involving Lys-99 are important for SAM-mediated EN polymerization and cellular transformation. We demonstrate that site-directed mutation of Lys-99 abrogates the ability of EN to transform NIH3T3 cells, impedes EN polymerization, and weakens SAM domain self-association. We also mutated other residues in the interface of SAM polymer that were predicted to participate in the salt bridge involving Lys-99 and tested the ability of these EN “salt bridge mutants” to transform NIH3T3 cells. Most notably, mutation of Asp-101, which is the Lys-99 intermolecular salt bridge partner, similarly blocks EN transformation. Both Lys-99 and Asp-101 mutants of EN exhibit defects in PTK activation, Akt, and Mek1/2 signaling as well as in tumor formation in nude mice. Moreover, using computational analysis of the SAM polymer structure, we identified two small molecule binding pockets at or near the salt bridge involving Lys-99 and Asp-101. Our results demonstrate the importance of salt bridge formation within the SAM polymer interface for SAM polymerization and EN transformation.
EXPERIMENTAL PROCEDURES
Plasmid Constructs and Site-directed Mutagenesis
Generation of full-length EN cDNA, ΔSAM-EN (SAM domain deleted EN), and K380N-EN (kinase dead EN) constructs were described previously (18). These cDNAs were cloned into the EcoRI sites of the MSCVpuro retroviral expression vector (Clontech) as described (33). Before site-directed mutagenesis, the full-length EN cDNA in MSCVpuro vector was amplified by PCR. Then these amplified PCR products with 5′-SalI and 3′-EcoRI restriction sites were subcloned into the pCR-Blunt II-TOPO vector using the Zero Blunt TOPO PCR Cloning kit (Invitrogen) according to the manufacturer's instructions. These constructs were designated as FLAG-EN-ZBTOPO or HA-EN-ZBTOPO. FLAG-EN-ZB-TOPO plasmid was used as template for the point mutations of Lys-99 and Arg-103 residues, and HA-EN-ZB-TOPO was used for Asp-101 and Glu-100 residues using the QuikChange site-directed mutagenesis kit (Stratagene). After confirming the correct mutations by sequencing, the cDNAs were cut from the pCR-Blunt II-TOPO vector using the SalI and EcoRI restriction enzymes (New England Biolabs) and cloned into the MSCVpuro vector precut with XhoI and EcoRI restriction enzymes.
SAM Domain Expression and Purification
The cDNA encoding residues 43–125 of the WT human ETV6-SAM domain, preceded by an N-terminal His6 affinity tag, was PCR-amplified from the full-length EN construct and cloned into the pET28a vector (Invitrogen). QuikChange site-directed mutagenesis (Stratagene) was performed to introduce the K99R, A93D, and V112E mutations. Leader sequences of either MGSSHHHHHHSSGLVPRGSHIH (for thrombin cleavage) or MHHHHHHSSGRENLYFQGHIH (for tobacco etch virus cleavage) were incorporated into the V112E and A93D variants, respectively. All resulting plasmids were confirmed by DNA sequencing. The SAM domain constructs were transformed into E. coli BL21 cells, grown at 37 °C to A600 of ∼0.6, induced with 1 mm IPTG, and grown at 30 °C overnight. LB media was used to produce unlabeled protein, and M9 minimal media supplemented with 1 g/liter 15NH4Cl was used for 15N labeling. Cells were collected by centrifugation, resuspended in 20 mm imidazole, 50 mm Na2HPO4, 500 mm NaCl, pH 7.5 buffer, and lysed by sonication and by passage through a French press. The soluble extract was applied to a 5-ml column of nickel-nitrilotriacetic acid-agarose (GE Healthcare). After washing with the same lysis buffer, the bound protein was eluted with 500 mm imidazole, 50 mm Na2HPO4, 500 mm NaCl, pH 7.5 buffer. The desired protein samples were pooled and dialyzed overnight into 20 mm MOPS, 50 mm NaCl, 0.5 mm EDTA, at pH 8.0. The His6 tags were cleaved only for samples characterized by NMR spectroscopy. Protein concentrations were determined using predicted molar absorptivities, ϵ280.
NMR Spectroscopy
Experiments were run at 25 °C on a 600 MHz Bruker Avance III NMR spectrometer. The protein samples were dialyzed into 20 mm MOPS, 50 mm NaCl, 0.5 mm EDTA at pH 8.0 and concentrated via Amicon centrifugal filters to 0.25–1 mm followed by the addition of 10% D2O for the signal lock.
Isothermal Titration Calorimetry
Isothermal titration calorimetry experiments were performed with an ITC200 instrument (GE Healthcare), and the resulting data were processed using Origin 7.0. Experiments were conducted with samples in 20 mm MOPS, 50 mm NaCl, 0.5 mm EDTA at pH 8.0 and 25 °C. The titrations consisted of 1.5–2.0-μl injections of A93D or A93D/K99R SAM domain constructs (0.16–0.4 mm) into a cell with 10-fold less concentrated V112E or K99R/V112E protein.
Cell Culture
NIH3T3 cells were obtained from the American Type Culture Collection and cultured in DMEM (Sigma) containing 10% calf serum (Sigma) and 1% penicillin/streptomycin (Invitrogen). Phoenix A (amphotropic) cells were maintained in DMEM with 10% FBS (Sigma) and 1% penicillin/streptomycin. For suspension cultures, 3 × 105 cells were seeded in poly(2-hydroxy methacrylate)-coated 6-well tissue culture plates and grown for 48 h.
Retrovirally Transduced Cell Lines
The retroviral plasmid DNAs in MSCVpuro vector (EN-MSCVp, FLAG-EN-MSCVp, HA-EN-MSCVp, FLAG-K99R-EN-MSCVp, FLAG-K99D-EN-MSCVp, HA-D101A-EN-MSCVp, HA-D101K-EN-MSCVp, FLAG-R103K-EN-MSCVp, FLAG-R103A-EN-MSCVp, HA-E100A-EN-MSCVp) were transfected into the Phoenix A retrovirus producer cell line using FuGENE 6 transfection reagent (Roche Applied Science). NIH3T3 cells were infected with the virus, and the successfully infected cells were selected with puromycin (Sigma) for 48 h.
Soft Agar Assays
Soft agar assays were performed as described previously (28, 34). 6-Well plates were covered with 4% agar in DMEM with 10% calf serum and 1% penicillin/streptomycin (bottom layer). 8 × 103 cells were resuspended in 2% agar in DMEM with 10% calf serum and 1% penicillin/streptomycin (top layer) and seeded in triplicate on top of the bottom layers. Two drops of media were added on the agar medium to feed the cells every other day. After 2 weeks, the pictures of the wells were taken (3 different sections/well) at 4× magnification, and the single cells and colonies were counted using ImageJ software. Results are given as percentages of colonies formed per total number of events counted.
Preparation of Cell Lysates, Immunoprecipitation, and Immunoblots
Subconfluent NIH3T3 cells expressing the various EN constructs were washed 2× with ice-cold PBS and lysed with the lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40, protease and phosphatase inhibitor cocktails (Roche Applied Science) and passed through a 23-gauge syringe for complete lysis of the cells. Protein concentration was determined using DC protein assay kit (Bio-Rad). For analysis of the total cell lysates, equal concentrations of cell lysates were mixed with 6× SDS sample buffer to a final concentration of 2× and run on a 9% SDS-PAGE. Proteins were then transferred onto nitrocellulose membranes (Bio-Rad) before immunoblot analysis with the indicated antibodies. For immunoprecipitation, lysates containing 1 mg of protein were mixed with 1 μg of NTRK3 antibody and 40 μl (50% slurry) of Protein A-Sepharose beads (GE Healthcare) and incubated at 4 °C overnight with gentle rotation. Beads were washed 3× with the wash buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40), and the immunoprecipitates were eluted by the addition of 40 μl of 2× SDS sample buffer and boiled for 10 min. Antibodies used (1:1000 in 1% milk) were: TrkC (C-15) (recognizes the C-terminal region of NTRK3) (Santa Cruz), total Akt, total PARP, phospho-Ser-473-Akt, phospho-Mek1/2, and 4G10 phosphotyrosine (Upstate).
Fast Performance Liquid Chromatography (FPLC)
NIH3T3 cells expressing non-mutated and mutant EN proteins were lysed in buffer containing 10 mm Tris, pH 7.4, 150 mm NaCl, 1.5 mm MgCl2, and 0.4% Nonidet P-40 and fractionated by FPLC using a Superose 6HR10/30 gel filtration column (GE Healthcare). Fractions were run on a SDS-PAGE and analyzed by immunoblots using NTRK3 antibody.
Tumor Growth in Nude Mice
Pathogen-free athymic nude mice, 6–8 weeks old, were purchased from Harlan Laboratories. One million NIH3T3 cells expressing EN, HA-EN, or EN mutants (K99R, D101K, and R103A) were injected subcutaneously at two sites/animal (n = 6 for D101K-EN, 5 mice/group for others) for a total of 10 monitored sites for each cell line (12 sites for D101K-EN). Animals were housed in laminar flow racks and microisolator cages under specific pathogen-free conditions and received autoclaved food and water. Nude mice were evaluated for tumor growth periodically until 30 days after injection. Tumor volume was estimated using the following formula: tumor length × (tumor width)2 × 0.5236. Kruskal-Wallis test was performed to compare tumor volume among the groups represented for the indicated day.
Prediction of Small Molecule Binding Pockets
The small molecule binding sites were predicted using the PocketFinder program (35) based on the ETV6-SAM domain polymer crystal structure (PDB ID 1JI7). This program uses a transformation of the Lennard-Jones potential calculated from a three-dimensional protein structure. The PocketFinder algorithm was validated as described previously (35).
RESULTS
Mutation of Lys-99 Abrogates Transformation of NIH3T3 Cells by EN
To determine if Lys-99 is important for transformation activity of EN, we replaced this residue with arginine (K99R-EN) by site-directed mutagenesis and tested its ability to transform NIH3T3 fibroblasts. We used a soft agar colony formation assay with EN-expressing cells as positive controls and empty MSCV vector-transduced cells as negative controls. As shown in Fig. 1A, unlike NIH3T3 cells stably expressing EN, cells expressing either K99R-EN or the control MSCV vector failed to form colonies in soft agar. We previously showed that NIH3T3 cells transformed by EN are resistant to detachment-induced apoptosis (anoikis) (36, 37), a well known characteristic of transformed cells (38). Therefore, we assessed detachment-induced cell death by Western blot analysis of cleaved PARP (a marker of apoptotic cell death (39)) levels in lysates of NIH3T3 cells expressing EN or K99R-EN. NIH3T3 cells expressing K380N-EN (kinase dead EN) and the ΔSAM-EN (lacking the SAM domain in the ETV6 portion of EN (18)) were included as non-transforming EN controls. As expected, PARP cleavage was not apparent in EN-expressing cells, whereas K99R-EN cells showed markedly high levels of cleaved-PARP, which were similar to those of non-transformed cells (MSCV, K380N-EN, ΔSAM-EN) (Fig. 1B). This indicates that K99R-EN is unable to suppress anoikis when expressed in NIH3T3 cells, in contrast to EN itself. Moreover, K99R-EN cells, along with the other three non-transformed cell lines, formed smaller sized multicellular spheroids as compared with EN-expressing cells in suspension cultures (Fig. 1C). Together, these results provide strong evidence that mutation of Lys-99 abolishes the transformation activity of EN.
FIGURE 1.

Mutation of Lys-99 abrogates the EN ability to transform NIH3T3 cells. A, shown is a soft agar colony formation assay. NIH3T3 cells expressing the indicated constructs were seeded in soft agar medium in triplicate. After 2 weeks of incubation, the single cells and colonies were counted. Results with S.E. are given as percentage of colonies formed per total number of events counted per sample. B, shown is immunoblot analysis of cleaved PARP to probe detachment-induced cell death. Equal numbers of cells were seeded in poly(2-hydroxy-methacrylate)-coated plates and grown as suspension cultures for 48 h. Spheroids were collected and analyzed by immunoblots. Total PARP antibody was used to detect cleaved PARP levels (lower band as indicated). NTRK3 antibody was used to detect EN expression and total Akt was used as loading control. Cleaved-PARP levels were quantified by ImageJ software and normalized to total Akt (loading control). C, pictures depict the spheroid sizes formed by the NIH3T3 cells expressing the indicated constructs in suspension cultures for 48 h. Spheroid sizes were measured using ImageJ software. A two-tailed Student's t test was used for statistical analysis. **, p < 0.005.
Mutation of Lys-99 Disrupts High Molecular Weight Complex Formation of EN
The SAM domain has been shown to mediate ETV6 polymerization (18). The crystal structure of the SAM domain polymer revealed that Lys-99 forms a salt bridge near the core hydrophobic interface (26). We previously reported that SAM-mediated higher order polymer formation is required for cellular transformation by EN (28). We, therefore, hypothesized that mutation of Lys-99 may abrogate EN transformation activity by disrupting salt bridge formation and consequently reducing polymerization of EN through its SAM domain. We have shown previously using size exclusion FPLC that, due to its ability to polymerize, EN is found in high molecular mass fractions of EN-expressing mammalian cell lysates, whereas the non-polymerizing mutants of EN (A93D-EN and V112E-EN) eluted in lower molecular mass fractions (28). We, therefore, employed a similar strategy to determine whether mutation of Lys-99 could limit formation of high molecular weight EN complexes. Using FPLC, we fractionated protein complexes of the lysates of NIH3T3 cells expressing EN, K99R-EN, V112E-EN, or MSCV vector control constructs based on their molecular sizes. Then we analyzed these fractions for EN content by SDS-PAGE and immunoblotting using anti-NTRK3 antibodies. As shown in Fig. 2A, K99R-EN is detected primarily in lower molecular weight FPLC fractions. This is similar to the previously characterized non-polymerizing V112E-EN and in marked contrast to EN itself, which is distributed over fractions corresponding to higher molecular weight complexes ranging from ∼400 to >2000 kDa. To support these findings, we compared the solubility of EN to K99R-EN. We lysed NIH3T3 cells expressing EN, K99R-EN, or MSCV vector controls using a lysis buffer containing 1% Triton X-100, separated the insoluble fraction (pellet) from the soluble fraction (supernatant) by centrifugation, and analyzed both fractions for EN content by SDS-PAGE and immunoblotting using anti-NTRK3 antibodies. We observed EN in both soluble and insoluble fractions, whereas K99R-EN appeared predominantly in the soluble fraction (Fig. 2B). These results suggest that mutation of Lys-99 within the SAM domain reduces polymerization of EN, rendering it more soluble.
FIGURE 2.

Mutation of Lys-99 inhibits high molecular weight complex formation of EN. A, immunoblot analysis of the NIH3T3 cell lysates fractionated by FPLC is shown. Fraction numbers are indicated above each lane, and retention volumes of molecular mass markers are shown with arrows. EN was eluted in high molecular mass fractions, whereas both K99R-EN and the non-polymerizing mutant V112E-EN were detected in lower molecular mass fractions. Although the expected mass of monomeric EN is ∼73 kDa, its domain structure should result in a larger apparent size. B, immunoblot analysis of TritonX-100 soluble and insoluble fractions of NIH3T3 cell lysates is shown. Total Akt and β-actin antibodies were used as soluble fraction controls. EN proteins were probed with the NTRK3 antibody.
Mutation of Lys-99 Reduces the Affinity of the Isolated SAM Domain for Self-association
To examine further the role of Lys-99, we used isothermal titration calorimetry (ITC) to thermodynamically characterize the self-association of the isolated ETV6-SAM domains. As demonstrated previously (26), A93D and V112E mutants are monomeric due to the presence of a charged residue in either the mid-loop and end-helix surface, respectively. However, the two species can still form a high affinity “SAM domain heterodimer” via their remaining unmodified surfaces, thus allowing ITC measurements of their equilibrium dissociation constants (Kd values). To carry out these studies, we expressed fragments (residues 43–125) of ETV6 encompassing the SAM domain preceded by an unstructured N-terminal sequence starting at Met-43. The latter was chosen as this methionine is an alternative start site for ETV6 translation (40). The K99R mutation was introduced in either the A93D or V112E SAM domain mutants to measure the effect of Lys-99 mutation on SAM domain heterodimer formation. Importantly, SAM-A93D, SAM-V112E, and SAM-A93D/K99R all yielded well dispersed NMR spectra, confirming that the mutations did not disrupt the tertiary structure of the SAM domain (Fig. 3).
FIGURE 3.
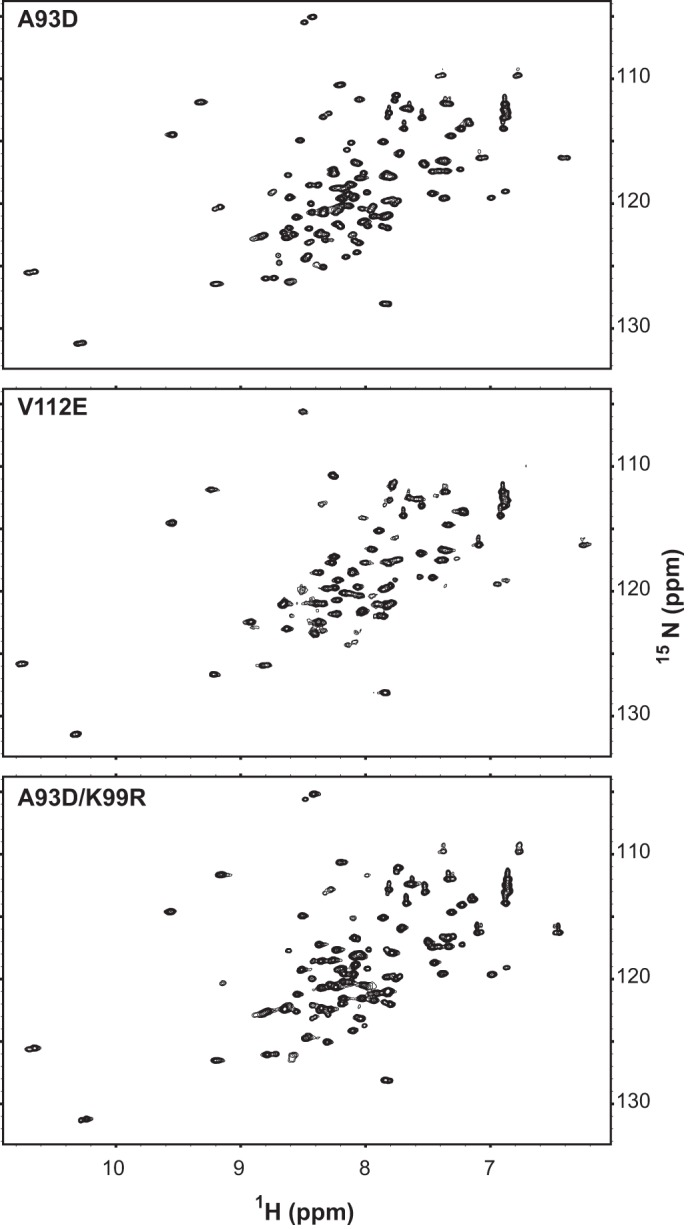
Mutation of Lys-99 does not disrupt the structure of the SAM domain. The well dispersed 15N HSQC (heteronuclear single quantum correlation) spectra of SAM-A93D, SAM-V112E, and SAM-A93D/K99R confirm that each ETV6 fragment (residues 43–125) adopts an independently folded structure. Thus, the reduction in EN self-association due to the K99R mutation does not arise indirectly from disruption of the SAM domain fold. Under these conditions, SAM-V112E showed some propensity to aggregate slightly, leading to a reduction in signal intensities. However, the relatively sharp spectral line widths indicate that each species is predominantly monomer. Based on partial main chain 15N and 13C resonances assignments, the ∼15 N-terminal residues preceding the helical SAM domain are conformationally disordered (not shown).
The results of ITC measurements on isolated SAM domains are presented in Fig. 4 and Table 1. Consistent with previously reported data (26), SAM-A93D and SAM-V112E formed a tight 1:1 complex with a low Kd value of 4.4 nm. A similar equilibrium dissociation constant of 6.6 nm was measured for dimerization of SAM-A93D and SAM-V112E/K99R. Based on the crystal structure of the ETV6-SAM domain polymer, residue 99 in this context is not involved in the WT dimer interface (26, 27). Thus, the lack of any significant change in Kd value supports the conclusion that the K99R mutation does not disrupt the tertiary structure of the SAM domain. In contrast, SAM-A93D/K99R bound SAM-V112E with a dramatically increased Kd value of 1900 nm. Thus, mutation of Lys-99 at the dimer interface to arginine weakened the association of the SAM domains by ∼400-fold. Furthermore, as generally expected for the disruption of a salt bridge, the mutation substantially reduced the magnitude of the enthalpy change accompanying dimer formation.
FIGURE 4.

Mutation of Lys-99 reduces the thermodynamic affinity of the ETV6-SAM domain for self-association. A, shown is an illustration of the WT SAM domain polymer structure of the locations of the interfacial residues in question. Shown are examples of ITC data for SAM-A93D titrated into SAM-V112E (B), SAM-A93D titrated into SAM-V112E/K99R (C), and SAM-A93D/K99R titrated into SAM-V112E (D). The raw data were buffer-blank-corrected (upper), integrated, and fit to a simple binding isotherm (lower) to yield the values reported in Table 1. The corresponding dimer structures of the isolated SAM domains with indicated mutations are depicted above each graph.
TABLE 1.
ETV6-SAM domain dimerization quantitated by ITC
ITC experiments were conducted with SAM domains in 20 mm MOPS, 50 mm NaCl, 0.5 mm EDTA at pH 8.0 and at 25 °C. The tabulated values for the stoichiometry (N), equilibrium dissociation constant (Kd), and enthalpy (ΔHo) and entropy (ΔSo) changes for dimerization are the mean and S.D. of three independent titrations.
| Protein in syringe | Protein in cell | N | Kd | ΔHo | ΔSo |
|---|---|---|---|---|---|
| nm | kcal/mol | cal/mol deg | |||
| A93D | V112E | 0.90 ± 0.03 | 4.4 ± 2.2 | −8.4 ± 0.2 | 10 ± 1.0 |
| A93D | V112E/K99R | 0.94 ± 0.05 | 6.4 ± 1.6 | −7.3 ± 0.3 | 13 ± 0.8 |
| A93D/K99R | V112E | 0.98 ± 0.19 | 1900 ± 1200 | −2.0 ± 1.0 | 19 ± 5 |
Mutations of Lys-99 and Asp-101, the Two Salt Bridge-forming Residues in the SAM Polymer Interface, Block Transformation Activity of EN by Inhibiting Tyrosine Phosphorylation of EN and Its Downstream Signaling Pathways
The above results indicate that mutation of Lys-99 blocks EN transformation activity at the level of polymer formation. Kim et al. (26) previously reported that an intermolecular salt bridge from Lys-99 in one SAM domain to Asp-101 in the neighboring SAM domain is consistently observed in the crystal structure of ETV6-SAM polymers. Therefore, we predicted that if Lys-99 mutation blocks EN transformation activity by disrupting the salt bridge in the interface of SAM polymer and inhibiting the association of SAM monomers, then mutation of Lys-99 to Asp as well as Asp-101 to either lysine or alanine should result in a similar effect. Accordingly, we generated K99D, D101A, and D101K mutants of EN and tested their ability to transform NIH3T3 cells using soft agar colony formation assays. We also generated the R103A, R103K, and E100A mutants of EN and included them in soft agar assays, as salt bridges from Arg-103 to Asp-101 and Glu-100 also appear in the crystal structure (Fig. 5A). Moreover, these residues are highly conserved in ETV6-SAM domains of different species, except that the homologous Drosophila Yan-SAM has an alanine and glycine instead of glutamate and arginine of the human ETV6-SAM domain (Fig. 5B). As shown in Fig. 5C, both D101A-EN and D101K-EN cells displayed markedly reduced colony formation in soft agar, similar to that of K99R-EN cells and K99D-EN cells. On the other hand, cells expressing E100A-EN formed colonies as efficiently as EN-expressing cells, indicating that this residue is dispensable for transformation. Interestingly, R103K-EN cells formed similar number of colonies as EN-expressing cells, whereas R103A-EN cells showed a modest reduction in colonies, implying that the charges contributed by Arg-103 residue to the salt bridge are only modestly important for EN transformation activity. Overall, it appears that the salt bridge formed by Lys-99 and Asp-101 is critical for EN transformation activity.
FIGURE 5.

Mutations of the critical residues involved in the salt bridge formation within the interaction face of SAM polymer abrogates EN transformation by inhibiting EN tyrosine phosphorylation and downstream signaling pathways. A, shown is a schematic illustration of a portion of the SAM domain polymer (PDB ID 1JI7) generated using PyMOL. The magnified inset shows a network of electrostatic interactions between Lys-99 and Arg-103 on one domain with Asp-101 and Glu-100 on the neighboring domain. B, shown is sequence alignment of the ETV6-SAM domain from different species. Alanine and the valine residues that form the hydrophobic core of the SAM polymer interface are shown in bold. The residues involved in the salt bridge were highlighted in gray. C, shown is soft agar colony formation assay. NIH3T3 cells stably expressing the indicated constructs were seeded in soft agar medium in triplicates and analyzed as described in Fig. 1A. A two-tailed Student's t test was used for statistical analysis. Each group was compared with MSCV control group. *, p < 0.05; **, p < 0.005. D, analysis of PTK activation status of the non-mutated and mutant EN proteins is shown. EN proteins were immunoprecipitated from the NIH3T3 cells expressing the indicated EN constructs or the MSCV control using the NTRK3 antibody, and EN tyrosine phosphorylation levels were determined by immunoblot analysis using the 4G10 phospho-tyrosine antibody. E, shown is a comparison of phospho-Akt and phospho-Mek1/2 levels in NIH3T3 cells expressing MSCV control vector or EN by immunoblot analysis after 36 h of serum starvation. F, shown is immunoblot analysis of the total cell lysates from the NIH3T3 cells expressing EN and mutant EN proteins. After 36 h of serum starvation, cells were lysed and analyzed by immunoblots. Akt and Mek1/2 activation was determined using the phospho-Akt and phospho-Mek1/2 antibodies. HC, heavy chain; P-Tyr, phosphotyrosine; P-Akt, phospho-Akt; P-Mek1/2, phospho-Mek1/2.
We previously reported that SAM-mediated polymerization of EN is required for optimum auto- or cross-phosphorylation of its PTK activation loop tyrosines and hence constitutive activation and downstream Ras-MAPK and PI3K-Akt signaling pathways (18, 19). We reasoned that if mutation of the salt bridge-forming residues within the SAM domain inhibits self-association of EN, then these mutations would also reduce EN tyrosine phosphorylation and block downstream signaling pathways. This would potentially explain the observed effects of these mutations on EN transformation activity. To test this hypothesis, we immunoprecipitated EN and mutant EN proteins from NIH3T3 cells using anti-NTRK3 antibodies and assessed their tyrosine phosphorylation status by immunoblotting using an anti-phosphotyrosine antibody. Consistent with the lack of transformation activity, Lys-99 and Asp-101 mutants of EN exhibited markedly lower levels of tyrosine phosphorylation (Fig. 5D). In contrast, the Arg-103 and Glu-100 mutant EN proteins, which retain transformation activity, were highly tyrosine-phosphorylated, as also seen with EN itself. EN-transformed NIH3T3 cells exhibit constitutive phosphorylation of Akt and Mek1/2 (Fig. 5E), which is consistent with our previously published results (18, 19). Therefore, we probed phospho-Akt and phospho-Mek1/2 levels in total cell lysates of NIH3T3 cells expressing EN and mutant EN proteins by immunoblotting to assess the ability of these mutant EN proteins to constitutively activate Akt and Mek1/2. In parallel with the reduced EN phosphotyrosine levels, cells expressing Lys-99 and Asp-101 mutants of EN had significantly lower levels of phospho-Akt and phospho-Mek1/2 as compared with cells expressing Arg-103 and Glu-100 mutants of EN as well as EN-expressing cells (Fig. 5F). Together with the above results, these findings strongly suggest that mutation of Lys-99 and Asp-101 weakens the self-association of EN and consequently inhibits constitutive activation of the PTK domain and key oncogenic signaling pathways downstream of EN.
Mutation of Lys-99 and Asp-101 Abolishes EN-induced Tumorigenicity of NIH3T3 Cells in Nude Mice
To determine the relative transformation activity of the EN mutants in vivo, we injected NIH3T3 cells expressing EN, HA-EN, K99R-EN, HA-D101K-EN, and FLAG-R103A-EN constructs subcutaneously into the flanks of nude mice (n = 6 for HA-D101K-EN, 5 mice/group for others, 2 sites/mouse) and monitored tumor formation over a period of 30 days (Fig. 6). Consistent with our previous reports (19, 28), EN-expressing cells rapidly formed large tumors reaching a size of ∼1000 mm3 per site after 16 days, at which point the mice bearing these tumors were sacrificed. In striking contrast, K99R-EN cells did not form any detectable tumors over the course of the study, and D101K-EN cells formed only very small tumors averaging below 100 mm3 at day 30. Interestingly, R103A-EN tumor growth was initially slow as compared with EN tumors; however, they eventually grew to ∼1000 mm3 at day 27, at which point these mice were also sacrificed. Consistent with the soft agar results above, these studies clearly demonstrate that the Lys-99 and Asp-101 residues are crucial for EN transformation activity in vivo.
FIGURE 6.
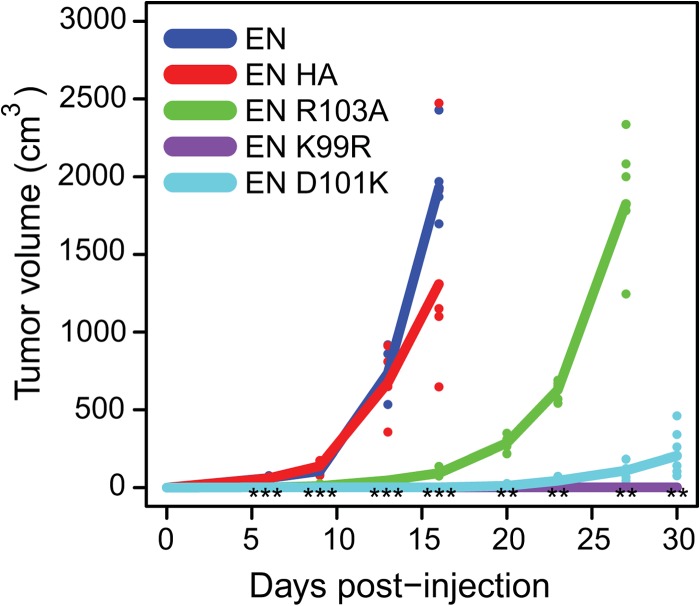
NIH3T3 cells expressing the Lys-99 and Asp-101 salt bridge mutants of EN fail to form tumors in nude mice. NIH3T3 cells expressing EN, HA-EN, K99R-EN, HA-D101K-EN, and FLAG-R103A-EN constructs were injected subcutaneously into the flanks of nude mice (n = 6 for HA-D101K-EN, n = 5 for all others, two sites/mouse), and tumor formation was monitored over a period of 30 days. At day 16, EN tumors grew to ∼1000 mm3, and therefore, mice bearing these tumors were sacrificed. K99R-EN did not form any detectable tumors over the course of the study, and D101K-EN formed only very small tumors averaging below 100 mm3 at day 30. R103A-EN tumors grew very slowly in the beginning but eventually reached ∼1000 mm3 at day 27. Statistical significance was determined using a Kruskal-Wallis test comparing tumor volumes among groups represented for the indicated day of monitoring. ***, p < 0.001; **, p < 0.01.
Prediction of Pockets for Small Molecule Binding at the Interface of SAM Domain Polymer
The above results demonstrate that mutation of salt bridge-forming residues Lys-99 and Asp-101 blocks EN transformation, likely by inhibiting polymerization. It was previously reported that there are no obvious binding pockets on the interaction interfaces of SAM monomers for small molecule inhibitors of polymerization (27). However, because the salt bridges are formed upon SAM domain association, we asked if any small molecule binding pocket(s) might be present at the interfaces in proximity to these salt bridges. Therefore, we analyzed the crystal structure of the ETV6-SAM domain polymer (PDB ID 1JI7) (26) using the PocketFinder program (35). Using this approach, we found two putative pockets located at the interfaces between the SAM domains (Fig. 7A). One of the pockets is directly formed by Lys-99 and Asp-101 (Fig. 7B). In addition, Val-112 and Arg-103 contribute to this ligand binding pocket. Interestingly we observed another cavity on the other interaction face of the SAM polymer, which is also located near the Lys-99–Asp-101 salt bridge and is formed by the electrostatic interactions of Arg-105 and Asp-111 from two individual SAM domains. Kim et al. (26) reported that the Arg-103-Asp109 salt bridge was also consistently observed in the three different interfaces of the SAM domain polymer crystal. These observations collectively suggest that salt bridges in the SAM domain interface may serve as binding pockets for small molecules, which could potentially modulate the formation of the EN polymer.
FIGURE 7.
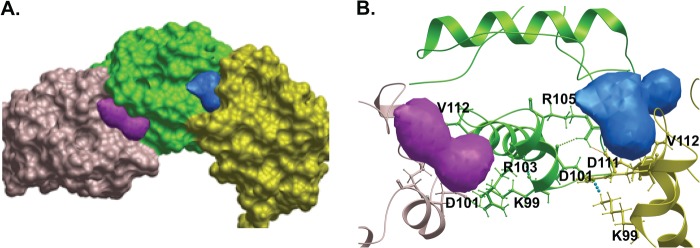
Prediction of two putative small molecule binding pockets adjacent to the salt bridges in the interface of SAM polymer. A, a surface representation of SAM polymer (PDB ID 1JI7) shows the predicted small molecule binding sites (colored in magenta and blue) at the two interfaces of SAM monomers. Three different colors were used to represent individual SAM monomers. B, the ribbon diagram of SAM polymer depicts the predicted pockets formed by the salt bridges between Lys-99–Asp-101 (magenta, left interface) and Arg-103–Asp-111 (blue, right interface). Arg-103 and Val-112 residues also appear to be part of the left pocket. PocketFinder was used to predict the possible small molecule binding sites.
DISCUSSION
The ETV6-SAM domain mediates polymerization of several chimeric oncoproteins, which is critical for their transformation activity (5, 7, 18, 23, 28). X-ray crystallographic structural studies of the isolated SAM domain polymer revealed two hydrophobic interfaces surrounded by salt bridges. Previously, the hydrophobic interactions between the SAM domains have been highlighted as major contributors of polymer formation (26, 27). In this study we demonstrate the importance of electrostatic interactions within the SAM domain polymer interface for EN-mediated cellular transformation. We show that mutation of the Lys-99, which is involved in a salt bridge with Asp-101 between neighboring SAM domains, reduces polymerization and formation of high molecular mass complexes and, in addition, blocks transformation activity of EN. Mutation of Asp-101, the intermolecular salt bridge partner of Lys-99, similarly blocks EN transformation. In line with these findings, both Lys-99 and Asp-101 mutants of EN exhibit reduced levels of tyrosine phosphorylation as compared with EN itself, and neither of the mutants can activate Akt or Mek1/2 signaling cascades in NIH3T3 cells. This correlates with the inability of the NIH3T3 cells expressing these EN mutants to form tumors in nude mice, which is in contrast with highly tumorigenic EN-expressing cells.
The salt bridge between the Lys-99 and Asp-101 is consistently observed in all of the interfaces found in the x-ray crystallographic structures of the ETV6-SAM domains (26, 27). This led us to hypothesize that these residues may be important in facilitating self-association. Indeed, Lys-99 mutation dramatically increased the Kd value for SAM domain dimerization by ∼400-fold as measured by ITC. It is intriguing that mutation of Lys-99 to a similarly charged arginine has such pronounced phenotype, implying that precise electrostatic and structural interactions involving this residue are critical for self-association of SAM domains and the transformation activity of EN. This indicates that, in addition to hydrophobic interactions, the specific electrostatic interactions surrounding the SAM polymer interface are also important for self-association. Consistent with this conclusion, mutation of the Lys-99 salt bridge partner Asp-101 similarly impedes oncogenic capacity of EN.
It is notable that Arg-103 and Glu-100 also appear to participate in the electrostatic interaction network involving Lys-99 and Asp-101 (Fig. 5A). Arg-103 is observed in all of the SAM polymer interfaces as part of the salt bridge, whereas Glu-100 is found only in one interface (26, 27). However, replacement of Arg-103 to lysine or Glu-100 to alanine has no significant effect on EN transformation activity. This is in sharp contrast to the Lys-99 and Asp-101 mutants, suggesting that these residues are not required for self-association. Accordingly, tyrosine phosphorylation of Glu-100 and Arg-103 mutants of EN is also comparable to EN, and these mutants efficiently activate Akt and Mek1/2. Interestingly, mutation of Arg-103 to alanine has a modest effect on EN-mediated colony formation in soft agar but not to the same extent as seen with the Lys-99 and Asp-101 mutants. Moreover, R103A-EN tumor growth was very slow at early time points, yet these tumors eventually grew to similar sizes as those observed with EN-expressing cells. This implies that both Lys-99 and Asp-101 are critical for SAM self-association and thus for EN transformation, whereas Arg-103 plays a more modest role and Glu-100 is not involved in these processes.
Akin to ETV6-SAM, the SAM domain of Yan, the Drosophila ortholog of ETV6, mediates its polymerization and hence transcriptional repressor activity (41, 42). Whereas the charge of the residues corresponding to Lys-99 and Asp-101 of the ETV6-SAM domain are conserved in the Yan SAM domain, Glu-100 and Arg-103 are replaced by an alanine and a glycine, respectively (Fig. 5B). This supports our proposed mechanism that Lys-99 and Asp-101 are the critical residues for the electrostatic interactions between SAM monomers, whereas Glu-100 and Arg-103 are dispensable. However, considering the striking effect of the K99R mutation on the ETV6-SAM domain self-association and EN polymerization, it is puzzling that an arginine occupies the corresponding position in the Yan SAM domain. This might be explained by differences in the structure and the electrostatic network surrounding these residues in the two SAM domains. For example, the ETV6-SAM domain has charged residues (Glu-100, Arg-103) in place of the neutral residues of Yan SAM domain (Ala-93, Gly-96) and thus may be less tolerant to the replacement of a lysine with an arginine side chain for maintaining a proper salt bridge that facilitates SAM domain self-association.
Because SAM domain-mediated polymerization is critical for the aberrant activity of many SAM domain-containing fusion oncoproteins, the discovery of small molecules that could effectively inhibit polymerization would potentially be of therapeutic value. Although there are no obvious cavities on the association interfaces of the SAM domain monomers, the algorithm PocketFinder detected two potential small molecule binding pockets located at the SAM domain dimer interface. One of these is directly formed by the Lys-99–Asp-101 salt bridge, and the other is located nearby. These predicted pockets may potentially serve as binding sites for small molecules, which could destabilize SAM domain polymers or alter their structure and prevent correct positioning of the PTK domains for maximal activation. Alternatively, Lys-99–Asp-101 salt bridge formation could generate docking sites for a yet unknown EN interaction partner that is critical for transformation. In this case, small molecule binding to these sites might prevent this interaction, inhibiting the downstream oncogenic events. It is also possible that small molecule binding to these pockets may stabilize the interactions between the flanking SAM domains and, therefore, enhance rather than inhibit the SAM-containing fusion oncoproteins. Regardless, this analysis strongly suggests that a high-throughput screen for molecules that interact with the ETV6-SAM domain is warranted and may yield interesting lead compounds that could provide further mechanistic insights into polymerization and reagents for modulating activity of fusion oncoproteins.
In summary, we show that disruption of an intermolecular salt bridge between neighboring SAM domains reduces EN polymerization and impairs EN tyrosine phosphorylation as well as its ability to activate PI3K-Akt and Ras-MAPK pathways and blocks EN transformation. Our studies provide an improved understanding of ETV6-SAM domain polymerization and EN transformation and may also be applicable to other SAM-containing fusion oncoproteins.
Acknowledgments
We thank Eric Escobar for preliminary NMR spectroscopic studies of the ETV6-SAM domain. Instrument support was provided by the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, the British Columbia Knowledge Development Fund, the UBC Blusson Fund, and the Michael Smith Foundation for Health Research.
This work was supported by Canadian Institutes for Health Research Grant MOP119599 and funds from the British Columbia Cancer Foundation through generous donations from Team Finn and Ride to Conquer Cancer (to P. H. S.) and Canadian Cancer Society Research Institute Grant 017308 (to L. P. M.).
- PTK
- protein-tyrosine kinase
- EN
- ETV6-NTRK3
- PARP
- poly(ADP-ribose) polymerase
- ITC
- isothermal titration calorimetry
- SAM
- sterile α-motif.
REFERENCES
- 1. De Braekeleer E., Douet-Guilbert N., Morel F., Le Bris M. J., Basinko A., De Braekeleer M. (2012) ETV6 fusion genes in hematological malignancies. A review. Leuk. Res. 36, 945–961 [DOI] [PubMed] [Google Scholar]
- 2. Bohlander S. K. (2005) ETV6. A versatile player in leukemogenesis. Semin. Cancer Biol. 15, 162–174 [DOI] [PubMed] [Google Scholar]
- 3. Golub T. R., Barker G. F., Lovett M., Gilliland D. G. (1994) Fusion of PDGF receptor β to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 77, 307–316 [DOI] [PubMed] [Google Scholar]
- 4. Golub T. R., Barker G. F., Bohlander S. K., Hiebert S. W., Ward D. C., Bray-Ward P., Morgan E., Raimondi S. C., Rowley J. D., Gilliland D. G. (1995) Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. U.S.A. 92, 4917–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golub T. R., Goga A., Barker G. F., Afar D. E., McLaughlin J., Bohlander S. K., Rowley J. D., Witte O. N., Gilliland D. G. (1996) Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol. Cell. Biol. 16, 4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papadopoulos P., Ridge S. A., Boucher C. A., Stocking C., Wiedemann L. M. (1995) The novel activation of ABL by fusion to an ets-related gene, TEL. Cancer Res. 55, 34–38 [PubMed] [Google Scholar]
- 7. Lacronique V., Boureux A., Valle V. D., Poirel H., Quang C. T., Mauchauffé M., Berthou C., Lessard M., Berger R., Ghysdael J., Bernard O. A. (1997) A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 278, 1309–1312 [DOI] [PubMed] [Google Scholar]
- 8. Kuno Y., Abe A., Emi N., Iida M., Yokozawa T., Towatari M., Tanimoto M., Saito H. (2001) Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12). Blood 97, 1050–1055 [DOI] [PubMed] [Google Scholar]
- 9. Yagasaki F., Wakao D., Yokoyama Y., Uchida Y., Murohashi I., Kayano H., Taniwaki M., Matsuda A., Bessho M. (2001) Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Res. 61, 8371–8374 [PubMed] [Google Scholar]
- 10. Lannon C. L., Sorensen P. H. (2005) ETV6-NTRK3. A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 15, 215–223 [DOI] [PubMed] [Google Scholar]
- 11. Knezevich S. R., McFadden D. E., Tao W., Lim J. F., Sorensen P. H. (1998) A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 18, 184–187 [DOI] [PubMed] [Google Scholar]
- 12. Knezevich S. R., Garnett M. J., Pysher T. J., Beckwith J. B., Grundy P. E., Sorensen P. H. (1998) ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 58, 5046–5048 [PubMed] [Google Scholar]
- 13. Eguchi M., Eguchi-Ishimae M., Tojo A., Morishita K., Suzuki K., Sato Y., Kudoh S., Tanaka K., Setoyama M., Nagamura F., Asano S., Kamada N. (1999) Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood 93, 1355–1363 [PubMed] [Google Scholar]
- 14. Tognon C., Knezevich S. R., Huntsman D., Roskelley C. D., Melnyk N., Mathers J. A., Becker L., Carneiro F., MacPherson N., Horsman D., Poremba C., Sorensen P. H. (2002) Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2, 367–376 [DOI] [PubMed] [Google Scholar]
- 15. Skálová A., Vanecek T., Sima R., Laco J., Weinreb I., Perez-Ordonez B., Starek I., Geierova M., Simpson R. H., Passador-Santos F., Ryska A., Leivo I., Kinkor Z., Michal M. (2010) Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene. A hitherto undescribed salivary gland tumor entity. Am. J. Surg. Pathol. 34, 599–608 [DOI] [PubMed] [Google Scholar]
- 16. Kazakov D. V., Hantschke M., Vanecek T., Kacerovska D., Michal M. (2010) Mammary-type secretory carcinoma of the skin. Am. J. Surg. Pathol. 34, 1226–1227 [DOI] [PubMed] [Google Scholar]
- 17. Forghieri F., Morselli M., Potenza L., Maccaferri M., Pedrazzi L., Paolini A., Bonacorsi G., Artusi T., Giacobbi F., Corradini G., Barozzi P., Zucchini P., Marasca R., Narni F., Crescenzi B., Mecucci C., Falini B., Torelli G., Luppi M. (2011) Chronic eosinophilic leukaemia with ETV6-NTRK3 fusion transcript in an elderly patient affected with pancreatic carcinoma. Eur. J. Haematol. 86, 352–355 [DOI] [PubMed] [Google Scholar]
- 18. Wai D. H., Knezevich S. R., Lucas T., Jansen B., Kay R. J., Sorensen P. H. (2000) The ETV6-NTRK3 gene fusion encodes a chimeric protein tyrosine kinase that transforms NIH3T3 cells. Oncogene 19, 906–915 [DOI] [PubMed] [Google Scholar]
- 19. Tognon C., Garnett M., Kenward E., Kay R., Morrison K., Sorensen P. H. (2001) The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res. 61, 8909–8916 [PubMed] [Google Scholar]
- 20. Ponting C. P. (1995) SAM. A novel motif in yeast sterile and Drosophila polyhomeotic proteins. Protein Sci. 4, 1928–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schultz J., Ponting C. P., Hofmann K., Bork P. (1997) SAM as a protein interaction domain involved in developmental regulation. Protein Sci. 6, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kyba M., Brock H. W. (1998) The SAM domain of polyhomeotic, RAE28, and scm mediates specific interactions through conserved residues. Dev. Genet 22, 74–84 [DOI] [PubMed] [Google Scholar]
- 23. Carroll M., Tomasson M. H., Barker G. F., Golub T. R., Gilliland D. G. (1996) The TEL/platelet-derived growth factor β receptor (PDGFβR) fusion in chronic myelomonocytic leukemia is a transforming protein that self-associates and activates PDGFβR kinase-dependent signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 93, 14845–14850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jousset C., Carron C., Boureux A., Quang C. T., Oury C., Dusanter-Fourt I., Charon M., Levin J., Bernard O., Ghysdael J. (1997) A domain of TEL conserved in a subset of ETS proteins defines a specific oligomerization interface essential to the mitogenic properties of the TEL-PDGFR β oncoprotein. EMBO J. 16, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peterson A. J., Kyba M., Bornemann D., Morgan K., Brock H. W., Simon J. (1997) A domain shared by the Polycomb group proteins Scm and ph mediates heterotypic and homotypic interactions. Mol. Cell. Biol. 17, 6683–6692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim C. A., Phillips M. L., Kim W., Gingery M., Tran H. H., Robinson M. A., Faham S., Bowie J. U. (2001) Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 20, 4173–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran H. H., Kim C. A., Faham S., Siddall M. C., Bowie J. U. (2002) Native interface of the SAM domain polymer of TEL. BMC Struct. Biol. 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tognon C. E., Mackereth C. D., Somasiri A. M., McIntosh L. P., Sorensen P. H. (2004) Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol. Cell. Biol. 24, 4636–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chakrabarti S. R., Sood R., Nandi S., Nucifora G. (2000) Posttranslational modification of TEL and TEL/AML1 by SUMO-1 and cell-cycle-dependent assembly into nuclear bodies. Proc. Natl. Acad. Sci. U.S.A. 97, 13281–13285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wood L. D., Irvin B. J., Nucifora G., Luce K. S., Hiebert S. W. (2003) Small ubiquitin-like modifier conjugation regulates nuclear export of TEL, a putative tumor suppressor. Proc. Natl. Acad. Sci. U.S.A. 100, 3257–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roukens M. G., Alloul-Ramdhani M., Vertegaal A. C., Anvarian Z., Balog C. I., Deelder A. M., Hensbergen P. J., Baker D. A. (2008) Identification of a new site of sumoylation on Tel (ETV6) uncovers a PIAS-dependent mode of regulating Tel function. Mol. Cell. Biol. 28, 2342–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Macauley M. S., Errington W. J., Schärpf M., Mackereth C. D., Blaszczak A. G., Graves B. J., McIntosh L. P. (2006) Beads-on-a-string, characterization of ETS-1 sumoylated within its flexible N-terminal sequence. J. Biol. Chem. 281, 4164–4172 [DOI] [PubMed] [Google Scholar]
- 33. Lannon C. L., Martin M. J., Tognon C. E., Jin W., Kim S. J., Sorensen P. H. (2004) A highly conserved NTRK3 C-terminal sequence in the ETV6-NTRK3 oncoprotein binds the phosphotyrosine binding domain of insulin receptor substrate-1. An essential interaction for transformation. J. Biol. Chem. 279, 6225–6234 [DOI] [PubMed] [Google Scholar]
- 34. Morrison K. B., Tognon C. E., Garnett M. J., Deal C., Sorensen P. H. (2002) ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene 21, 5684–5695 [DOI] [PubMed] [Google Scholar]
- 35. An J., Totrov M., Abagyan R. (2005) Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell Proteomics 4, 752–761 [DOI] [PubMed] [Google Scholar]
- 36. Martin M. J., Melnyk N., Pollard M., Bowden M., Leong H., Podor T. J., Gleave M., Sorensen P. H. (2006) The insulin-like growth factor I receptor is required for Akt activation and suppression of anoikis in cells transformed by the ETV6-NTRK3 chimeric tyrosine kinase. Mol. Cell. Biol. 26, 1754–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ng T. L., Leprivier G., Robertson M. D., Chow C., Martin M. J., Laderoute K. R., Davicioni E., Triche T. J., Sorensen P. H. (2012) The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 19, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khwaja A., Rodriguez-Viciana P., Wennström S., Warne P. H., Downward J. (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu S. W., Wang H., Poitras M. F., Coombs C., Bowers W. J., Federoff H. J., Poirier G. G., Dawson T. M., Dawson V. L. (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263 [DOI] [PubMed] [Google Scholar]
- 40. Poirel H., Oury C., Carron C., Duprez E., Laabi Y., Tsapis A., Romana S. P., Mauchauffe M., Le Coniat M., Berger R., Ghysdael J., Bernard O. A. (1997) The TEL gene products. Nuclear phosphoproteins with DNA binding properties. Oncogene 14, 349–357 [DOI] [PubMed] [Google Scholar]
- 41. Qiao F., Bowie J. U. (2005) The many faces of SAM. Sci. STKE 2005, re7. [DOI] [PubMed] [Google Scholar]
- 42. Qiao F., Song H., Kim C. A., Sawaya M. R., Hunter J. B., Gingery M., Rebay I., Courey A. J., Bowie J. U. (2004) Derepression by depolymerization. Structural insights into the regulation of Yan by Mae. Cell 118, 163–173 [DOI] [PubMed] [Google Scholar]