

Background: MEKK2 is an important kinase involved in the activation of MAPK pathways.
Results: MEKK2 binds to 14-3-3 in a phosphorylation-dependent manner. This reduced its phosphorylation at an activating site.
Conclusion: MEKK2 remains inactive until it is dephosphorylated at Thr-283, which releases 14-3-3 and allows autophosphorylation and activation of JNK.
Significance: Binding of 14-3-3 to MEKK2 provides a mechanism for reducing activity and directing signal specificity.
Keywords: Cell Proliferation, Gene Expression, Jun N-terminal Kinase (JNK), MAP Kinases (MAPKs), Signal Transduction, MEKK2
Abstract
MEKK2 (MAP/ERK kinase kinase-2) is a serine/threonine kinase that belongs to the MEKK/STE11 family of MAP kinase kinase kinases (MAP(3)Ks). MEKK2 integrates stress and mitogenic signals to the activation of NF-κB, JNK1/2, p38, and ERK5 pathways. We have found that MEKK2 is regulated through a phosphorylation-dependent association with 14-3-3, a group of adapters that modulate dimerization and association between proteins. We found that MEKK2 was phosphorylated at Thr-283, which resulted in decreased activation loop phosphorylation at Ser-519 and consequently reduced activity. Mechanistically, we found that MEKK2 associated with inactive MEKK2 in the absence of 14-3-3 binding, which led to trans-autophosphorylation of Ser-519. Enforced binding with 14-3-3 reduced Ser-519 trans-autophosphorylation. Expression of T283A MEKK2 within a MEKK2−/− background enhanced stress-activated c-Jun N-terminal kinase activity while elevating IL-6 expression, but also reduced ERK activation with a corresponding reduced proliferation rate. These results indicate that Thr-283 phosphorylation is an important regulatory mechanism for MEKK2 activation.
Introduction
In yeast, stress and nutrient signals are relayed through the Ste11 family of protein kinases to activate transcription. In mammals, a similar pathway exists that utilizes Ste11-like MAP(3)Ks2 MEKK2 and MEKK3 to sense growth factor, stress, and pro-inflammatory signals (1–8). For example, MEKK3 contributes to NF-κB activation in macrophages by signaling through the NF-κB axis (9–12). In addition, MEKK2 and MEKK3 also activate a number of MAPKKs including MKK6 and MKK7, which in turn activate MAPK members such as p38 and JNK (2, 6, 10, 13–15). Thus, the stimuli that utilize signaling through MEKK2, MEKK3, and other MAP(3)Ks are diverse, and the precise mechanism for their activation has remained difficult to fully understand.
MEKK2 and MEKK3 are assembled in a pre-activation complex with other receptor-proximal signaling proteins and utilize conserved protein interaction domains including PB1 domains (11, 16–22). Upon activating signals, MEKK2/3 undergoes phosphorylation within the activation loop Ser-519/Ser-526 (23), which is critical for kinase activity. Dimerization of MEKK2/3 is necessary (24, 25), suggesting that Ser-519/Ser-526 undergoes a mechanism of trans-autophosphorylation. Furthermore, MEKK3 is phosphorylated at a site that promotes binding to 14-3-3 proteins (9). This interaction appears to regulate activation as mutation results in constitutive activation of MEKK3 pathways, leading to enhanced NF-κB activation (9).
In the current study, we have investigated the mechanism of 14-3-3 regulation of MEKK2 and the activation of MEKK2-mediated c-Jun N-terminal kinase (JNK) following stress and mitogen stimulation. We show that Thr-283 phosphorylation of MEKK2 is necessary for 14-3-3 binding, and disrupting this interaction leads to elevated activation of JNK, increased c-Jun phosphorylation, and elevated transcriptional activity. Mechanistically, 14-3-3 interaction alters MEKK2 activity by limiting MEKK2 dimerization and Ser-519 phosphorylation, leading to inactivation. In mouse embryonic fibroblasts devoid of MEKK2, add-back of T283A MEKK2 led to slower growth rates and reduced ERK phosphorylation/activation, but displayed greater TNF-α induced IL-6 expression. Therefore, phosphorylation of Thr-283 and binding of 14-3-3 appear to allow control over MEKK2 input to several MAPK pathways.
EXPERIMENTAL PROCEDURES
Plasmids and Mutagenesis
pCMV5-HA-MEKK2 was purchased from Addgene (Boston, MA; plasmid 12182). To generate FLAG-WT MEKK2, primers were designed to introduce a NotI restriction site at the 5′ end of HA-MEKK2 and an NheI site at the 3′ end by PCR. PCR-generated fragments were cloned into pCMV10-3×FLAG vector to introduce 3×FLAG epitope tags at the N terminus. Site-directed mutagenesis of pCMV5-HA-MEKK2 and pCMV10-3×FLAG-MEKK2 was performed using QuikChange® mutagenesis kit (Stratagene), and mutations were sequence-verified.
To generate the R18 DE/KK MEKK2, we removed the amino acid sequence surrounding Thr-283 of MEKK2 from both WT and kinase-dead K385M MEKK2. To excise these regions, BstBI was introduced in the 5′ end, and ApaI was introduced at the 3′ end of MEKK2 by site-directed mutagenesis. Oligonucleotides representing the R18 DE peptide (PHCVPRDLSWLDLEANMCLP) and KK peptide (PHCVPRDLSWLDLEANMCLP) with the relevant overhangs synthesized and annealed to ApaI and BstBI digested WT and kinase-dead MEKK2.
Retroviral Infection of MEKK2−/− MEF Cells
MEKK2−/− MEF cells were a kind gift of Dr. Bing Su (Yale University). Various forms of MEKK2 were cloned into the viral expression plasmid pQCXIP and transfected into the Ecopack2-293 packaging cell line. Supernatant was harvested 24 h later, and virus was concentrated using Retro-X concentrator by Clontech, according to manufacturer's protocol. MEKK2−/− MEF cells were plated at 60% confluency in 4 μg/ml Polybrene for 4 h, at which point 50-μl aliquots of virus were added directly to cells and incubated for 24 h at 37 °C. Cells were then selected in 2 μg/ml puromycin for 5 days.
Production of Phosphoantibodies
Peptides were generated to correspond to amino acids surrounding Ser-519 of MEKK3 and Thr-283 of MEKK2. CMSGTGIRpSVTGTPY and CMSGTGIRSVTGTPY (where pS denotes phosphoserine) were synthesized to correspond to amino acids 511–524 of MEKK2. YNDGRKpTFPRARR and YNDGRKTFPRARR (where pT denotes phosphothreonine) were synthesized to correspond to amino acids 277–289 of MEKK2. Production of antibodies was performed by Open Biosystems.
Cell Lysis, Immunoprecipitation, and Immunoblotting
Cells were lysed in 50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 25 mm NaF, 25 mm β-glycerophosphate, 5 mm EDTA, 0.05% SDS, 100 nm okadaic acid, and protease inhibitors. Anti-FLAG M2 antibody conjugated to agarose beads (Sigma) or anti-HA antibody conjugated to agarose beads (Roche Applied Science) was added to lysates and incubated overnight at 4 °C. Beads were washed three times with lysis buffer, and proteins were eluted with 200 μl of 1× lithium dodecyl sulfate sample buffer and heated to 70 °C for 10 min. Portions of the lysates prior to immunoprecipitation were also boiled with lithium dodecyl sulfate-containing sample buffer. Lysates and immunoprecipitations were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a low fluorescence polyvinylidene difluoride (PVDF) membrane (Millipore Immobilon-FL PVDF), blocked in 5% skim milk for 30 min, and probed with the appropriate antibody overnight at 4 °C. Secondary decoration with IRDye-680 and IRDye-800 was performed at room temperature for 1 h. Proteins were visualized by direct fluorescence using an Odyssey infrared laser scanner, and fluorescence intensities were quantified by the ImageJ software.
MEKK2 In Vitro Kinase Assay
HEK293 cells were transfected with WT, T284A, or K385M MEKK2. Cells were lysed in solubilization buffer containing 20 mm Tris (pH 7.6), 0.5% Nonidet P-40, 0.25 m NaCl, 1 mm PMSF, 2 mm Na3VO4, 1 μg/ml aprotinin, 250 mm β-glycerophosphate, 250 mm NaF, 100 nm okadaic acid. MEKK2 was immunoprecipitated with FLAG M2 monoclonal antibody conjugated to agarose (Sigma-Aldrich) for 3 h. Immunoprecipitates were pelleted, and beads were washed three times in lysis buffer and twice in kinase buffer containing 25 mm Tris (7.4), 25 mm MgCl2, 1 mm EGTA, 25 mm β-glycerophosphate, 2 mm dithiothreitol, and 1 μg/ml microcystin-LR. Beads were resuspended in 30 μl of kinase buffer, and the reactions were initiated by adding 10 μm cold/unlabeled ATP and 1 μCi of [γ-32P]ATP. Samples were incubated for 20 min at 30 °C, and the reactions were terminated by adding an equal volume of 2× lithium dodecyl sulfate sample buffer and boiling at 70 °C for 10 min. The proteins were resolved by SDS-PAGE, transferred to polyvinylidene difluoride, and immunoblotted with anti-FLAG M2 antibody (Sigma). γ-32P-radiolabeled MEKK2 was visualized by autoradiography using a Personal Molecular Imager (Bio-Rad Laboratories). In some reactions, a peptide corresponding to the target site of MKK6 was added (50 nm), and incorporated 32P was measured by liquid scintillation counting.
In Vitro JNK Assays
Mouse embryonic fibroblast cells were co-transfected with 500 ng of HA-tagged WT, T283A, K385M MEKK2, or empty vector along with 500 ng of FLAG-JNK1a where indicated following transfection protocol as outlined above. Cells were lysed in solubilization buffer containing 20 mm Tris (pH 7.6), 0.5% Nonidet P-40, 0.25 m NaCl, 1 mm PMSF, 2 mm Na3VO4, 1 μg/ml aprotinin, 250 mm β-glycerophosphate, 250 mm NaF, 100 nm okadaic acid. HA-tagged MEKK2 constructs or FLAG-tagged JNK constructs were immunoprecipitated from cell lysates with 25 μl of HA-conjugated agarose beads (anti-HA affinity matrix, Roche Applied Science) or 25 μl of FLAG M2 monoclonal antibody conjugated to agarose (Sigma-Aldrich), respectively. Immunoprecipitates were washed three times in lysis buffer and twice in kinase buffer containing 25 mm Tris (7.4), 25 mm MgCl2, 1 mm EGTA, 25 mm β-glycerophosphate, 2 mm dithiothreitol, and 1 μg/ml microcystin-LR, and finally, beads were resuspended in 30 μl of kinase assay buffer containing 10 μm ATP and 1 μCi of [γ-32P]ATP. GST-c-Jun (0.5 μg) was used as a substrate. Samples were incubated for 15 min at 30 °C, and reactions were terminated by the addition of 2× lithium dodecyl sulfate sample buffer and resolved by SDS-PAGE. MEKK2 autophosphorylation was visualized by autoradiography, and [32P]c-Jun was visualized using a Personal Molecular Imager (Bio-Rad Laboratories).
Luciferase Reporter Assay
HELA cells were transiently co-transfected with 500 ng of WT-MEKK2, T283A MEKK2, or empty vector along with 400 ng of Gal4-c-Jun together with 50 ng of Gal4-luciferase and 200 ng of pCMV-β-galactosidase as indicated. After 24 h, the cells were stimulated with 50 ng of anisomycin for 4 h where indicated. Cellular extracts were prepared using lysis buffer (Promega) and cleared by centrifugation at 12,000 × g for 10 min. 5 μg of protein was used to measure luminescence, and all values were normalized to β-galactosidase activity.
Quantification of IL-6
Stable puromycin-resistant pools of MEKK2−/− MEF cells expressing various forms of MEKK2 were plated in triplicate in 24-well dishes at 30,000 cells/well. TNF-α (1 ng/ml) was added for 18 h. Supernatants were harvested, and IL-6 was quantified using the Quantikine immunoassay kit (R&D Systems).
XTT Assay
MEKK2−/− MEF pools expressing various forms of MEKK2 were plated in triplicate at 4,000 cells/well in 96-well dishes. On each day of the assay, wells were washed twice with PBS and incubated in 100 μl of XTT solution (1 mg/ml XTT containing 0.03 mg/ml PMS (N-methylphenazonium methyl sulfate) in PBS) for 2 h, and then absorbance at 490 nm was measured.
RESULTS
Thr-283 Is a Site of MEKK2 Phosphorylation and 14-3-3 Binding
Of the family of MAP(3)Ks, MEKK2 is most closely homologous to MEKK3, with 91% amino acid identity in the C-terminal kinase domain and 61% identity in the N-terminal regulation domain. We previously reported that the phospho-binding protein 14-3-3 associates with MEKK3 at Thr-294 within the sequence RRT294FPRI (9). Phosphorylation of this site could occur by autophosphorylation because Thr-294 phosphorylation was abolished for the kinase-dead mutant. Furthermore, disruption of 14-3-3 binding by introducing an alanine at this site increased NF-κB activation, suggesting that 14-3-3 suppresses MEKK3-NF-κB pathway signaling through an as yet unknown mechanism.
We examined MEKK2 and found that the corresponding region of the kinase contained a site that was highly similar: RKT283FPRA. This similarity prompted us to consider that Thr-283 of MEKK2 could be phosphorylated and might bind to 14-3-3 to alter MEKK2 activity. When expressed in cells, we found that both MEKK2 and MEKK3 bound to similar amounts of 14-3-3 (Fig. 1A). MEKK2 also captured ectopically expressed 14-3-3ϵ (Fig. 1A), which agrees with Fanger et al. (26), who detected ϵ interaction with MAP3K proteins.
FIGURE 1.

MEKK2 associates with 14-3-3 at Thr-283. A, FLAG-tagged MEKK3, MEKK2, or MEKK2 + HA-14-3-3ϵ was co-expressed in 293 cells for 24 h. MEKKs were immunoprecipitated (IP) with anti-FLAG conjugated to agarose and fractionated by SDS-PAGE. MEKK2 or MEKK3 and endogenous co-immunoprecipitated 14-3-3 and HA-14-3-3ϵ were detected by anti-FLAG and pan-14-3-3 immunoblotting. B, FLAG-tagged MEKK2, or various mutants, were expressed in HEK293 cells and immunoprecipitated with anti-FLAG-conjugated agarose. MEKK2 and co-immunoprecipitated endogenous 14-3-3 were detected as in A. Fluorescent secondary antibodies were detected by the Odyssey infrared imager; the density of each band was determined using the ImageJ software, and the numbers beneath the lower panel represent the ratio of 14-3-3 signal to FLAG-MEKK2 signal. T283A, threonine 283 substituted with alanine; K282R, lysine 282 substituted with arginine; K385M, kinase-dead, lysine 385 substituted with methionine; P, phosphorylated. C, HA-MEKK2, T283A MEKK2, K385M MEKK2, or empty vector was expressed in HEK293 cells for 24 h. Proteins were immunoprecipitated with anti-HA-conjugated agarose, fractionated by SDS-PAGE, and immunoblotted with anti-phospho-Thr-283, anti-phospho-Ser-519, anti-HA, and anti-14-3-3. Imaging by the Odyssey infrared scanner was performed as in B. D, MEKK2 was immunoprecipitated from lysates generated from RAW264 cells or mouse embryonic fibroblasts using a monoclonal anti-MEKK2 antibody (BD Biosciences), fractioned by SDS-PAGE, and immunoblotted with anti-MEKK2, anti-phospho-Thr-283, or anti-14-3-3. Results are representative of three independent sets of experiments.
To test whether binding involved Thr-283, we probed MEKK2 with a phosphospecific antibody we had generated for MEKK3. This antibody reacted very weakly with MEKK2; however, the weak signal was abolished upon mutation of Thr-283 to alanine (Fig. 1B). MEKK2 differs from MEKK3 at Lys-282, which is an arginine in MEKK3 (RKTFPRI versus RRTFPRI), so we mutated the MEKK2 Lys-282 residue to arginine and immunoblotted again with anti-Thr-294 antibody. K282R strongly immunoreacted with anti-phospho-Thr-294. Furthermore, a compound mutation of the kinase domain Lys-385 to methionine, which renders the kinase inactive, resulted in a large decrease in anti-Thr-294 immunoreactivity (Fig. 1B).
These results prompted us to generate a phosphospecific antibody to Thr-283 of MEKK2. This reagent showed that wild-type MEKK2, but not T283A MEKK2, was reactive with the anti-Thr-283 antibody, and again this was dependent on kinase activity (Fig. 1C). Furthermore, we noticed that the activation loop of MEKK2 was phosphorylated at Ser-519 in wild-type MEKK2, but not K385M MEKK2, as had been reported previously (27). Interestingly, Ser-519 phosphorylation was higher in T283A MEKK2 (Fig. 1C), suggesting that Thr-283 phosphorylation and 14-3-3 binding might play a role in restricting Ser-519 phosphorylation, which we investigate further below. In all cases where we quantified immunoblot signals, we used direct immunofluorescence and report the phosphorylation signal as the ratio of total protein.
To test whether endogenous MEKK2 bound to endogenous 14-3-3, we immunoprecipitated MEKK2 from both RAW264 cells and mouse embryonic fibroblasts (Fig. 1D). In both cell types, MEKK2 was phosphorylated at Thr-283, with corresponding association to 14-3-3. Control antibody did not precipitate either Thr-283 signal or 14-3-3, and a second antibody raised against MEKK2 produced identical results (not shown).
14-3-3 Binding to Thr-283 Restricts Dimer Formation and Ser-519 trans-Autophosphorylation
To understand how Thr-283 phosphorylation could impact on MEKK2 function, we measured MEKK2 activity in vitro following expression and immunoprecipitation from cells. Mutation of MEKK2 Thr-283 to alanine increased both autophosphorylation (Fig. 2A) and its activity toward a peptide substrate (Fig. 2B). This suggested that Thr-283 phosphorylation might restrict MEKK2 activation, consistent with our earlier observations with MEKK3 (9). Thus, we set out to understand the mechanism of this activity modulation.
FIGURE 2.

Thr-283 reduces MEKK2 activity in vitro. A, WT MEKK2, T283A MEKK2, or K385M MEKK2 was expressed in HEK293 cells for 24 h and immunoprecipitated using anti-HA-agarose. The immunoprecipitates were subjected to in vitro kinase reaction as described under “Experimental Procedures.” 32P-incorporated MEKK2 was visualized by a Personal Molecular Imager (Bio-Rad Laboratories), and total protein was visualized by anti-HA immunoblotting. Error bars represent the S.D. of triplicate measurements. B, similar experiment as A, except that the MKK6 peptide was used as a substrate and measured by liquid scintillation counting. Results are averages of duplicate samples from three independent experiments. Error bars represent the S.D. of triplicate measurements.
To begin, Fig. 1C indicates that Ser-519 phosphorylation and activity are higher in MEKK2 harboring the T283A mutation, suggesting that trans-autophosphorylation of Ser-519 could be regulated by 14-3-3. Work by Cheng et al. (24, 25) has shown that the kinase domain of MEKK2 dimerizes when inactive, so we hypothesized that dimer formation and subsequent autophosphorylation could be altered upon 14-3-3 binding. We set out to test this hypothesis by conducting a series of experiments in which we co-expressed FLAG- and HA-tagged MEKK2 proteins harboring various mutations and monitored dimerization, Thr-283 phosphorylation, and Ser-519 phosphorylation. First, we co-expressed kinase-dead FLAG-K385M MEKK2 with HA-MEKK2, HA-MEKK2 T283A, and HA-MEKK2 K385M, immunoprecipitated with anti-HA antibodies, and probed for the co-immunoprecipitation of FLAG-K385M MEKK2, which was quantified by fluorescent detection using the Odyssey infrared scanner (Fig. 3A). FLAG-K385M MEKK2 co-immunoprecipitated to a greater extent with HA-T283A MEKK2 than with wild-type HA-MEKK2 (almost 3-fold; Fig. 3A). Furthermore, this interaction appeared to be stronger than the co-immunoprecipitation between kinase-dead forms (HA-K385M MEKK2 precipitating FLAG-K385M MEKK2), which was marginally greater than wild-type HA-MEKK2. These results show that the optimal dimer formation is between an active, but non-Thr-283 phosphorylated MEKK2, and the kinase inactive form.
FIGURE 3.
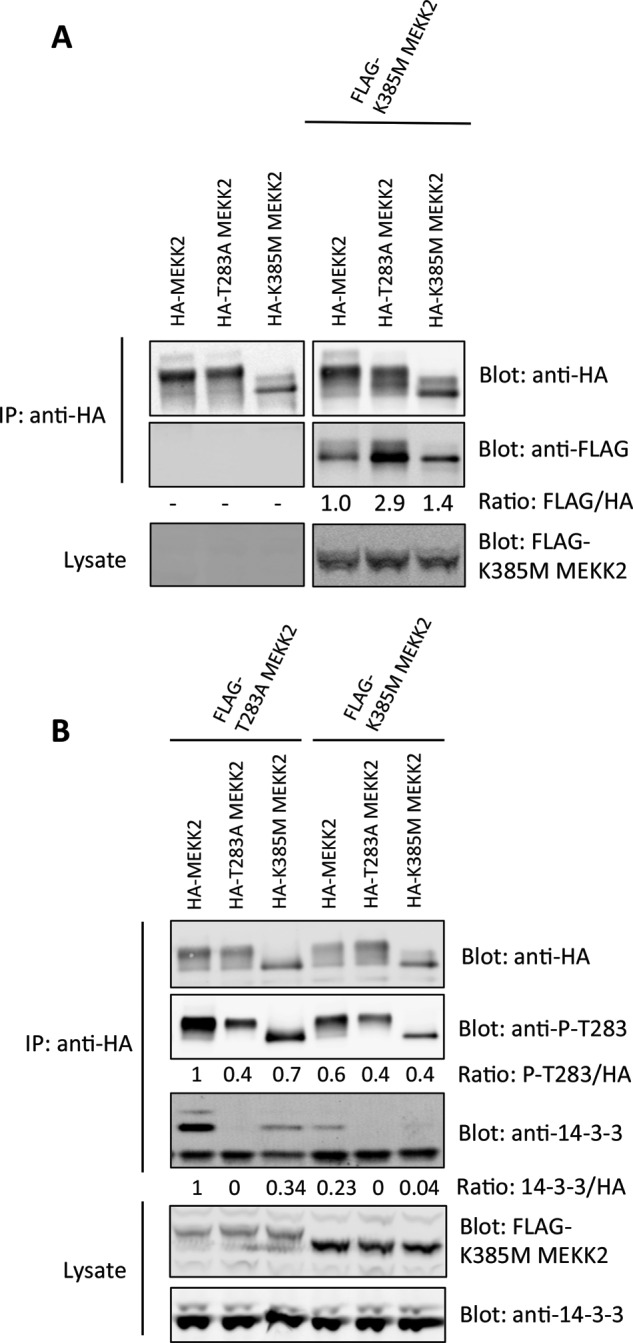
Increased dimerization and trans-autophosphorylation at Thr-283 by MEKK2. A, HA epitope-tagged MEKK2 proteins were expressed either alone or with FLAG-K385M-MEKK2 in HEK293 cells. After 24 h, HA-MEKK2 was immunoprecipitated (IP), and the co-precipitating FLAG-MEKK2 was determined by immunoblotting. B, wild-type HA-MEKK2 or HA-MEKK2 with various mutations was co-expressed with FLAG-T283A-MEKK2 or FLAG-K385M-MEKK2. HA-tagged proteins were immunoprecipitated, and the degree of Thr-283 phosphorylation and bound 14-3-3 was determined by immunoblotting and detection with the Odyssey Infrared scanner. Results are representative of three independent experiments. P, phosphorylated.
We showed in Fig. 1 that Thr-283 phosphorylation was dependent upon kinase activity, suggesting a mechanism of autophosphorylation. We therefore rationalized that if MEKK2 dimerizes in its inactive state, this could allow for trans-autophosphorylation at Thr-283 and increased 14-3-3 association. To test this, we co-expressed FLAG-T283A MEKK2 or the kinase-dead FLAG-K385M MEKK2 together with HA-MEKK2, HA-T283A MEKK2, or HA-K385M MEKK2 and immunoprecipitated the HA-tagged proteins to evaluate their phosphorylation at Thr-283 and association with 14-3-3 (Fig. 3B). Co-expression of FLAG-T283A MEKK2 led to higher Thr-283 phosphorylation of HA-MEKK2. More importantly, the HA-K385M MEKK2 showed detectable Thr-283 phosphorylation and 14-3-3 binding when co-expressed with FLAG-T283A MEKK2, but not when co-expressed with FLAG-K385M MEKK2. This result showed that the HA-K385M MEKK2 underwent trans-autophosphorylation by T283A MEKK2, which then enabled binding to 14-3-3.
This led us to ask how Thr-283 phosphorylation and 14-3-3 binding could influence Ser-519 phosphorylation. For these experiments, we used a kinase-dead MEKK2 as a substrate for phosphorylation in cells by an active MEKK2 with Thr-283 intact or not. In addition, we engineered MEKK2 to bind to 14-3-3 independently of Thr-283. To do this, we substituted the 18 amino acids surrounding Thr-283 with the R18 peptide (28). This peptide was isolated from a phage display screen that selected for peptides with strong 14-3-3 interaction (28). The mode of interaction depends upon two acidic residues (DE) within the peptide and therefore binds independent of phosphorylation. A control peptide was also used that harbors lysine substitutions at the acid residues (termed KK) and does not bind to 14-3-3. As expected, the HA-DE-MEKK2 precipitated more 14-3-3 than wild-type HA-MEKK2, whereas the HA-KK-MEKK2 did not bind to 14-3-3 (Fig. 4A). Importantly, kinase-dead HA-DE-K385M MEKK2 bound to 14-3-3 equally well as active HA-DE-MEKK2, indicating that 14-3-3 association to this protein was now independent of activity.
FIGURE 4.
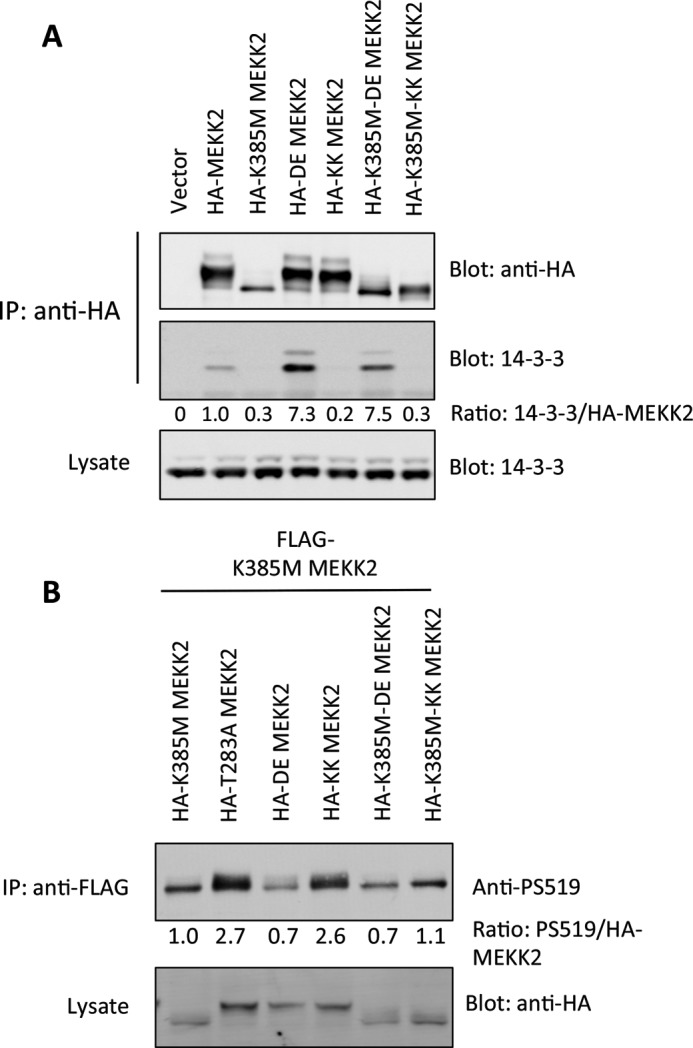
14-3-3 binding reduces Ser-519 phosphorylation. A, HA-MEKK2 harboring the 14-3-3 binding R18 peptide in the position of Thr-283 (DE-MEKK2) or the negative control R18 peptide (KK-MEKK2), either in an active or in a kinase-dead background. Proteins were immunoprecipitated (IP), and endogenous 14-3-3 was detected by immunoblotting and quantified using the Odyssey infrared scanner, with the ratio of 14-3-3 to total MEKK2 shown beneath the panel. B, HA-MEKK2 or MEKK2 with various mutations including the DE and KK R18 peptide inserts were co-expressed with kinase-dead FLAG-K385M-MEKK2. After 24 h, FLAG-K385M-MEKK2 was immunoprecipitated with anti-FLAG-agarose and immunoblotted with anti-Ser-519 antibody using the Odyssey infrared scanner. The numbers beneath the panel represent the ratio of phospho-Ser-519 to total HA-MEKK2. P, phosphorylated.
We then used FLAG-K385M MEKK2 as a substrate in cells with the various HA-MEKK2 forms and probed for Ser-519 phosphorylation. The HA-T283A MEKK2 provided the strongest input for Ser-519 phosphorylation, correlating with the highest level of dimerization (Fig. 4B). In contrast, the HA-DE MEKK2, which bound very well to 14-3-3, provided the weakest input to Ser-519. The HA-KK-MEKK2, on the other hand, did result in Ser-519 phosphorylation, about as well as HA-T283A MEKK2. Together these results provide evidence that Thr-283 phosphorylation and 14-3-3 binding influence dimerization and Ser-519 trans-autophosphorylation. In contrast to the DE and KK mutations, conversion of Thr-283 to single aspartic acid or glutamic acid residues did not promote binding to 14-3-3, and thus, these minimally altered proteins could not be used to characterize MEKK2 function. This was similar to our observations with MEKK3 (9). Furthermore, although R18 DE and KK substitutions appeared to permit MEKK2 autophosphorylation, downstream substrate phosphorylation was impaired by these modifications. Therefore, DE and KK MEKK2 had limited utility for probing MEKK2 pathway activation.
14-3-3 Association Controls MEKK2 Activation of JNK
MEKK2 has been reported to be necessary for JNK activation in various cell models in response to some agonists. We asked whether Thr-283 phosphorylation and the binding to 14-3-3 could control MEKK2 signaling to JNK. To test this, we co-expressed JNK1 together with WT or T283A MEKK2, and measured JNK1 activity in vitro (Fig. 5). In serum-starved cells, the basal level of JNK activity was higher in T283A MEKK2-expressing cells when compared with WT MEKK2 (Fig. 5A). Upon stimulation with FGF2, JNK1 activity increased to similar levels (Fig. 5B). When cells were growing in the presence of 10% serum, anisomycin did not activate JNK1 above basal levels in cells expressing ectopic WT MEKK2 (Fig. 5C). However, T283A MEKK2 co-expression led to robust activation of JNK1 following anisomycin treatment, which returned to basal levels by 30 min (Fig. 5C). Expression of both WT MEKK2 and T283A MEKK2 also led to increased endogenous phospho-c-Jun localized in the nucleus (Fig. 5D). These results indicate that anisomycin can utilize T283A MEKK2 to activate JNK1, but not WT MEKK2. Finally, we measured the transcriptional activity of a GAL4-c-Jun fusion protein on a gal-luciferase reporter. Both WT MEKK2 and T283A MEKK2 could elevate c-Jun activity above expression of GAL4-c-Jun alone; however, T283A MEKK2 led to a great increase in both the basal and the anisomycin-stimulated transcriptional response when compared with WT MEKK2 (Fig. 5E).
FIGURE 5.

MEKK2 activation of JNK. A, FLAG-JNK1α was co-expressed with empty vector, wild-type MEKK2, or T283A-MEKK2 in HEK293 cells for 24 h. FLAG-JNK1α was immunoprecipitated, and an in vitro kinase assay using GST-c-Jun(1–186) as substrate was performed. Labeled proteins were detected by a Personal Molecular Imager (Bio-Rad). Total MEKK2 expression in lysates was detected by anti-MEKK2 immunoblotting. The numbers beneath the image represent the -fold increase when compared with vector alone. B, mouse embryonic fibroblasts were transfected with FLAG-JNK1α with either empty vector or wild-type MEKK2 or T283A MEKK2. After 24 h, cells were starved of serum for 4 h followed by stimulation with FGF2 for the indicated times. JNK was immunoprecipitated, and activity was measured in vitro using GST-c-Jun(1–186) as substrate. The error bars represent the S.D. of triplicate measurements and are representative of two experiments. Asterisks indicate p < 0.05. C, similar as B, except that MEFs cultured in serum-containing medium (no starvation period) were treated with anisomycin for the indicated times. The error bars represent the S.D. of triplicate measurements and are representative of two experiments. Number signs indicate p < 0.05. D, HEK293 cells were transfected with either WT MEKK2 or T283A MEKK2 on glass coverslips. After 24 h, cells were fixed with 3% paraformaldehyde, membrane was solubilized with 0.25% Triton X-100, and cells were stained with mouse anti-FLAG, rabbit anti-phospho-c-Jun, and DAPI. Fluorescence images were visualized at 40× magnification on an Olympus inverted microscope and captured using the Q-Capture software. E, HELA cells were transfected with gal4-Luc reporter and either Gal4-c-Jun or Gal4-c-Jun + WT MEKK2 or T283A MEKK2. After 24 h, cells were treated with anisomycin for 4 h, and luciferase activity was determined relative to control a β-galactosidase activity. Error bars represent the S.D. of triplicate measurements.
IL-6 Expression Is Modulated by Thr-283 Phosphorylation
Our results probing the activation of JNK and phosphorylation of c-Jun by WT MEKK2 and T283A MEKK2 suggested that downstream signaling events might be altered in cells expressing these forms. To test this, we established stable retrovirus-induced pools of MEKK2−/− MEFs expressing either WT or T283A MEKK2 and measured the generation of IL-6 in response to TNFα. The mouse IL-6 promoter contains several cis-regulatory sites that govern both inducible and constitutive expression, including cAMP-response element-binding protein (CREB), NF-κB, C/EBP-1, and AP1. T283A MEKK2 caused a significant potentiation of IL-6 expression following stimulation with TNFα when compared with WT MEKK2, despite similar levels of expression (Fig. 6).
FIGURE 6.
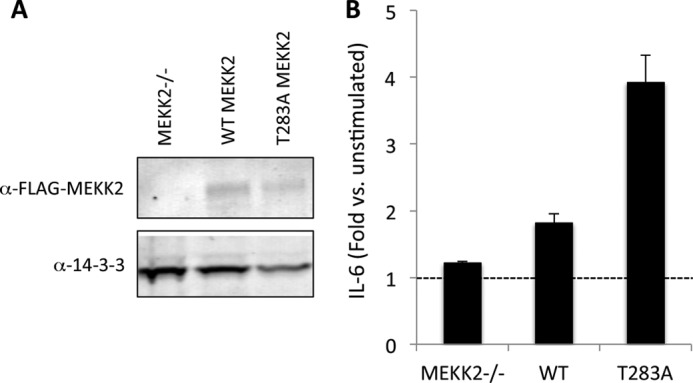
MEKK2 regulation of IL-6 expression. A, MEKK2−/− mouse embryonic fibroblasts were infected with retrovirus containing empty vector, FLAG-WT MEKK2, or FLAG-T283A MEKK2 cDNA. Following 5 days of selection in puromycin, resistant pools were isolated and immunoblotted for expression of MEKK2. B, stable puromycin-resistant pools of MEKK2−/− MEFs expressing various forms of MEKK2 were plated in triplicate in 24-well dishes at 30,000 cells/well and then treated with TNFα (1 ng/ml) for 18 h. Supernatants were harvested, and IL-6 was quantified using the Quantikine immunoassay kit (R&D Systems). Error bars represent S.D.
Proliferation Is Reduced by the T283A MEKK2
We also measured the proliferation rate of MEKK2 knock-out MEFs expressing WT, T283A, or kinase-dead K385M MEKK2. Under low serum conditions, the vector alone and K385M MEKK2-expressing MEFs displayed the highest proliferation, whereas WT MEKK2 proliferated slower and T283A MEKK2 MEFs displayed the slowest proliferation (Fig. 7A). The addition of 2% serum elevated the response of MEFs expressing WT MEKK2, but not T283A MEKK2 (Fig. 7B).
FIGURE 7.

MEKK2 expression regulates ERK phosphorylation and proliferation. A, stable puromycin-resistant pools of MEKK2−/− MEFs expressing empty vector or various forms of MEKK2 were plated in triplicate at 4,000 cells/well in 96-well dishes in medium containing 0.5% serum. On each day of the assay, wells were washed twice with PBS and incubated in 100 μl of XTT solution for 2 h, and then absorbance at 450 nm was measured. OD, optical density. B, similar to A, except serum concentration was raised to 2%. C, stable puromycin-resistant pools of MEKK2−/− MEFs expressing empty vector or WT and T283A MEKK2 were starved of serum for 4 h and then stimulated with FGF2 for the indicated times. ERK phosphorylation within the activation loop site was immunoblotting with a phosphospecific antibody (Cell Signaling Technology). D, MEFs expressing empty vector, kinase-dead, WT, and T283A MEKK2 growing in medium containing 10% serum were isolated, and ERK phosphorylation was immunoblotted. The ratio of total phospho-ERK (pERK) and total ERK was determined by direct fluorescence using the Odyssey infrared scanner.
To understand this effect, we monitored the activation state of ERK within these cells following either growth factor starvation or stimulation with FGF2. The level of serum-starved ERK phosphorylation at its activating site was lower in MEKK2-expressing MEFs when compared with vector alone-expressing MEFs (Fig. 7C). Stimulation with FGF2 led to a robust activation of ERK, indicating that the basal status of ERK was affected by MEKK2 presence but not by the acute stimulation by FGF2 activation (Fig. 7C). Cells growing continuously in serum were also analyzed; WT, kinase-dead, and empty vector-expressing MEFs had ∼3-fold higher ERK phosphorylation when compared with T283A MEKK2-expressing MEFs (Fig. 7D). Together these results indicate that MEKK2 activation by T283A results in reduced ERK phosphorylation accompanying a reduced proliferation rate.
DISCUSSION
We show here that MEKK2 is phosphorylated at Thr-283 through autophosphorylation and that this leads to association with 14-3-3 proteins. This observation is similar to our previous findings with the closely related MAP(3)K, MEKK3, which is phosphorylated at the homologous site Thr-294 (9). For MEKK3, phosphorylation of Thr-294 reduced the activation of NF-κB, a primary target of MEKK3 signaling downstream of TLR4 receptors. Thus, we considered whether a similar mode of negative regulation could occur for MEKK2.
Mutation of Thr-283 led to higher levels of activity in vitro and stronger activation of JNK and c-Jun transcriptional activity in cells. We extend these observations by providing explanations as to the mechanism of Thr-283 phosphorylation-dependent 14-3-3 regulation. The nonphosphorylated T283A MEKK2 was able to dimerize with inactive MEKK2 to a greater extent than WT MEKK2. It was also able to stimulate a higher level of Ser-519 phosphorylation of a kinase-dead substrate. Furthermore, enforced binding of 14-3-3 by substituting the R18 peptide at the Thr-283 site resulted in diminished trans-Ser-519 phosphorylation, an effect that could be reversed by substituting the two critical 14-3-3 binding residues in the R18 peptide to lysine. The resulting KK-MEKK2 was able to phosphorylate kinase-dead MEKK2 at Ser-519 severalfold better than DE-MEKK2. In each of these experiments, we utilized a model in which an active form of MEKK2 phosphorylated a kinase-dead substrate MEKK2 in cells. Each kinase was differentially epitope-tagged and could be purified from the other. This prevented the complication of cis-autophosphorylation; we normally found that active MEKK2 was highly phosphorylated at both Thr-283 and Ser-519 in cells. The K385M MEKK2 was not, and only experienced phosphorylation upon co-expression of active MEKK2.
These results point to a mechanism whereby MEKK2 dimerizes in an inactive state and processes Thr-283 autophosphorylation, which then facilitates binding to 14-3-3. Inputs that dephosphorylate Thr-283 result in increased autophosphorylation of Ser-519 and elevated activity. Our hypothesis is strengthened by previous findings that inactive MEKK2 dimerizes (24) and that Ser-519 phosphorylation increases following stimulation with agonist that activates the MEKK2 pathway (27). 14-3-3 could play a number of potential roles in this regard; it could maintain MEKK2 in a state that restricts autophosphorylation of Ser-519, or it could facilitate interaction with a Ser-519-specific phosphatase. It may also restrict MEKK2 access to a membrane-proximal signal activation complex.
If Thr-283 phosphorylation is a mechanism for MEKK2 deactivation, dephosphorylation by phosphatases could represent a reciprocal mechanism to specifically activate MEKK2. A candidate may be the JNK stimulatory phosphatase (JSP)-1 identified by Shen et al. (29), who reported that expression of JSP-1 activated the JNK pathway.
Loss of Thr-283 phosphorylation had significant impact on pathways downstream of MEKK2. We found that JNK activation was higher in cells expressing T283A MEKK2, which was further amplified by either serum starvation or treatment with the translation inhibitor anisomycin. Furthermore, expression of a TNFα-target gene, IL-6, was significantly elevated in T283A MEKK2-expressing cells. Finally, we found that the proliferation rate of cells expressing T283A MEKK2 was diminished under reduced serum conditions. Although it remains to be precisely understood how activated MEKK2 affects cell cycle progression, these results support previous biochemical and genetic studies suggesting that chronic activation of the JNK pathway leads to deactivation of ERK (30–32), possibly through the JNK-dependent expression of dual specificity phosphatases (31). This prompted us to probe ERK phosphorylation levels in MEFs expressing T283A MEKK2; phosphorylation of ERK at an activating site did not change significantly in a background of vector alone, WT, or kinase-dead MEKK2, whereas T283A caused a reduction of phospho-ERK signal by 70%.
The observations we have made with MEKK2 closely parallel those of MEKK3 (9). With MEKK3, we also noted transient Thr-294 phosphorylation (the analogous site of Thr-283) and association with 14-3-3, and this reduced MEKK3-dependent activation of NF-κB following stimulation with TNFα and LPS. Given that MEKK2 and MEKK3 share significant homology both within their kinase domains and in the region surrounding 14-3-3 binding, we conclude that 14-3-3 is a common modulator for both kinases with the likely role to coordinate specific signals to appropriate subcellular targets.
Acknowledgments
We thank Michael De Marco for technical assistance. We also thank Dr. Bing Su (Yale University) for the kind gift of MEKK2−/− MEFs.
This work was supported by an operating grant from the Canadian Institute of Health Research (CIHR) (to M. P. S.).
- MAP(3)K
- MAP kinase kinase kinase
- MEF
- mouse embryonic fibroblast
- XTT
- sodium 3′-1-(phenylaminocarbonyl)-3,4-tetrazolium-bis(4-methoxy-6-nitro)benzene sulfonic acid.
REFERENCES
- 1. Blank J. L., Gerwins P., Elliott E. M., Sather S., Johnson G. L. (1996) Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3: regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J. Biol. Chem. 271, 5361–5368 [DOI] [PubMed] [Google Scholar]
- 2. Deacon K., Blank J. L. (1999) MEK kinase 3 directly activates MKK6 and MKK7, specific activators of the p38 and c-Jun NH2-terminal kinases. J. Biol. Chem. 274, 16604–16610 [DOI] [PubMed] [Google Scholar]
- 3. Chao T. H., Hayashi M., Tapping R. I., Kato Y., Lee J. D. (1999) MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J. Biol. Chem. 274, 36035–36038 [DOI] [PubMed] [Google Scholar]
- 4. Zhao Q., Lee F. S. (1999) Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-κB through IκB kinase-α and IκB kinase-β. J. Biol. Chem. 274, 8355–8358 [DOI] [PubMed] [Google Scholar]
- 5. Garrington T. P., Ishizuka T., Papst P. J., Chayama K., Webb S., Yujiri T., Sun W., Sather S., Russell D. M., Gibson S. B., Keller G., Gelfand E. W., Johnson G. L. (2000) MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J. 19, 5387–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chayama K., Papst P. J., Garrington T. P., Pratt J. C., Ishizuka T., Webb S., Ganiatsas S., Zon L. I., Sun W., Johnson G. L., Gelfand E. W. (2001) Role of MEKK2-MEK5 in the regulation of TNF-α gene expression and MEKK2-MKK7 in the activation of c-Jun N-terminal kinase in mast cells. Proc. Natl. Acad. Sci. U.S.A. 98, 4599–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang J., Lin Y., Guo Z., Cheng J., Huang J., Deng L., Liao W., Chen Z., Liu Z., Su B. (2001) The essential role of MEKK3 in TNF-induced NF-κB activation. Nat. Immunol. 2, 620–624 [DOI] [PubMed] [Google Scholar]
- 8. Uhlik M. T., Abell A. N., Johnson N. L., Sun W., Cuevas B. D., Lobel-Rice K. E., Horne E. A., Dell'Acqua M. L., Johnson G. L. (2003) Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 9. Matitau A. E., Scheid M. P. (2008) Phosphorylation of MEKK3 at threonine 294 promotes 14-3-3 association to inhibit nuclear factor κB activation. J. Biol. Chem. 283, 13261–13268 [DOI] [PubMed] [Google Scholar]
- 10. Huang Q., Yang J., Lin Y., Walker C., Cheng J., Liu Z. G., Su B. (2004) Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat. Immunol. 5, 98–103 [DOI] [PubMed] [Google Scholar]
- 11. Schmidt C., Peng B., Li Z., Sclabas G. M., Fujioka S., Niu J., Schmidt-Supprian M., Evans D. B., Abbruzzese J. L., Chiao P. J. (2003) Mechanisms of proinflammatory cytokine-induced biphasic NF-κB activation. Mol. Cell 12, 1287–1300 [DOI] [PubMed] [Google Scholar]
- 12. Blonska M., You Y., Geleziunas R., Lin X. (2004) Restoration of NF-κB activation by tumor necrosis factor α receptor complex-targeted MEKK3 in receptor-interacting protein-deficient cells. Mol. Cell. Biol. 24, 10757–10765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garner A. P., Weston C. R., Todd D. E., Balmanno K., Cook S. J. (2002) ΔMEKK3:ER* activation induces a p38 α/β2-dependent cell cycle arrest at the G2 checkpoint. Oncogene 21, 8089–8104 [DOI] [PubMed] [Google Scholar]
- 14. Su B., Cheng J., Yang J., Guo Z. (2001) MEKK2 is required for T-cell receptor signals in JNK activation and interleukin-2 gene expression. J. Biol. Chem. 276, 14784–14790 [DOI] [PubMed] [Google Scholar]
- 15. Qin J., Yao J., Cui G., Xiao H., Kim T. W., Fraczek J., Wightman P., Sato S., Akira S., Puel A., Casanova J. L., Su B., Li X. (2006) TLR8-mediated NF-κB and JNK activation are TAK1-independent and MEKK3-dependent. J. Biol. Chem. 281, 21013–21021 [DOI] [PubMed] [Google Scholar]
- 16. Cheng J., Yang J., Xia Y., Karin M., Su B. (2000) Synergistic interaction of MEK kinase 2, c-Jun N-terminal kinase (JNK) kinase 2, and JNK1 results in efficient and specific JNK1 activation. Mol. Cell. Biol. 20, 2334–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun W., Vincent S., Settleman J., Johnson G. L. (2000) MEK kinase 2 binds and activates protein kinase C-related kinase 2: bifurcation of kinase regulatory pathways at the level of an MAPK kinase kinase. J. Biol. Chem. 275, 24421–24428 [DOI] [PubMed] [Google Scholar]
- 18. Lee C. M., Onésime D., Reddy C. D., Dhanasekaran N., Reddy E. P. (2002) JLP: A scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc. Natl. Acad. Sci. U.S.A. 99, 14189–14194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamark T., Perander M., Outzen H., Kristiansen K., Øvervatn A., Michaelsen E., Bjørkøy G., Johansen T. (2003) Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 278, 34568–34581 [DOI] [PubMed] [Google Scholar]
- 20. Nakamura K., Johnson G. L. (2003) PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 278, 36989–36992 [DOI] [PubMed] [Google Scholar]
- 21. Sun W., Wei X., Kesavan K., Garrington T. P., Fan R., Mei J., Anderson S. M., Gelfand E. W., Johnson G. L. (2003) MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell. Biol. 23, 2298–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakamura K., Uhlik M. T., Johnson N. L., Hahn K. M., Johnson G. L. (2006) PB1 domain-dependent signaling complex is required for extracellular signal-regulated kinase 5 activation. Mol. Cell. Biol. 26, 2065–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fritz A., Brayer K. J., McCormick N., Adams D. G., Wadzinski B. E., Vaillancourt R. R. (2006) Phosphorylation of serine 526 is required for MEKK3 activity, and association with 14-3-3 blocks dephosphorylation. J. Biol. Chem. 281, 6236–6245 [DOI] [PubMed] [Google Scholar]
- 24. Cheng J., Yu L., Zhang D., Huang Q., Spencer D., Su B. (2005) Dimerization through the catalytic domain is essential for MEKK2 activation. J. Biol. Chem. 280, 13477–13482 [DOI] [PubMed] [Google Scholar]
- 25. Cheng J., Zhang D., Kim K., Zhao Y., Zhao Y., Su B. (2005) Mip1, an MEKK2-interacting protein, controls MEKK2 dimerization and activation. Mol. Cell. Biol. 25, 5955–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fanger G. R., Widmann C., Porter A. C., Sather S., Johnson G. L., Vaillancourt R. R. (1998) 14-3-3 proteins interact with specific MEK kinases. J. Biol. Chem. 273, 3476–3483 [DOI] [PubMed] [Google Scholar]
- 27. Zhang D., Facchinetti V., Wang X., Huang Q., Qin J., Su B. (2006) Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 25, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petosa C., Masters S. C., Bankston L. A., Pohl J., Wang B., Fu H., Liddington R. C. (1998) 14-3-3ζ binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 273, 16305–16310 [DOI] [PubMed] [Google Scholar]
- 29. Shen Y., Luche R., Wei B., Gordon M. L., Diltz C. D., Tonks N. K. (2001) Activation of the Jnk signaling pathway by a dual-specificity phosphatase, JSP-1. Proc. Natl. Acad. Sci. U.S.A. 98, 13613–13618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Black E. J., Walker M., Clark W., MacLaren A., Gillespie D. A. (2002) Cell transformation by v-Jun deactivates ERK MAP kinase signalling. Oncogene 21, 6540–6548 [DOI] [PubMed] [Google Scholar]
- 31. Shen Y. H., Godlewski J., Zhu J., Sathyanarayana P., Leaner V., Birrer M. J., Rana A., Tzivion G. (2003) Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J. Biol. Chem. 278, 26715–26721 [DOI] [PubMed] [Google Scholar]
- 32. Friedman A., Perrimon N. (2006) A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 444, 230–234 [DOI] [PubMed] [Google Scholar]