

Abstract
As compared to the adult, the developing fetus and newborn infant are at much greater risk for dysregulation of cerebral blood flow (CBF), with complications such as intraventricular and germinal matrix hemorrhage with resultant neurologic sequelae. To minimize this dysregulation and its consequences presents a major challenge. Although in many respects the fundamental signal transduction mechanisms that regulate relaxation and contraction pathways, and thus cerebrovascular tone and CBF in the immature organism are similar to those of the adult, the individual elements, pathways, and roles differ greatly. Here, we review aspects of these maturational changes of relaxation/contraction mechanisms in terms of both electro-mechanical and pharmaco-mechanical coupling, their biochemical pathways and signaling networks. In contrast to the adult cerebrovasculature, in addition to attenuated structure with differences in multiple cytoskeletal elements, developing cerebrovasculature of fetus and newborn differs in many respects, such as a strikingly increased sensitivity to [Ca2+]i and requirement for extracellular Ca2+ for contraction. In essence, the immature cerebrovasculature demonstrates both “hyper-relaxation” and “hypo-contraction”. A challenge is to unravel the manner in which these mechanisms are integrated, particularly in terms of both Ca2+-dependent and Ca2+-independent pathways to increase Ca2+ sensitivity. Gaining an appreciation of these significant age-related differences in signal mechanisms also will be critical to understanding more completely the vulnerability of the developing cerebral vasculature to hypoxia and other stresses. Of vital importance, a more complete understanding of these mechanisms promises hope for improved strategies for therapeutic intervention and clinical management of intensive care of the premature newborn.
Keywords: Cerebral vasculature, fetus, newborn, calcium signaling
INTRODUCTION
Cerebral blood flow (CBF) is the complex, multifeedback, integrated response to numerous regulatory influences including: transmural pressure gradients, shear stress, and perivascular neuronal activity as well as chemical, endocrine, and metabolic factors originating in the brain and circulating blood [1–5]. The “holy grail” of cerebrovascular biology and essence of homeostasis is the hierarchy of regulatory mechanisms that couple CBF to tissue metabolism. In contrast to the idea that the fetus and newborn are simply small adults, it is critical to remember that not only do stimuli reaching immature cerebral arteries differ from those of the adult, so too the responses of these vessels to those stimuli differ dramatically [6–19].
The maintenance of well-regulated cerebral vascular tone and blood flow is essential to the developing organism as well as adult. For the developing fetus and newborn infant, in part, this is because the brain is uniquely susceptible to a broad variety of injuries and insults, the majority of which typically culminate in diverse patterns of encephalopathy. A major pathogenic factor in these disorders is dysregulation of cerebral blood flow with intracerebral hemorrhage. Hemorrhage into the germinal matrix and periventricular region, which occurs in about 2 to 5 per 1000 live births, is associated with the development of cerebral palsy, convulsive disorders, and other neurological diseases [20]. Among very preterm and very low birth weight (<32 weeks gestation; <1500 g) and extremely preterm and extremely low birth weight (<28 weeks gestation; <1000 g) infants the prevalence of brain damage is particularly high [21–26]. The consequent pathology can result in severe neurological sequelae with lifelong personal, social, and economic consequences. Such infants exhibit spastic motor deficits associated with cerebral palsy [27–29] and/or with cognitive attention deficiencies [22, 23, 25, 30–36].
Whereas recent advances in neuroimaging and cell biology are revealing a growing number of factors that contribute to neonatal brain injury, these are only slowly displacing the long-held, but limited, views that such injury is primarily asphyxic in origin and distributed homogeneously throughout the immature brain [21, 37]. To the contrary, it is becoming increasingly evident that strokes and other cerebrovascular pathologies not associated with asphyxia, many of which occur long before parturition, are important in the genesis of neonatal neurological morbidity [38, 39]. Contributing to neonatal morbidity and mortality, although more than 95 percent of low birth weight infants are born in the developing world, this remains a problem in the U.S., which has the greatest rate of low birth weight (>10 percent), and infant mortality (0.7 percent, from birth to one year of age) in the developed world [40]. These conditions underscore the importance of well-regulated CBF during the period of perinatal development. A growing appreciation of the cerebrovascular involvement in neonatal brain injury has motivated new interest in the details concerning cerebrovascular maturation both structurally and functionally, the role of the neurovascular unit, and the manner in which maturation influences overall regulation of CBF to match metabolism.
Because of their major clinical relevance, cerebrovascular responses to hypoxia, hypercapnia, and other pathophysiological stresses have attracted considerable investigative effort. Nonetheless, many fundamental issues regarding CBF regulation in response to increased neuronal activity and other physiological stimuli remain uncertain. Indeed, regardless of age, the basic mechanisms whereby CBF is matched to neuronal activity and metabolic rate are highly controversial, uncertainties complicate understanding of how this regulation changes with development.
During the past several decades, numerous studies have revealed important aspects in the fundamental signaling transduction mechanisms that regulate cerebrovascular contractility in the course of maturational development [9, 14–16, 18, 19, 41, 42]. Some of these important mechanistic differences include unique features of the relation of function to structure [10], the role of endothelium-mediated prostacyclin and eicosanoid, nitric oxide (NO) and cyclic nucleotide relaxation mechanisms [43, 44], the role of calcium (Ca2+)-dependent receptor-second messenger coupling with plasma membrane potassium K+- and Ca2+ -channels, and the virtual dependence of the immature organism on extra-cellular Ca2+ (as opposed to intracellular Ca2+ stores in adult) for Ca2+-dependent thick (myosin) filament regulation [45–47]. Additionally, many elements of the non-Ca2+-dependent pathway of protein kinase C (PKC) to specific enzymes such as the mitogen-activated protein kinase (MAPK) cascade, extracellular regulated kinases (ERK1/2), downstream effectors [Rho A, Rho kinase, myosin light chain20 (MLC20) and others] differ dramatically in the fetus, compared to adult [9, 16, 41]. Taken together, these studies demonstrate profound differences in cerebral artery relaxation/contraction mechanisms as a function of developmental age, and emphasize the need to understand the biochemical and molecular basis of these changes. Further complicating cerebrovasculature maturation, is the marked heterogeneity observed among vessels from different species, arteries of different size, and among similarly sized arteries from different vascular beds. Nutritional history and health status also have an important bearing, not only on vascular characteristics per se, but also on the rate at which these characteristics change with postnatal age. The emerging picture is one of a highly dynamic vascular phenotype that changes in concert with its environment.
As the cerebrovascular smooth muscle cells (SMC) and vessels are smaller with less connective tissue [10], commonly the age-related differences in cerebrovascular contractile mechanisms have been attributed to vascular structural and functional immaturity [48, 49]. This is the case, particularly as it relates to the relative inability of vessels to constrict with enough force to prevent propagation of transient arterial blood pressure increases to the microcirculation, i.e., the “hypo-contractile” hypothesis. Alternatively, increased likelihood of cerebral artery rupture may result from a greater abundance and/or potency of relaxant mechanisms in less mature vessels, the “hyper-relaxation” hypothesis [15, 17, 47, 50]. On one hand, reduced tone and contractility of cerebral arteries may be appropriate for the relatively low perfusion pressures typical of fetal life in utero. On the other hand, we know little of how this may be a consequence of important differences from the adult in relation to the fine structure and vascular reactivity, as well as differences in patterns of stimuli received. Unfortunately, little is known about the basis of this vulnerability in terms of cerebrovascular structure and function. This review attempts, in part, to address these issues.
In the present review, we place in perspective the cellular/subcellular mechanisms of electro- and pharmaco-mechanical coupling in the relaxation and contraction of cerebral artery SMCs, with emphasis on those elements we believe of most importance in terms of development. In an attempt to clarify key features of signal transduction in the immature cerebral arteries, we explore and compare several aspects of fundamental mechanisms in fetal, newborn, and adult cerebral arteries. For heuristic purposes we believe it essential to identify functional compartments within the vascular smooth muscle cells, and categorize the essential features that distinguish these during the course of development. We review these differences in relation to vascular development, composition and structure, endothelial-mediated relaxation mechanisms, cell surface contractile associated ion channels, cell surface receptors, second messenger systems, and effector proteins. For each of these topics, we consider not only its relation to the regulation of vascular tone, but its importance in the coupling of blood flow to metabolism. Thus, a major purpose of this review is to synthesize recent evidence illuminating the developmental aspects, cellular and subcellular mechanisms whereby cerebral arteries of the immature organism rely on extracellular Ca2+ and much greater Ca2+ sensitivity (CaS), in contrast to vessels in the mature adult that rely on the intracellular Ca2+, [Ca2+]i and lower CaS. Rather than stress complexity, our goal is to present unifying principles. Finally, we would hope that this synthesis with increased mechanistic understanding will provide a basis for a rational approach to the prevention and treatment of cerebrovascular dysregulation in the prematurely born infant.
CEREBRAL BLOOD FLOW IN THE FETUS AND NEWBORN INFANT
Basal Levels of Blood Flow and Metabolism
The physiologic transitions which occur at the time of birth constitute the single most dramatic events in the life of an individual. In these few moments of parturition, the central circulatory pattern must change from one based on placental transfer of respiratory gases to one of pulmonary ventilation. Systemic vascular resistance increases dramatically, as does arterial blood pressure, while pulmonary vascular resistance and pressure fall. Cardiac (i.e., left ventricular) output initially increases, and then decreases slowly over succeeding days. Despite these dramatic changes in cardiac function and vascular resistance, blood flow to the brain increases only slightly to maintain optimal cerebral oxygenation and metabolism [51].
To set the stage for the regulation of cerebrovascular tone, first we may consider CBF. In the adult, arterial PO2, PCO2, Hydrogen ion concentration (H+), and cerebral perfusion pressure play key roles in the regulation of CBF [52]. Because these major physiological factors (except pH) vary considerably as a function of developmental age, it might be anticipated that their roles would be of particular relevance to the fetus and newborn infant. In the human and several other species, global CBF in the near-term fetus and newborn does not differ significantly from that of the adult on a per Kg brain weight basis, e.g., ~100 to 150 ml·min−1·100 g tissue−1 [53–67]. Additionally, the near-term fetus appears to have a cerebral metabolic rate for O2 (CMRO2) similar to that of the adult [68, 69] (Table 1). Nonetheless, under many circumstances the relation of CBF to metabolic rate remains poorly documented. Also, compared to adult, the fetus in utero has a relatively low mean arterial blood pressure (46±2 mmHg) [70, 71]. Thus, in the presence of a lower mean arterial pressure, but a similar CBF value, fetal overall cerebrovascular resistance equals about one-half that of the adult (e.g., 0.40 versus 0.80 mmHg·min−1·ml−1, respectively) (Table 1).
Table 1.
Ovine Physiologic Values with Development
| Physiologic Variable | Fetus | Newborn | Adult | Reference |
|---|---|---|---|---|
| PaO2 (Torr) | 25±1a | 70±10b | 100±5 | [70, 72–74] |
| [Hgb] (g·dl−1) | 10.1±0.7b (116) | 14.0±0.5a (161) | 8.7±0.3 | [70, 74, 75] |
| Arterial [HbO2] (%) | 70 to 80 | 80 to 100 | 100 | Many sources, see text |
| O2 Content (ml·dl−1) | 7.7±0.5a (59) | 15.7±1.2 (121) | 13±1.0 | [70, 74] |
| PaCO2 (Torr) | 44±2 (120) | 40±4 (114) | 35±2 | [70, 74] |
| pH | 7.36±0.01 (‡) | 7.38±0.01 (‡) | 7.40±0.01 (‡) | [70, 74] |
| Mean Arterial Blood Pressure (mmHg) | 46±2b (57) | 75±3 (94) | 80±4 | [70, 73] |
| Cerebral Blood Flow (ml·100g−1·min−1) | 100±20 | 100±20 | 100±20 | Many sources, see text |
| Cerebrovascular Resistance (mmHg·min−1·ml−1) | 0.40±0.04a (50) | 0.75±0.10 (94) | 0.80±.10 | Many sources, see text |
Values are mean ±SE of 5 or more in each group; Values in parentheses represent % of adult value;
Because these are logarithmic functions, percent change is not an accurate or appropriate representation;
Significantly different from adult value:
P<0.001;
P<0.01
In contrast to the air breathing newborn and adult, the fetus develops in an environment of relatively low arterial oxygen (O2) tension (~25±2 Torr, 70 to 80% oxyhemoglobin saturation [HbO2], Table 1), and its hormonal milieu differs from adult. Additionally, following birth maturation of contractile responses to many receptor agonists change dramatically. Following birth the PO2 rises to 70±10 Torr, and then continues to rise to an adult value of ~100±5 Torr [72]. In terms of cerebral O2 delivery, because of relatively higher values of hemoglobin concentration (~21g/dl) and a lower value of P50 (O2 tension at which hemoglobin is 50% saturated, ~20 Torr), the fetus has a similar value of arterial O2 content, despite its significantly lower arterial O2 tension [76] (Table 1). The fetus also has not only a relatively higher CO2 tension (44±2 versus 35±2 Torr) [70, 74], but a 30% greater mass of total CO2 in the blood, compared to the adult [77]. In the near-term ovine fetus (140 gestational days, GD), cerebral vasodilatory reactivity to CO2 is similar in the fetal and adult cerebral circulations [78], although in the premature infant CO2 reactivity is decreased [79–81]. (Table 1 summarizes several of these and other physiologic variables). In addition to differences in blood gases and pH, the concentrations of many other circulating vasoactive substances (including adenosine, cyclic nucleotides, prostanoids and eicosanoids, and others) differ significantly in fetus and adult. These factors undoubtedly influence resting cerebrovascular tone, and also may attenuate reactivity to other contractile influences (For review see Vannucci & Vannucci, 1998 [69]). It is at birth, a time of major adjustments in vascular reactivity, particularly in the immature organism, that the cerebral circulation is most vulnerable to hypoxiaischemia, loss of homeostatic mechanisms, and hemorrhage [25, 57, 82–84].
Autoregulation and Myogenic Tone
In brain and most other tissues, blood flow is adjusted to the metabolic activity of that tissue. Importantly, changes in perfusion pressure (e.g., arterial blood pressure independent of neurogenic or metabolic influences) are associated with vascular resistance changes that tend to maintain constant flow. This autoregulation occurs as a result of pressure-sensitive myogenic mechanical and electrocoupling mechanisms in the cerebral arteries [85–88]. Myogenic tone is the process by which vascular smooth muscle alters force production in response to transmural pressure in order to maintain appropriate diameter, and thus contribute to autoregulation [85, 88–92]. Although myogenic mechanisms per se have not been well studied in the immature cerebral vasculature, several groups have examined autoregulation.
In the adult, cerebral vascular resistance is effectively autoregulated over a wide range of perfusion pressures (e.g., 60 to 180 mmHg) [93], and in the middle cerebral artery (MCA) this has been shown to vary with vessel caliber (from 60 to 100 mmHg in the main vessel to 20 to 60 mmHg in the penetrating arterioles) [94]. In contrast, in near-term fetal sheep (e.g., 135 to 140 GD) autoregulation occurs over a more abbreviated range ~30 to 90 mmHg [54, 95–98]. In those instances in which it was examined, autoregulatory efficiency did not vary significantly by brain region. In the preterm fetus (e.g., 0.76 gestation; 110 GD; [96, 99]; and at 0.83 gestation; 120 GD), this autoregulatory range is somewhat lower (e.g., 20 to 50 mmHg). A limited study in human newborns showed a similar relationship of variation of mean CBF velocity as a function of gestational age [100]. Autoregulation in the fetus also may be modulated by the α-adrenergic system [101], which, however, is relatively poorly developed at 0.63 gestation (e.g., 92 GD) [95]. When comparing the “autoregulatory index” (an assessment of “pressure reactivity” calculated as the change in blood flow (ml·min−1·100g−1) divided by the change in perfusion pressure), autoregulatory sensitivity is much less for fetal than adult arteries. It also is more fragile, as in fetal sheep acute hypoxia (PO2=16±1 Torr; [HbO2]=46±3%) [98], or asphyxia (elevated arterial PCO2 with pH ~7.0) [102] can compromise fetal cerebral autoregulation. By use of 133Xe clearance in human newborns that had experienced asphyxia or had respiratory distress syndrome, autoregulation was impaired to a significant degree [103]. Intraventricular hemorrhage in the newborn resulted in a similar failure of autoregulation [104]. As mean arterial blood pressure increases with advancing gestational age, so do the autoregulatory limits. Unknown are the mechanisms by which perfusion pressure is coupled to vascular resistance, that coordinate changes to maintain appropriate matching between mean arterial blood pressure in the autoregulatory range to metabolism.
Studies from the Pearce laboratory demonstrate that although the cerebrovascular myogenic response is highly conserved with development, stretch-induced modulation of thin filament reactivity may be of greater importance in the fetus [105]. Although changes in cerebral autoregulatory capacity have been demonstrated with development in sheep, little is known about the variability of the associated myogenic responses with vessel size. In pressurized endothelium-intact neonatal (4 to 8 days of age) mouse cerebral arteries (<150 m in diameter), active intrinsic tone was evident as low as 10 mmHg intraluminal pressure, whereas in adult vessels pressures needed to be 60 mmHg or greater [106]. Also in pressurized resistance sized (~150 μm) cerebral arteries from the 95- and 140-GD fetal sheep, forced dilatation (i.e., an acute increase in vascular diameter in response to intramural pressure) occurred at significantly lower pressures than in adult vessels [107]. This also was true for cerebral arteries in mice [106]. This forced dilatation response was not affected by removal of endothelium [107], and paralleled changes in mean systemic pressure noted above. Under no-flow conditions, isolated human cerebral arteries (200–500 μm) from preterm (24–37 weeks gestation) fetuses exhibited a degree of vascular tone similar to that observed in ovine preterm (95 GD) vessels [108]. As near-term human vessels were not examined in these studies, the relation between basal tone at term in human and sheep arteries remains unknown. Regardless, the combined data infer important developmental differences in myogenic properties. Clearly, some of these age-related changes may be due to differences in artery diameter. This would be anticipated as according to Poiseuille's law relatively small decreases in vascular diameter result in a significant increase in vascular resistance, and the Law of LaPlace predicts that the smaller diameters and arterial pressures typical of fetal arteries must yield far lower values of wall tension than observed in the adult. Because wall tension has been suggested to be the main determinant of myogenic tone [109], more detailed investigations into the age-related differences in both myogenicity and autoregulatory behavior in the fetus and neonate would be of clinical importance.
DEVELOPMENT OF THE CEREBRAL VASCULATURE AND ITS STRUCTURE
Embryogenesis
As a highly specialized organ involved in a number of critical physiologic functions, including the transport of oxygen, glucose, and other nutrients to the brain, the cerebral vasculature architecture is unique, complex, and heterogeneous in its structure, organization, and function. Its maturational changes parallel complementary development of the brain itself. Embryonic cerebrovascular development involves a complex series of events during which endothelial cells differentiate, proliferate, migrate, and undergo maturation into an organized vascular network. During early embryogenesis, the essential and rate-limiting developmental steps of cerebrovascular formation may be divided into several stages [110–113]. Although believed for many years to be dominated by terminal “end-arteries”, evidence demonstrates a more anastomotic vascular network [114, 115]. The complex cerebrovasculature architectural and functional nature is reflected in intricate and involved embryogenesis that involves elaborate transcriptional, post-transcriptional, and post-translational mechanisms that modulate the signaling pathways. As in other tissues and organs, this occurs by a combination of several mechanisms: vasculogenesis, the de novo formation of new vessels; angiogenesis, the growth of new vessels by sprouting from those pre-existing; and, according to some, arteriogenesis, the increase in arterial diameter in response to increased blood flow or shear stress [116, 117]. Each of these processes is extremely complex, mediated by the precise coordination of multiple cell types and a host of signaling molecules that interact with specific receptors. In turn, these involve the genesis and differentiation of smooth muscle cells (SMCs) with their unique repertoire and expression of genes that determine their phenotype and function. The Owens group have made remarkable contributions to this field, elucidating the regulation of and roles of a number of proteins, including: α-actin, smooth muscle myosin heavy chain (MHC), calponin, desmin, Krüppel-like factor 4, palladin, and others [118–123]. These include several pathways and transcription factors that regulate genes essential to this process [120, 124–128].
Receptor Tyrosine Kinases and Development of Cerebrovasculature
During the past several decades, remarkable progress has been made in elucidating the molecular regulators and their orchestration needed for development of the cerebrovasculature. Important among these factors are receptor tyrosine kinases (RTK)s, high-affinity cell surface receptors for a number of polypeptide growth factors and hormones [129]. These protein families include: vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), platelet derived growth factor (PDGF), placental growth factor, insulin-like growth factor (IGF), insulin receptor (IR), brain-derived neurotrophic factor (BDNF), the Trk and Eph receptor, and others. For the most part, these are single subunit receptors, although some exist as multimeric complexes. Functionally, tyrosine kinases transfer phosphate groups from high-energy donor molecules such as ATP to specific substrate target molecules. Phosphorylation of specific tyrosine residues within the activated receptor creates binding sites for Src homology 2 (SH2) domain- and phosphotyrosine binding (PTB) domain- containing proteins [130]. Proteins containing these domains include Src and phospholipase Cγ, with phosphorylation leading to initiation of signaling. In addition, the hedgehog family of morphogens, neuropilins, notch signaling, and many other elements and pathways are essential. Despite the vital role of the RTKs in cellular signaling, relatively little is known of their role in the cerebrovasculature, much less of potential developmental changes [131].
Vascular Endothelial Growth Factor
A major inducer of vascular endothelial and smooth muscle development, including both vasculogenesis and angiogenesis, is VEGF which binds either of two RETS at the cell membrane, VEGF-R1 (flt-1) and VEGF-R2 (KDR/flk-1) [132]. The acronym VEGF includes a number of proteins, from two families, that result from alternative mRNA splicing from a single, 8 exon vegf gene. The major isoform present in most mammalial tissues is VEGF165, a 45 KD heparin binding glycoprotein [133]. VEGF-A, the most important member of this family, binds to both flt-1 and flk-1, the latter of which appears to mediate most VEGF-initiated cellular responses [134]. Effectively, VEGF proteins stimulate cellular responses by binding to RTK receptors causing them to dimerize and become active through transphosphorylation. Discovered in the mid-1970s, VEGF initially was given the name vascular permeability factor [135]; however, subsequent studies demonstrated its ability to promote endothelial cell transformation and angiogenesis [136, 137]. Soon thereafter, the common identity of vascular permeability factor and VEGF was established [138]. Following its binding to flk-1 receptors in endothelial cells, VEGF triggers mitotic and migratory processes [139–142] including SMC proliferation [143]. Following brain injury [144], the VEGF receptors flt-1 and flk-1 have been shown to be important in astroglial and vascular remodeling of the neurovascular unit.
In terms of cerebrovascular development, in endothelium-denuded carotid arteries VEGF depressed smooth muscle α-actin 15% in adult, but not fetal vessels [145]. In contrast, in fetal arteries VEGF increased myosin light chain20 (MLC20) expression up to 28%, while this decreased 17% in adult. Further, VEGF increased myosin light chain kinase (MLCK) expression up to 140% in fetal, but not adult, arteries (see Table 2). For both flt-1 and flk-1 in fetal and adult vessels, mRNA measurements verified VEGF receptor transcript expression [145]. These studies support the hypothesis that, compared to adult, the role of VEGF differs significantly during maturational development. As an aside, these studies also support the thesis of differential effects of growth factors in the differing lamina across the vascular wall. Following MCA occlusion in 10-day old rat pups (which demonstrate maturity similar to that of a human infant born at term), VEGF-R2 (flk-1) inhibition aggravated stroke with increased cleavage of spectrin, and increased areas of the apoptotic factor caspase-3 staining, with a shift from apoptosis to necrosis [146].
Table 2.
Transmission Electron Microscopy Measurements and Stereological Analysis of Media, Medial Smooth Muscle Cells, and Other Components of Ovine Middle Cerebral Artery with Development
| Parameter | Fetus | Adult | |
|---|---|---|---|
| 95 GD | 140 GD | ||
| Lumen | |||
| Diameter (μm) | 623 ± 80a, e (46) | 941 ± 73b (70) | 1351 ± 68 |
| Vessel Wall | |||
| Media thickness (μm) | 13.7 ± 2.3a, f (47) | 21.3 ± 0.9c (73) | 29.2 ± 2.3 |
| Media cross-section area (μm2 · 103) | 26.0 ± 3.2a, d (20) | 67.1 ± 6.8b (52) | 128.7 ± 12.0 |
| Number of SMC cell layers in media | 3.3 ± 0.5b, d (45) | 5.6 ± 0.2 (76) | 7.4 ± 1.0 |
| SMC volume fraction in media | 0.72 ± 0.01 (111) | 0.69 ± 0.01 (106) | 0.65 ± 0.04 |
| SMC numerical density in media (μ−3 · 10−3) | 0.65 ± 0.20 (181) | 0.36 ± 0.02c | 0.36 ± 0.06 |
| SMC number per unit arterial length (N1), (μ−1) | 15.3 ± 2.9b, f (33) | 23.6 ± 1.5c (51) | 46.3 ± 7.6 |
| Smooth Muscle Cell | |||
| Length (lc), (μ) | 102 ± 16c, e (67) | 171 ± 14 (113) | 151 ± 15 |
| Cross-section area (ac), (μ2) | 13.8 ± 2.3 | 11.4 ± 0.4 (83) | 13.8 ± 2.3 |
| Volume (vc), (μ3) | 1407 ± 271 (69) | 1937 ± 105 (95) | 2042 ± 427 |
| Nucleus length (ln), (μ) | 15.2 ± 2.2c (63) | 20.3 ± 2.6 (84) | 24.2 ± 2.2 |
| Nucleus as % SMC (mid-length) cross section area | 42.6 ± 2.0a, f (203) | 36.6 ± 1.2a (174) | 21.0 ± 0.6 |
| Nuclear heterochromatin as % nuclear area | 31.3 ± 2.0 a, f (63) | 38.5 ± 2.2b (77) | 49.9 ± 0.3 |
| Dense body profiles as % SMC cross section area | 3.4 ± 0.7a, d (33) | 6.8 ± 0.6b (67) | 10.2 ± 0.4 |
| Basal lamina thickness (nm) | 75.3 ± 4.0b (51) | 69.2 ± 3.8b (47) | 148.0 ± 14.8 |
| Extracellular Matrix | |||
| Electron dense structures as % ECM cross section area | 44.7 ± 3.9a, e (48) | 61.8 ± 2.5b (81) | 75.4 ± 2.5 |
| Other Variables | |||
| Base soluble protein (% Dry Wt)º | 24.5 ± 2.2 (102) | 23.9 ± 2.7 | |
| Base soluble protein (μg protein · mg wet wt−1) | 23 ± 3+ (70) | 33±4 | |
| DNA Content (relative to tissue wet weight)º | 5.0 ± 0.3 (147) | 3.4 ± 0.3 | |
| α-Actin (% of adult value) | 105 ± 10 (105) | 100 | |
| α-Tubulin (% of adult value) | 240 ± 40b (240) | 100 | |
| Water (% Wet Wt)º | 80.0 ± 0.8 (105) | 76.5 ± 0.7 | |
Values are mean ± SE of 5 or more in each group;
Newborn; Values in parentheses represent % of adult value;
Significantly different from adult value:
P<0.001;
P<0.01;
P<0.05;
Significantly different from GD 140:
P<0.001;
P<0.01;
P<0.05 (From Goyal et al, 2012 [10]);
Elliott & Pearce, 1995 [147]
Other Receptor Tyrosine Kinase Growth Factors
In terms of cerebrovascular development, the potent mitogen PDGF also plays a key role in vasculogenesis and angiogenesis. Its absence in the mouse results in various anomalies from embryonic lethality to abnormal cerebral vascularization with loss of neuroependymal integrity depending on breed [148]. Cerebral hypoxia-ischemia has been shown to induce IGF-1 [149] and other growth factors in the developing cerebral vasculature. The major cytokine brain-derived neurotrophic factor is a member of the family of several neurotrophic factors. As its name implies, BDNF participates in development of the nervous system [150], but also is found in SMCs of large conduit arteries [151]. Importantly, it plays a role in regulating the interaction among neurons, glial cells, and vasculature in the neurovascular unit. Here, it is a vital element in oxidative stress-induced neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons [152]. Among its many functions, BDNF may activate arachidonic acid (AA) metabolism, leading to production of prostaglandin I2 (PGI2, prostacyclin), a primary mediator of endothelium-dependent relaxation in the cerebral circulation of newborn infants [153]. During the course of aging, PGI2's contribution to cerebral endothelial regulation of vasomotor function decreases, so that in the adult, nitric oxide (NO) becomes the dominant endothelium-derived vasodilator [154, 155]. For an extended survey of these growth factors, the reader can consult reviews of this field given above.
Vascular Smooth Muscle Structure
Increasingly, it is becoming evident that cellular biochemical signal transduction mechanisms are organized in complex and highly interactive systems which involve multiple molecular components. These include those elements formerly considered to be primarily structural, and many other molecules involved in diverse cell activities [156–159]. Thus, for developing cerebral vasculature the relation of structure to function is both unique and of particular importance. From a morphologic standpoint, in middle cerebral arteries the transition from preterm fetus (95±2 GD) to term fetus (140±2 GD) to adult involves significant increases in resting lumen diameter and media thickness, media cross-section area (CSA), and number of cell layers in media. An important observation is that of the development of fascicles or bundles of SMCs, which presumably play a critical role in coordinated maintenance of myogenic tone and contraction (See Fig. 1 and Table 2) [10]. In addition, SMCs are embedded in a stroma comprised of a more sparse and loosely arranged collagen matrix, as compared to the closely packed collagen fibrils that surround SMCs in the adult. Equally important, the fraction of medial extracellular matrix (ECM) occupied by electron dense structures (collagen fibrils, elastin, basal lamina-like material) increases significantly as % SMC (midlength) CSA with maturation. In addition to the SMCs being significantly smaller, they also exhibited a much greater ratio of nucleus to cytoplasm than those of the adult (Table 2). This fits with a greater DNA content relative to tissue weight in vasculature of immature sheep [147], and rat and rabbit [160]. In many other respects, the ultrastructure of the near-term fetal SMCs is qualitatively similar to that of the adult (Table 2). These changes were associated with a major increase in basal (external) lamina thickness, and expression of smoothlin (a marker of contractile phenotype) and decrease in proliferating cell nuclear antigen (a marker of synthetic SMC phenotype). Although caveolae appear well developed in fetal cerebral SMCs, caveolin-3 levels in these arteries were significantly less than in the mature vessel. In addition, significant increases were observed in expression of other structural proteins: collagen III, focal adhesion kinase, integrin α-2, integrin α-v, β1, and β3, and α-tubulin. In contrast, significant decreases were observed in expression of fibronectin [10], and tissue compliance (inverse of stiffness) decreased significantly [17].
Fig. (1).
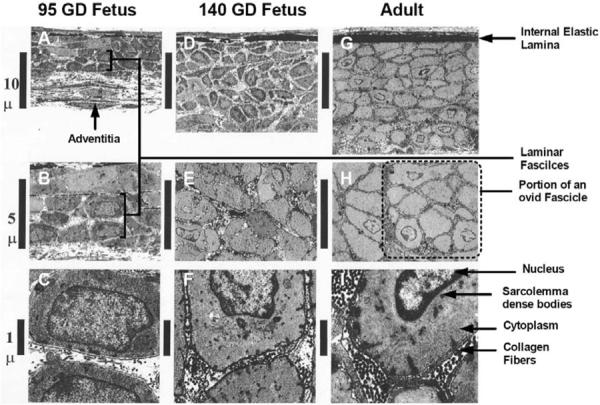
Transmission electron microscopy images of arterial walls of main branch middle cerebral arteries of preterm 95-gestational day fetus, near-term 140-gestational day fetus, and adult sheep. Original magnification of axial longitudinal section: ×1,600 (A, D, and G), ×8,000 (B, E, and H), and ×10,000 (C, F, and I) [10].
Overall, these changes reflect a progression from synthetic and poorly connected SMCs at 95 GD to more functionally linked cells at term (140 GD), and robustly linked cells in the adult. Undoubtedly, these structural differences are of vital importance to the development and integrity of the mature cerebral artery and its role in the regulation of tone, blood pressure, and flow. Nonetheless, elucidation of the mechanisms whereby structure influences smooth muscle activation presents a significant challenge, particularly in light of the large number of structural and contractile proteins typically found in vascular SMCs [161, 162].
A related consideration of relevance to the cerebral vasculature, is development of the blood–brain barrier (BBB) composed of a continuous layer of endothelial cells connected by intercellular tight junctions [163, 164]. This specialized endothelial layer serves as a relatively impermeable interface between the circulating blood and the brain cells, isolating brain tissue from blood constituents. Thus, it helps to maintain homeostasis within the central nervous system by preventing the entry of substances that might be invidious to CNS neuronal and other cell function. By measuring the BBB permeability to α-aminoisobutyric acid, Stonestreet and colleagues have demonstrated developmentally-associated permeability decreases in the ovine fetus from 82 days (0.6) gestation [165, 166] to 112 days (0.8) [167] to 128 days (0.9) gestation [166], with the BBB becoming relatively impermeable in the newborn at term and the adult [165].
Additionally, a number of important questions remain. For instance, how do growth factors, transmural pressure, trophic input from perivascular nerves, and other unknown factors combine to orchestrate arterial development? To what extent do changes in extracellular matrix composition and SMC organization into fascicles contribute to vascular function? An issue of particular relevance is the relation of the caveolae to elements of the signal transduction cascade, how these change with developmental maturation, and the extent to which these influence artery reactivity and contractility. By what cellular and subcellular mechanisms is this maturational development regulated? Exploration of these important topics offers many exciting opportunities for improving understanding of fetal vascular biology.
NEUROEFFECTOR MECHANISMS
Recent studies in the adult give evidence of the role of the neurovascular unit, with its pericytes, astrocytes, microglia, and other nonneuronal cells in proximity to neurons and arterioles [168], play a key role in this regulation [169, 170]. Because of their vulnerability to hypoxia-ischemia and systemic infection-inflammation, these structures with the developing oligodendrocyte are of particular relevance as key cellular targets in brain injury for the premature newborn [26]. Few studies have examined the developmental aspects of this regulation. Neurovascular units in the cerebrovasculature are multicellular complexes that consist of neurons, nonneuronal astrocytes and microglia, as well as pericytes and vascular endothelial cells [171]. These differing cell types are immersed by the extracellular space containing a milieu of extracellular matrix proteins [172–175]. In contrast to the vascular system as a static network of plumbing, recent studies of this complex suggest that the interaction of nerves and the microvasculature plays a vital role in its growth, development, and regulation of flow. Thus, in addition to the vasculature providing O2, glucose, and other nutrients to meet CNS metabolic demands, the neurons and astrocytes secrete neurotransmitters, growth factors, signaling proteins, and other compounds that regulate the formation and function of the microvasculature [169, 176–179], as well as regulate the blood brain barrier [172, 180]. In terms of signaling proteins, a host have been identified including: members of the integrin family of ECM adhesion receptors, vascular endothelial growth factor [181], Wnts and related ECM proteins, semiphorins, and ephrins [182], the Rho family of proteins [183], and various transcription factors [174]. Koehler and colleagues have presented a provocative schema for the role of astrocytes in the synthesis and release of epoxyeicosatrienoic acids (EETs), prostaglandins, arachidonic acid, K+, and other molecules as candidate mediators of communication between astrocyte end-feet, pericytes, and vascular SMCs [184, 185].
In the developing brain, neurogenesis is prominent in the subventricular zone of the lateral ventricle and the subgranular layer of the hippocampal dentate gyrus [186, 187]. Within these regions, neural stem cells associate intimately with special neuro-vascular structures termed “vascular niches” [188]. These neural stem cells associate preferentially with capillary regions that display increased blood brain barrier permeability, suggesting a role for circulating proteins in modulating neural stem cell function [189]. In turn, ECM adhesion molecules, basic fibroblast growth factor, and other proteins interact with neural stem cells to modulate their role in the neurovascular unit [190].
Considering the vital aspects of regulation, and the potential consequences of dysregulation, of vascular tone and CBF in the developing fetus and newborn infant, one might imagine the neurovascular unit to be a topic of great interest to those physiologists and others who concentrate on development. Surprisingly, that is not quite the case. In the germinal matrix of human fetuses (16 to 22 weeks gestation, 23 to 27 weeks, and near-term 36 to 40 weeks) pericyte density and coverage of microvasculature were significantly less than in cerebral cortex and white matter [191]. Associated with the paucity of pericytes were significant decreases in expression of transforming growth factor-β1 (TGF- β1), whereas expression levels of sphingosine-1-phosphate 1 and N-cadherin (which assist vascular maturation) were higher in germinal matrix than cortex or white matter. The authors propose that low TGF- β1 expression may be a basis of reduced pericyte density with, in turn, the relative paucity of pericytes contributing to the propensity of hemorrhage in the immature germinal matrix [191]. A related review has described fragility of the germinal matrix vasculature with paucity of pericytes, immaturity of the basal lamina, and deficiency of glial fibrillary acidic protein in end-feet of ensheathing astrocytes [192]. These findings may have considerable relevance to the germinal matrix and intraventricular hemorrhage with development of periventricular leukomalacia in the premature infant.
Glutamate and NMDA-Mediated Responses in Neurovascular Unit
A prevalent neurotransmitter in the brain, glutamate, can activate a number of ionotropic receptors on neurons and astroglia [193]. One of these receptors, named for its activating synthetic analogue, is that of N-methyl-D-aspartate (NMDA), which plays a critical role in regulating synaptic plasticity, learning, and memory. NMDA receptor activation leads to Ca2+ entry, membrane depolarization, cytosolic signaling pathway activation, and subsequent production and release of vasoactive agents. These can diffuse to vascular SMCs decreasing tone and producing vasodilatation [193, 194]. Linkage between neuronal-glial activated release and actions of glutamate, and the associated decrease in cerebral vascular resistance, provide for well-regulated coupling between cerebral metabolism and blood flow. Nonetheless, some controversy has concerned the chemical nature of the vasodilators, as well as the relative roles of neurons, microglia, astrocytes, and endothelium in mediating these responses [194–200].
On the basis of a number of studies of these interactions, a prevailing view is that glutamate- and/or NMDA-mediated vasodilator responses are a consequence of adenosine-mediated [201] stimulation of neuronal NMDA-receptors, stimulation of nNOS and vasodilatation by nitric oxide (NO). As reviewed by Busija and colleagues, two major controversies have challenged this concept [196]. One is the idea that NMDA elicits electrophysiological responses with cortical spreading depression [195], thus complicating specific mechanistic details. Another is the finding that glutamate and NMDA receptor responses may be mediated by generation of the vasorelaxant carbon monoxide (CO, [197]). In fact considerable evidence, particularly from the Leffler laboratory, supports the role of CO in the regulation of vascular tone [198, 199, 202, 203]. Busija and colleagues have concluded that “… NO appears to be the predominant, perhaps initiating component of the cascade of events leading to cerebrovascular dilator responses following NMDA-receptor activation of cortical neurons…” [196]. In part because many of the studies were performed in piglets, these authors concede that a combination of CO, adenoside, cytochrome P450 monooxygenase products, and other factors mediate these responses, particularly in the newborn infant [196].
Perivascular Innervation
In cerebral arteries of the human [204] and mouse [205], antedating functional innervation is the ability to inactivate neuronally released neurotransmitters. Although several features of cerebral perivascular autonomic innervation and maturation have been described, many aspects of functional significance remain unknown. Numerous studies have established that neuronal functional activation is associated with increases in both cerebral energy metabolism and blood flow [206, 207]. The mechanisms that mediate CBF increases with functional activation remain, however, poorly defined [208]. Postnatally, vascular maturation is characterized by a reduction in reliance upon circulating amines, which characterize fetal life, with an increased dependence upon neuronally released vasoactive hormones [7]. The specific changes vary with species, vascular bed, and neurotransmitter type [209–211]. Postnatal maturation also is associated with an increased density of autonomic innervation [8, 212], and with enhanced reactivity to autonomic neurotransmission [212, 213]. Cranial nerves supply an abundant perivascular innervation that includes adrenergic, cholinergic, and peptidergic components [214].
Adrenergic Innervation
As compared with cerebral arteries of the adult, those of preterm and term fetal sheep [15, 17, 215], baboon [213], and beagle pup [216] show relatively greater reactivity to agonists such as norepinephrine (NE). This suggests a developmental denervation supersensitivity, as seen in ovine renal arteries [217]. In the fetal sheep, this robust contractile response of MCA to NE decreased significantly during maturation from 0.75 gestation to term, being quite low in the newborn lamb [218]. With pretreatment by the β-adrenergic antagonist propranolol, contractility increased 2-fold in newborn lamb. Addition of the nitric oxide (NO) inhibitor L-NAME markedly increased the NE-induced contractile response in near term fetus, but not pre-term nor newborn. For the near-term fetus, but not the other age groups, these findings suggest that endothelium-derived NO functionally antagonizes sympathetic vasoconstriction [218]. In terms of adrenergic-neurotransmitter-mediated contractile response to transmural nerve stimulation at 16 Hz, this averaged 9 % of Kmax in adult MCA, but only 1% for the fetus [18] (Table 3). Although both total NE content and cocaine-sensitive NE uptake were similar in the two age groups, stimulation-induced NE release in fetal vessels was twice that of the adult, as was the stimulation-evoked fractional NE release [18] (Table 3). Because these responses were obtained in endothelium-denuded arteries during blockade of receptors for acetylcholine, sensory neuronal peptides, and nitric oxide, it is unlikely that the responses were attenuated. That fetal cerebral arteries responded in such a desultory fashion to transmural nerve stimulation agrees with observations that such responses increase with maturation [7]. This contrasts, however, with reports of the opposite trend in ovine pial [215], monkey basilar and middle cerebral [49], canine coronary [216], and mesenteric [219], ovine renal [217, 220], and rat tail [221] arteries. Also in newborn piglets, denervation of the superior sympathetic ganglion was associated with a loss of autoregulation, CBF becoming pressure dependent [222]. Again, these contrasting responses emphasize the importance of species, vascular bed, and methodological approach [223]. Nonetheless, the development and role of perivascular innervation in the cerebral vasculature remains uncertain, as does the extent to which trophic and motor influences of these nerves are balanced. Together, these studies suggest the relative inability of sympathetic nerves to promote vasoconstriction in immature arteries (while they may suggest an important role in tropic function). The former may compromise autoregulation [224], particularly during hypertensive transients when adrenergic vasoconstriction typically is recruited to protect arteries from rupture.
Table 3.
Ovine Cerebral Artery Perivascular Innervation with Development
| Physiologic Variable | Fetus | Adult | Reference |
|---|---|---|---|
| Vessel NE Content (10−9 g/10−3 g tissue) | 32±7 (94) | 34±7 | [225] |
| Stimulation-Evoked NE Release (10−9 g/10−3 g tissue) | 820±90b (195) | 420±70 | [225] |
| Basal NE Release (10−9 g/10−9 g content) | 4.7±0.5b (188) | 2.5±0.5 | [226] |
| Stimulation-Evoked Fractional NE Release Following Uptake Blockade |
0.51±0.1c (47) | 0.8±0.1 | [226] |
| Stimulation-Induced Adrenergic Contractions (% Kmax at 16 Hz) |
1.1±0.6b (12) | 9.5±3.7 | [18] |
Values are mean ± SE of 5 or more in each group; Values in parentheses represent % of adult value;
Significantly different from adult value:
P<0.01;
P<0.05
Non-Adrenergic Innervation
Cholinergic, neuropeptide-Y, and peptidergic nerves have been demonstrated in mature arteries [227–229]. Although there exists some controversy as to their functional importance, one would speculate that among other actions these exert trophic effects. Little is known, however, regarding the roles of cerebrovascular innervation with development. In fact, isolated cerebral artery strips of the premature (0.75 gestation, 180 days term) and newborn baboon demonstrated robust contractile responses to acetylcholine (EC50=4×10−7 M), in contrast to only a negligible response in the adult [213]. In several strains of rats, cerebral arteries displayed both nitric oxide synthase and vasoactive intestinal peptide containing nerves; however, essentially nothing is known of these from a developmental standpoint [230]. This suggests that investigation of the possible presence and influences of these nerves in developing cerebral arteries would be a fruitful area to explore.
CYTOSKELETAL COMPONENTS
Vascular SMC internal scaffolding cytoskeleton consists of three classes of filamentous assemblies: actin microfilaments (~70 Å diameter), intermediate filaments (70 to 110 Å diameter), and microtubules (~300 Å diameter). These elements participate in numerous functions including coordination of processes as varied as contraction, migration, cell polarity and mitosis. In addition, there exists a well-developed membrane skeleton, which provides the interface between the extracellular matrix, plasma membrane, and the contractile structure within the cells [231]. As with other elements, these cytoskeletal proteins may differ in a species-tissue- and age-dependent manner [156, 232, 233].
Actin and Thin Filament Regulation
An important aspect of understanding the relation of function to structure in vascular SMCs is that of the role of the thin filament actin in contraction/relaxation responses [159, 234]. In part, such understanding requires knowledge of the relative abundance of this and other cytoskeletal components. In different parts of SMCs and in response to different stimuli, actin filaments assemble from a common pool of monomers. To date, a number of assembly factors have been identified, including multiple isoforms of formin; however little is known of how these factors are regulated, or their possible change with developmental maturation [235]. As noted earlier, a crucial component of the contractile apparatus, actin “thin” filaments, with smooth muscle myosin “thick” filaments interact to generate contractile force.
To test the hypothesis that the expression levels of several of these proteins change significantly during the course of development, and that these changes contribute to age-related changes in contractile responses, in cerebral arteries from 95-GD and 140-GD fetus, newborn lambs, and adult sheep, by Western immunoblot we quantified the relative expression of α-actin, α-tubulin, cyclophilin A, and proliferating cell nuclear antigen (PCNA) [236]. In addition, we examined middle cerebral artery (MCA) tension responses to phenylephrine (PHE) stimulation in the absence or presence of inhibitors of polymerization of α-actin (cytochalasin D). During the course of development from 95-GD fetus to adult expression levels of α-actin and cyclophilin A remained essentially constant [236]. Also as expected, PCNA expression was several-fold greater in cerebral arteries of the 95-GD fetus (×8), 140-GD fetus (×5), and newborn (×3) compared to adult. In both adult and fetal MCA cytochalasin D-inhibition of actin polymerization decreased PHE-induced contraction to ~60% and ~40% of control, respectively, despite no significant change in expression level. Thus, α-actin, the expression of which varies little with SMC maturation, appears to be an important component of cerebral artery contractility, but not age-related changes. Because analysis of protein abundance requires an internal control or norminative standard, the relatively constant developmental expression levels of α-actin and cyclophilin A suggest that these are useful internal standards for studies of cytosolic protein expression [236]. In addition, the actin binding proteins caldesmon and calponin, the actin anchoring proteins talin, paxillin, and others, as well as the several integrins all play important roles in actin function and modulation of SMC contraction [159, 162, 232, 233]. Nonetheless, essentially nothing is known of the changes of these other proteins with development.
Intermediate Filaments
In essentially all animal cells including SMC, intermediate filaments constitute a major structural element, being essential in both the cytoplasm and nucleus. In general, these function as an absorber of mechanical stress, and an integrating device for the entire cytoskeleton. Little is known of the role of these filaments in developing SMCs, however [237].
Microtubules
Contraction as well as movement of elements such as chromosome, mitotic spindles and other organelles within the cell require microtubules that can elongate or shorten and their attachment to specific cellular structures. Regulated, in part, by GTP hydrolysis, microtubules form a distinct target for binding by specific proteins. Again, little is known of the regulation of these structures within developing SMCs [238]. As noted above in our studies of actin, in the several age groups we measured expression levels of α-tubulin and PHE contractile responses to inhibition of its polymerization with nocodazole [236]. In contrast to α-actin, α-tubulin expression was ~2.5-fold greater in both fetal age groups, compared to adult (Table 2). Again in contrast to α-actin, α-tubulin inhibition by nocodazole showed little effect on PHE-induced tension, in spite of its age-related decrease in protein expression [236]. Thus, α-tubulin, which is important in intracellular protein trafficking and which reflects cell developmental maturation, appears not to be related closely to contractility per se [236]. This suggests that other cytoskeletal structural proteins and/or elements of pharmaco-mechanical coupling are important to developmental differences in cerebrovascular contractility. During the past decade or so, advanced ultrasound and Doppler modalities have allowed sophisticated evaluation of development of the cerebrovascular circulation in the fetus and newborn infant [239].
VASORELAXATION PATHWAYS/RESPONSES
Numerous reports support the role of the local metabolic regulation of CBF, which serves to couple flow to metabolic demand [3, 52]. Mechanisms proposed include the release of vasoactive metabolites by neural cells for action on local resistance arterioles, or release of vasodilator metabolites from hypoxic or acidotic vascular SMCs. With its high resting metabolic rate for O2, these mechanisms would appear appropriate for the brain. A number of substances have been suggested to mediate these cerebrovascular responses, and cerebrovascular SMCs relax in response to numerous circulating and locally released compounds. These include adenosine, prostaglandins and other eicosanoids, NO, CO, calcitonin gene-regulated peptide, opioid peptides, and many others [240]. Maturational development is associated with significant changes in the metabolism and role of essentially each of these compounds. As noted above, important functional differences among fetal, newborn and adult cerebral arteries may be a consequence of the relative inability of the less mature vessels to constrict, i.e., the “hypo-contractile” hypothesis. Alternatively these vessels may possess a greater variety of potent vasorelaxant mechanisms which follows the predictions of the “hyper-relaxation” hypothesis. Consistent with this potential balance between hypo-contractile and hyper-relaxant mechanisms, (Fig. 2) presents a schema of pharmaco-mechanico-coupling for vasorelaxation.
Fig. (2).

Endothelium-dependent vasodilatory pathways and mechanisms. Ca2+, calcium; calmodulin (41), DAG, diacylglycerol, eNOS, endothelial nitric oxide synthase, abundance (263), specific activity (~60); NO, nitric oxide (significantly less); sGC, soluble guanyl cyclase, abundance (~500), specific activity (~300); cGMP, synthesis rate (376), activity (954); prostaglandins (significantly greater), PDE, phosphodiesterase (193), PKG, protein kinase G, PL, phospholipids. (Numbers in parentheses represent fetal values, as percent of those of adult).
Cerebrovascular Endothelium
As noted in discussion of the neurovascular unit, lining the luminal surfaces of the cerebral vasculature, endothelial cells interconnected with tight junctions constitute the blood-brain barrier. These specialized endothelial cells appear early in fetal life and continue to develop and differentiate throughout the postnatal period. Although early work suggested that the BBB is not fully functional in early development, more recent studies indicate that in the near-term fetus and newborn it is functionally specialized and relatively impermeable [241]. Also, the expression of tight junction proteins continues throughout early postnatal life [242]. Also as noted above, an additional vital role of the developing endothelium is that of angiogenesis, arteriogenesis, and regulation of cerebral SMC phenotype [122, 243]. A third essential role of vascular endothelium is that of synthesis and release of many vasodilatory molecules including NO, eicosanoids, and the elusive endothelium-derived hyperpolarizing factor [86]. Recently, in cultured embryonic mouse aorta endothelial cells, Nauli and coworkers demonstrated the role of mechanosensitive molecules polycystin-1 (required for cilia function) and polaris (required for cilia structure) for sensing shear stress and NO production [244]. A number of critical questions, and about which little is known, concern the extent to which these vasodilating factors and functions differ in the developing, as opposed to mature, cerebral vasculature.
Endothelium-based relaxation responses in cerebral arteries of the fetus and newborn differ significantly from those of the adult, and some of these are summarized in (Table 4). For instance, in precontracted arteries, in response to the calcium ionophore A-23187 (a receptor-independent stimulant to release endothelium-derived relaxing factor), 140-GD fetal MCA relaxed ~22% more than the adult [245] (Table 4). Also in precontracted arteries, the relaxation response of fetal arteries to the NO donor S-nitroso-N-acetylpenicillamine (SNAP; a vasorelaxant acting directly on SMC independent of endothelium), was ~16% greater than that for adult [245]. This somewhat greater propensity of immature vessels to relax may account for their being less capable of resisting vascular injury and rupture associated with the markedly elevated cerebral perfusion that accompanies asphyxia and/or transient hypertension.
Table 4.
Cerebrovascular Relaxation Pathways with Development
| Variable | Fetus | Newborn | Adult | Reference |
|---|---|---|---|---|
| Intrinsic tone at 40 mmHg with endothelium intact (Mouse; %) | 21±3 (210) | 10±1 | [106] | |
| A-23187-Induced Relaxation (%) | 35 (35) | 100 | [245] | |
| Constriction to indomethacin at 40 mmHg (Mouse) | 12±1 (46) | 26±1 | [106] | |
| Constriction to L-NAME at 40 mmHg (Mouse) | 17±1 (49) | 35±2 | [106] | |
| pD2 of SNAP-Induced Relaxation | 6.3±0.1 | 5.9±0.1 | [246] | |
| eNOS Relative Abundance | 3.1±0.2 (135) | 2.3±0.2 | [247] | |
| eNOS abundance (ng eNOS · mg protein−1)* | 29±3 (263) | 11±2 | [248] | |
| Maximum eNOS Specific Activity Slope NO (10−12 M)·eNOS (10−6 g)·min−1 | 3.8±0.5b (67) | 5.7±1.0 | [248] | |
| eNOS abundance in microvessels (% newborn value)++ | 100 (36) | 280±30 | [249] | |
| eNOS specific activity (nmol NO · μg eNOS−1 · min−1)* | 97±36 (55) | 177±44 | [248] | |
| Slope of relation of eNOS specific activity to fluid shear stress (nmol NO · μg eNOS−1 · min−1 · dyne−1·cm2 −1)* | 2.9±0.1 (43) | 6.8±0.1 | [248] | |
| Basal eNOS activity in microvessels (14C-citriline incorporation, cpm · mg protein−1)++ | ~3,500±200 (47) | ~7,500±400 | [249] | |
| sGC abundance (% that in adult kidney) | 0.37±0.6 (276) | 0.17±0.03 | [250] | |
| sGC abundance (ng · mg protein−1)† | 7.1±0.9 (132) | 5.4±1.5 | [251] | |
| sGC specific activity (pmol · μg sCG−1 · min−1)† | 60±2c (35) | 171±19 | [251] | |
| sGC specific activity (pmol · cGMP−1 · mg−1 · min−1) | 3.1±0.31 (214) | 1.45±0.08 | [250] | |
| sGC activity, Vmax as determined by cGMP formation (pmol cGMP · mg protein−1 · min−1) | 4.48±1.13 (954) | 0.47±0.13 | [252] | |
| sGC abundance (ng sGC · mg protein−1) | 17.6± 1.6a (1036) | 1.7±0.3 | [252] | |
| Basal cGMP (10−9 M·10−3 protein−1) | 3.3±0.5b (194) | 1.7±0.2 | [253] | |
| Basal cGMP (10−6 M)* | 0.59±0.11 (536) | 0.11±0.02 | [254] | |
| cGMP Synthesis rate (10−6 M·1 cell water−1·min−1)* | 0.31 ±0.06b (207) | 0.15 ±0.04 | [254] | |
| cGMP synthesis rate (pmol cGMP · mg protein−1 · min−1) | 29.5±5.6b (356) | 8.3±1.4 | [251] | |
| cGMP-induced relaxation with 8-pCPT-cGMP (% maximal)† | 99.2±0.8 (111) | 89.3±4.6 | [251] | |
| cGMP Degradation (10−6 M·1 cell water−1·min−1)* | 106±6c (136) | 78±6 | [254] | |
| cGMP (pm · mg protein−1) | 1.0±0.1 (333) | 0.3±0.1 | [253] | |
| cGMP (10−9 M·10−3 g Soluble Protein) | 0.35±0.1 | [250] | ||
| cGMP (10−9 M·sGC (10−6 g)·min− | 145±20 (−) | [250] | ||
| ΔpD2 for 5-HT-induced contraction with 8-pCPT-cGMP* | To 6.30±0.08 from 6.72±0.02 | NSC | [47] | |
| Δ pD2 for 5-HT calcium sensitivity ΔTension · Δ [Ca2+]i (%)* | To 176±27 from 128±17b | NSC | [47] | |
| Δ pD2 for K+-induced contraction with 8-pCPT-cGMP in α-toxin permeabilized vessels* | −0.4±0.1b | NSC | [47] | |
| Δ maximum 5-HT-induced rise in [Ca2+]i with 8-pCPT-cGMP (%)* | To 64±9 (150) from 88±6c | To 82±7 from 98±14 | [47] | |
| Basal ratio of cGMP to cAMP | ~6 | ~3 | [255] | |
| Prostaglandin I2 NO |
> < |
[154, 155] | ||
| COX-2 in microvessels (% newborn value)++ | 100 | 128±10 | [249] | |
| Basal COX activity in microvessels (PG production, pg · μg protein−1)++ | 2.2±0.4 | 2.2+-0.5 | [249] | |
| COX activity in microvessels (PGE2 production, pg · μg protein−1)++ | 1.6±0.3 (133) | 1.2±0.3 | [249] | |
| Hypoxic-Induced CBF (% Control) | +43 | [256] | ||
| Hypoxic-Induced CBF (% Control) | +50 | [257] | ||
| Above with L-NAME inhibition | + | [256] | ||
| Carbon Monoxide as measured by [HbCO] | 2 to 3† (200 to 300) | 1 | [258] |
Values are mean ± SE of 5 or more in each group;
Values in parentheses represent % of adult values;
Significantly different from adult value:
P<0.001;
P<0.01;
P<.05;
Basilar artery;
CCA;
Pigs; NSC, no significant change
Endothelial Nitric Oxide Synthase, Nitric Oxide, and Soluble Guanylate Cyclase
NO relaxes vascular SMCs by interacting with the heme group of soluble guanylate cyclase (sGC) to produce the second messenger guanosine 3, 5′-cyclic monophosphate (cGMP). In turn, this activates protein kinase G (PKG) to decrease tone in many vascular beds [259]. cGMP also binds and modulates the activities of several phosphodiesterases, which can affect adenosine, 3′, 5′-cyclic monophosphate (cAMP) levels. Additionally, cGMP modulates cyclic nucleotide gated (CNG) channels [260], although the role of these channels in the developing organism is relatively unexplored.
Considerable evidence supports the role of NO, produced by both endothelial and neuronal/astroglial nitric oxide synthase (eNOS and nNOS, respectively), in CBF regulation [261′264], although this role is disputed in response to somatosensory activation [265]. In perfused CCA from near-term fetal and adult sheep, White and colleagues demonstrated several aspects of NO synthesis [248]. eNOS abundance, when calculated in terms of endothelial surface area, was similar in the two age groups, as were mRNA levels. When normalized relative to eNOS protein, the specific activity was considerably greater in adult than in fetal vessels. Similarly, the slope of the relation of eNOS specific activity to fluid shear stress was significantly greater in adult than fetal arteries [248] (Table 4). These results were reinforced by further studies demonstrating with maturation significant decrease in eNOS abundance, suggesting independent mechanisms for their regulation [247] (Table 4). In cerebral microvessels of pigs, both eNOS abundance and specific activity were much greater in adult than in newborn. As an aside, expression and activity of cyclooxygenase was similar in the two age groups [249] (Table 4). These results demonstrate that in several species in which it has been examined, endothelial vasodilatory capacity is a function of age-dependent increases in eNOS-specific activity with NO release. Nonetheless, in terms of development, NO's precise role in either maintenance of basal CBF or the response to acute hypoxia is unclear, as are the relative roles of eNOS and nNOS (although nNOS abundance was similar in MCA of the two age groups [266]). For instance, in the adult rat NO has been reported to play a minor role in the hypoxic-induced increase in CBF [267, 268], although others have challenged this view [262, 269, 270]. In the adult, although NO may to contribute little to the maintenance of basal CBF, it is a most important mediator of hypoxic-induced vasodilatation. As with so many other stresses, this response, however, varies with species, vascular bed, duration and intensity of hypoxia, brain region studied, activation of neuronal NMDA receptors [271], and other factors [208, 262, 272′274].
In fetal, newborn, and adult ovine cerebral arteries and CCA sGC abundance and specific activities differed widely. In newborn ovine cerebral arteries, both sGC abundance and specific activity were double that of the adult. Similar findings were seen in CCA [250] (Table 4). While sGC protein expression was 30% greater in fetal arteries, specific activity was only a third that of the adult [251] (Table 4). In turn, the sGC-mediated cGMP synthesis rate was 3-fold greater in fetal arteries as compared to adult. cGMP-induced relaxation also was significantly greater in fetal CCA, as compared to adult [251] (Table 4). In MCA abundance was greater in fetus as compared to adult [252] (Table 4). This fits with data demonstrating that in ovine newborn CCA, cGMP levels were three times that of adult. Nonetheless, the time to reach peak values in response to SNAP were more rapid in adult than in newborn, as were the time to return to baseline values [253]. These results suggest that maturation from newborn to adult is associated with both decreased capacity of the cGMP pathway to produce relaxation, in concert with accelerated rate of cGMP turnover [253]. cGMP plays a critical role in cerebrovascular vasorelaxation mechanisms, its basal levels and rate of synthesis are much greater in fetal than adult cerebral arteries [253, 254] (Table 4). In concert with these findings, for both 5-HT- and K+-induced contractions in fetal, but not adult, basilar arteries the non-metabolizable cGMP analog 8-pCMP-cGMP produced significant decreases in the pD2, suggesting increased calcium sensitivity [47]. In a subsequent publication, these authors confirmed the 8-pCPT-cGMP decreases in K+- and 5-HT-induced maximum contractility and CaS [275] (Table 4). The ratio of basal levels of cGMP to cAMP also were considerably greater in newborn than adult MCA [255] (Table 4). In addition, in newborn CCA, the Km value for phosphodiesterase, which metabolizes cGMP to GMP, was twice that of adult [254].
Generally, it is agreed that in the developing fetus NO is involved in the regulation of resting tone and thus basal CBF, and in mediating the hypoxic-induced increase in CBF [276′279]. For instance in the near-term (140-GD) ovine fetus, basal CBF, measured by laser Doppler flowmetry, decreased ~14% following infusion of the NO antagonist Nω-nitro-L-arginine methylester (L-NAME), despite a significant increase in arterial blood pressure with increased cerebrovascular resistance [256, 279]. In normoxic control fetuses in response to acute hypoxia, relative CBF increased ~43% and cortical cGMP levels (an index of NOS activity) increased ~26%. In contrast, in L-NAME infused fetuses, relative CBF increased only 15% above baseline and cortical cGMP release was unchanged [277]. A complicating factor in this study, however, was the failure to maintain isocapnea, so that the arterial PCO2 value was ~7 Torr less than that of control. In a subsequent study in which a similar degree of hypoxia was maintained isocapnic, fetal cortical CBF increased ~50% above baseline [71, 257]. In agreement with findings of a major role for NO in developing, as compared to adult, MCA, the efficacy of NO donors [255] as were basal levels of both cAMP and cGMP, were generally greater in newborn than adult MCA (Table 4).
By use of the cranial window technique to measure changes in pial artery diameter, in the anesthetized newborn piglet, β1-adrenergic receptor-induced vasodilatation with dobutamine was significantly blunted in presence of the NOS inhibitor NG-nitro-L-arginine (L-NNA). Additionally, L-NNA blocked dobutamine-induced increase in periarachnoid cerebrospinal fluid (CSF) levels of cGMP (but not cAMP) [280]. Both the β2-agonist salbutamol and nonselective β-agonist isoproterenol followed by L-NNA showed a similar response. Forskolin, a direct activator of adenyl cyclase, also induced vasodilatation that was attenuated by LNNA [280]. These data suggest that in the piglet, cAMP, cGMP, and NO each contribute to β-adrenergic receptor-mediated vasodilatation. Further studies along this line have demonstrated the roles of prostaglandins, both PGE2 and PGI2 increased CSF levels of cGMP and cAMP (as well as methionine-enkephalin and leucine-enkephalin) [281]. Substances other than NO also appear to be involved in hypoxic-induced calcium-activated potassium (KCa) channel activation with pial artery dilatation in the piglet [272]. As a caveat, although an important physiological method, the pial window technique may alter proximal nerve and astroglia function, thus not necessarily reflecting the immediate action of applied agents. Also problematic for developmental studies, because of the lack of adult controls, the age-dependence of these responses is unknown.
In endothelium intact pressurized resistance sized cerebral arteries (~150 μm), at a given pressure, total contractile tone was significantly less in the near-term ovine fetus, than that of the adult (and also that of the 95-GD fetus) [107]. This difference probably is a consequence of the smaller vessels with less media, as the fetal, neonatal, and adult CA display similar maximum active force [17]. The pressurized near-term fetal myocytes also showed significantly less basal [Ca2+]i, compared to the adult. Removal of endothelium showed essentially the same overall relationships, except that lumen diameter was reduced (greater tone) and Ca2+ sensitivity increased. Although selective NOS inhibition with L-NAME decreased vessel diameter (increased tone) as a function of pressure in each age group, [Ca2+]i increased in relation to pressure only in the near-term fetus [107]. Thus, endothelium removal resulted in a marked decrease in vessel Ca2+ sensitivity. In this system, the cyclooxygenase (COX) inhibitor indomethacin showed similar responses with greater effects in near-term fetal vessels, as well as interaction with the NOS-mediated relaxation effects [107]. Taken together, these data suggest that development from the preterm to near-term fetus is associated with increasing modulation of cerebrovascular tone by CaS-dependent mechanisms. By way of contrast, during postnatal life CaS decreases and NO-dependent modulation of tone appears to occur via mechanisms independent of Ca2+ sensitivity.
Arachidonic Acid, Cyclooxygenase and Eicosanoids
Several arachidonic acid metabolites also play important roles in the regulation of cerebrovascular tone, and these involve both endothelium and the vascular SMCs. The metabolites, which are limited by AA availability, include those generated by the cyclooxygenase pathway, (prostaglandins; PGs), those produced by the lipoxygenase (hydroxyeicosatetraenoic acid; HETEs), and those generated by cytochrome P-450 (CYP) enzymes (epoxyeisatrienoic acid; EETs, and HETEs). Because of the complexity of specific enzymes and their interactions, a comprehensive survey of this field is beyond the scope of this review. As noted above, in porcine cerebral microvessels, while expression and activity of cyclooxygenase is relatively great in the newborn, these values are similar in adult [249]. In a host of studies in cerebral microvascular endothelial cells of the newborn piglet, the Leffler group also has demonstrated the role of prostanoids and eicosanoids in the regulation of vascular tone and CBF, both under control conditions [282–284], and in response to hypoxia and hypercapnia [44, 155, 285–287]. In contrast, in pressurized endothelium-intact mouse cerebral arteries, administration of the cyclooxygenase (COX) inhibitor indo-methacin (10−5M) produced no effect in either neonatal or adult [106]. In both age groups, subsequent administration of the NOS inhibitor L-nitro-arginine methyl ester (L-NAME, 10−4M) produced significant increase in tone. In adult, but not fetal, arteries, in turn, administration of L-NAME alone significantly increased tone and this was increased further with addition of indomethacin [106]. Compared to the neonate, these studies demonstrate the significant interaction of the NOS and COX pathways in the mature vessel. Of importance, when the endothelium was removed tone was similar in both age groups at all pressures [106]. Also in the newborn piglet, pial artery studies via a cranial window have demonstrated that for both PGE2 and PGI2 vasodilatation is mediated by increased activities of cAMP as well as cGMP, with higher concentrations of these second messengers in cortical periarachnoid CSF [281, 288]. Also in the piglet CSF, PGE2 is abundant and appears to contribute to resting cerebrovascular tone as well as the CBF response to hemorrhagic hypotension [285] and hypercapnia [289].
A related consideration regards developmental aspects of the relative roles and interactions of vasodilator prostanoids and NO in cerebrovascular regulation. In newborn pig cerebral circulation, dilator prostanoids, but not NO, contribute to vasodilatation of pial arterioles in response to hypoxia, hypercapnia, and other stress [155, 249, 290–292]. In contrast, in the near-term fetal sheep, although infusion of diclofenac (prostanoid synthesis inhibitor), blunted the expected hypoxic-induced increase in CBF as measured by laser-Doppler, this was accompanied by a concomitant decrease in arterial blood pressure with no change in cerebral vascular resistance [293]. As noted above, NO plays a vital role in the hypoxic-mediated decrease in vascular resistance in the fetal sheep [256, 277]. NO also appears to play a more important role in the newborn and older animals [155, 249, 287, 290, 294]. In affirmation of this view, in porcine cerebral arteries, microvessels, and cultured endothelial cells, the expression of endothelial cyclooxygenase, the functional role of COX-2 and COX-1 isoforms, and the production of vasodilator prostanoids remain relatively constant with age. In contrast, eNOS expression in this species is upregulated with developmental age [249]. In cultured endothelial cells from both small (<300 μm) and large (>300 m) vessels from human newborn (22 to 26 week gestation) cerebral cortex and cerebellum, basal, as well as proinflammatory cytokine IL-1β stimulated, COX activity was inhibited by NS-398 [284]. Overall, these data indicate that both COX-1 and COX-2 contribute to endothelial prostanoid synthesis in the neonatal cerebrovasculature. These and other studies attest to the vital role of eicosanoids in modulating cerebrovascular tone in the fetus and newborn.
An example of the role development plays in vascular reactivity is illustrated further by that of NOS-mediated vasodilatation in response to topical acetylcholine in cerebral and other vascular beds. In the adult rat, this results in arteriolar dilatation that can be blocked by NOS inhibition [295]. In the newborn piglet in contrast, acetylcholine stimulated pial artery constriction [296, 297], a response blocked by the PGH2 synthase inhibitor indomethacin [298]. Also in the piglet, NOS inhibition failed to alter the indomethacin response [298] or acetylcholine-induced vasoconstriction [290], suggesting a minor role for NO in this regard. Nonetheless, in piglets in which cyclooxygenase had been chronically inhibited for 6 to 8 days, NOS activity (but not eNOS expression) increased [299]. This suggests that chronic COX inhibition may result further in increased contribution of NO to cerebrovascular regulation. With the caveats noted earlier, the cranial window technique presents several complicating issues. While it provides an “integrated” picture of response to a given change in CSF composition, one does not know which cell types are involved. In addition, in some instances the responses may not agree with those of isolated vessel, perhaps because the agent is administered to the serosal, rather than luminal, surface.
Adenosine and Cerebrovascular Relaxation
Adenosine (ADO), an endogenously produced purine nucleoside, has been proposed as an important regulator of CBF [300–303]. In instances of diminished local O2 supply, as occurs with severe hypoxia and/or ischemia, or when the tissue O2 requirement is increased, as may occur with seizures, cellular ATP is metabolized to 5′-AMP which then is converted to adenosine. Intracellular and interstitial ADO concentration increase to cause relaxation of local resistance arterioles and decrease the rate of tissue metabolism. Thus, it has been referred to as a “retaliatory” metabolite. In response to hypoxia ischemia, brain ADO levels have been shown to increase in a number of species [300, 303, 304]. Although these studies support a role for adenosine in CBF responses to stress, studies of blockade of ADO or increasing its metabolism by adenosine deaminase have been controversial.
With respect to development, several groups have attempted to identify age-related differences in ADO vascular sensitivity and the role of its receptors. For instance, cerebrovascular response to hypoxia was reported to be greater in young (6 months) rats as compared to those that were older (24 months), and this was attributed to ADO [304]. Also, ADO receptor blockade by therapeutic levels of theophylline inhibited cerebrovascular smooth muscle tone [305]. In 0.6 to 0.7 gestation and full-term fetal sheep equipped in utero with a closed cranial window, pial arteries dilated in response to two different ADO receptor agonists, and this was attenuated by an ADO receptor blockade [306]. ADO infusion in the near-term fetal lamb, at levels approximating those observed during hypoxia, produced an increase in oxidized cerebral cytochrome oxidase, indicating a reduction in electron flow down the mitochondrial electron transport chain and fall in metabolic rate [307]. By use of laser Doppler flowmetry probes implanted in near-term fetal sheep parietal cortex, ADO-receptor blockade (theophylline) had no significant effect on basal CBF; however it resulted in a failure of the normal hypoxic-induced increase in CBF [308]. In a follow-up of these studies, these authors demonstrated increased cerebral O2 delivery mediated by adenosine A2 receptors, and a decrease in cerebral metabolic rate mediated by adenosine A1 receptors within the blood brain barrier [309]. In a further report in near term fetal sheep, these investigators demonstrated that the adenosine A1-receptor mediates the suppression of neural activity in response to asphyxia produced by umbilical cord occlusion [277]. On a cautionary note, in preterm (0.7 gestation) fetal sheep the Auckland, New Zealand group demonstrated the two-edged nature of A1-receptor antagonist infusion in selectively impairing renal perfusion, presumably by affecting the glomerular arteriole [310].
Heme Oxygenase and Carbon Monoxide-Mediated Vasorelaxation
Analogous to NO, the endogenously produced gasotransmitter carbon monoxide (CO) has been shown to be a potent physiological vasodilator of CBF [43, 311–315], and recently Leffler and colleagues have reviewed this rapidly developing field in terms of the neurovascular unit [179]. CO is produced by the degradation of heme by heme oxygenase (HO) [316], chiefly the HO-2 isoform expressed constitutively in vascular endothelium and SMCs [317]. One might assume that CO, as NO, acts chiefly by stimulating the formation of cGMP [318]; however, evidence suggests that CO also can activate K+ channel activity independently of cGMP elevation [314]. Specifically, CO activates KCa channels [319], in some cases by enhancing the coupling of Ca2+ sparks to BK channels [320]. In newborn piglet cerebral artery SMCs, CO acts on the BK channel α-subunit to increase Ca2+ sensitivity [321]. In addition, because CO inhibits the activity of cytochrome P-450 and other heme containing enzymes, its vasodilatory action also may be due to inhibition of 20-hydroxyeicosatetraenoic acid formation [322]. Nonetheless, in the newborn piglet, AA and PGE2, but not reactive oxygen species, mediate CO-induced cerebral vasodilatation [323–325]. In addition in piglets, the astrocytes of the microvascular unit have been shown to play a key role in glutamate-induced Ca2+ signals to stimulate CO production [198, 202, 203, 326]. Although different mechanisms account for CO- and NO-mediated BK channel stimulation [327], considerable evidence supports interactions between HO/CO and NOS/NO systems in vasoregulation [179, 312, 328, 329].
With somewhat higher concentrations of hemoglobin and shorter erythrocyte half-life, (both dependent upon species and gestational age), in general, fetal carboxyhemoglobin levels [HbCO] are almost double that of the adult [258] (Table 4). This undoubtedly reflects a significantly greater CO production rate in the fetus, as compared to the adult [330]; however, this does not appear to be due to greater heme oxygenase activity in the fetal vasculature [331]. Although considerable attention has been devoted to the relation of fetal oxygenation to blood HbCO levels [330], little is known of the developmental role of CO in CBF regulation. L-glutamic acid (glutamate), the principal excitatory neurotransmitter in the central nervous system, can exert its effects via several receptors [332], and acts as a vasodilator, a response mediated by CO [43, 44]. In both isolated, pressurized newborn pig middle cerebral arterioles (~200 μm diameter) [197] and pial arterioles (60 to 80 μm), CO plays a key role in glutamate-mediated vasodilatation [333]. As noted, in view of the relatively higher [HbCO], compared to the adult, as well as for a more complete understanding, these findings raise the important issue of the role of CO in CBF regulation in the developing fetus and newborn. In concert with CO, hydrogen sulfide (H2S), also is becoming appreciated to be an important signaling molecule in regulation of microvascular tone and CBF [178, 311].
Glutamate and the Role of Astrocytes
As noted in consideration of the neurovascular unit, in addition to traditional neurotransmitters such as NE and 5-HT, glutamate produced by perivascular neurons can activate N-methyl-D-aspartate receptors. Elevation of [Ca2+]i, activates NOS to release NO, which upon diffusing into adjacent cerebral arteriole SMCs, decreases tone [334]. In addition, glutamate can activate its receptor on astrocytes, to increase [Ca2+]i, thereby activating phospholipase A2 (PLA2) to generate arachidonic acid [200]. In turn, AA can activate the enzyme cytochrome P-450 subtype CYP-4A to generate the vasoconstrictor 20-hydroxyeicosatetroenoic acid (20-HETE). Alternatively, by activating COX it can generate the vasodilator prostaglandin PGE2. In addition, upon binding astrocyte α1-AR, NE can elevate [Ca2+]i to activate PLA2 thus activating either a vasodilator (PGE2) or vasoconstrictor (20-HETE) response [169]. As noted, recent studies on the role of astrocytes, present strong evidence for a paradigm shift in our thinking to include these cells as critical to CBF regulation. A key element here is the role of astrocytes, and the effect of increased [Ca2+]i leading to production of both vasoconstricting CYP-generated derivatives of arachidonic acid (20-HETE), and vasodilating COX-generated derivatives (PGE2). By elevating cGMP levels, NO also can inhibit 20-HETE synthesis [322]. An additional component of the role of astrocytes is that, rather than NE-producing neurons signaling directly to vascular SMCs as has been assumed, their action may be mediated indirectly by astrocytes. This is consistent with the observation that many neuronal NE-release sites are located on or near this cell type [168]. Unfortunately, in terms of developmental regulation, essentially nothing is known of the relative roles of these vital yet competing agents and pathways.
Opioids in the Cerebrovasculature
Opioids also contribute to the regulation of cerebral hemodynamics [289, 335, 336], and under basal conditions are in the vasoactive range in newborn pig CSF [337]. In the piglet, prostaglandin-mediated elevation of cAMP and cGMP levels is associated with increased CSF levels of these cyclic nucleotides. In turn, these are associated with increased concentrations of the opioids methionine enkephalin and leucine enkephalin [281, 288], which contribute to cerebral vasodilatation [337, 338]. Under basal conditions, the influence of opioids appears to be rather subtle, becoming more prominent in pathophysiologic states such as hypoxia and hypotension [269].
Additionally, the recently described opioid, nociception/orphanin FQ, elicits vasodilatation in piglet pial arteries [339]. In response to global cerebral ischemia-reperfusion, CSF levels of these opioids increase, and are associated with vasodilatation. In contrast, in response to combined hypoxia-ischemia-reperfusion, CSF levels of these opioids decrease in association with vasoconstriction [340]. Nociception/orphanin FQ-associated pial artery dilatation appears to be mediated via cAMP acting upon both KATP and KCa channels [339]. Following hypoxia with ischemia and reperfusion, impaired nociception/orphanin FQ vasodilatation results from altered cAMP and activation of both KCa- and KATP-channels [341]. Such impaired vascular reactivity is also associated with altered nociception/orphanin FQ modulation of NMDA receptor-induced pial artery dilation [340]. Thus, the roles of glutamate, opioids, and glial cells adds a new dimension to CBF regulation and changes with development. Unfortunately, at this time we have little understanding of the complexity of these interactions.
VASOCONTRACTION PATHWAYS/RESPONSES
On the other side of the coin are the factors and their interactions that regulate cerebrovascular contraction. In this regard, a number of reports have demonstrated that the concentration of cytoplasmic Ca2+ (determined by its entry from the extracellular Ca2+ pool, as well as entry and release from intracellular stores) acts as a multifaceted messenger to regulate the contractile state of smooth muscle cells [240, 342], as well as a diverse array of other cellular processes. An issue not to be ignored is the need to integrate the contractile electro-mechanical and pharmaco-mechanical pathways. Unfortunately, this would be enormously complex, and beyond the scope of this review. For this reason, investigators have separated these. Electro-mechanical transduction deals almost exclusively with the regulation of cytosolic [Ca2+]i. In contrast, pharmaco-mechanical coupling deals not only with how receptor activation increases cytosolic [Ca2+]i, but also how activation of the several pathways alter thick and thin filament reactivity and calcium sensitivity. (Fig. 3) presents in schematic form some elements of Ca2+ dependent contractile mechanisms. As noted, there exists considerable “crosstalk” between these systems as electromechanical-induced changes in [Ca2+]i alter kinase and phosphatase activities, which alter G-protein and other signaling. Not only may transmembrane Ca2+ flux induce Ca2+ release from the sarcoplasmic endoplasmic reticulum (so called Ca2+-induced Ca2+ release [343]), but in turn, G-protein receptor activation can lead to desensitization of plasmalemmal Ca2+ channels, pump activation, et cetera. Although integration of the several pathways is a desirable goal, we simply do not have enough data to accomplish this in a logical and/or meaningful manner. In fetal, newborn, and adult CCA maximum tension in response to K+ depolarization (1.22×10−1M) increase significantly with maturational age. This increase in maximum response also was seen in MCA, but less so [17] (Table 5). When contractile force was normalized to vessel cross-sectional area to give maximum active stress, however, these age-dependent differences disappeared, except in the case of the newborn which increased ~20%. Also across all age groups, maximum wall stress was less in larger, more proximal arteries, than in the smaller, more distant vessels [17].
Fig. (3).

Calcium-dependent vasocontraction pathways and mechanisms. Vasoactive substances include Norepinephrine, Serotonin, or other; Receptor, specific receptor for agonist, G protein, G protein complex; Ca2+ sensitivity (200–300); Maximum K+-induced tension (~80); maximum NE-induced tension (82); maximum amine-induced tension (~90); maximum 45Ca2+ uptake in response to K+ depolarization (152); Maximum Ins(1,4,5)P3 (inositol 1,4,5 trisphosphate) response to norepinephrine (97); Ins(1,4,5)P3-receptor density (116); MLC20, myosin light chain20 concentration (119); MLCK, myosin light chain kinase concentration (22), maximum velocity (585); MLCP, myosin light chain phosphatase concentration (56); calmodulin concentration (41). (Numbers in parentheses represent fetal values, as percent of those of adult).
Table 5.
Cerebrovascular Contractile Pathways and Electro-Mechanical Coupling with Development
| Physiologic Variable | Fetus | Newborn | Adult | Reference |
|---|---|---|---|---|
| Ca2+ Sensitivity | >> | >> | Numerous reports Please see text | |
| Maximum tension (g)* | 6±1 (40) | 10±2 (67) | 15±2 | [17] |
| Maximum tension (g) | 2.4±0.5 (83) | 2.8±0.5 (96) | 2.9±1 | [17] |
| K+max Tension (g) | 1.0±0.1 (55) | 1.8±0.1 | [344] | |
| L-type Ca2+ Channel density (Bmax, fmol · mg protein−1) | 141±12 (243) | 58±8 | [45] | |
| Membrane Potential (mV) | −38.1±3.4c (84) | −45.4±2.5 | [345] | |
| Basal [Ca2+]i (10−9 M) in arterial segments | 130±5 (at 45 mmHg) (72) | 180±5 (at 75 mmHg) | [107] | |
| Basal [Ca2+]i in arterial segments† | 87±17 (81) | 107±15 | [346] | |
| Basal [Ca2+]i in arterial segments (10−9 M)† | 84±3 (153) | 55±2 | [347] | |
| Intracellular [Ca2+]i (10−9 M)† | 145±19 (95) | 157±15 | [345] | |
| Maximal 45Ca uptake in response to K+ depolarization (μM · g dry wt−1 · min−1) | 231±19a (152) | 152±13 | [348] | |
| % Inhibition Tension by Nifedipine | 100±10 | 100±10 | [46] | |
| pIC50 | 7.3±0.1 (‡) | 6.6±0.1 | [46] | |
| % Inhibition Fluorescence Ratio by Nifedipine | 100 ±10 | 100±10 | [46] | |
| K+ Stress(10−6 Dynes/cm2) | ||||
| pIC50 | 7.2±0.1 (‡) | 6.9±0.1 | [344] | |
| KCa Channel (% Inhibition Tension by NS-1619) | 100±10a (238) | 42±9 | [344] | |
| pIC50 | 8.2±0.1 (‡) | 7.6±0.1 | [344] | |
| KCa Channel (% Inhibition Fluorescence Ratio by NS-1619 | 37±6 (−) | unchanged | [344] | |
| pIC50 | 7.3±0.1 (‡) | – | ||
| KATP Channel (% Inhibition Tension by Pinacidil) | 100±10 | 100±10 | [344] | |
| pIC50 | 5.0±0.1 (‡) | 5.1±0.1 | [344] | |
| KATP Channel (% Inhibition Fluorescence Ratio by Pinacidil) | 72±8c (1600) | 4.5±0.5 | [344] | |
| pIC50 | 4.6±0.1 (− ‡) | – | ||
| Total Outward Current Density at 60 mV (pA/pF)† | 57.9 ±6.6a (153) | 37.9 ±1.8 | [345] | |
| Calcium Set Point (10−6 M)† | 4.7±0.1a (53) | 8.8±0.1 | [345] | |
| Native V1/2 Control (mV)† | 10.5±8.1a (31) | 34.0±36 | [349] | |
| Right-Shift by Dephos-phorylation State, ΔV1/2 (mV)† | 52.3±3.9a (173) | 30.3±3.4 | [349] | |
| V1/2 of Dephosphorylated Channels (mV)† | 62.8±5.2 (98) | 64.3±3.4 | [349] | |
| Endogenous Activated Kinase Shift, ΔVmax (mV)† | 38.8±4.6c (69) | 56.5±7.9 | [349] | |
| Channel-Associated PKA Shift, ΔVmax (mV)† | 36.4±2.8a (67) | 53.9±3.9 | [349] | |
| Channel-Associated PKG Shift, ΔVmax (mV)† | 32.1±3.2 (90) | 35.6±1.5 | [349] |
Values are mean ± SE of 5 or more in each group;
Values in parentheses represent % of adult value;
Significantly different from adult value:
P<.0.01;
P<0.05;
Basilar artery;
CCA;
Because these are logarithmic functions, percent change is not an accurate or appropriate representation
PLASMA MEMBRANE ION CHANNELS
L-type Calcium Channels
A critical feature of late fetal and early postnatal development is the high rate of calcium accretion characteristic of neonates [350, 351]. Indeed, if sufficient dietary calcium is not available during this perinatal period, a variety of bone disorders can result, many of which have consequences reaching into adolescence and adult life [352]. Of relevance to the cerebrovasculature, evidence suggests that the rapid rate of neonatal calcium accretion affects vascular smooth muscle as well [45, 353, 354]. Compared to arteries in the adult, those of the immature organism appear to have poorly developed intracellular Ca2+ stores, and their contractility is far more dependent on the entry of extracellular Ca2+ [45, 346, 353, 355]. Reports of basal [Ca2+]i in cerebral vessels vary with method. In patch-clamped individual myocytes that value was only slightly lower in fetal, compared to adult, basilar artery [345] (Table 5). This relationship also was reported in ovine arterial segments [107]. In another report, based on Fura-2 fluorescence that value in fetal arterial segments was one and a half times that of adult [347]. Of considerable importance, myofilament calcium sensitivity plays a paramount role in developing cerebral arteries, declining with maturation [107, 353, 356, 357]. Thus, the uptake and deposition of dietary calcium, a hallmark of neonatal metabolism, has critical consequences on cerebrovascular Ca2+ metabolism, and ultimately, on regulation of vascular tone and contractility. We will examine this recurrent theme on maturational effects on cerebral artery contractility in several contexts.
An important aspect of age-related differences in cerebral artery contractility concerns differences in the manner in which the SMCs handle and distribute Ca2+. This differs considerably in relatively immature “synthetic”, as opposed to the mature “contractile”, SMC phenotypes [122, 358, 359]. As emphasized by Owens and colleagues, both phenotypes are present at all ages, so that although at a given age each may predominate, their plasticity and response to growth factors belies attempts to make them age-specific [122]. In view of the marked age-related differences in cerebral arteries contractility, the question arises as to the role developmental modulation of L-type Ca2+ channels in agonist-mediated contraction.
Intracellular Ca2+ concentration is a fundamental determinant of vascular SMC contractile state, and represents the dynamic integration of multiple mechanisms governing Ca2+ influx, sequestration, release, and extrusion [342, 360]. Owing to sparse or poorly developed sarcoplasmic reticulum, for developing cerebral arteries in particular, calcium entry through L-type Ca2+channels constitutes the main fraction of contractile Ca2+ [46, 353, 355, 361]. For instance, following 2 min in Ca2+-free buffer in response to NE stimulation, adult MCA showed a robust increase in [Ca2+]i (measured as the Fura-2 fluorescence ratio 340/380). In contrast, in fetal MCA the tension response was barely detectable [46]. A similar contrast in responses is seen following 2 min exposure to the plasmalemmal Ca2+ channel blocker lanthium ion. In addition, inhibition of NE-induced contraction by several L-type Ca2+ channel blockers (e.g., the dihydropyridine nifedipine, the phenylalkylamine verapamil, and the benzothiazepine diltiazem) is much greater in fetal cerebral, compared to adult arteries. In contrast to these results, receptor-operated Ca2+ channel blockade showed no significant effect on NE-induced vessel contractility or [Ca2+]i [46]. Again, this emphasizes the greater contractile dependence of fetal SMC on extracellular Ca2+ entry.
With this almost total reliability of fetal SMCs on [Ca2+]o (as well as their greater CaS), the question arises as to the extent to which this is associated with mechanisms for ample Ca2+ influx across the plasma membrane? On the basis of specific antagonist (3[H] PN200–110) binding, L-type Ca2+ channel density in fetal and newborn cerebral arteries was more than twice that of adult [45] (Table 5). This dramatic age-related difference in Ca2+ channel density also was observed in the CCA, but not aorta [45]. Using a monoclonal antibody against the L-type Ca2+ channel α1C-subunit, Western immunoblot analysis showed a similar relationship [45]. In ovine plasma, fetal values were significantly high for both total and ionized Ca2+ [45]. Also in fetal MCA, the increases in tension and [Ca2+]i induced by the L-type Ca2+ channel agonist Bay K-8644 were similar to that of NE or other agonists. In contrast, adult vessel contractile and [Ca2+]i responses to Bay K-8644 were significantly less than that to NE [45]. These results support further the idea that, for contraction, fetal cerebrovascular SMCs rely almost exclusively on extracellular Ca2+ flux, as opposed to the adult that exhibits more well-developed SR, and perhaps better Ca2+ buffering as well. This is not to suggest that mitochondrial and nuclear Ca2+ pools are unimportant; however, evidence does not suggest that they play a major role in terms of readily releasable Ca2+ stores. That fetal cerebral arteries are so completely dependent upon extracellular Ca2+ (as well as greater CaS) for contraction should not be unexpected. Such appears to be the case for developing smooth muscle of the trachea [362], stomach [363], bladder [364], and myocardium [365, 366]. Maximum 45Ca uptake in ovine newborn and adult middle cerebral arteries, in response to either K+ depolarization or serotonin stimulation, also showed a significant age-related difference, being ~50% greater in newborn than in adult [348].
Potassium Channels
In vascular smooth muscle cells, electro-mechanical coupling describes the relation between membrane potential and contractile tone. Important components of this coupling are both the potassium (K+) and the L-type calcium channels, the latter of which, by virtue of voltage-dependent conductivity, directly couples changes in membrane potential to the rate of Ca2+ influx [367], and thus intracellular calcium concentration [Ca2+]i [368]. As noted, resting membrane potential is a primary determinant of voltage-gated Ca2+-channel activity, [Ca2+]i and thus vascular tone and contractility. In turn, this is determined chiefly by plasma membrane K+ channel activity, which in part, is a function of sodium-potassium-ATPase (Na+-K+-ATPase) activity. The adult vasculature demonstrates a high degree of specialization of this pump [369] in terms of localization [370], and a relatively selective inhibitor is helping to clarify some issues (Mir et al, 1987). Cerebrovascular membrane potential differs significantly with developmental age that for fetal SMCs being −38.1 ±3.4mV, while that for adult equals −45.4 ±2.5mV [345] (See Table 5).
Of particular relevance to the present discussion is the role of developmental maturation of membrane potential, and changes in K+ channel activity. In cerebral and other arteries, because of the ionic properties of the cell membrane at rest, activity change of only a few K+ channels is sufficient to alter membrane potential, and thereby tone [372]. In general, the relation of vessel diameter to membrane potential is linear, e.g., a decrease in membrane potential (less negative) is associated with increased tone and contraction [373]. Four major K+ channel families have been identified, each of which is present in cerebral arteries: Ca2+-activated (KCa), ATP sensitive (KATP), voltage dependent (KV), and inward rectifier (KIR) [86, 372, 374]. A variety of vasoactive stimuli have been shown to alter cerebral artery K+-channel activity, including α-adrenergic- and serotonergic-agonists, which, in addition to their other effects, act to inhibit K+ ef-flux, thereby depolarizing the plasma membrane and opening voltage-gated Ca2+ channels [86, 372].
A common approach to assess electro-mechanical coupling is to monitor contractile tone produced by high concentrations of extracellular potassium. This reduces the transmembrane K+ gradient, depolarizes the cell, and increases Ca2+ influx via L-type Ca2+ channels. In response to KCl-induced membrane depolarization, cerebral arteries of the fetus and newborn produce 50 to 80% contractile tension, compared to adult [17, 46, 107]. (This relation also holds for receptor-mediated responses, i.e., pharmaco-mechanical coupling; see below). Nonetheless, when normalized relative to arterial wall cross-sectional area, and expressed as active stress (10−6 dynes·cm−2), the Kmax (peak height as a fraction of the maximum response to K+ depolarization; a measure of “efficacy”) for fetus, newborn, and adult do not differ significantly [17]. Additionally, the pD2 value (negative logarithm of the half-maximal concentration for agonist; a measure of “sensitivity” or “potency”) for K+ depolarization, is significantly greater in immature than in mature arteries [17, 46, 147, 375].
KCa Channels
Differences in activity of plasma membrane K+ channels, and their interaction with L-type Ca2+ channels, appear to account for important differences in agonist-induced responses of fetal and adult cerebral arteries [345, 349, 375]. For instance, in the presence of the KCa channel activator NS-1619, NE-induced tension in fetal MCA was greatly inhibited, as compared to adult, supporting the idea of greater dependence on extracellular Ca2+ [375] (Table 5). Administration of the KCa channel blocker iberiotoxin followed by ryanodine totally eliminated the increase in tension and [Ca2+]i seen with ryanodine alone (see below). With other evidence, this strongly suggests coupling of the sarcoplasmic reticulum (SR) ryanodine receptor to the KCa channel [46]. KCa channels also play an important role in modulating (5-HT)-induced MCA contractility. Also in adult, the KCa channel antagonist iberiotoxin enhanced maximum serotonin-induced responses to 82% from 59% [376].
To explore further subtleties in cerebral artery voltage-gated K+ channel function, we performed patch-clamp electrophysiologic studies in both whole SMCs and excised membrane patches. Because “big” or “maxi” KCa (BKCa) channels respond to changes in both [Ca2+]i and membrane potential, each of which varies considerably during development, these are prime candidates for developmental regulation. In adult ovine basilar artery, perforated whole cell voltage-dependent normalized outward K+ current densities were similar to that of the rat [345, 377]. In contrast, near-term fetal myocytes showed a 30% greater net outward current density, primarily from iberiotoxin-sensitive BKCa channel currents [345] (Table 5). For each age group, excised, inside-out membrane patches revealed nearly identical BKCa channel unitary currents and Hill coefficients. Additionally, for both fetus and adult, the plot of [Ca2+]i versus voltage for half-maximal activation (V½) yielded similar and parallel relationships, as did the change in V½ for a 10-fold change in [Ca2+]i, and the increase in channel activity for a given degree of depolarization. In contrast, however, at varying [Ca2+]i the relation between BKCa channel open probability and membrane potential was left-shifted for fetal myocytes, compared to adult. Thus, the [Ca2+]i for half-maximal activation, i.e., calcium “set-point” at 0 mV for fetal myocytes was significantly less than that for the adult, i.e., greater sensitivity to [Ca2+]i (Table 5); and probably accounts for their increased BKCa current density [345]. These findings raise important issues regarding the role of various K-channels in regulating L-type Ca2+ channel activity, and how regulation of these channels differs as a function of developmental maturation.
As noted above, cerebrovascular plasma membrane potential differed significantly with age, that for fetal SMCs (Table 5). Because both BKCa− and L-type Ca2+ channels are voltage dependent, a more depolarized membrane potential in the less mature organism should correspond to elevated [Ca2+]i. As noted above, in contrast to this prediction pressurized fetal cerebral arteries had a [Ca2+]i almost 30% less than that of adult [107] (Table 5). Acting as a negative feedback mechanism, however, BKCa channels should repolarize the membrane potential. In relation to this more depolarized state, immature rat myocytes also display a significantly less Ca2+ “spark” activity, and a lower number of spontaneously transient outward current events [378, 379]. This relative absence of spontaneously transient outward currents, presumably resulting from a failure of functional coupling between Ca2+ sparks and BKCa channel activity, would tend towards a more depolarized state with lower membrane potential and greater basal activation of voltage-dependent Ca2+ entry. Thus, the finding that the fetal vascular SMCs have a more depolarized membrane potential, yet lower [Ca2+]i and greater BKCa channel current density may seem counterintuitive, but as noted above probably result from the lower Ca2+ set point. These maturational differences also may result from intrinsic dissimilarities between adult and fetal SMC in terms of expression levels of several proteins such as calcium buffers (calbindin, calreticulin, calsequestrin, et cetera), and plasma membrane and/or SR-associated channels and pumps. Few of these factors have been examined, however, and further studies are required to explain these intriguing findings.
Considerable evidence supports the role of protein kinases in regulating BKCa channel activity [380]. To test the hypothesis that age-related differences in Ca2+ set-point result from developmentally regulated channel phosphorylation, in excised, inside-out membrane patches we examined the responses to selective activation and inhibition of channel-associated protein phosphatases and kinases. In fetus, control V½ values were one-third of those of adult, while by 20 min following patch excision the voltage activation of the BKCa channel had right shifted (i.e. toward a more positive V½ value) to be equal [349] (Table 5). In patches from both age groups, at 20 min adding either the PKA catalytic subunit or purified PKG shifted BKCa channel voltage activation back to the left. Adding ATP alone also left-shifted the BKCa channel voltage activation for both groups, suggesting the presence of endogenous protein kinase activity in the isolated patch preparations [349]. Also, in fetal myocyte patches, the rate of the right-shift (i.e., dephosphorylation) was three times more rapid than that seen in adult, suggesting that fetal myocytes have significantly more associated phosphatase activity. Rate measurements also suggested that PKG left-shifted (i.e., phosphorylated) the fetal BKCa channel activity much more rapidly than in the adult. In contrast, the rate of PKA left-shift was much lower in fetal than in adult membrane patches. For both fetus and adult, simultaneous addition of the PKA inhibitor KT-5720 and PKC inhibitor KT-5823 prevented the effect of added ATP in left-shifting BKCa channel activation curves. However, neither of these inhibitors prevented the ATP-associated left-shift of these activation curves, suggesting the presence of endogenous PKA and PKG activity [349]. These findings demonstrate that BKCa channel activity is modulated during development by differential phosphorylation, and that with development the activities of channel-associated kinases and phosphatases change substantially. They also suggest that the functional stoichiometry of the channel-associated kinases and phosphatases varies significantly during development, and that such variation may be a hitherto unrecognized mechanism of ion channel regulation [349].
To test further the hypothesis that the developmental change in Ca2+ set point results from differences in channel phosphorylation and dephosphorylation, in excised, inside-out membrane patch preparations of basilar arterial SMCs from adult and fetal sheep, we measured BK channel activity in four states of phosphorylation; i.e., 1) native, 2) dephosphorylated, 3) PKA phosphorylated, and 4) PKG phosphorylated. BK channels from both age groups exhibited similar voltage activation curves and Ca2+ set points for the dephosphorylated, the PKA phosphorylated, and PKG phosphorylated states. In contrast, in the native phosphorylation state, fetal BKCa channels exhibited voltage-activation curves significantly (19.3±3.1 mV; P<0.05) to the left of those of the adult, as well as a Ca2+ set point much less than that of adult. When plotted as a function of [Ca2+], the voltage at half-maximal BKCa channel activation for the two age groups at each of the phosphorylation states showed a linear relationship [381]. Associated PKG activity was 3-times greater in BKCa channels from fetal than adult myocytes, whereas associated PKA activity was 3-times greater in channels from adult than fetal myocytes [381]. These studies add further support to the idea that BKCa channel Ca2+ set point can be differentially modulated by channel phosphorylation and dephosphorylation, and that compared to adult, the lower BKCa channel Ca2+ set point of fetal myocytes can be explained by a much greater extent of channel phosphorylation [381].
In cerebral artery SMCs, BKCa channel activity is modulated by endogenous protein kinases and phosphatases, and in the regulation of vascular tone these change with development. In comparison to adult, fetal SMCs appear to have more associated phosphatase activity, as well as endogenous PKG activity, but significantly less channel-associated PKA activity [349]. These age-related differences in protein kinase and phosphatase activities suggest strongly that differential modulation by phosphorylation accounts for the increased Ca2+ sensitivity of fetal, compared to adult, vascular SMCs [381]. They also raise vital questions regarding the mechanisms of these differences and their biologic/physiologic meaning in terms of lower resting [Ca2+]i, a more depolarized resting membrane potential, and so forth. These data suggest that in association with their lower levels of spontaneously transient outward current events and Ca2+ “spark” activity [378, 379], differential phosphorylation may allow the fetal BK channel, acting as a Ca2+ sensor, to detect unsynchronized Ca2+ release from ryanodine receptors and thus modulate L-type Ca2+ channel activity and vascular tone.
KATP, KV, and KIR Channels
The case for KATP channel activation is less clear. Following NE-induced contraction, in both fetus and adult tension was inhibited to a similar extent by the KATP channel activator pinacidil; however, [Ca2+]i decreased to a much greater extent in the fetus [375] (Table 5). In contrast, in the presence of pinacidil NE-induced tension was inhibited ~40% in adult, but not fetal, MCA; and in neither age group were [Ca2+]i responses to NE significantly altered [375]. Additionally, in adult but not fetal MCA the KATP channel blocker glibenclamide showed a modest effect in inhibiting NE-induced increase in tension and [Ca2+]i [375]. In partial agreement with these observations, in second- and fourth-order MCA following contraction with 5-HT (10−6M) and in response to the KATP channel activator lemakalim, relaxation with decreased tension was much greater in the adult than that of newborn [382]. As with NE-induced responses [375], following KATP channel inhibition by glibenclamide, serotonin (5-HT) stimulation resulted in a significant decrease in sensitivity in fetal, but not adult, arteries [376]. These results suggest significant increases in KATP channel activity with maturational development.
In regard to KV and KIR channel function, although cerebral arteries from the two age groups showed slightly differing responses in the presence of channel activators and blockers, these were not dramatic [375]. In a related study in adult ovine second-order branch MCA, the Kv antagonist 4-aminopyrine produced stretch-dependent contractions at 100 and 145% of optimal length (as did the KCa channel antagonist iberiotoxin). By way of contrast, in fetal MCA these contractions were quite small, and KIR channels did not appear to contribute to resting vascular tone [376]. Also in contrast to the effect on NE-induced responses, the KV channel antagonist 4-AP enhanced serotonin-induced responses in adult but not fetal arteries. In addition, 4-AP resulted in the maximum serotonin-induced responses increasing 60 to 80% in arteries of both age groups [376]. An important question regards the abundance and distribution of the several K+ channels in the cerebral vasculature.
In vivo Studies of K+ Channels
Relatively few studies have examined the role of K+ channels in the regulation of CBF in vivo. In the anesthetized newborn (<7 days) piglet, by use of a cranial window, several groups have examined the in vivo regulation of K+ channel activity as measured by pial artery diameter, in response to various agents. For instance, Armstead and colleagues have shown that KCa channels are involved in hypoxia-induced vasodilatation [269], and both KCa and KATP channels contribute to hypotension-induced [339] vasodilatation. Again, the caveats noted above regarding the cranial window technique are germane.
Non-Specific Cation and Anion Channels
These channels have received deserved attention in the pulmonary and other vasculature, however their role in cerebral arteries, much less changes with development, are relatively unexplored.
PLASMA MEMBRANE RECEPTORS
G-protein Coupled Receptors (GPCRs)
These receptors provide a common structure and mechanism for pharmaco-mechanical coupling of information transfer between the extracellular and intracellular environments [383, 384]; and these receptor proteins share the overall structural plan of an extracellular N terminus, seven transmembrane domains, and an intracellular C terminus. When activated, the receptor molecule undergoes a conformational change that facilitates its interaction with, and activation of, specific intracellular signaling proteins such as heterotrimeric guanine nucleotide binding proteins (G proteins), which then relay signals to intracellular signaling networks [383]. Biogenic amines, such as catecholamines (norepinephrine and epinephrine), and serotonin (5-hydroxytryptamine, 5-HT), activate their receptors by entering into the transmembrane core. Several regulatory processes, collectively known as receptor desensitization (which occurs by G protein-coupled receptor kinase (GRK)-mediated phosphorylation and βarrestin binding [385]), can attenuate GPCR-regulation to modulate these signal transduction pathways. In addition, β-arrestins can serve as signaling intermediates in their own right, connecting the receptors to downstream effectors such as mitogen activated protein kinases (MAPK) [386]. Also, as emphasized by Pearce and colleagues, receptor-G protein coupling is a function of agonist-receptor affinity, receptor density, and gain of the coupling mechanism [17, 252, 355]. The G protein complex of α, β, and γ subunits may be either stimulatory or inhibitory, depending on the specific isoforms and related factors [383]. Despite the importance of G proteins in signal transduction, we are aware of no studies on this mechanism in developing vascular SMCs.
Alpha-Adrenergic-Mediated Contractile Responses
An example of G-protein-mediated pharmaco-mechanical coupling involves the influences of membrane receptor activation by the binding of the catecholamine and sympathetic neurotransmitter norepinephrine to adrenergic receptors, second messenger signaling, contractile protein phosphorylation state, and contractile tone. This coupling is highly specialized for each of the many receptor types and second messenger systems present in vascular SMCs; and is in contrast to electro-mechanical coupling, which chiefly governs the relations between membrane potential, Ca2+ influx, [Ca2+]i, and contraction. In general, α-adrenergic-mediated pharmaco-mechanical coupling operates via both Ca2+-dependent and Ca2+-independent mechanisms. The cerebral vasculature is rich in adrenergic innervation, which arises predominantly from the superior cervical ganglion [1, 3, 387–390]. In addition to their trophic function [387], adrenergic nerves appear to play a particularly important role under conditions of hypoxia, hypertension, and other stress [3, 391].
Considerable evidence supports the importance of the α-adrenergic system in ovine and porcine cerebrovascular reactivity [14, 15, 392–394]. Cerebral arteries contract in response to NE via stimulation of postjunctional α1- and/or α2-adrenergic receptors (AR) [392, 395], and this reactivity varies with both vessel size [14, 15, 393] and developmental age [15, 17, 213, 393, 394, 396]. For instance, as measured by pD2 value (−log EC50), smaller cerebral arteries appear to be more sensitive to α-adrenergic agonists than are larger vessels [393, 397]. One would anticipate that, for the most part, adrenergic-mediated responses will occur via neuronally released neurotransmitters, rather than those circulating which must cross the BBB. Given the higher pD2 values in fetal arteries, the effect of perivascular stimulation should be proportionally greater in fetal than adult arteries. An understanding of the ontogeny of cerebrovascular adrenergic-mediated contraction and its regulatory factors should help to define the normal physiologic responses mediated by these receptors.
In several artery types, the relative increase in agonist response with developmental age is greater than the increase in response to K+ depolarization, suggesting increased efficiency of coupling between the agonist receptor and contraction [13]. This also appears to be the case in the cerebral arteries [344, 353, 375] (Table 6). For intracellular Ca2+ levels, the relative responses of fetal and adult cerebral arteries to adrenergic agonists were of a similar magnitude. Nonetheless, when NE-induced tension was plotted as a function of [Ca2+]i, the fetal MCA showed markedly lower Ca2+ sensitivity (i.e., lower slope) than that of adult [344]. As an example of the varying response to NE with developmental age, in several species including fetal sheep [215, 398], and fetal and newborn baboons, the MCA in vitro response was more sensitive to NE stimulation than was seen in the adult [213]. Also in fetal and newborn sheep, in vivo pial arteriolar responses to both NE and electrical stimulation of the superior cervical ganglion were greater than those for adult [215]. For both newborn and adult sheep MCA, the artery required less stretch to attain optimal length, i.e., greater vascular compliance [393]. In both 2nd- and 4th-order cerebral arteries the Hill coefficient (the slope of the agonist concentration-response relation) increased significantly with age [393]. As an aside, in rat aorta and small mesenteric arteries the contractile response to NE at a given [Ca2+]i was decreased in aged animals (70 to 100 weeks), as compared to adults (12 to 14 weeks), suggesting further decreased Ca2+ sensitivity with ageing [399].
Table 6.
Alpha Adrenergic-Mediated Calcium-Dependent Pharmaco-Mechanical Coupling Contractile Mechanisms with Development
| Physiologic Variable | Fetus | Adult | Reference |
|---|---|---|---|
| NE-Induced Tension | 1.4±0.1 (82) | 1.7±0.1 | [344] |
| pD2 | 6.1±0.1 | 6.1±0.1 | |
| NE/K+max (% max) | 139±15 (131) | 106±10 | |
| NE-Induced Ratio* | 0.17±0.03 (121) | 0.14±0.02 | [344] |
| pD2 | 6.5±0.1 (‡) | 6.2±0.1 | |
| NE/K+max (% max) | 85±10 (90) | 94±8 | |
| Slope Tension vs Fluorescence Ratio | 4.1±0.4† (56) | 7.3±0.6 | [344] |
| PHE-induced Tension with Cytochalasin D (% PHE max) | 40±5 (64) | 62±8 | [236] |
| PHE-induced Tension with Nocardazole (%PHE max) | 102±10 (106) | 96±10 | [236] |
| α1-AR Subtype | |||
| Dissociation constants (KB) of antagonist inhibition | |||
| α1-AR (Prozosin, 10−8 M) | 1.1 × 10−10 M‡ | 6.4 × 10−10 M | [42] |
| α1A-AR (5-MU, 10−8 M) | 0.2 × 10−9 M | 1.1 × 10−9 M | [42] |
| α1B-AR (CEC, 10−5 M) | NSI | 5.3 × 10−10 M | [42] |
| α1D-AR (BMY-7378, 10−7 M) | NSI | 6.7 × 10−9 M | [42] |
| Subtype Protein Abundance | |||
| α1A-AR (% of α-actin) | 10±1%† (9.1) | 110±10% | [42] |
| α1B-AR (% of α-actin) | 15±2%† (14) | 105±10% | [42] |
| α1D-AR (% of α-actin) | 20±2%† (20) | 100±10% | [42] |
| α2-AR Density (fmol·mg Protein−1) | 20±18† (386) | 52±6 | [396, 407] |
| Ins(1,4,5)P3 Basal | 40±10 (111) | 36± | [408] |
Values are mean ± SE of 5 or more in each group;
Values in parenlheses represent % of adult value;
Significantly different from adult value:
P<0.01;
P<0.05;
Basilar artery;
Because these are logarithmic functions, percent change is not an accurate or appropriate representation;
CCA; NSI – No Significant Inhibition
α1-Adrenergic Receptors and their Subtypes
Cerebral blood vessels are sensitive to NE via stimulation of α1- and/or α2-receptors [392, 400], and this sensitivity varies with species. α1-ARs appear to play a key role in cerebrovascular contraction in humans and monkeys [400] and sheep [14, 15, 393], whereas α2-ARs appear to be more important in dogs [400] and pigs [401]. In contrast, one report suggests that α1-AR but not α2-AR are important in the newborn piglet [402]. Several lines of evidence suggest that adrenergic innervation plays a critical role in vascular trophic function [6, 403]. These subtypes also appear to vary with arterial size, e.g., the receptor mediating responses to NE shifts from the α1-AR subtype in large arteries to α2-AR in smaller vessels [404, 405]. Variations in vascular agonist sensitivity may result from a change in either receptor density or affinity, as detailed in both the variable receptor hypothesis, and the concept of receptor “reserve”, i.e., the number of receptors present being in excess of the number required to elicit a maximal contractile response [6]. In turn, these caveats suggest species and/or maturational differences in receptor structure per se, receptor-membrane microenvironment, and/or intracellular receptor-linked mechanisms.
In light of the age-related differences in response to α-adrenergic stimulation, the question arises as to the extent to which these result from differences in α1-adrenergic receptor (α1-AR) density and/or affinity. In ovine main branch cerebral arteries the receptor density (βmax) values of the near-term fetus were only a fraction of the values of the newborn and adult [42]. Nonetheless, these arteries showed no significant relation of NE maximal contractile response to α1-AR density, and α1-AR affinity did not differ with developmental age [15]. As an aside, α1-AR density values in the fetal and newborn common carotid artery were double those of CA, again demonstrating the independent regulation of α1-ARs as a function of developmental age, which may relate to the development of noradrenergic nerve innervation [6, 394, 406].
In view of several aspects of differing responses of cerebral arteries to adrenergic agonists as a function of developmental age, and in the role of inositol 1,4,5-trisphosphate (Ins(1,4,5)P3)-Ca2+-dependent contraction (see below), we examined the extent to which these differences resulted, in part, as a consequence of different α1-adrenergic receptor (α1-AR) subtypes (i.e., differing densities and/or activities of α1A-, α1B-, and α1D-AR). In essence, in main branch fetal MCA the relatively selective α1A-receptor blockers 5-MU, or WB-4101, each showed significantly greater inhibition of phenylephrine (PHE)-induced tension and [Ca2+]i, and increased sensitivity to tension inhibition, compared to adult [42]. In contrast, whereas in adult MCA the α1B-AR blocker chloroethylclonidine (CEC), and the α1D-AR blocker BMY-7378 produced significant inhibition, this was not the case in fetal MCA (Table 6). Compatible with these results, in adult vessels western immunoblot analysis demonstrated an abundance of protein of α1-, α1A-, α1B-, and α1D-AR, but only modest level of the three subtypes in immature arteries (Table 6). Of interest, in fetal CA the α1D-AR was expressed significantly greater (essentially double) that of the other two subtypes [42]. In addition, in both fetal and adult MCA the α1B-AR inhibitor 5-MU and the α1B-AR inhibitor CEC each resulted in a 30 to 40% reduction of Ins(1,4,5)P3 response to PHE (3×10−5 M) [42]. In contrast, whereas the α1D-blocker BMY-7378 (10−5 M) resulted in a 15% reduction in adult, there was in Ins(1,4,5)P3 responses to PHE no significant decrease in fetal MCA [42]. Also of importance, in fetal, but not adult, CA, PHE induced a significant increase in activated (phosphorylated) extracellular regulated kinase 1 and 2 (ERK½). This increase in phosphorylated ERK½ was blocked by α1B-AR (CEC) and α1D-AR (BMY-7378) inhibitors, but not by the α1A-AR inhibitors (5-MU or WB-4101). This suggests that in addition to the fetal α1-AR subtypes playing a critical role in contractile responses, the α1B-AR and α1D-AR may be critically associated with cerebrovascular growth [42].
That fetal cerebral arteries showed much greater sensitivity than those of adult to α1-AR subtype specific inhibitors, probably reflects the relatively low abundance of subtype receptor protein in these vessels. Despite these low protein levels, in both age groups α1A- and α1B-, but not α1D-AR, subtypes appeared to mediate α1-adrenergic Ins(1,4,5)P3 responses. Also, α1D-AR, and to a lesser extent α1B-AR, appeared to function by Ins(1,4,5)P3-independent mechanisms. In addition, the blockade of PHE-induced activated ERK responses by α1B- and α1D-AR antagonists, but not those for α1A, suggests discordance in the α1-AR mediated responses to Ins(1,4,5)P3, [Ca2+]i and tension. This also suggests the possibility that in the immature organism, relative inefficiency of α1-AR coupling, or that the coupling may occur somehow in a different manner, may be a factor associated with dysregulation of cerebral blood flow [42].
At this time, the role of the α1-AR subtypes in SMC signal transduction pathways is unclear, particularly in terms of development. The complex and age-dependent pattern of developmental effects reveals that α-adrenergic pharmacomechanical coupling is regulated closely by multiple physiological mechanisms. The manner in which maturation affects these mechanisms, whether by changes in gene expression, in efficiency of message translation, through posttranslational modifications, or turnover of key signaling proteins, remains undetermined. Given that the α-adrenergic pathway is critically important for cardiovascular regulation during embryonic and fetal development, this is a promising field for future investigation.
α2-Adrenergic-Mediated Contractile Responses
Cerebrovascular α2-AR, which may be prejunctional as well as postjunctional, have been shown to play an important role in the human [409, 410], monkey [410], cat [411], pig [392], and perhaps the dog [412]. α2-AR exist in at least three subtypes, defined pharmacologically and by molecular cloning, and each has been shown to inhibit adenylate cyclase activation, and thus reduce cAMP and PKA activities [413, 414]. In sheep, α2-AR density values were three- to four-fold greater in cerebral arteries and microvessels of the near-term fetus, compared to the adult [396] (Table 6). Also, α2-AR density values were significantly greater in these smaller vessels than in the larger extracranial CCA. In fetal MCA, in response to NE in the presence of the α2-AR antagonist yohimbine, neuronal NE blockade by cocaine, blockade of extraneuronal NE reuptake by deoxycorticosterone, and minimization of sympathetic nerve-associated pre-synaptic α2-AR by tetrodotoxin, the pD2 was decreased several orders of magnitude. Adult arteries showed no such effect. Also in fetal, but not adult, vessels in response to PHE in the presence of the α2-AR agonist UK-14304, the sensitivity was shifted to the right [396]. Stimulation-evoked NE release from fetal cerebral arteries also was significantly greater than from the adult. Nonetheless, when prejunctional α2-AR were inhibited by idazoxan, thereby blocking NE negative feedback on further NE release, the fractional NE release increased to a similar extent in vessels of both age groups [396]. This suggests that in fetal cerebral arteries, a significant α2-AR component is postjunctional, while in the adult the majority of functional α2-AR are prejunctional. Some have argued that α2-AR play a greater role in vascular contraction in vivo [415], as it has been difficult to demonstrate the functional role of α2-AR in vitro [226, 416]. The difficulty may lie in the fact that α2-AR agonists do not produce vasoconstriction directly, but provide an ancillary drive to the vasoconstrictor stimulus produced by α1-AR agonists [414, 417]. Thus, α2-AR appear to play a role in mediating adrenergic contractions of fetal SMCs, but their role in the adult is less clear. α2-AR also may play a role in regulating cerebrovascular tone under conditions of hypoxia or other stress; however, evaluation of this possibility await in vivo studies.
Serotonergic-Mediated Responses
Another important pharmaco-mechanical coupling pathway in cranial arteries is that activated by the binding of serotonin, a monoamine neurotransmitter, to its receptors. Serotonin has a number of effects on the cerebral vasculature of importance, including its role in hemostasis, platelet aggregation, and cerebral vasospasm following intracranial hemorrhage [418]. In this regard, 5-HT may play a role in coupling perfusion and metabolism via serotonergic perivascular innervation [419]. Although the precise role of serotonin in cerebrovascular responses is unclear, it is unquestionably of importance. Serotonin receptors are of several families and subtypes, of which the 5-HT types 1 and 2 (5-HT1 and 5-HT2, respectively) appear to be associated with vascular SMC [420–422].
In fetal, newborn, and adult sheep, in relation to the K+-induced response, the maximum contractile response to combined 5-HT (10−5M) and histamine (2×10−5M) decreased with maturation dramatically in CCA, but much less so in MCA [17] (Table 7). Based upon serotonin antagonist dissociation constant (pKb), the receptors mediating response to 5-HT shift from predominantly 5-HT2α in CCA to a mixed 5-HT1/5-HT2 population in MCA to a predominantly 5-HT1 subtype in second branch MCA [423]. Ovine 5-HT2 receptor density is much greater in larger arteries with more abundant SR. In turn, this receptor density is much less in smaller arteries, in which 5-HT1 expression is greater [423]. Cerebral artery responses to serotonin and other amines vary as a function of both artery size and type, and for both CCA and MCA these were similar in newborn and adult sheep [348]. In both MCA from newborn lambs and CCA, 45CA uptake in response to 5-HT (10−4 M) was one and one-half times that of adult vessels [348]. In cranial arteries of sheep permeabilized with β-escin, 5-HT-induced contractile responses were significantly greater in the fetal CCA, and both main branch and 2nd-order MCA, than that for the adult [356]. This Ca2+ sensitivity was G protein-dependent, being significantly increased by the GTP analogue GTPγS, and attenuated by the G protein inhibitor GDPβS [356]. Despite the age-related difference demonstrated in these responses, there was no significant difference among the several artery types [356]. Also in ovine fetal and adult basilar arteries, in relation to K+, 5-HT-induced tension was one-third greater in the fetus, while in relation to the Δ [Ca2+]i the fetal artery was only one-half that of adult [424] (Table 7). These results gave values of Ca2+ sensitivity for fetal vessels greater than twice that of adult. This greater CaS in developing arteries also was evident in the slopes of 5-HT-induced tension to Δ [Ca2+]i [424].
Table 7.
Serotonergic-Mediated Calcium-Dependent Pharmaco-Mechanical Coupling Contractile Mechanisms with Development
| Physiologic Variable | Fetus | Newborn | Adult | Reference |
|---|---|---|---|---|
| Amine to K+ Response Ratio (10−5 5-HT, 1.22×10−1 K)* | 103±10 (343) | 50±6 (167) | 30±4 | [17] |
| Amine to K+ Response Ratio (10−5 5-HT, 1.22×10−1 K)* | 90±10 (129) | 75±8 (107) | 70±8 | [17] |
| Amine-Induced Tension (g) (10−5 M 5-HT and 2×10−5 M Histamine) | 2.0±0.2 (91) | 2.2±0.2 | [245] | |
| Amine/K+max (%) | 85±5a (224) | 38±5 | [245] | |
| 5-HT · K−1 induced tension† | 2.0±0.3 (133) | 1.5±0.2 | [346] | |
| 5-HT · K−1 induced Δ [Ca2+]i† | 40±5 (53) | 75±7 | [346] | |
| 5-HT · K−1 tension · [ΔCa2+]i† | 0.05 (250) | 0.02 | [346] | |
| pD2 for 5-HT-induced contr†‡ | 6.7±0.1 | 6.3±0.1 | [47] | |
| Maximum 45Ca uptake in response to 10−5 M 5-HT (μM · g dry wt−1 · min−1) | 201±15 (156) | 129±23 | [348] | |
| Maximal response to 5-HT | - | To 82% from 59% | [376] | |
| pD2 for 5-HT response with BK channel blockade‡ | To 7.51 from 7.05 | To 7.75 from 7.15 | [376] | |
| Enhance 5-HT response with BK channel blockade (also %) | NSC | To 82 from 59 | [376] | |
| pD2 for 5-HT response with Kv channel blockade | - | To 7.49 from 7.15 | [376] | |
| Enhance 5-HT response with Kv channel blockade (also %) | To 85 from 67 (100) | To 79 from 59 | [376] | |
| pD2 for 5-HT response with KATP channel blockade‡ | To 6.71 from 7.02 | NSC | [376] | |
| pD2 for 5-HT response with ERK1/2 inhibition‡ | 10-fold | 2-fold | [19] | |
| Emax with ERK1/2 inhibition (% decrease) | ~37 | ~17 | [19] |
Values are mean ± SE of 5 or more in each group;
Values in parentheses represent % of adult value;
Significantly different from adult value:
P<0.001;
Basilar artery;
Because these are logarithmic functions, percent change is not an accurate or appropriate representation;
CCA;
NSC – No Significant Change
In adult, but not fetal, MCA, the Kv sensitive channel blocker 4-aminopyridine (5×10−3 M) enhanced the pD2 for adult, but not fetal vessels, the BK channel blocker iberiotoxin (10−7 M) enhanced maximal 5-HT contraction. Also, in contrast in fetal but not adult vessels, the KATP sensitive channel blocker glibenclamide (10−4 M) attenuated the pD2 for 5-HT. Coupled through Ins(1,4,5)P3 synthesis to Ca2+ release, these appear to increase myofilament Ca2+ sensitivity. In response to ERK1/2 inhibition, the pD2 values for fetal and adult CCA fell ~10- and 2-fold, respectively, while Emax values decreased ~37% and 17%, respectively [19]. As with the α-adrenergic system, development clearly affects multiple components of the serotonergic pathway; however, the mechanisms through which these influences are mediated remain unexplored.
Cholinergic-Mediated Responses
The cerebral arteries of a number of species have been shown to be innervated by nerves containing acetylcholine (ACh), neuropeptide Y, vasoactive intestinal polypeptide, calcitonin gene-related peptide, and other vasodilators [425–427]. Based on anatomic and physiologic studies in the early 1930s, cerebral cholinergic nerves were suggested to mediate vasodilatation [428]. A wealth of evidence from subsequent studies supports this idea, and that ACh is an important transmitter in these nerves [429–431]. The enzyme choline acetyltransferase (ChAT), which generates ACh, has been identified as an appropriate marker for these nerves [432]. Several reports have demonstrated that these nerves originate in the sphenopalatine ganglion and several other ganglia, and traverse to the cerebral cortex, hippocampus, and other areas [426, 427]. Of importance the enzyme neural NOS (nNOS) is colocalized with ChAT in most of these neuronal ganglia, suggesting that these nerves be named cholinergic nitric oxidergic [426]. Based on these and other studies, evidence supports the idea of ACh activating the release of NO to cause vasodilatation [426, 433, 434]. Nonetheless, some investigators question the physiologic role of these nerves, as cholinergic blockade fails to alter resting CBF [435], and muscarinic receptor inhibition suggests that the cotransmitters NO or VIP probably account for any increase in CBF [427, 436].
In terms of the role of cholinergic, nitric oxidergic neurons in the developing cerebral vasculature, not a great deal is known. In isolated cerebral arteries from immature baboons [213] and pial arteries in anesthetized newborn piglets [296, 297] acetylcholine induced contraction rather than dilation. In contrast, in anesthetized near-term fetal and newborn sheep, ACh resulted in significant vasodilatation of pial arteries [437]. Based on these and other studies, these opposing observations probably represent a species difference, rather than a lack of endothelium-dependent vasodilatory mechanisms. Additionally, in middle cerebral arteries from premature fetal (0.8 and 0.9 gestation), term fetal, and newborn lambs, no relaxation to ACh (10−10 to 10−4 M) was observed, despite ACh producing relaxation in the isolated adrenal, femoral, and renal arteries [438]. In contrast, over a period of years in our in vitro studies of endothelium-intact fetal and newborn MCA, ACh administration (10−5 M) routinely results in 30 to 40% relaxation. This is not the case in endothelium-denuded vessels, and is the test we perform to insure that the endothelium is, in fact, completely removed [14, 16, 375, 439, 440]. Thus, it would appear that final judgment with regard to the physiological role of ACh must await in vivo studies in unanesthetized, endothelium-intact sheep or other laboratory animals, and their comparison with the adult of that species.
CALCIUM-DEPENDENT SECOND MESSENGER SYSTEMS
Phospholipase Cβ–Inositol(1,4,5)-Trisphosphate-Mediated Responses
From intracellular organelles, agonist-induced Ca2+ release that induces SMC contraction is mediated by phospholipase Cβ (PLCβ) synthesis and release of the second messenger Ins(1,4,5)P3 [441, 442]. Intracellular Ca2+ release by activation of ryanodine-sensitive receptors (RYN-R) on the sarcoplasmic reticulum also can be promoted by ion channel-mediated Ca2+ influx with Ca2+-induced Ca2+ release [443]. The SR appears to be the major intracellular source for both Ins(1,4,5)P3- and ryanodine-sensitive Ca2+ release [444]. Nonetheless, the relation between receptor activation and Ca2+ release is poorly defined, suggesting that coupling between receptor or ion channel activation and Ca2+ release may be regulated by a number of factors including variable receptor number and receptor reserve. An additional consideration is the distribution of Ca2+ within SMCs, which is highly heterogenous and dynamic, varying with artery type, size, and other factors [445–447]. A recent e-conference considered the multidimensional aspects and complexity of intracellular Ca2+ signaling [342, 360].
In view of the less robust contractile response of fetal CA compared to the adult, despite two-fold greater α1-AR density values, one might argue that other components of the pharmaco-mechanical coupling pathway limit contractile response. Consistent with the idea of greater receptor reserve, CCA basal Ins(1,4,5)P3 levels and their mobilization following α1-AR activation were similar of both age groups (Table 7). Thus, the ratio of the NE-induced Ins(1,4,5)P3 signal to α1-AR density was significantly lower in the fetal vessels [15]. In CCA NE-mediated increases in Ins(1,4,5)P3 were negligible in the fetal and newborn vessels, as compared to robust responses in the adult [14]. This difference in Ins(1,4,5)P3 responses to agonist stimulation between cerebral arteries and the CCA is, in part, reflected in the basal levels of Ins(1,4,5)P3 in the two vessel types, which were three- to four-fold greater in cerebral arteries [14, 15]. Nonetheless, neither artery type showed a significant relation of NE-induced maximum contraction to the Ins(1,4,5)P3 responses [15]. The idea that different components of the cerebral artery excitation-contraction pathway are independently regulated was supported by comparing α1-AR densities and NE-induced Ins(1,4,5)P3 responses. For main branch cerebral arteries, Ins(1,4,5)P3 response per α1-AR density increased progressively from fetus to newborn to adult [15].
The relative lack of NE-induced Ins(1,4,5)P3 response in fetal or newborn CCA contrasts with the magnitude of the NE-induced contraction in this vessel [15]; and may reflect a relatively greater NE-induced Ca2+ influx via voltage-gated Ca2+ channels [45, 344] or greater dependence upon Ca2+ pathways. In β-escin-permeabilized fetal and adult ovine CCA, contractile responses to exogenous Ins(1,4,5)P3 equaled 19±4 and 42±5%, respectively, that of the Ca2+-induced maximum (Ca2+max; i.e., that response to 10−5 M Ca2+). For vessels of both age groups, Ins(1,4,5)P3–induced contractile response decreased as a function of vessel size equaling only 9±2% and 26±5%, respectively in main branch MCA (Table 8), and 8±2% and 18±3% in second branch MCA [356, 357].
Table 8.
Related Calcium-Dependent Contractile Mechanisms with Development
| Physiologic Variable | Fetus | Newborn | Adult | Reference |
|---|---|---|---|---|
| Gqα expression (in rat portal vein, % adult value) | 400 to 500 | 100 | [448] | |
| Ins (1,4,5)P3-induced contraction (% Ca2+-induced maximum) | 9±2 (35) | 26±5 | [357] | |
| Ins(1,4,5)P3 Response to Norepinephrine (% of Basal) | 345±27 (97) | 355±23 | [407] | |
| Ins(1,4,5)P3 ·α1-AR−1 | 5.2±1.0c (45) | 11.5±1.0 | [407] | |
| Ins(1,4,5)P3-Receptor Density (fmol·mg protein−1) | 115±15 (116) | 99±5 | [449] | |
| Ca2+ Mass (nmol·mg dry wt−1) | 12±2b (400) | 3±1 | [354] | |
| Ca2+ Mass of Ins(1,4,5)P3-Sensitive pool | 1.6±0.2 (94) | 1.7±0.2 | [354] | |
| Ca2+ Mass of Ryanodine-Sensitive Pool | 1.2±0.1b (1200) | 0.1±0.1 | [354] | |
| Calcium “sparks” | Significantly less | Significantly less | Control value | [378] |
| MLCK concentration (10−6M)* | 1.8±0.1b (22) | 8.2±0.6 | [450] | |
| (normalized to fetal value) | 1.0 (26) | 3.8±0.7 | [451] | |
| MLCK maximum velocity (ng MLC phosphorylated · ng MLCK−1)* | 1.52±0.2b (585) | 0.26±0.01 | [450] | |
| MLCK In situ Fractional Activation* | 9.1±0.8b (535) | 1.7±0.2 | [450] | |
| Total MLCK activity (% MLC20 phosphorylated · sec−1)* | 7.39±0.53 (89) | 6.56±0.29 | [450] | |
| MLCP Concentration (normalized to fetal value) | 1.0 (56) | 1.8±0.2 | [451] | |
| MLC20 concentration (10−6M)* | 236±44 (119) | 198±28 | [450] | |
| Phosphorylated/Total MLC20 (% Adult Value) | (64) | 100 | [9] | |
| CPI-17 – Total (% Adult Value) | (15) | 100 | [9] | |
| Calmodulin concentration (10−6M)* | 12.4±1.3b (41) | 30.6±3.7 | [450] | |
| (normalized to fetal value) | 1.0 (31) | 3.2±0.7 | [451] | |
| HSP-27 (normalized to fetal value) | 1.0 (50) | 2.0±0.2 | [451] |
Values are mean ±SE or 5 or more in each group;
Values in parentheses represent % of adult value;
Significantly different from adult value:
P<0.01;
P<0.05;
CCA
An issue not to be ignored is that cellular constituent values for Ins(1,4,5)P3, or other second messenger, must be normalized to a standard tissue characteristic. In most studies, this standard is per mg protein. Because of differences in cell size or composition, however, one might argue that intracellular constituents should be normalized in terms of intracellular water volume, this being smaller in fetal vessels [147]. Nonetheless, when calculated by three measures: per min per mg wet weight, picomoles per mg wet weight, or micromoles per liter water, the expression of Ins(1,4,5)P3 values in these different ways did not alter interpretation of the data [15].
Sarcoplasmic Reticulum and Ca2+ Stores
As detailed above, vascular SMC contraction is dependent on an increase in cytosolic free Ca2+ concentration as a result of both rapid Ca2+ release from intracellular stores, chiefly sarcoplasmic reticulum, and Ca2+ influx via plasmalemmal Ca2+ channels. Although the vascular SMC Ca2+ dependence on signaling is unquestioned, irrespective of developmental age the intracellular distribution of Ca2+, the several Ca2+ pools and their relative size, and the regulatory mechanisms for these dynamic changes are poorly understood. Vascular smooth muscle SR is a membranous tubular system with junctional components closely approximating the plasma membrane, as well as deeper more central portions contiguous with rough endoplasmic reticulum and the nuclear membrane [452]. In addition to actively transporting Ca2+ from the cytoplasm into its lumen via Ca2+-ATPase (SERCA) and thereby enhancing relaxation, upon stimulation the SR rapidly releases its luminal Ca2+ into the cytosol. Ca2+ then binds to calmodulin and other Ca2+ binding proteins, and the Ca2+-calmodulin complex activates myosin light chain kinase (MLCK) to effect contraction [453, 454]. In addition to buffering Ca2+ entry from the extracellular space into the cell [455, 456], in mature SMCs, the SR serves as the principal Ca2+ store and contributes significantly to Ca2+ release and intracellular signaling [360, 441, 445, 456–458]. Functionally, vascular SMC SR Ca2+ stores may be divided into two [459], three [460], or four [354] compartments. The Ins(1,4,5)P3-releasable store is activated by that second messenger, the receptor of which is a major Ca2+ channel mediating signal transduction [461]. The ryanodine-sensitive Ca2+ store releases Ca2+ in response to ryanodine (RYN), as well as to caffeine, and appears to be involved in Ca2+-induced Ca2+ release (CICR) [462, 463]. SR Ca2+-ATPase is believed to fill both of these stores [464, 465]. Several lines of evidence suggest that Ca2+ compartmental heterogeneity allows vascular SMCs to generate spatially and temporally distinct Ca2+ signals to regulate specific Ca2+-dependent processes [360, 441, 466]. The role of intra-cellular Ca2+ stores and their sensitivity to various agents may differ greatly as a function of species, vessel type, and size. In light of age-related differences in vascular SMC Ca2+ dynamics in relation to Ins(1,4,5)P3- and ryanodine-release mechanisms, the question arises as to the nature of SR Ca2+ stores, their number and relative size, and the kinetics of their Ca2+ release with development. In one-month-old rabbit and rat aortic SMCs, the SR volume appeared smaller than that of adult [160].
An important consideration is the role of SR in release of minute amounts of Ca2+ as “sparks” via ryanodine receptors. As noted, such sparks activate plasma membrane BK channels to hyperpolarize the cell [372]. From a developmental standpoint, in neonatal rat basilar artery SMCs the Ca2+ spark frequency is a thousand-fold less than that activity in the adult [378, 379]. Despite such very low Ca2+ spark activity, with differential phosphorylation the much greater Ca2+ sensitivity (i.e., low Ca2+ set-point) may allow fetal BK channels to act as a Ca2+ channel sensor to modulate L-type Ca2+ channel activity, and thus vascular tone [345, 349, 381].
As noted, in contrast to those in the adult, fetal cerebral arteries are exquisitely sensitive to extracellular Ca2+ concentration, [Ca2+]o, with both [Ca2+]i and tension being markedly attenuated by exposure to either zero [Ca2+]o or to pharmacological blockade of plasma membrane L-type Ca2+ channels [45, 375]. In addition, for contraction newborn MCA require more transmembrane Ca2+ uptake than the adult [348], and fetal arteries rely less on Ins(1,4,5)P3- mediated contractile mechanisms [15, 449]. In addition, the size and releasability of fetal intracellular Ca2+ stores [160, 353, 354, 467] emphasize further the dependence of fetal cerebral arteries on extracellular Ca2+.
To summarize a number of studies in regard to cerebral artery SR Ca2+ stores, and their role in Ca2+-dependent contraction, in the adult these stores play a significant role in agonist-induced contractility. For the fetus, in contrast, that role appears to depend upon the agonist used [46, 354]. As a corollary, although both adult and fetal cerebral arteries show considerable dependence on extracellular Ca2+ for sustained contraction, for the fetus that dependence is essentially complete. For instance, in both permeabilized and non-permeabilized ovine basilar artery, total Ca2+ mass, as measured by 45Ca washout, was several-fold greater in fetal arteries, compared to adult [354] (Table 8). The total Ca2+ mass of the Ins(1,4,5)P3-sensitive pool was similar for both age groups. In contrast, the relative size of the ryanodine-sensitive Ca2+ pool in the fetal artery was much greater than adult. Because the Ca2+ fraction in the ryanodine Ins(1,4,5)P3 pool was small in both fetal and adult arteries, these pools (Ins(1,4,5)P3 and ryanodine) appear to be separate. An additional Ca2+ pool, sensitive to both Ins(1,4,5)P3 and ryano-dine, was of similar size in the two age groups [354]. In addition, a Ca2+ pool sensitive to neither Ins(1,4,5)P3 nor ryanodine was ten-fold greater in fetal, than in adult, arteries. These results, with those of contractility studies in the rabbit mesenteric artery [468], suggest that although total intracellular Ca2+ content may be greater in fetal than adult vessels, the age-related changes in both Ins(1,4,5)P3- and RYN-receptor function must involve factors other than pool size alone, although the releasable fraction is less.
A related study demonstrated the agonist specific nature of these SR Ca2+-ATPase blockade by thapsigargin (TSG) significantly inhibited the response to norepinephrine (10−5 M), in the fetal vessel such inhibition was negligible. Again, this emphasizes their dependence on [Ca2+]o [353], and this dependence became particularly evident when [Ca2+]o = zero in the presence of SERCA blockade [353]. In response to RYN stimulation of Ca2+ release, fetal cerebral arteries were more sensitive than those of the adult by an order of magnitude. This increased RYN sensitivity, and the elimination of the RYN response by either nifedipine-mediated L-type Ca2+ channel blockade or zero extracellular Ca2+, also fits with the almost complete dependence of the fetal arteries on extracellular Ca2+. Importantly, in view of the lack of response to RYN in either Ca2+-free media or in the presence of a L-type Ca2+channel blocker, it would appear that in cerebral arteries of both age groups the RYN receptor is closely linked with the plasma membrane L-type Ca2+ channel [353, 375]. In addition, the lack of response to RYN in the presence of the KCa channel blocker iberiotoxin, suggests coupling of the RYN receptor to the KCa channel. In turn, this is linked to the L-type Ca2+ channel [375].
Adult cerebral arteries showed a robust response of tension and [Ca2+]i to caffeine release of SR Ca2+ stores [353]. These responses were markedly attenuated in fetal arteries, again, which were totally dependent upon extracellular Ca2+. In addition, while fetal cerebral arteries were more sensitive than those of the adult to RYN, the reverse was the case for caffeine. This suggests differing Ca2+ pools sensitive to these agents, or that caffeine acted as an inhibitor of phosphodiesterase, thus affecting cAMP and cGMP metabolism to a greater extent in fetal than in adult arteries. In both age groups, neither nifedipine blockade of L-type Ca2+ channels nor zero extracellular Ca2+ markedly attenuated the NE- induced peak contractile responses or Fura-2 fluorescence ratios [46]. Thus, in fetal cerebral arteries the elimination of agonist-induced increase in [Ca2+]i and tension in the presence of SR Ca2+ store blockers, blockade of plasma membrane L-type Ca2+ channels, or zero [Ca2+]o, further demonstrate the critical role of extracellular Ca2+, as opposed to SR Ca2+ stores, in NE-induced contraction, in these vessels. In addition, the coupling of the RYN receptor to plasma membrane BK and L-type Ca2+ channels would appear to be important in terms of the regulation of CICR and Ca2+ sparks [46, 375]. As noted, both ionized and total plasma Ca2+ concentrations were significantly greater in the ovine fetus than in the ewe [45].
In summary, although the fetal cerebral artery total SR Ca2+ store appears to be greater than that of the adult, its relative fraction of releasable Ca2+ was far less than in the adult (while the agonist-resistant pool was much greater in the fetus) [354]. In agreement with this, despite its relatively large Ca2+ stores, the fetal SR Ca2+ appears to play a relatively minor role in NE-mediated contraction [353], and as compared to the adult, its [Ca2+]i is critically dependent upon [Ca2+]o. Thus, the actual location and functional role of this large ryanodine + Ins(1,4,5)P3-resistant store remains an enigma. Related questions of importance are the relation of these Ca2+ pools to SMC phenotype, the role of coupling of Ins(1,4,5)P3- and ryanodine-receptor Ca2+-induced Ca2+ release in these SMCs, the role Ca2+ pools in overall cellular Ca2+ homeostasis, and how these changes with development.
Inositol (1,4,5)P3-Receptor
Ins(1,4,5)P3-receptor (Ins(1,4,5)P3-R) serves as a gated SR calcium channel to release Ca2+ from stores. The question thus arises as to the extent to which this receptor density and/or affinity might change with developmental age, and its importance. In MBC of near-term fetal, newborn, and adult sheep, Ins(1,4,5)P3-R density values did not differ significantly [449] (Table 8). In contrast, in adult CCA Ins(1,4,5)P3-R density was several-fold greater, as compared to fetus or newborn. Nonetheless, in neither MCA nor CCA did Ins(1,4,5)P3-R density correlate with the NE-induced maximal contraction as a percent of Kmax. Also in these studies, Ins(1,4,5)P3-R affinity showed no significant differences with developmental age or vessel type [449]. In fetal cerebral vessels expression of high affinity type 1 Ins(1,4,5)P3-R was quite low in contrast to adult. In contrast, the opposite was true for low affinity type 3 receptors [469]. Thus, by their role in the regulation of [Ca2+]i, the Ins(1,4,5)P3–R and their subtypes may play a major role in the developmental-associated modulation of vascular tone.
Sarcoplasmic Reticulum Ca2+-ATPase
Several isoforms of the SR Ca2+-ATPase exist; however, we are aware of no data on comparative expression of these isoforms with development in cerebral arteries. In aorta of both WKY and SHR rats, the level of SERCA 2a mRNA increased from 5 to 17 weeks of age, whereas that for the 2b isoform remained constant [470]. In ovine fetal myocardium, SERCA abundance was only ~40% that of adult, and its activity was two-thirds that of the adult [471]. Also, phospholamban, the integral membrane protein postulated to interact with and regulate the Ca2+ pump in fetal myocardium, was one-half that of adult [471]. A possible developmental increase in SERCA in cerebral arteries certainly fits with the results described above. In keeping with these findings, as determined by western immunoblot the density of SR Ca2+-activated ATPase in fetal cerebral arteries was less than one-half that of the adult (Zhao & Longo, unpublished)1.
Adenylate Cyclase and cAMP-mediated Responses
Soluble Adenylate cyclase (sAC) and adenosine 3′, 5′-cyclic monophosphate (cAMP) play a critical role in cerebrovascular vasorelaxation.
Guanylate Cyclase and cGMP-mediated Responses
An important pathway for vasorelaxation is that mediated by soluble guanylate cyclase (sGC), cGMP, and protein kinase G. Significant increases in cGMP follow NO release from both perivascular nerves [230] and vascular endothelium [472, 473]. The sensitivity of guanylate cyclase to NO may increase with maturation, as shown in pulmonary arteries from 3- to 30-day-old pigs [474]. An obvious question is the extent to which alterations of sGC basal levels and activity account for these differences in the cerebral vasculature in ovine neonatal cerebral arteries. By western immunoblot, sGC abundance (relative to that of kidney) exceeded that in the adult by 50% or more, and in response to NO-heme activation the apparent maximal rate of sGC activity was about twice that of the adult [250], which supports the hyper-relaxant hypothesis.
Increases in cGMP synthesis have been shown to mediate vasodilatory responses to atrial natriuretic peptide, various nitrites, as well as NO released from perivascular neurons, endothelial cells, and vascular SMCs per se [472, 473, 475]. cGMP also may be involved in cerebrovascular responses to both hypoxia [476] and hypercapnia [268]. By activating protein kinase G, cGMP can attenuate vasoconstriction by decreasing [Ca2+]i, phospholamban phosphorylation, and thus stimulating SERCA-mediated Ca2+ uptake in the SR [477], or by inhibiting Ca2+ release [478–480]. In addition, cGMP and PKG can promote vasorelaxation by decreasing myofilament Ca2+ sensitivity [481, 482], probably by activation of myosin light chain phosphatase (MLCP) [483, 484]. As noted above, PKG can activate K+ channels [485, 486], or inhibit L-type Ca2+ channels [365] to produce hyperpolarization with vasodilatation. In view of the several-fold greater activity of PKG with activation of BK channels in fetal cerebral vessels described above [349], this may be an important mechanism in the developmental regulation of vascular tone. A further class of possible substrates for PKG are proteins that regulate myofilament Ca2+ sensitivity [481], such as MLCP [487].
The effects of maturation on cGMP-mediated vasodilatation in endothelium-denuded basilar and common carotid arteries of newborn and adult sheep also have been examined. In precontracted vessels of both types exposed to either of the NO donors nitroglycerine (10−10 to 10−3 M) or S-nitroso-N-acetylpenicillamine (SNAP), for both vasorelaxants the maximum relaxation efficacy was 20 to 50% greater in the newborn than in adult [253]. Basal cGMP levels were about twice as great in the newborn basilar and CCA, as compared to the adult [253]. In a related study, basal cGMP levels in fetal CCA were several-fold greater than that of adult, and these were not altered by endothelium removal [254] (Table 4). In a further study along this line, in ovine newborn and adult CCA, the maximal rates of cGMP synthesis (10−6 M·1 cell water−1·min−1) were significantly greater in newborn than in adult, as were the maximal rates of degradation [254]. Taken together, these data suggest that the basis of the elevated cGMP levels in the newborn is, in part, a result of the higher rate of synthesis. Nonetheless, determination of the time course of cGMP-mediated relaxation responses did not vary with developmental age, the rate of rise and fall in cGMP levels were significantly slower in the newborn arteries. Theoretically this could be a consequence of lower phosphodiesterase activity in these vessels, however, in fact, the levels were greater [254], suggesting higher rates of cGMP synthesis and degradation with increased reactivity of the system. In both vessel types and in response to both vasodilators, at the peak cGMP levels relaxation was significantly more complete in newborn than in adult arteries [253]. Thus, the mechanisms necessary for cGMP-dependent relaxation appear to be fully functional in the neonatal cerebral circulation. These studies also suggest that maturational improvements in endothelium-dependent vasodilation in these and other vascular beds [474, 488] likely result from changes in function of endothelium, rather than that of the vascular SMCs. Importantly, this greater tendency of newborn cerebral arteries to relax may contribute to the greater incidence of intracranial hemorrhage observed in the immature organism [31, 34].
In a related study, using the membrane permeable and non-metabolizable cGMP analogue 8-para-chlorophenylthio-cGMP (8-pCMT-cGMP [489]) to determine the effects of cGMP-dependent processes on [Ca2+]i, contractile tension, and Ca2+ sensitivity of the ovine basilar artery, vessels of the fetus were much more sensitive to cGMP vasorelaxant effects than those of adult [47]. Also, in fetal, but not adult arteries, cGMP promoted vasorelaxation by attenuating both basal and 5-HT-induced myofilament Ca2+ sensitivity ~27% [47]. The intracellular effects of cGMP follow from the activation of cGMP-dependent PKG. Nonetheless, the target substrates of these kinases, particularly those proteins involved in [Ca2+]i regulation are not well identified. Another consideration is the extent to which the G Kinases vary in isoform and function with maturation, as reported for the rabbit myocardium [490].
Myosin and Thick Filament Regulation - Calcium Sensitivity
Importantly, and as noted above, in pressurized resistance sized (~150 μm) cerebral arteries basal intracellular Ca2+ concentration increased significantly both with age from 95-GD fetus to 140-GD to adult, and as a function of intravascular pressure [107]. In addition to [Ca2+]i per se, the sensitivity of myofilaments and the entire vascular contractile apparatus to Ca2+ plays a key role in the regulation of cerebrovascular contractility [442, 491, 492]. Several studies have shown a proportionally smaller change in [Ca2+]i for a given force production in response to receptor-mediated agonists, as compared to K+ depolarization [105, 353, 375, 393, 493]. This supports the idea that modulation of Ca2+ sensitivity of the contractile apparatus is a key mechanism governing vascular reactivity [442, 491]. The precise mechanisms which govern Ca2+ sensitivity involve not only G proteins [494], and coupling of G protein subunits to PKC [495], but also myosin light chain20 (MLC20), a number of kinases, phosphatases, and other factors. These influence the phosphorylation state of actin and related contractile proteins involved in the regulation of MLC20 phosphorylation and dephosphorylation [496]. In addition, proteins that interact with actin such as calponin, caldesmon, and so forth, play a vital role in this regard [234, 442, 491, 497]. The biochemical mechanisms by which Ca2+ sensitivity changes with development from fetus to newborn to adult is poorly understood.
We and others have addressed this issue with several approaches. Of note, in fetal and adult developing CCA, although MLC20 concentration was similar, calmodulin concentrations were significantly less [450] (Table 8). Again, while MLCK concentration was significantly less in fetal arteries [450, 451], both MLCK maximum velocity and fractional activation were much greater in the immature vessels, compared to adult [450] (Table 8). Additionally, the concentrations of MLCP, calmodulin, and heat shock protein-27 were significantly less in fetal vessels [451] (Table 8). In MCA in which we measured simultaneously vascular tension and [Ca2+]i in response to NE (10−9 – 10−4 M), the slopes of these relations for near-term fetal and adult vessels were 4.1±0.4 and 7.3±0.6, respectively [46]. Additionally, in simultaneous measurements of diameter and [Ca2+]i at different intravascular pressures (10 to 80 mmHg) in 95-GD, 140-GD, and adult 2nd- or 3rd-order MCA branches, the relation of vascular tone to [Ca2+]i (normalized to zero extracellular Ca2+ plus ethylene glycol-bis(β-aminothyl ether)-N,N,N',N'-tetraacetic Acid (EGTA)) was much less for the near-term fetus than that for either 95-GD fetus or adult. Removal of the endothelium magnified these age-related tone versus [Ca2+]i differences [107]. Additionally, fetal and adult cranial vessels exhibit significant differences in length-tension relationships (stretch ratio diameter/resting diameter). For instance, in fetal CCA percent maximum response was 1.70±0.06, compared to 2.58±0.07 in adult [450]. Undoubtedly, these reflect the developmental differences in arterial wall thickness and compliance [17].
In a further study of mechanisms underlying maturational changes in the pressure-evoked myogenic response, we tested the hypothesis that development alters the sensitivity of pressure-evoked myogenic constrictions of cerebral arteries via differential regulation of Ca2+ influx and Ca2+ sensitivity. We measured simultaneously pressure-mediated myogenic tone and changes in wall [Ca2+]i in endothelium denuded MCA from adult (6 month) and pup (P14) Sprague-Dawley rats [498]. Increases in hydrostatic pressure from 20 to 80 mmHg produced pressure-dependent myogenic tone that was significantly greater in pup, compared to adult, MCA. At each pressure step, vascular wall [Ca2+]i also was significantly greater in pup than that in adult MCA. While the L-type Ca2+ channel blocker nifedipine significantly attenuated pressure-evoked constrictions in the adult, these were essentially eliminated in the pup. Although both adult and pup MCA demonstrated pressure-dependent increases in Ca2+ sensitivity, there was no significant different in this Ca2+ sensitivity between adult and pup vessels. These results suggest that both Ca2+ influx and Ca2+ sensitivity play important roles in the regulation of cerebral artery myogenic tone. The greater cerebral myogenic response in early development is likely due to increased Ca2+ influx, resulting in higher [Ca2+]i, as well as greater myofilament Ca2+ sensitivity [498].
In agreement with these results, in β-escin-permeabilized cranial (common carotid and basilar) arteries of 140-GD fetal and adult sheep, Ca2+ sensitivity as reflected by % Ca2+max at varying [Ca2+]i, the pD2 values (CCA) were 6.4 ±0.1 and 6.0 ±0.1, respectively [357]. For neither main branch nor 2nd-order branch MCA did pD2 values vary with age [357]. In a further study in several vessels of β-escin-permeabilized rabbit cranial and femoral arteries, Ca2+ sensitivity was greater in vessels from 8 to 9 day-old, as opposed to either 24 to 25 day-old or adult [346]. Of course it must be recognized that these studies included differences in experimental conditions such as: vessel size; wire-mounted versus pressurized; mediation by different receptors (NE versus 5-HT), intact versus permeabilized SMCs, and so forth. In addition, studies have documented differences in agonist sensitivity and calcium handling in isometric versus isobaric preparations [499, 500]. Furthermore, as noted earlier vessel reactivity to pressure varies with diameter, which also can confound interpretation among studies [85]. In fact, when larger (250 μm) endothelium-intact and denuded cerebral arteries from near-term fetal and adult sheep were used in similar experiments, the tension to [Ca2+]i relationship was unaffected by development [107]. Collectively, these studies suggest that [Ca2+]i sensitivity of the sheep cerebral vasculature varies with both vessel diameter and developmental age.
A related issue is that of intracellular Ca2+ responses to pressure per se. These have been examined in isolated arteries from a number of vascular beds [85, 501], and consistently have shown that both [Ca2+]i and vascular tone increase with elevation of transmural pressure. In cerebral arteries of 140-GD fetal and adult sheep, [Ca2+]i increased in response to pressure in endothelium-intact arteries [107]. Removal of extracellular Ca2+ from the superfusate produced a sharp reduction in [Ca2+]i, and abolished tone in cerebral arteries of both age groups. These data strongly suggest that the maintenance of tone requires Ca2+ entry, most likely through voltage-operated (L-type) and nonselective stretch-operated Ca2+ channels, as observed in both adult [501] and fetus [45, 46].
CALCIUM-INDEPENDENT SECOND MESSENGER SYSTEMS
Protein Kinase C-Mediated Pathways
In addition to mobilization of Ins(1,4,5)P3 via G proteins and PLCβ, adrenergic, serotonergic, and other receptor-mediated agonist stimulation is associated with the synthesis of diacylglycerol (DAG), which results in the activation of protein kinase C (PKC) [502]. A dual function kinase, the PKC family of isoforms is integral part of the agonist-induced, Ca2+-dependent signaling pathway. In addition, it can play a key role in non-Ca2+-dependent signaling via mitogen-activated protein kinase (MAPK), which, in turn, mediates the extracellular regulated kinases 1 and 2 (ERK1/2) and the heat-shock protein (HSP) 27-linked p38 kinase pathway. Even in this latter role, PKC requires Ca2+, but not necessarily a change in its concentration [240, 503]. Maturational development is associated with changes in several aspects of these pathways. (Fig. 4) illustrates some of these non-Ca2+ dependent pathways and mechanisms.
Fig. (4).

Non-calcium-dependent contraction pathways, showing both thick and thin filament regulation. PKC, protein kinase C, basal activity (117); PKCε abundance (30); MAPK, mitogen activated protein kinase; ERK1/2, extracellular regulated kinase abundance (100); Phosphorylated ERK1/2 (70); ERK1/2 negative feedback on PKC (significantly greater); Rho A abundance (100); CPI-17 (significantly less). (Numbers in parentheses represent fetal values, as percent of those of adult).
A phospholipid-dependent serine/threonine kinase, the PKC family includes at least 11 isoforms with differing structures of the regulatory domain. “Conventional”, “classic”, or group A PKCs (α, βI, βII, and δ) are activated by cytosolic-free Ca2+, DAG, and phosphatidylserine, a membrane phospholipid. “Novel”, “new”, or group B PKCs (δ, ε, η, θ) also are activated by DAG, but are Ca2+ independent. “Atypical” PKCs (ζ and λ) require neither Ca2+ nor DAG for activation, but respond to phosphatidylserine [504, 505]. In general, PKC activation is associated with its translocation from cytoplasm to plasma membrane, although several isoforms translocate to the nucleus [502, 506]. DAG binding to the PKC regulatory domain (C1 region) dramatically increases Ca2+ affinity and enzyme activity. The PKC family of isoenzymes play key regulatory roles in a wide variety of cell functions including: gene expression, cell growth and differentiation, membrane function, and so forth [502]. Although many of these isoforms have been reported in vascular SMCs [240, 503, 504, 507], their roles in modulating contraction are poorly defined. Additionally, vascular contraction/relaxation may be mediated, in part, by tyrosine protein kinase, and related enzyme pathways [234, 240, 442].
In cerebral and other SMCs, PKC isoforms play dual roles in the regulation of vascular tone. By negative feedback, PKC may inhibit PLC, thereby attenuating agonist-induced increases in Ins(1,4,5)P3, [Ca2+]i, and contraction [16, 508, 509]. In addition, PKC activation per se can increase vascular tone [16, 439, 510–512] via activation of CPI-17 or other mechanisms to inhibit MLCP [440]. PKC also appears to modulate SMC Ca2+ sensitivity to α-adrenergic and other agonists [16, 513], although the mechanism of this effect is unclear.
The role of PKC in modulating cerebral artery tone, and its regulation during development, remains poorly understood. In main branch cerebral arteries, basal PKC levels were similar in fetal and adult sheep, both in total absolute value, and as percent membrane bound (i.e., the active form) [16] (Table 9). The extent to which the several PKC isoforms are expressed probably accounts for differences in agonist-induced responses. For instance, by western immunoblot vessels of both fetus and adult showed abundant levels of the class A and class B isoforms, with the exception of low levels of PKCε expression in the fetal arteries compared to adult [16]. Arteries in both age groups responded to PKC activation by phorbol 12,13-dibutyrate (PDBu), indolactam V, or NE, with a 35 to 75% increase in both membrane-bound fraction and activity [16]. Nonetheless, the role of PKC activation in the α-adrenergic-induced Ins(1,4,5)P3 responses differed considerably as a function of developmental age. For instance, in adult cerebral arteries NE stimulation resulted in a dose-dependent doubling of the Ins(1,4,5)P3 concentration. Stimulation of PKC activity by PDBu or indolactam V resulted in Ins(1,4,5)P3 levels decreasing to ~50% control, and showed no response to NE. By comparison, in these adult vessels PKC inhibition by either staurosporine or calphostin C resulted in markedly increased sensitivity of Ins(1,4,5)P3 response to NE. In contrast in fetal cerebral arteries, although Ins(1,4,5)P3 showed a robust dose-response increase to NE stimulation and marked inhibition by PDBu, following PKC inhibition by staurosporine, the NE-induced Ins(1,4,5)P3 increase did not differ significantly from that response seen to NE alone [16]. The basis of this difference in response awaits further studies (see below).
Table 9.
Non Calcium-Dependent Contractile Mechanisms with Development
| Physiologic Variable | Fetus | Adult | Reference |
|---|---|---|---|
| PKC (% Membrane Bound) | 38±4 | 32±4 | [16] |
| PKC Basal Activity (pmol·min−1·mg−1) | 239±31 (117) | 204±15 | [16] |
| PKCε Expression (% adult value) | <30b (30) | 100 | [16] |
| ERK1/2 – Total (% Adult Value) | 1.0±0.1 | 1.0 | [236] |
| Phosphorylated ERK1/2 | 0.7±0.1c (70) | 1.0 | [236] |
| PHE-induced Change in Phosphorylated ERK1/2 (% control) | 50 to 100c (50 to 100) | – | [439] |
| Change in PHE-induced Tension with U-0126 (Emax; K+ max) | 0.4±0.1c (400) | 0.1±0.1 | [439] |
| Change in PHE-induced Ratio with U-0126 (Emax; %Kmax) | −0.05 ±0.03b (−55) | (no signal) | [439] |
| Rho-A (% Adult Value) | 100 | 100 | [9] |
| ROCK-mediated response | Significantly greater | [9] | |
| CPI-17-mediated response | Significantly less | [9] |
Values are mean ± SE of 5 or more in each group;
Values in parentheses represent % of adult value;
Significantly different from adult value:
P<0.01;
P<0.05
A related issue concerns intracellular interactions of PKC with Ca2+. In adult and fetal MCA, PDBu inhibited the NE-induced increase of both [Ca2+]i and tension in a dose-dependent manner [16]. Nonetheless, PKC inhibition produced contrasting responses in the two age groups. In adult MCA, PKC inhibition resulted in a marked shift to the left of both the NE-induced [Ca2+]i and tension dose-response (i.e., increased sensitivity), as well as a significant increase in maximal tension. In fetal MCA, in contrast, staurosporine pretreatment resulted in a major decrease in both NE-induced [Ca2+]i and tension.
A further issue concerns the changes in PKC-induced vascular contraction with development. In adult MCA with increasing concentrations of PDBu, tension increased to ~65% Kmax, with no change in [Ca2+]i, i.e., Ca2+ sensitivity increased marked ly. Removal of ex tracellular Ca2+ ([Ca2+]O=0) did not affect significantly the PDBu-induced tension increase. In response to PDBu, fetal MCA also showed a dose-dependent increase in tension to ~65% Kmax, however, in contrast to the adult, [Ca2+]i also increased significantly to ~30% Kmax. This emphasizes further the role of extracellular Ca2+ for the fetal SMC, for in the presence of zero [Ca2+]o PDBu administration failed to produce a significant increase in either [Ca2+]i or tension [16]. Overall, these studies demonstrate the dual role of PKC in modulating agonist-induced responses in developing cerebrovasculature, and important differences in these mechanisms between fetal and adult vessels. Nonetheless as noted, PKC, ERKs, and probably other substrates, interact by both positive and negative feedback mechanisms. Thus, an understanding of the role of PKC requires an appreciation of the ERK1/2, and several other downstream effector enzymes/elements (see below). In the adult, the Ins(1,4,5)P3-dependent pathway appears to be the major mechanism for regulation of NE-induced contraction. That is, PKC mediates NE-induced contraction via negative feedback on agonist-induced Ins(1,4,5)P3 release and modulation of myofilament Ca2+ sensitivity. In fetal vessels in contrast, PKC inhibition failed to augment NE-induced contraction or increase [Ca2+]i. Rather, with PKC inhibition both the NE-induced increase in tension and [Ca2+]i were significantly decreased. Again, a substantial developmental difference is that in the fetus, as opposed to adult, NE-induced contraction is almost totally dependent on Ca2+ influx via the L-type Ca2+ channel. We thus speculate that in the immature organism PKC plays a more important role in regulating channel activity to modulate agonist-induced contraction. This idea is supported by our patch-clamp studies that PKC activation inhibits BK channel opening [349], releasing that channel's inhibitory effect on the L-type Ca2+ channels, thus allowing increased Ca2+ influx. Additionally, PKC activation increases L-type Ca2+ channel activity, further augmenting Ca2+ influx. Thus, for NE-induced contraction the adult SMCs utilize the Ins(1,4,5)P3 pathway, as has been well described, whereas the fetal SMCs rely more on the Ca2+ influx pathway [16].
Mitogen-Activated Protein Kinases
As noted, in mature vascular SMCs the main contraction pathway is via Ca2+-calmodulin-mediated phosphorylation of the myosin light chain [240, 514]. Accumulating evidence also suggests that activation of the MAPK cascade, including the extracellular signal-regulated kinases ERK1 (p44) and ERK2 (p42) [515, 516] play key roles in modulating contraction/relaxation [517–519]. ERK activation is dependent upon dual phosphorylation of a tyrosine (Try185) and a threonine (Thr187) residue [520]. Although the relation of ERK1/2 activation to cell proliferation and differentiation is well established [521, 522], the role of ERKs in modulating SMC contraction and relaxation remains unclear. Several agonists that produce vascular smooth muscle contraction simultaneously phosphorylate ERKs, thereby modifying contraction [439, 440, 442, 514, 517, 519, 523–529]. However, the exact nature of these mechanisms and associated signaling pathways have remained unknown.
Given that cerebral arteries show significant vessel-specific and age-related developmental differences in agonist-induced contraction, and that ERKs appear to play a major role in the regulation of vascular tone, questions arise as to their function in modulating cerebrovascular reactivity and their temporal activation pattern in response to α-adrenergic and other agonists. Also of importance is the extent to which ERKs play a prominent role in developing vascular SMCs, in which one would expect a “synthetic”, as opposed to the more mature “contractile”, phenotype seen in the adult [122, 530]. Basal levels of total ERKs 1 and 2, as measured by Western immunoblots, were similar in the cerebral arteries of fetus and adult, while the phosphorylated ERK levels in fetal vessels were only three-quarters that of adult [439] (Table 9). In adult MCA, the PHE dose-response was shifted one-half log unit to the right, as compared to control (i.e., decreased sensitivity) in the presence of ERK inhibition by U-0126. Even more striking in adult MCA, despite a maximal tension increase, U-0126 totally eliminated the normal PHE-induced [Ca2+]i increase [439]. In fetal MCA, in contrast, following ERK inhibition, the PHE-induced contraction was augmented ~40%, while the [Ca2+]i response was inhibited ~50%, but not completely eliminated [439].
In fetal, but not adult, cerebral arteries PHE-stimulation increased phosphorylated ERK1/2 levels each ~50 to 100% above control in a dose-dependent manner, with the maximal increase at 5 min (Table 9). Also in the fetal, but not adult, arteries the ERK antagonist U-0126 of itself resulted in a significant reduction in the basal phosphorylated ERK1/2 levels, and PHE + U-0126 produced no significant increase in phosphorylated ERK1/2 levels above that seen with U-0126 alone [439]. K+ depolarization failed to alter phosphorylation levels of ERK1/2 in cerebral arteries of either age group. One might argue that phosphorylated ERK1/2 levels, as determined by western immunoblot, do not accurately reflect ERK1/2 activity. To rule out this possibility, in agreement with the immunoblots, biochemical assay of phosphorylated ERK1/2 disclosed about one-half the activity in fetal cerebral arteries, compared to the adult. Also in response to PHE, activity in the fetal arteries increased ~140%, while that increase in adult vessels was negligible [439].
PKC-ERK1/2 Interactions
A further question concerns the mechanism of the ERK-mediated effect on Ca2+ sensitivity. Studies in ferret and rat aorta have reported PKC-mediated Ca2+-independent SMC contraction [531], and differences in the [Ca2+]i responses to PKC stimulation in comparison with PHE. PKC may phosphorylate caldesmon and/or calponin to mediate this response [532]. PKC-dependent modulation of myosin phosphorylation also can effect such a contractile response in the presence of only a small increase in [Ca2+]i [533]. PKC also increases myofilament Ca2+ sensitivity in rat cerebral arteries [534]. In a manner similar to our studies in ovine cerebral arteries, in the vascular smooth muscle type II knockout mouse, PKC induced a sustained contraction of cerebral arteries, with no significant change in [Ca2+]i [535]. Such PKC-induced, Ca2+-independent contraction of cerebral arteries, with important differences between fetus and adult, may be related to the paucity of PKCε in early development [16].
As noted, in adult MCA ERK inhibition by U-0126 totally eliminated PHE-induced increase in [Ca2+]i, despite no appreciable attenuation of the tension increase [439]. This was not unlike the increase in tension in the absence of a change in [Ca2+]i seen in response to PDBu stimulation of PKC, we reported earlier [16]. To explore the mechanistic basis for this phenomenon, and possible role of PKC and ERK1/2 in modulating agonist-induced increase in [Ca2+]i and tension, we examined PHE dose-response relations in the absence or presence of U-0126 following PKC inhibition by staurosporine. In adult MCA in the presence of staurosporine, both the tension and [Ca2+]i response to PHE were significantly attenuated. In contrast, with staurosporine administration prior to ERK inhibition, PHE-induced contraction and [Ca2+]i were markedly increased in a dose-dependent manner [439]. In a similar manner in fetal MCA, staurosporine inhibition of PKC resulted in marked attenuation of both PHE-induced tension and [Ca2+]i. However, following PKC inhibition by staurosporine, and in the presence of ERK inhibition by U-0126, PHE-induced tension showed greater sensitivity, although the [Ca2+]i rise was not significantly greater than that with staurosporine alone. Taken together with the data on the effect of PKC inhibition on modifying the U-0126-mediated modulation of PHE-induced [Ca2+]i response, these results suggest an interaction between ERK and PKC signaling pathways in the regulation of agonist-mediated [Ca2+]i and tension. They suggest strongly that in adult cerebral vessels, ERKs mediate agonist-induced Ca2+ release but suppress Ca2+ sensitivity, at least in part, by modulating PKC activity.
In neither fetal nor adult cerebral arteries has a causal relationship between ERK1/2 phosphorylation and the increase in vascular tension or [Ca2+]i been established. In part, this is compatible with the idea that ERKs play a role in the maintenance of sustained tone, rather than its induction [536]. Alternatively, tension may be totally independent of phosphorylated ERK levels. Furthermore, in fetal cerebral arteries, the relatively selective inhibitor U-0126 eliminated agonist-induced ERK1/2 phosphorylation, while at the same time enhancing PHE-induced tension [439]. In the fetus, but not adult, in addition to the usual increases in tension and [Ca2+]i the selective α1-AR agonist PHE produced a sustained increase in ERK1/2 phosphorylation. These results are consistent with studies in the rabbit femoral artery [526] and sheep uterine artery [529]. α1-adrenergic receptor stimulation thus may have activated a second signaling cascade which, in association with increased [Ca2+]i, resulted in increased ERK1/2 phosphorylation [526]. These developmental differences in cerebral artery PHE-induced responses of phosphorylated ERK1/2 levels, tension, and [Ca2+]i may help to explain the lack of agreement regarding the role of ERKs in vascular reactivity [537, 538].
A related consideration is that in addition to PKC activation of ERK1/2 and other substrates, by negative feedback ERKs may attenuate PKC-induced contraction, and these interactions modulate both thick and thin myofilament pathways and Ca2+ sensitivity. In adult MCA, in addition to PDBu-induced increase in Ca2+ sensitivity (augmented isometric tension without concomitant increase [Ca2+]i), PDBu also increased phosphorylated ERK1/2 levels, a response blocked by U-0126. In turn, U-0126 augmented PDBu-induced vascular tension. As shown by western immunoblot, PDBu administration also was associated with significant increases in levels of phosphorylated caldesmonSer789 and myosin light chain20 (MLC20) levels, each of which peaked at 5 to 10 min. These changes also were associated with increased phosphorylated CPI-17 levels, which peaked at 2 to 3 min [440]. Importantly, in the adult vessel administration of neither the MLCK inhibitor ML-7 nor the Rho kinase inhibitor Y-27632 altered PKC-induced contraction. These results support the idea that PKC can increase CPI-17 phosphorylation, thereby decreasing myosin light chain phosphatase activity, and thus increasing MLC20 phosphorylation independent of either MLCK or Rho kinase activation. By this mechanism, PKC can increase Ca2+ myofilament sensitivity. In turn, not only may ERK1/2 feedback to inhibit PKC activity, but ERK-dependent phosphorylation of caldesmonSer789 appears to attenuate the thin filament regulatory pathway, thereby inhibiting PKC-mediated contraction and decreasing myofilament Ca2+ sensitivity [440]. Finally, ERK1/2-dependent phosphorylation of caldesmonSer789 was not required for PDBu-induced contraction, and appears not to be involved in the reversal of calmodulin inhibitory effect on actin-myosin ATPase [440].
Conversely, in fetal cerebral arteries the PKC-ERK1/2 interactions differ considerably from those of the adult. For instance, although in response to PKC activation by PDBu, MCA tension increased to about 20% Kmax (significantly less than the 35% increase in adult), following U-0126 inhibition of ERKs the tension increase was to a value similar to that of the adult (e.g., ~45% Kmax). Also, in the fetus the level of basal phosphorylated CPI-expression was only one-third that of the adult; however as with the adult the levels increased in response to PDBu, and were not affected by ERK1/2 inhibition. Also in contrast to adult, ERK inhibition was associated with an increase of [Ca2+]i in response to PKC activation by PDBu. Additionally, in contrast to adult, fetal vessels demonstrated a significant effect of Rho A kinase inhibition on PDBu-induced contraction. Thus, in fetal cerebral arteries, PKC would appear to increase MLC20 phosphorylation via stimulation of both CPI-17 and Rho A kinase [9].
In terms of non-Ca2+-dependent pathways for PKC-mediated thin filament contraction, activated phospholipase C (PLC)-mediated production of DAG stimulates PKC. In turn, activated PKC phosphorylates CPI-17, which by inhibiting myosin light chain phosphatase, promotes MLC20 phosphorylation. In the fetus, but not the adult, PKC can activate Rho A and Rho kinase and thus further modulate MLC20 phosphorylation by inhibition of MLCP [9]. In the adult artery, PKC thus can increase myofilament Ca2+ sensitivity via its phosphorylation of CPI-17, while in the fetus this is augmented by activation of Rho A/Rho kinase. PKC activation also can lead to phosphorylation of ERK1/2 and CaDSer789. ERK1/2 activation with phosphorylation of CaDSer789 may have a negative role in the regulation of PKC-mediated caldesmon phosphorylation on site(s) other than Ser789.
Overall, in developing cerebral arteries it would appear that there exists a fine balance of several elements in the regulation of myofilament Ca2+ sensitivity and vascular tone. These include the balance between thick and thin filament regulatory pathways, and the balance between MLCP activity in determining the degree of MLC20 phosphorylation, and that of the thin filament binding proteins (e.g. caldesmon) in affecting myofilament Ca2+ sensitivity. In essence, in regards to cerebrovascular regulation, the fetus and newborn rely on increased myofilament Ca2+ sensitivity while the more mature organism depends upon increases in [Ca2+]i per se (see below).
Importantly, in vivo studies in the newborn piglet also have demonstrated a role for the MAPK-ERK pathway in cerebrovascular regulation. In studies using the cranial window technique, preadministration of the protein tyrosine kinase antagonist genistein, or the ERK1/2 inhibitor U-0126 prevented such O2−-mediated impairment. In contrast, neither of these inhibitors prevented O2−-mediated vasoconstriction in vessels pretreated with the KATP channel agonist cromakalim or the KCa channel agonist NS-1619. Protein tyrosine kinase and ERK activation also were important in the attenuation of O2−-mediation of decreased response to N-methyl-D-aspartate (NMDA)-induced vasodilatation [539]. Pathways competing for attention in this regard include the classical receptor, G-protein, PLCβ, Ins(1,4,5)P3, Ca2+, calmodulin pathway involved in activation of MLCK and MLC20 phosphorylation. Nonetheless, the Ca2+-independent pathways that involve PKC, MAPKs, ERK1/2, calmodulin, CPI-17, and other effectors also are of vital importance, and in concert, this myriad of elements interact to determine Ca2+ sensitivity and vascular tone. As additional elements of these cascades are discovered and their relative importance established, we will construct a more clear and understandable picture of this complex of regulatory mechanisms. In addition, as we learn more of the nuances of developmental regulation of these multiple factors and elements we can approach an understanding of their relative importance, and how these pathways interact in contraction, transcription, and other elements of cell regulatory control.
PKC-Rho A-Rho Kinase Interactions
In the Ras superfamily of proteins, Ras homolog gene family member A (Rho A), is a small GTPase. Involved in the regulation and timing of the formation of stress fibers of the actin cytoskeleton, Rho A interacts with a number of substrates in the regulation of cell growth division, and other functions. Rho-associated protein kinase (ROCK) is a serine/threonine kinase involved in regulation of cell shape and movement, by acting on the cytoskeleton. ROCKs can act on a number of substrates, including LIM (Lin I of C. elegans, Isl of rats, MEC3 of C. elegans, where the LIM domain was discovered) domain, kinase, myosin light chain (MLC20) and MLC phosphatase (MLCP) to inhibit actin filament depolymerization. This occurs by phosphorylation of MLC20 and increase myosin II ATPase activity, and other functions [540]. In VSMCs, ROCK activation increases myofilament Ca2+ sensitivity by inhibiting MLCP activity, thus enhancing MLC20 phosphorylation [234, 541].
To explore the role of Rho A and ROCK in PKC-mediated contraction in fetal and adult ovine middle cerebral artery, we quantified Rho A expression and responses to ROCK inhibition. The basal expression levels of total Rho A (measured by Western Immunoblot) were similar, while in contrast to adult, measurable levels of the active phosphorylated Rho A were negligible in the fetus. In response to PDBu-mediated PKC stimulation, however, while in adult arteries phosphorylated Rho A remained unchanged, in fetal vessels by 5 and 10 min, the level rose many-fold (perhaps 50) [9]. Also of importance in adult MCA, administration of the ROCK inhibitor Y-27632 failed to attenuate the PDBu-mediated contractile response. In marked contrast, in the fetal cerebral arteries, Y-27632 reduced the contractile response to PDBu about 50%. These responses occurred in the absence of changes in [Ca2+]i. This evidence strongly supports the role of the Rho A/Rock pathway as playing a vital role in cerebral artery Ca2+ sensitivity and contraction in the fetus, but not the adult [9]. In a subsequent study, these results were confirmed [41].
Summary of Calcium Sensitization Mechanisms
As noted above, SMC myofilament Ca2+ sensitivity is represented by the increase in vascular tension for a given change in [Ca2+]i [234]. A composite characteristic, CaS is a function of a number of physiologic mechanisms that regulate both thick (myosin) and thin (actin) filament phosphorylation and dephosphorylation [234, 542, 543]. Patterns of CaS regulation vary as a function of SMC phenotype [42, 357, 544], vascular bed [399], pregnancy [545, 546], chronic hypoxia [41, 347], and various pathologic states including diabetes [547, 548], ageing [399] and hypertension [549, 550]. As postulated by Murphy and colleagues [542, 543], myofilament CaS is a function of the integrated sum of the relation between [Ca2+]i and MLK20 phosphorylation with modulation by cyclic nucleotides to develop contractile force. In addition, of critical importance is the ability of actin to interact with phosphorylated MLC20 in producing force [347].
As documented in this review, because a given increase in fetal cerebral artery tension is associated with significantly less increase in [Ca2+]i, myofilament CaS of the fetal/neonatal arteries is much greater than that of the adult [346, 353]. The relative roles of these various factors and mechanisms are highly age dependent with maturation [9, 15, 16, 46, 47, 275, 346, 353, 356, 357, 439, 448, 451, 551]. Mechanisms which account for the greater CaS in immature vessels include the roles of MLCK [347], and MLCP via PKC and CPI-17 [9, 346], PKC and the ERKs [9], and ROCK [41, 355] in the phosphorylation and dephosphorylation of MLC20, and interactions with actin. In terms of stretch-induced myogenic response, modulation of thin filament reactivity with increased CaS may be much more important in cerebral vessels of the fetus than in the adult [451]. As noted by the Pearce group, failure of the vasculature to shift to a significantly lower CaS with maturation may be associated with vascular hyper-reactivity in the adult [346].
GENE REGULATION IN DEVELOPING CEREBROVASCULATURE
Maturation and Gene Expression in Cranial Arteries
Classical studies of physiologic mechanisms, combined with biochemical assays, have contributed greatly to an understanding of fundamental aspects of biologic functions and their pathophysiology. More recently, molecular biology has given us the ultimate in reductionist data. As biology moves into the postgenomic era, a central problem that remains is how cells use their complements of structural, enzymatic, and signaling proteins to translate extracellular stimuli into highly specific responses. Within this context, a major challenge for cerebrovascular physiology is to understand the manner in which cell and tissue behaviors emerge from the dynamic interactions among the elaborate and parallel bio-molecular networks that maintain homeostatic cellular, tissue, and organ dynamics. In the cerebrovasculature, the mechanisms by which endothelial and vascular smooth muscle cells integrate responses among multiple pathways are slowly emerging. However, relatively little is known about the manner in which these patterns of response change with developmental maturation, or the impact these changes have on overall regulation of the cerebrovasculature, its tone, and blood flow.
In light of the body of evidence presented above, it is clear that these maturational changes in cerebral artery signaling mechanisms require elucidation of their molecular basis. Unfortunately and of critical importance, however, our current understanding of the role of gene expression that underlies cerebrovascular homeostasis, and its maintenance during maturation, is extremely limited. To address this vital issue, by use of oligonucleotide microarrays and signal pathway analysis, we tested the hypothesis that the profound age-related differences in cerebral artery reactivity are associated with fundamental changes in gene expression. Thus, we examined highly significant expression changes in CCA from four age groups: premature fetus (95±2 GD), near-term fetus (140±3 GD), newborn (1 to 5 day old), and adult (18–24 months of age). Labeled cRNA was prepared and applied to an ovine gene expression microarray. Each gene was annotated using one of several databases, and these were analyzed by pathways. To validate the results of the microarray analysis, we used real time-polymerase chain reaction (RT-PCR) of a half dozen genes that were highly expressed [11].
Our results demonstrated profound changes in CCA gene expression profiles with developmental maturation from premature fetus to mature fetus, and to newborn, compared to adult. For a 2-fold change (p<0.05) 2,570, 1,212, and 2,371 genes were up-regulated in premature fetus, near-term fetus, and newborn lamb, compared to adult. For a > 4-fold change (p<0.001) compared to adult, 373, 110, and 304 genes were up-regulated in the three age groups, respectively. For down-regulated genes, the number was one-half to two-thirds that of the up-regulated values for each group [11]. In a striking manner, changes in expression profiles in arteries from premature fetus and newborn lamb, differed to a similar extent compared to adult. To our surprise, compared to adult, in the near-term fetus fewer genes showed differential regulation. A similar pattern of increased changes in gene expression in premature fetus and newborn lamb carotid arteries, with far fewer changes in near-term fetus compared to adult also was observed in several functional and canonical gene pathways/networks. Striking is the “U” shaped pattern of these gene expression responses [11].
Table 10 enumerates the top 20 up-regulated genes in the cell proliferation, growth, and assembly pathways from premature fetus, near-term fetus, and newborn lamb, compared to adult [11]. Perhaps of no surprise, many genes involved in cell cycle regulation, DNA replication, chromosome assembly, cell proliferation and other components of carotid artery cell replication, growth, and assembly were up-regulated. However of importance, the fold-change of these genes was to a significant degree greater in the premature fetus and newborn than in near-term fetus. The finding emphasizes the complex changes which occur during the perinatal period. Similarly, Table 11 the top genes in down-regulated pathways from specific age groups. These include the integrin, actin cytoskeleton, and PKC-Rho Kinase-mitogen activated protein kinase (MAPK) pathways [11]. Fig. 5 depicts CCA functional gene regulation pathways that change significantly with development from premature fetus to term fetus and to newborn, as compared to adult.
Table 10.
Cellular Proliferation, Growth, and Assembly Pathway was Up-regulated in Early Life
| Symbol | Entrez Gene Name | Premature fetus Vs Adult | Near-term fetus Vs Adult | Newborn Vs Adult | |||
|---|---|---|---|---|---|---|---|
| Fold Change | p-value | Fold Change | p-value | Fold Change | p-value | ||
| ASPM | asp (abnormal spindle) homolog, microcephaly associated | 37.38 | 9.41E-06 | 19.21 | 2.04E-05 | 16.29 | 5.45E-05 |
| AURKA | aurora kinase A | 11.78 | 9.23E-05 | 5.694 | 3.68E-04 | 8.833 | 1.46E-04 |
| AURKB | aurora kinase B | 58.15 | 4.34E-07 | 30.03 | 3.66E-06 | 27.24 | 1.35E-04 |
| BIRC5 | baculoviral IAP repeat containing 5 | 177.4 | 6.28E-06 | 33.53 | 2.65E-04 | 82.05 | 1.81E-05 |
| CCNA2 | cyclin A2 | 54.41 | 6.46E-06 | 21.72 | 1.93E-05 | 39.31 | 7.80E-06 |
| CCNB2 | cyclin B2 | 29.65 | 2.08E-07 | 11.42 | 1.67E-04 | 19.98 | 2.42E-05 |
| CDK1 | cyclin-dependent kinase 1 | 51.19 | 1.61E-04 | 15.16 | 1.97E-04 | 41.47 | 1.14E-05 |
| CDK2 | cyclin-dependent kinase 2 | 5.852 | 2.12E–05 | 5.064 | 3.75E–03 | 3.454 | 1.71E–04 |
| CDT1 | chromatin licensing and DNA replication factor 1 | 7.823 | 1.16E–04 | 11.94 | 1.01E–02 | 3.098 | 7.26E–03 |
| CHEK2 | CHK2 checkpoint homolog (S. pombe) | 8.841 | 3.65E–05 | 3.24 | 1.27E–01 | 5.385 | 1.81E–04 |
| CENPE | centromere protein E, 312kDa | 61.96 | 1.10E-05 | 42.58 | 9.18E-06 | 42.73 | 3.05E-06 |
| CENPH | centromere protein H | 12.61 | 3.67E-06 | 5.277 | 1.21E-04 | 7.478 | 9.92E-06 |
| CKAP2 | cytoskeleton associated protein 2 | 30.63 | 1.84E-04 | 19.23 | 1.88E-04 | 19.94 | 8.83E-05 |
| CKS2 | CDC28 protein kinase regulatory subunit 2 | 73.8 | 4.67E-08 | 30.33 | 1.50E-06 | 57.91 | 3.18E-06 |
| DIAPH3 | diaphanous homolog 3 (Drosophila) | 35.37 | 4.90E-06 | 27.37 | 1.28E-06 | 46.56 | 9.83E-07 |
| ESPL1 | extra spindle pole bodies homolog 1 (S. cerevisiae) | 28.7 | 1.49E-04 | 12.95 | 7.42E-05 | 15.62 | 2.57E-05 |
| GATA6 | GATA binding protein 6 | 33.72 | 8.69E-05 | 8.271 | 2.27E-04 | 16.16 | 1.27E-05 |
| KNTC1 | kinetochore associated 1 | 25.58 | 2.16E-04 | 9.572 | 0.00145 | 14.78 | 5.03E-04 |
| MXD3 | MAX dimerization protein 3 | 31.4 | 4.21E-06 | 26.09 | 3.17E-06 | 14.69 | 8.09E-05 |
| NCAPG | non-SMC condensin I complex, subunit G | 100.5 | 8.00E-07 | 46.93 | 6.68E-06 | 74.72 | 1.24E-06 |
| PRC1 | protein regulator of cytokinesis 1 | 23.2 | 5.61E-05 | 21.77 | 5.18E-04 | 17.77 | 5.21E-05 |
| PCNA | proliferating cell nuclear antigen | 10.78 | 1.03E–04 | 3.439 | 4.35E–04 | 9.182 | 1.43E–05 |
| RFC1 | replication factor C (activator 1)1, 145kDa | 2.064 | 4.06E–03 | 1.066 | 6.71E–01 | 2.092 | 3.06E–03 |
| RFC3 | replication factor C (activator 1)3, 38kDa | 4.476 | 8.78E–04 | 1.371 | 1.98E–01 | 3.577 | 4.97E–04 |
| RFC4 | replication factor C (activator 1)4, 37kDa | 2.799 | 1.31E–02 | 1.198 | 7.25E–01 | 1.878 | 6.80E–02 |
| RFC5 | replication factor C (activator 1)5, 36.5kDa | 2.486 | 2.22E–07 | 1.348 | 1.68E–02 | 2.065 | 1.55E–03 |
| SKA1 | spindle and kinetochore associated complex subunit 1 | 26.35 | 3.44E-06 | 12.14 | 6.36E-06 | 17.75 | 7.18E-05 |
| SOX9 | SRY (sex determining region Y)-box 9 | 8.645 | 2.50E-05 | 9.4 | 0.0017 | 8.419 | 3.40E-04 |
| STMN1 | stathmin 1 | 73.07 | 5.69E-05 | 20.7 | 9.17E-04 | 27.51 | 2.71E-05 |
| TYMS | thymidylate synthetase | 15.49 | 6.62E-06 | 11.37 | 4.44E-05 | 7.212 | 5.09E-05 |
| UBE2C | ubiquitin-conjugating enzyme E2C | 259.3 | 1.89E-07 | 66.4 | 7.52E-05 | 146.5 | 3.72E-07 |
See Goyal & Longo, 2012 [11]
Table 11.
Top Three Down-regulated Pathways
| Symbol | Entrez Gene Name | Premature fetus Vs Adult | Near-term fetus Vs Adult | Newborn Vs Adult | |||
|---|---|---|---|---|---|---|---|
| Fold Change | p-value | Fold Change | p-value | Fold Change | p-value | ||
| Integrin Signaling Pathway | |||||||
| CAPN1 | calpain 1, (mu/I) large subunit | −4.608 | 1.29E–04 | −1.131 | 5.36E–01 | −5.263 | 8.63E–05 |
| FNBP1 | formin binding protein 1 | −7.143 | 4.46E–05 | −1.543 | 5.59E–02 | −4.000 | 1.57E–04 |
| ITGA5 | integrin, alpha 5 (fibronectin receptor, alpha polypeptide) | −39.062 | 7.94E–07 | −6.579 | 8.73E–03 | −13.405 | 9.38E–05 |
| ITGA8 | integrin, alpha 8 | −12.225 | 1.20E–04 | −2.410 | 1.54E–02 | −2.320 | 3.95E–02 |
| TLN1 | talin 1 | −12.469 | 3.97E–05 | −2.070 | 7.29E–02 | −7.143 | 1.41E–04 |
| VASP | vasodilator-stimulated phosphoprotein | −8.197 | 1.02E–03 | −5.155 | 8.11E–02 | −3.817 | 2.87E–03 |
| ZYX | zyxin | −22.936 | 3.75E–04 | −2.625 | 5.61E–02 | −13.175 | 1.43E–04 |
| Actin Cytoskeleton Pathway | |||||||
| ACTA2 | actin, alpha 2, smooth muscle, aorta | −4.292 | 4.03E–04 | −1.168 | 5.14E–01 | −2.114 | 4.97E–03 |
| ACTC1 | actin, alpha, cardiac muscle 1 | −23.529 | 1.40E–03 | −3.584 | 6.02E–02 | −6.944 | 1.03E–02 |
| ACTG2 | actin, gamma 2, smooth muscle, enteric | −5.291 | 1.37E–03 | −2.347 | 9.19E–03 | −2.387 | 1.24E–02 |
| ACTN1 | actinin, alpha 1 | −11.274 | 1.46E–04 | −3.030 | 1.35E–03 | −7.634 | 3.48E–05 |
| ACTN3 | actinin, alpha 3 | −5.525 | 1.83E–03 | −6.173 | 1.43E–01 | −3.226 | 4.49E–03 |
| ACTR2 | ARP2 actin-related protein 2 homolog (yeast) | −4.255 | 7.40E–05 | −2.809 | 2.69E–02 | −2.188 | 8.49E–04 |
| CFL1 | cofilin 1 (non-muscle) | −6.536 | 5.74E–04 | −1.314 | 5.58E–01 | −6.536 | 1.11E–04 |
| FLNA | filamin A, alpha | −16.807 | 4.21E–06 | −2.577 | 1.28E–02 | −5.587 | 3.82E–05 |
| FNBP1 | formin binding protein 1 | −7.143 | 4.46E–05 | −1.543 | 5.59E–02 | −4.000 | 1.57E–04 |
| GSK3A | glycogen synthase kinase 3 alpha | −8.772 | 6.13E–05 | −1.916 | 1.33E–02 | −6.061 | 1.49E–03 |
| WASF2 | WAS protein family, member 2 | −4.386 | 8.08E–06 | −3.774 | 8.74E–03 | −3.413 | 2.60E–04 |
| TESK1 | testis-specific kinase 1 | −7.463 | 2.31E–03 | −3.922 | 2.25E–01 | −4.926 | 1.12E–02 |
| TGFB1I1 | transforming growth factor beta 1 induced transcript 1 | −9.524 | 2.50E–04 | −2.227 | 9.38E–03 | −6.098 | 6.52E–05 |
| PKC-ERK-ROCK Pathway | |||||||
| PLCB2 | phospholipase C, beta 2 | −5.714 | 4.01E–04 | −1.456 | 3.35E–01 | −3.175 | 7.21E–04 |
| PLCD1 | phospholipase C, delta 1 | −9.259 | 3.64E–03 | −7.463 | 1.73E–02 | −4.367 | 3.41E–05 |
| PIK3CD | phosphoinositide-3-kinase, catalytic, delta polypeptide | −8.264 | 4.17E–06 | −5.319 | 1.01E–01 | −6.452 | 2.43E–04 |
| PRKACA | protein kinase, cAMP-dependent, catalytic, alpha | −7.463 | 2.59E–05 | −1.546 | 1.86E–01 | −6.211 | 6.85E–05 |
| PRKAG2 | protein kinase, AMP-activated, gamma 2 non-catalytic subunit | −4.695 | 6.43E–05 | −3.125 | 9.67E–04 | −2.809 | 8.77E–04 |
| PRKCD | protein kinase C, delta | −8.850 | 4.45E–06 | −4.950 | 8.82E–05 | −3.937 | 4.98E–05 |
| CAMK2G | calcium/calmodulin-dependent protein kinase II gamma | −12.516 | 2.99E–07 | −5.747 | 4.65E–04 | −5.376 | 2.92E–05 |
| ITPR1 | inositol 1,4,5-triphosphate receptor, type 1 | −4.831 | 1.66E–02 | 1.567 | 3.27E–01 | −4.484 | 1.77E–02 |
| PPP1R11 | protein phosphatase 1, regulatory (inhibitor) subunit 11 | −5.155 | 1.82E–04 | −1.299 | 1.09E–01 | −5.747 | 2.55E–04 |
| CAPN1 | calpain 1, (mu/I) large subunit | −4.608 | 1.29E–04 | −1.131 | 5.36E–01 | −5.263 | 8.63E–05 |
| ARHGAP1 | Rho GTPase activating protein 1 | −9.346 | 1.30E–04 | −2.545 | 6.02E–02 | −6.061 | 3.72E–04 |
| RHOB | ras homolog gene family, member B | −5.128 | 6.26E–03 | −1.340 | 4.00E–01 | −4.630 | 6.78E–03 |
| RHOG | ras homolog gene family, member G (rho G) | −5.747 | 9.13E–05 | −1.366 | 2.46E–01 | −11.507 | 2.06E–04 |
| RRAS | related RAS viral (r-ras) oncogene homolog | −13.423 | 9.08E–06 | −2.457 | 1.55E–02 | −5.848 | 1.06E–05 |
| MAP2K2 | mitogen-activated protein kinase kinase 2 | −3.436 | 1.33E–04 | −1.499 | 3.18E–02 | −3.876 | 3.15E–06 |
| MAPK3 | mitogen-activated protein kinase 3 | −3.846 | 4.37E–03 | −2.421 | 3.23E–02 | −5.076 | 5.44E–04 |
| MKNK1 | MAP kinase interacting serine/threonine kinase 1 | −2.183 | 1.55E–04 | −1.715 | 4.39E–02 | −2.331 | 1.09E–04 |
| MKNK2 | MAP kinase interacting serine/threonine kinase 2 | −3.906 | 7.11E–04 | −1.653 | 1.03E–01 | −3.390 | 2.19E–03 |
| MYL9 | myosin, light chain 9, regulatory | −18.832 | 1.80E–06 | −3.003 | 2.30E–02 | −5.181 | 4.87E–05 |
| MYLK | myosin light chain kinase | −11.050 | 2.61E–05 | −3.155 | 9.59E–02 | −3.030 | 1.78E–03 |
See Goyal & Longo, 2012 [11]
Fig. (5).

Functional gene regulation pathways altered with development. Bar graph demonstrates specific functional pathways. N was 4 in each experimental group, and all groups were significantly different compared to adult values (P<0.05) [11].
As noted, the deviations of gene expression were much greater (either up- or down-regulated) in premature fetus and newborn lamb than in the near-term fetus. Changes in gene expression profiles (both up- and down-regulation) follow a U-shaped curve with developmental maturation from preterm fetus, to near-term fetus, to newborn. We are not aware of any previous report demonstrating this pattern of expression. These findings underscore the immense changes in gene expression which occur during the perinatal period, and how these differ in a unique and unexpected manner. Of critical importance, several parameters associated with cerebral blood flow such as PCO2, Hemoglobin g%, heart rate, follow a similar U-shaped pattern during preterm, near-term and newborn age group [97]. Although it may be no surprise that gene expression follows a similar pattern, at present, the importance of this finding or its meaning in a deep sense are not clear [11]. In terms of gene expression during early life, there are other significant events, such as complete de-methylation and re-methylation of genome, transcriptional silencing of ovum, histone acetylation of spermatic DNA, for which the explanations are not clear. Our present findings add to this list.
In terms of some specific changes of interest, this study demonstrated that collagen triple helix repeat containing protein 1 (Cthrc1) was up-regulated ~ 450-fold in premature, however, its expression decreased 47-fold in near-term fetus, and 35-fold in newborn cranial arteries. A gene product with novel biochemical activities, by inhibition of Smad2/3 activation Cthrc1 reduces collagen deposition and plays a major role in vascular development, repair, and fibrosis [552], as well as increasing cellular migration [553]. As noted earlier, premature fetal arteries are significantly more fragile than those of near-term fetus [10]. Cthrc1 up-regulation may be responsible for reduced collagen content and, thus increased fragility, leading to higher propensity of the premature fetus for germinal matrix hemorrhages and other intracerebral bleeds. Further studies are needed to examine its role during fetal life, however. Similarly, extracellular superoxide dismutase (SOD) was significantly down-regulated during early life, compared to adult. Vascular tissues express three distinct isoforms of SOD: cytosolic or copper-zinc SOD (CuZn-SOD; SOD-1), manganese SOD (Mn-SOD) localized in mitochondria (mitochondrial SOD or SOD-2), and an extracellular form of CuZn-SOD (EC-SOD; SOD-3) [554]. SOD plays a crucial role in conversion of superoxide anion (O2˙−) to H2O2; which is further converted to H20 by the actions of glutathione peroxidases and peroxiredoxins. The functional importance of individual SOD isoforms has been difficult to define as no selective pharmacological inhibitors of these are known. Nonetheless, the present finding of significant down-regulation of specifically EC-SOD3 suggest a distinct role in early life. Of importance, we observed that along with down-regulation of SOD-3, there was a significant up-regulation of both glutathione peroxidase (GPX-8) and peroxiredoxin4 (PRDX-4) [11]. Thus, the system is geared towards reduced production and rapid clearance of H2O2. At present, a clear rationale for this is not known.
This study also demonstrated significant up-regulation during early life of aurora kinase A and B, several cyclins, centromere proteins and replication factors. By hyperphosphorylation of normal cell cycle targets and aberrant phosphorylation of cytoplasmic targets, Aurora A and Aurora B overexpression can lead to genetic instability (gain or loss of whole chromosomes) [555]. The resultant chromosomal instability is a common feature of many cancer types [556]. Moreover, ubiquitin conjugating enzyme e2c (UBE2C) was up-regulated almost 260-fold in premature fetus and its expression fell to ~66-fold in near-term fetus. Importantly, excessive UBE2C is known to disrupt normal chromosome segregation, or even lead to mis-separation of the chromosomes [557] and may lead to malignancies [558]. At present, the precise role of these kinases during early life is not known and needs further investigation.
In summary, significant alterations of gene expression occur in concert with maturation of the cranial vasculature. Compared to more active gene expression of several signaling pathways in the premature fetus or newborn organism, in the near-term fetus differential gene expression is attenuated significantly. Although this may be an important step for the preparation of the fetus for birth, its rationale is unclear. These findings agree with the concept of molecular mechanisms acting in an integrated manner to regulate both phenotypic and mechanical plasticity in vascular SMCs [559]. In addition to raising a number of questions regarding developmental regulation of changes in vascular gene expression and their biological significance, perhaps of most importance, these findings also provide a basis for future studies to explore the importance of changes in the major signal transduction pathways during early vascular development and their function. In concert, it suggests avenues in which to target the developing cerebral vasculature for gene therapy [560].
DEVELOPMENTAL CEREBRAL BLOOD FLOW IN RESPONSE TO ACUTE HYPOXIA: A CASE STUDY
With this background, it may be instructive to consider the case of a not uncommon stress, that of hypoxia. As in the adult, in the developing fetus and newborn a range of protective cardiovascular and cerebral circulatory mechanisms come into play in response to decreased O2 in blood or tissue. Oxygenation of the fetus is achieved via the uteroplacental and umbilical circulations, with delivery of oxygenated blood to the organs and tissues of the fetal body [72]. The regulation of the cardiovascular system and distribution of blood flow have been the focus of much clinical interest and physiologic research. Compromised fetal oxygenation can have a number of sequelae of clinical importance, including intrauterine growth restriction, neurodevelopmental handicaps, and others [561]. Hypoxic-induced activation of carotid body chemoreceptors initiates an increase in vagal efferent activity to the heart producing bradycardia (which is augmented by the release of vasopressin [562]). This is associated with an increase in sympathetic activity, which with the release of catecholamines [563], produces vasoconstriction in the carcass and a marked rise in systemic blood pressure.
In the fetal lamb, in contrast to the hypoxia-induced decrease in blood flow to the carcass, that to the brain, heart, and adrenal glands increase significantly [563–566]. As noted above, this CBF increase appears to be mediated by local vasodilatory agents such as NO [256, 277, 279] and adenosine [306, 308, 309, 567]. In a similar manner, hypoxic-induced redistribution of blood flow to the developing brain has been observed in preterm fetal sheep (0.6 and 0.7 gestational age [568]), as well as in the chick embryo [569], llama [570, 571], and rhesus monkey [572]. A similar increase in CBF has been reported in response to isovolemic decrease in the fetal hematocrit [573]. In fetal sheep, gestational age had a major influence on hormonal responses, those to neuropeptide Y and arginine vasopressin increasing dramatically from 0.9 gestation to full term [574].
The magnitude of this CBF increase is 30 to 100 percent, depending upon the degree of hypoxia and the maintenance of isocapnia, with a preservation of O2 delivery to the brain. An exception, and in contrast to sheep, is the fetal llama in which hypoxia is associated with intense peripheral vasoconstriction that is unaffected by section of carotid sinus nerves, and only a minor increase in CBF [571]. Thus, this was associated with a significant decrease in cerebral O2 consumption. In addition, the increase in the vasoconstrictors catecholamines, neuropeptide Y, and arginine vasopressin were much greater than in sheep. Furthermore, as in sheep, administration of an α-adrenergic antagonist to the fetal llama resulted in a profound decrease in blood pressure, and carotid artery blood flow, rapidly leading to fetal death [570, 571, 575, 576].
In the fetal sheep, the acute hypoxia-induced increase in CBF was not uniform to brain regions, with a hierarchy of responsiveness, so that flow to the brainstem and subcortex, significantly exceeded that to the cerebral cortex [53]. With hypercapnia and acidosis this pattern of blood flow distribution was maintained [54]. In another study, while the hypoxia-induced increase in CBF showed the regional distribution noted above, flow to the pituitary gland and choroid plexus decreased [566].
Although the mechanisms are as yet unclear, in sheep acclimatized to high altitude, the hypoxic-induced CBF increase was only ~20 percent, as opposed to almost 50 percent in sea level controls. In both groups, tissue PO2 values decreased similarly, as did the cerebral metabolic rate for O2 [71]. In the presence of acute hypoxemia at either altitude, the fetal electrocorticographic activity switched from the low voltage high frequency to the high voltage low frequency state [257].
A further consideration of hypoxic-mediated changes in CBF, is that of the role of corticosteroids. Commencing in the late 1960s, the value of corticosteroids in maturing the lungs of the preterm fetus was recognized [577, 578]. Although maturation of the lungs with their surfactant is critical for such premature infants, it was several decades before the two-edged sword of such therapy was recognized. By performing adrenalectomy and infusing cortisol in the ovine fetus, Nathanielsz and coworkers determined the essential nature of the adrenal gland in the maintenance of blood pressure [579]. They also demonstrated in the near-term (0.9 gestation) fetal lamb that infusion of either betamethazone or dexamethazone increased vascular resistance and blood pressure [580]. Multiple mechanisms were demonstrated to account for these changes [581], including endothelin-induced vasoconstriction and reduced endothelium-dependent relaxation [582]. These workers also demonstrated that following 48 hr betamethasone infusion cerebrovascular resistance was increased significantly, with a 25 to 30% decrease in total CBF. In the thalamus and hindbrain flow was reduced 35 to 40% and overall cerebral O2 delivery was reduced 30 to 45%. The suggestion was made that in the newborn infant such a CBF decrease might be protective against intracerebral hemorrhage [583]. The Nathanielsz group also reported on the role of dexamethasone administered in three weekly doses, beginning at day 103 (0.73) of gestation, in producing endothelial dysfunction as manifest by increased sensitivity to endothelin-1 and abolishment of the L-NAME suppression of vasodilatory response [584]. Following a study in late gestation fetal sheep [585], they also showed that during the last third of gestation these effects were independent of fetal age [586]. Following ligation of one umbilical artery in fetal sheep that produced intrauterine growth restriction, betamethasone treatment produced a doubling of CBF [587].
As noted in the section on development of the cerebrovasculature, in the fetal lamb Stonestreet and colleagues have demonstrated increasing impermeability of the blood-brain barrier (BBB) from 0.6 to 0.9 gestation [165, 166]. In part, BBB permeability is regulated by corticosteroids [588], and in the premature lamb this group also showed that antenatal dexamethazone administration significantly decreased BBB permeability [166, 167]. As noted, these findings are of relevance to the beneficial effects of antenatal corticosteroids in lowering the risk of early onset intraventricular hemorrhage [589, 590].
Importantly, the Giussani group has been examining these corticosteroid-induced cerebrovascular effects in the presence of hypoxia. In the 0.8 gestation sheep fetus they demonstrated the role of adenosine in mediating hypoxic-induced responses [591], and reported that despite antenatal dexamethasone treatment decreasing CBF, it enhanced glucose delivery to the brain [592].
Finally, a consideration of the clinical applications in the human fetus and newborn is appropriate. With the introduction of Doppler ultrasound imaging, to measure vascular blood velocity as a surrogate for flow, it was not long before it was applied to the fetal middle cerebral artery. The absence of end-diastolic flow (abnormal pulsatility index) in the umbilical artery or aorta has been shown to be evidence of fetal distress necessitating immediate delivery [593, 594]. In an effort to predict fetal hypoxia or intrauterine growth restriction (IUGR), several groups have examined this principle in the MCA [595–597]. In early reports, Doppler velocimetry of MCA among fetuses with signs of increased umbilical artery resistance failed to indentify those that could withstand the stress of labor [598]. Nonetheless, with persistence, experience, and improved methodology, neurobehavioral impairment has been reported among preterm newborns who demonstrated abnormal MCA pulsatility index with IUGR [599]. It probably is too early to generalize in this regard, however, confounding factors such as fetal gestational age and birthweight necessitate caution in drawing causal effects from associations with the neurobehavioral scoring system used [600–602].
CONCLUSIONS AND PERSPECTIVE
A unique aspect of biology is represented by an organism's development during embryonic and fetal life, the events of parturition, with continued growth and maturation during the neonatal period through adolescence and on to adulthood. Somewhat surprisingly, despite its importance and the seemingly miraculous orchestration of critical events and processes, the formative aspects of life have attracted relatively modest interest. Here, we have attempted to gain insight into one small facet of this developmental continuum, that of regulation of relaxation/contraction mechanisms of the cerebral vasculature. As reviewed, these mechanisms are extremely complex and involve multiple pathways, “cascades”, or networks with considerable interaction and “crosstalk”. Thus, the subject defies any overly simplistic synthesis. Nonetheless, the last few years have witnessed enormous progress in sorting out this virtual “spiders web” of receptors, chemical second and third messengers, enzymes, and other proteins involved in the mechanistic intricacies of electro-mechanical and pharmaco-mechanical coupling.
Fig. (6) attempts to place in perspective some of the major differences in cerebrovascular relaxation/contraction mechanisms in the fetus and newborn, as opposed to the adult. A particularly striking feature of the observations reported, is the degree to which certain specific elements and signal transduction pathways and their interactions differ as a function of developmental age. As is evident, there exist three basic developmental patterns of cerebrovascular reactivity: i.e., those elements that remain essentially constant over the course of maturation, those that increase to play a more prominent role, and those that decrease. Clearly, important differences in the manner in which these mechanisms function distinguish the developing cerebral vasculature from that of the adult. Thus, one might be tempted to ask, are these developmental changes merely an epiphenomenon or are they of fundamental biologic significance. Considered from one point of view, cerebral artery relaxation and contraction mechanisms in the fetal and newborn organism work amazingly well. Nonetheless, the significant differences in mechanistic coupling and interaction may be a key factor in the vulnerability of the cerebral vasculature to dysregulation in response to hydrostatic pressure surges, hypoxia and other stress.
Fig. (6).

Calcium sensitivity pathways in fetal and adult cerebral arteries. Developing fetal vessels are characterized by having a significantly greater requirement for Ca2+ from extracellular source, much greater Ca2+ sensitivity, contraction mediated by prostaglandins and the Rho kinase and ERK1/2 pathways, a paucity of Ca2+ sparks to activate the BK channel, the important role of PKG in mediating BK channel activity, and up-regulation of a large number of genes and pathways. In turn, adult cerebral arteries are characterized by Ca2+ from intracellular sarcoplasmic reticulum stores, lower Ca2+ sensitivity, relaxation mediated by the eNOS-NO pathway, CPI-17, and caldesmon which play a major role in vasoreactivity, abundant Ca2+ sparks to activate the BK channel, and the role of PKA in mediating BK channel activity.
That elements of the cerebral artery agonist-mediated, presynaptic and postsynaptic, relaxation and contraction mechanisms should be regulated independently and show differing responses during the course of maturation should come as no surprise. This only furthers the view that homeostatic responses differ in the immature as opposed to the more mature animal. These include the structural transition from immature “synthetic” to the more mature “contractile” SMC phenotype, accompanied by major structural changes of essentially every element. For developing cerebral arteries, the character of many of the responses are similar to those of the adult, with the trend that the magnitudes of changes are smaller for contractility and larger for changes in receptor density. Maturation of many of the SMC mechanisms also illustrates the concept of “developmental plasticity.” An issue not considered here is that, whereas such homeostatic responses are important during fetal development, clearly multiple secondary responses are of critical importance during the process of birth and in the newborn infant. These include: levels of circulating catecholamines, 5-HT, prostaglandins, cortisol, other stress-related hormones, growth factors, and their related cellular responses. Thus overall, multiple mechanisms are recruited in response to normal development, both to promote relaxation (hyper-relaxation hypothesis) and to attenuate contraction (hypo-contractile hypothesis). Within this framework, the multiple independent mechanisms are heterologous and unique for each signal transduction pathway. In terms of relaxation, considerable evidence supports the “hyper-relaxation” hypothesis, with many factors playing a critical role in this regard, including active roles for prostaglandins.
In terms of contractile mechanisms, the relatively immature organism is characterized by the unique roles that L-type Ca2+ channels and K+ channels (in particular the BK channel) play in regulating Ca2+ entry into the fetal cerebral SMC. The BK channel in the fetus shows greater conductance, and is most sensitive to PKA phosphorylation, in contrast to the adult which is more sensitive to PKG. Additionally, Ca2+ stores in fetal SMC sarcoplasmic reticulum demonstrate much less releasable Ca2+ than adult, either en masse or as “sparks”, and the developing cerebrovasculature being more dependent on extracellular Ca2+ for contraction. This contrasts with the adult, in which intracellular Ca2+ store in sarcoplasmic reticulum play a more important role in Ca2+ signaling. In the developing fetus, the relative density of alpha1-adrenergic receptor subtypes (A,B,D) are much less than that of the adult and differ strikingly in their responses, the B and D subtypes being responsible for trophic responses. Also, compared to adult cerebrovascular SMCs, which show little activity of Rho kinase-mediated mechanisms, fetal arteries show great dependence on ROCK. In turn, developing SMC lack the adult sensitivity to CPI-17 inhibition of MLCP. Of particular significance, compared to the more mature vessels, those of the developing organism display much greater Ca2+ sensitivity. Thus, in terms of overall regulation one may postulate a “ying and yang”, wherein a combination of elevated Ca2+ sensitivity and reliance on extracellular Ca2+ characterize early development, whereas the mature adult cerebrovasculature is dependent on intracellular Ca2+ stores with decreased CaS. Also compared to adult SMCs, those of the developing fetus and newborn demonstrate manifold differences in gene regulation of critical signal transduction pathways.
A caveat of most of these studies is that a limited number of age groups were examined, e.g., preterm fetus, term fetus, newborn, and adult. Thus, the true developmental profiles of variable changes with maturation are either unknown or poorly described. Nonetheless, it is the late fetal and newborn period, that phase upon which most studies have focused, that has high relevance to dysregulation and disease. An additional caveat is that much of the data in the present review is based on studies in the middle cerebral arteries. This is not necessarily negative, as several lines of evidence in the adult cerebral circulation demonstrate it to be unique in that, in contrast to many vascular beds, relatively large arteries such as these main branch vessels (>150 μm in diameter) account for 25 to 30% of total cerebrovascular resistance [6, 86, 603]. In the developing organism these cerebral vessels may play an even greater regulatory role, as one of their key functions is to provide collateralization and shunting of blood flow in several directions, depending upon pressure gradients within the system. Nonetheless, the extent to which small resistance artery function differs from that of larger vessels, and the role of innervation in the neurovascular unit in these responses, is of vital importance but not well defined. An additional caveat which has not been addressed is that of heterogeneity of vascular responses, not only within different vascular beds, but also in the several segments of a given vessel. Additionally, and importantly, we have little data on the extent to which most of the in vitro changes reported here are reflected in the in vivo regulation of vascular tone and CBF. Considerable current effort is directed toward using the reductionist approach as a basis for “translational” studies relevant to the clinical standpoint. Finally, while the present studies apply to cerebral arteries, the extent to which such changes occur in developing myocardial, pulmonary, renal, or other vital vessels remains to be determined.
SPECULATION AND CHALLENGES FOR THE FUTURE
In light of the above, one might ask what do the present findings mean for future research? A number of major challenges lie ahead. Biocomplexity poses perhaps the greatest challenge, to understand the manner in which cell and tissue functions emerge from the interactions within the manifold array of biochemical and molecular networks. During recent years, linear models of signal transduction have provided useful information on the role of various receptors, second messengers, enzymes, and other cascade elements in terms of development of cerebrovascular function and dynamics. Nonetheless, as we progress a more encompassing network model incorporating interactions among a host of molecules will be required to understand the emergent properties of activity and function of the diverse cerebrovascular cells. Thus, in our effort to unravel the underlying basis of bio-complexity we must move beyond the phenomenology of signaling pathways, to consider their interactions, feedback mechanisms, and temporal regulation in health as well as disease [156–158]. In terms of development, an important lesson is the plasticity of signal transduction mechanisms in a given cell type and/or vessel. This suggests that continued exploration of these regulatory mechanisms is likely to be rewarded with unexpected, important new findings and insights. Although the general principles of signaling are becoming clarified, a challenge is to elucidate the details of the regulatory mechanisms of information flow, signal amplification, feedback regulation, and crosstalk among different pathways.
In terms of CBF dysregulation in the fetus and newborn infant, many factors have profound implications as the basis of hypo- and hyper-reactivity, vessel rupture, intraventricular hemorrhage, and long-term neurological sequelae. In addition and of consequence, these studies have implications for the developmental “programming” during fetal and neonatal life of cerebrovascular, cardiovascular, and other disease in the adult. Thus, one must ask, what do these findings mean in terms of the clinical care of premature and other newborn infants and other patients? It is hoped that the present synthesis will have vital implications for diagnosis and potential therapy of those infants with dysregulation of CBF, as well as relevance to the broad field of preventive medicine and public health. Recent advances are promising in that they point the direction for moving beyond descriptive phenomenology to investigation of fundamental cellular and molecular regulatory mechanisms, and understanding in a deeper sense. Of unprecedented opportunity and great promise, the rapidly growing diversity and power of new investigative tools and technology offer the opportunity for further insights into the role of development in regulation of the cerebrovasculature. Hopefully, a new generation of studies of gene regulation, protein-protein interactions, and related mechanisms will yield key information that will add to, and stimulate further, rapid advancement in this field of critical importance. The translational application of this knowledge to the prevention and amelioration of cerebrovascular complications in the fetus and newborn infant constitutes one of our greatest challenges for the future.
ACKNOWLEDGMENTS
We thank Brenda Kreutzer and Jimin Suh for preparing the manuscript. We also thank William J. Pearce for critiquing an early version of this manuscript. In addition, we thank the late Charles J.C. Kean, DVM, Ph.D., and David Wolff, DVM, Ph.D., Directors of the Animal Care Facility, Loma Linda University for their invaluable assistance in many of the studies reported.
Grants. This work was supported, in part, by United States Public Health Service grants HD-03807 and HD 312226 to LDL.
ABBREVIATIONS
- AA
Arachidonic acid
- α1-AR
α1-adrenergic receptors
- ADO
Adenosine
- BBB
Blood brain barrier
- BKCa
“Big” calcium-activated potassium channel
- cAMP
Adenosine 3′;5′-cyclic monophosphate
- CaS
Calcium sensitivity
- CBF
Cerebral blood flow
- CCA
Common carotid artery
- cGMP
Guanosine 3′;5′ cyclic monophosphate
- CNS
Central nervous system
- CO
Carbon monoxide
- COX
Cyclooxygenase
- CPI-17
PKC-potentiated inhibitor protein of 17 kDa
- CSF
Cerebrospinal fluid
- ED50
Effective dose for half-maximal response
- EET
Epoxyeicosatrienoic acid
- ERK1/2
Extracellular regulated kinase 1 and 2
- GD
Gestational days
- HbCO
Carboxyhemoglobin
- HETE
Hydroxyeicosatetraenoic acid
- HO
Heme oxygenase
- HSP
Heat-shock protein
- Ins(1,4,5)P3
Inositol 1,4,5-trisphosphate
- KCa
Calcium-activated potassium channel
- Kmax
Peak height as a fraction of the maximum response to K+ depolarization
- L-NAME
Nw-nitro-L-arginine methyl ester
- L-NNA
NG-nitro-L-arginine
- MAPK
Mitogen-activated protein kinase
- MBC
Main branch cerebral artery
- MCA
Middle cerebral artery
- MLC20
Myosin regulatory light chain of 20 kDa
- MLCK
Myosin light chain kinase
- MLCP
Myosin light chain phosphatase
- Na+-K+-ATPase
Sodium-potassium ATPase
- NE
Norepinephrine
- NO
Nitric oxide
- NOS
Nitric oxide synthase
- NMDA
N-methyl-D-aspartate
- PCNA
Proliferating cell nuclear antigen
- pD2
Negative logarithm of the half-maximal response concentration
- PDBu
Phorbol 12,13-dibutyrate
- PGE2
Prostaglandin E2
- PHE
Phenylephrine
- pIC50
Negative logarithm of the half-maximal inhibition concentration
- PKA
Protein kinase A
- pKb
Antagonist dissociation constant
- PKC
Protein kinase C
- PKG
Protein kinase G
- PLA2
Phospholipase A2
- PLCβ
Phospholipase Cβ
- PO2
Partial pressure of oxygen
- RYN
Ryanodine
- SERCA
Sarcoplasmic-endoplasmic reticulum Ca2+-ATPase
- sGC
Soluble guanylate cyclase
- SMC
Smooth muscle cell
- SNAP
S-nitroso-N-acetylpenicillamine
- SR
Sarcoplasmic reticulum
- V1/2
Voltage for half-maximal activation
- Vmax
Maximal rate of activity
- 5-HT
Serotonin
Footnotes
CONFLICT OF INTEREST The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Bevan JA, Bevan RD. Developmental influences on vascular structure and function. Ciba Found Symp. 1981;83:94–107. doi: 10.1002/9780470720653.ch5. [DOI] [PubMed] [Google Scholar]
- [2].Bevan RD, Bevan JA. The human brain circulation: functional changes in disease. Humana Press; Totowa, NJ: 1994. [Google Scholar]
- [3].Busija DW, Heistad DD. Factors involved in the physiological regulation of the cerebral circulation. Rev Physiol Biochem Pharmacol. 1984;101:161–211. doi: 10.1007/BFb0027696. [DOI] [PubMed] [Google Scholar]
- [4].Ursino M. Regulation of the circulation of the brain. In: Bevan RD, Bevan JA, editors. The human brain circulation. Humana Press; Totowa, NJ: 1994. pp. 291–318. [Google Scholar]
- [5].Wahl M, Schilling L. Regulation of cerebral blood flow – a brief review. Acta Neuroschir Suppl (Wien) 1993;59:3–10. doi: 10.1007/978-3-7091-9302-0_1. [DOI] [PubMed] [Google Scholar]
- [6].Bevan JA, Bevan RD, Duckles SP. Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle. American Physiological Society; Bethesda, MD: 1980. Adrenergic regulation of vascular smooth muscle; pp. 515–66. [Google Scholar]
- [7].Duckles SP, Banner W. Changes in vascular smooth muscle reactivity during development. Ann Rev Pharmacol Toxicol. 1984;24:65–83. doi: 10.1146/annurev.pa.24.040184.000433. [DOI] [PubMed] [Google Scholar]
- [8].Fleisch JH. Age-related changes in sensitivity of blood vessels to drugs. Pharmacol Ther. 1980;8:477–87. doi: 10.1016/0163-7258(80)90055-8. [DOI] [PubMed] [Google Scholar]
- [9].Goyal R, Mittal A, Chu N, Shi L, Zhang L, Longo LD. Maturation and the role of PKC-mediated contractility in ovine cerebral arteries. Am J Physiol Heart Circ Physiol. 2009;297:H2242–52. doi: 10.1152/ajpheart.00681.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Goyal R, Henderson DA, Chu N, Longo LD. Ovine middle cerebral artery characterization and quantification of ultrastructure and other features: changes with development. Am J Physiol Regul Integr Comp Physiol. 2012;302:R433–45. doi: 10.1152/ajpregu.00519.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Goyal R, Longo LD. Gene expression in sheep carotid arteries: major changes with maturational development. Pediatr Res 2012 Pediatr Res. 2012;72:137–46. doi: 10.1038/pr.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hayashi S, Park MK, Kuehl TJ. Relaxant and contractile responses to prostaglandins in premature, newborn and adult baboon cerebral arteries. J Pharmacol Exp Ther. 1985;233:628–35. [PubMed] [Google Scholar]
- [13].Seidel CL, Ross B, Michael L, Freedman J, Burdick B, Miller T. Maturational changes in the pharmacological characteristics and actomyosin content of canine arterial and venous tissue. Pediatr Res. 1987;21:152–8. doi: 10.1203/00006450-198702000-00009. [DOI] [PubMed] [Google Scholar]
- [14].Longo LD, Ueno N, Zhao Y, Zhang L, Pearce WJ. NE-induced contractions, α1-adrenergic receptors, and Ins(1,4,5)P3 responses in cerebral arteries. Am J Physiol. 1996;270:H915–23. doi: 10.1152/ajpheart.1996.270.3.H915. [DOI] [PubMed] [Google Scholar]
- [15].Longo LD, Ueno N, Zhao Y, Pearce WJ, Zhang L. Developmental changes in α1-adrenergic receptors, IP3 responses, and NE-induced contraction in cerebral arteries. Am J Physiol. 1996;271:H2313–9. doi: 10.1152/ajpheart.1996.271.6.H2313. [DOI] [PubMed] [Google Scholar]
- [16].Longo LD, Zhao Y, Long W, et al. Dual role of PKC in modulating pharmacomechanical coupling in fetal and adult cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1419–29. doi: 10.1152/ajpregu.2000.279.4.R1419. [DOI] [PubMed] [Google Scholar]
- [17].Pearce WJ, Hull AD, Long DM, Longo LD. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol. 1991;261:R458–65. doi: 10.1152/ajpregu.1991.261.2.R458. [DOI] [PubMed] [Google Scholar]
- [18].Pearce WJ, Duckles SP, Buchholz J. Effects of maturation on adrenergic neurotransmission in ovine cerebral arteries. Am J Physiol. 1999;277:R931–7. doi: 10.1152/ajpregu.1999.277.4.R931. [DOI] [PubMed] [Google Scholar]
- [19].Pearce WJ, Williams JM, Chang MM, Gerthoffer WT. ERK inhibition attenuates 5-HT-induced contractions in fetal and adult ovine carotid arteries. Arch Physiol Biochem. 2003;111:36–44. doi: 10.1076/apab.111.1.36.15143. [DOI] [PubMed] [Google Scholar]
- [20].Sheth RD. Trends in incidence and severity of intraventricular hemorrhage. J Child Neurol. 1998;13:261–4. doi: 10.1177/088307389801300604. [DOI] [PubMed] [Google Scholar]
- [21].Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–95. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- [22].Nelson KB. Can we prevent cerebral palsy? N Engl J Med. 2003;349:1765–9. doi: 10.1056/NEJMsb035364. [DOI] [PubMed] [Google Scholar]
- [23].Stoll BJ, Hansen NI, Bell EF, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–56. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vannucci RC, Vannucci SJ. Perinatal brain metabolism. In: Polin RA, Fox WW, Abman SH, editors. Fetal and neonatal physiology. 3rd ed vol 2. Saunders; Philadelphia, PA: 2004. pp. 1713–25. [Google Scholar]
- [25].Volpe JJ. Brain injury in the premature infant: neuropathology, clinical aspects, pathogenesis, and prevention. Clin Perinatol. 1997;24:567–87. [PubMed] [Google Scholar]
- [26].Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. Reprint of “The developing oligodendrocyte: key cellular target in brain injury in the premature infant”. Int J Devl Neuroscience. 2011;29:565–82. doi: 10.1016/j.ijdevneu.2011.07.008. [DOI] [PubMed] [Google Scholar]
- [27].Doyle LW, Roberts G, Anderson PJ. Victorian Infant Collaborative Study Group. Outcomes at age 2 years of infants <28 weeks' gestational age born in Victoria in 2005. J Pediatr. 2010;156:49–53. e1. doi: 10.1016/j.jpeds.2009.07.013. [DOI] [PubMed] [Google Scholar]
- [28].Groenendaal F, Termote JU, van der Heide-Jalving M, van Haastert IC, de Vries LS. Complications affecting preterm neonates from 1991 to 2006: what have we gained? Acta Paediatr. 2010;99:354–8. doi: 10.1111/j.1651-2227.2009.01648.x. [DOI] [PubMed] [Google Scholar]
- [29].Munck P, Haataja L, Maunu J, et al. Cognitive outcome at 2 years of age in Finnish infants with very low birth weight born between 2001 and 2006. Acta Paediatr. 2010;99:359–66. doi: 10.1111/j.1651-2227.2009.01589.x. [DOI] [PubMed] [Google Scholar]
- [30].Aarnoudse-Moens CS, Weisglas-Kuperus N, van Goudoever JB, Oosterlaan J. Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics. 2009;124:717–28. doi: 10.1542/peds.2008-2816. [DOI] [PubMed] [Google Scholar]
- [31].Del Toro J, Louis P, Goddard FJ, Finegold J. Cerebrovascular regulation and neonatal brain injury. Pediatr Neurol. 1991;7:3–12. doi: 10.1016/0887-8994(91)90098-6. [DOI] [PubMed] [Google Scholar]
- [32].Johnson S, Hollis C, Kochhar P, Hennessy E, Wolke D, Marlow N. Autism spectrum disorders in extremely preterm children. J Pediatr. 2010;156:525–31. e2. doi: 10.1016/j.jpeds.2009.10.041. [DOI] [PubMed] [Google Scholar]
- [33].Msall ME. Central nervous system connectivity after extreme prematurity: understanding autistic spectrum disorder. J Pediatr. 2010;156:519–21. doi: 10.1016/j.jpeds.2009.12.035. [DOI] [PubMed] [Google Scholar]
- [34].Pape KE. Etiology and pathogenesis of intraventricular hemorrhage in newborns. Pediatrics. 1989;84:382–5. [PubMed] [Google Scholar]
- [35].Perlman JM, Rollins NK, Evans D. Neonatal stroke: Clinical characteristics and cerebral blood flow velocity measurements. Pediatr Neurol. 1994;11:281–4. doi: 10.1016/0887-8994(94)90002-7. [DOI] [PubMed] [Google Scholar]
- [36].Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res. 1990;27:317–26. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- [37].Volpe JJ. Neurology of the newborn. 5th ed. Elsevier; Philadelphia: 2008. [Google Scholar]
- [38].Ozduman K, Pober BR, Barnes P, et al. Fetal stroke. Pediatr Neurol. 2004;30:151–62. doi: 10.1016/j.pediatrneurol.2003.08.004. [DOI] [PubMed] [Google Scholar]
- [39].Paul RH, Yonekura ML, Cantrell CJ, Turkel S, Pavlova Z, Sipos L. Fetal injury prior to labor: does it happen? Am J Obstet Gynecol. 1986;154:1187–93. doi: 10.1016/0002-9378(86)90697-6. [DOI] [PubMed] [Google Scholar]
- [40].Martin JA, Hamilton BE, Ventura SJ, et al. Births: Final data for 2009. National vital statistics reports. vol 60, no 1. National Center for Health Statistics; Hyattsville, MD: 2011. [PubMed] [Google Scholar]
- [41].Goyal R, Mittal A, Chu N, Arthur RA, Zhang L, Longo LD. Maturation and long-term hypoxia-induced acclimatization responses in PKC-mediated signaling pathways in ovine cerebral arterial contractility. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1377–86. doi: 10.1152/ajpregu.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goyal R, Mittal A, Chu N, Zhang L, Longo LD. Alpha(1)-adrenergic receptor subtype function in fetal and adult cerebral arteries. Am J Physiol Heart Circ Physiol. 2010;298:H1797–806. doi: 10.1152/ajpheart.00112.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Leffler CW, Nasjletti A, Yu C, Johnson RA, Fedinec AL, Walker N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am J Physiol. 1999;276:H1641–6. doi: 10.1152/ajpheart.1999.276.5.H1641. [DOI] [PubMed] [Google Scholar]
- [44].Leffler CW, Nasjletti A, Johnson RA, Fedinec AL. Contribution of prostacyclin and nitric oxide to carbon monoxide-induced cerebrovascular dilation in piglets. Am J Physiol Heart Circ Physiol. 2001;280:H1490–5. doi: 10.1152/ajpheart.2001.280.4.H1490. [DOI] [PubMed] [Google Scholar]
- [45].Blood AB, Zhao Y, Long W, Zhang L, Longo LD. L-type Ca2+ channels in fetal and adult ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2002;282:R131–8. doi: 10.1152/ajpregu.00318.2001. [DOI] [PubMed] [Google Scholar]
- [46].Long W, Zhao Y, Zhang L, Longo LD. Role of Ca2+ channels in NE-induced increase in [Ca2+]i and tension in fetal and adult cerebral arteries. Am J Physiol. 1999;277:R286–94. doi: 10.1152/ajpregu.1999.277.1.R286. [DOI] [PubMed] [Google Scholar]
- [47].Nauli SM, Ally A, Zhang L, Gerthoffer WT, Pearce WJ. Maturation attenuates the effects of cGMP on contraction, [Ca2+]i and Ca2+ sensitivity in ovine basilar arteries. Gen Pharmacol. 2000;35:107–18. doi: 10.1016/s0306-3623(01)00100-8. [DOI] [PubMed] [Google Scholar]
- [48].Bevan JA, Su C. Sympathetic mechanisms in blood vessels: nerve and muscle relationships. Annu Rev Pharmacol. 1973;13:269–85. doi: 10.1146/annurev.pa.13.040173.001413. [DOI] [PubMed] [Google Scholar]
- [49].Toda N. Age-related changes in responses to nerve stimulation and catecholamines in isolated monkey cerebral arteries. Am J Physiol. 1991;260:H1443–8. doi: 10.1152/ajpheart.1991.260.5.H1443. [DOI] [PubMed] [Google Scholar]
- [50].Pryds O. Control of cerebral circulation in the high-risk neonate. Ann Neurol. 1991;30:321–9. doi: 10.1002/ana.410300302. [DOI] [PubMed] [Google Scholar]
- [51].Donegan JH, Traystman RJ, Koehler RC, Jones MD, Jr, Rogers MC. Cerebrovascular hypoxic and autoregulatory responses during reduced brain metabolism. Am J Physiol. 1985;249:H421–9. doi: 10.1152/ajpheart.1985.249.2.H421. [DOI] [PubMed] [Google Scholar]
- [52].Heistad DD, Kontos HA. Cerebral circulation. In: Shepherd JT, Abboud FM, Geiger SR, editors. The handbook of physiology, The cardiovascular system. vol 3, part 2. American Physiology Society; Baltimore: 1983. pp. 137–82. [Google Scholar]
- [53].Ashwal S, Majcher JS, Vain N, Longo LD. Patterns of fetal lamb regional cerebral blood flow during and after prolonged hypoxia. Pediatr Res. 1980;14:1104–10. doi: 10.1203/00006450-198010000-00003. [DOI] [PubMed] [Google Scholar]
- [54].Ashwal S, Dale PS, Longo LD. Regional cerebral blood flow: studies in the fetal lamb during hypoxia, hypercapnia, acidosis, and hypotension. Pediatr Res. 1984;18::1309–16. doi: 10.1203/00006450-198412000-00018. [DOI] [PubMed] [Google Scholar]
- [55].Bishai JM, Blood AB, Hunter CJ, Longo LD, Power GG. Fetal lamb cerebral blood flow (CBF) and oxygen tensions during hypoxia: a comparison of laser Doppler and microsphere measurements of CBF. J Physiol. 2003;546:869–78. doi: 10.1113/jphysiol.2002.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gagnon R, Lamb T, Richardson B. Cerebral circulatory responses of near-term ovine fetuses during sustained fetal placental embolization. Am J Physiol. 1997;273:H2001–8. doi: 10.1152/ajpheart.1997.273.4.H2001. [DOI] [PubMed] [Google Scholar]
- [57].Greisen G. Effect of cerebral blood flow and cerebrovascular autoregulation on the distribution, type and extent of cerebral injury. Brain Pathol. 1992;2:223–8. doi: 10.1111/j.1750-3639.1992.tb00695.x. [DOI] [PubMed] [Google Scholar]
- [58].Harris AP, Koehler RC, Gleason CA, Jones MD, Jr, Traystman RJ. Cerebral and peripheral circulatory responses to intracranial hyper-tension in fetal sheep. Circ Res. 1989;64:991–1000. doi: 10.1161/01.res.64.5.991. [DOI] [PubMed] [Google Scholar]
- [59].Jones MD, Jr, Sheldon RE, Peeters LL, Makowski EL, Meschia G. Regulation of cerebral blood flow in the ovine fetus. Am J Physiol. 1978;235:H162–6. doi: 10.1152/ajpheart.1978.235.2.H162. [DOI] [PubMed] [Google Scholar]
- [60].Jones MD, Jr, Traystman RJ. Cerebral oxygenation of the fetus, newborn, and adult. Semin Perinatol. 1984;8:205–16. [PubMed] [Google Scholar]
- [61].Kjellmer I, Karlsson K, Olsson T, Rosén KG. Cerebral reactions during intrauterine asphyxia in the sheep. I. Circulation and oxygen consumption in the fetal brain. Pediatr Res. 1974;8:50–7. doi: 10.1203/00006450-197401000-00009. [DOI] [PubMed] [Google Scholar]
- [62].Koehler RC, Jones MD, Jr, Traystman RJ. Cerebral circulatory response to carbon monoxide and hypoxic hypoxia in the lamb. Am J Physiol. 1982;243:H27–32. doi: 10.1152/ajpheart.1982.243.1.H27. [DOI] [PubMed] [Google Scholar]
- [63].Koehler RC, Traystman RJ, Zeger S, Rogers MC, Jones MD., Jr Comparison of cerebrovascular response to hypoxic and carbon monoxide hypoxia in newborn and adult sheep. J Cereb Blood Flow Metab. 1984;4:115–22. doi: 10.1038/jcbfm.1984.16. [DOI] [PubMed] [Google Scholar]
- [64].Mueller SM, Heistad DD, Marcus ML. Total and regional cerebral blood flow during hypotension, hypertension and hypocapnia. Circ Res. 1977;41:350–6. doi: 10.1161/01.res.41.3.350. [DOI] [PubMed] [Google Scholar]
- [65].Rosenberg AA, Jones MD, Jr, Traystman RJ, Simmons MA, Molteni RA. Response of cerebral blood flow to changes in PCO2 in fetal, newborn, and adult sheep. Am J Physiol. 1982;242:H862–6. doi: 10.1152/ajpheart.1982.242.5.H862. [DOI] [PubMed] [Google Scholar]
- [66].Rosenberg AA, Harris AP, Koehler RC, Hudak ML, Traystman RJ, Jones MD., Jr Role of O2–hemoglobin affinity in the regulation of cerebral blood flow in fetal sheep. Am J Physiol. 1986;251:H56–62. doi: 10.1152/ajpheart.1986.251.1.H56. [DOI] [PubMed] [Google Scholar]
- [67].Tuor UI. Local cerebral blood flow in the newborn rabbit: an autoradiographic study of changes during development. Pediatr Res. 1991;29:517–23. doi: 10.1203/00006450-199105010-00020. [DOI] [PubMed] [Google Scholar]
- [68].Jones MD, Jr, Rosenberg AA, Simmons MA, Molteni RA, Koehler RC, Traystman RJ. Oxygen delivery to the brain before and after birth. Science. 1982;216:324–5. doi: 10.1126/science.6801768. [DOI] [PubMed] [Google Scholar]
- [69].Vannucci SJ, Vannucci RC. Brain metabolism in the fetus and neonate. In: Cowett RM, editor. Principles of perinatal–neonatal metabolism. 2nd ed Springer; New York: 1998. pp. 537–50. [Google Scholar]
- [70].Kitanaka T, Alonso J, Gilbert RD, Siu BL, Clemons GK, Longo LD. Fetal responses to long-term hypoxemia in sheep. Am J Physiol. 1989;256:R1348–54. doi: 10.1152/ajpregu.1989.256.6.R1348. [DOI] [PubMed] [Google Scholar]
- [71].Pena JP, Tomimatsu T, Hatran DP, McGill LL, Longo LD. Cerebral blood flow and oxygenation in ovine fetus: responses to superimposed hypoxia at both low and high altitude. J Physiol. 2007;578:359–70. doi: 10.1113/jphysiol.2006.119925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dawes GS. Foetal and neonatal physiology. A comparative study of changes at birth. Year Book Medical Publishers; Chicago: 1968. [Google Scholar]
- [73].Kamitomo M, Alonso JG, Okai T, Longo LD, Gilbert RD. Effects of long-term, high-altitude hypoxemia on ovine fetal cardiac output and blood flow distribution. Am J Obstet Gynecol. 1993;169:701–7. doi: 10.1016/0002-9378(93)90646-z. [DOI] [PubMed] [Google Scholar]
- [74].Kitanaka T, Gilbert RD, Longo LD. Maternal responses to long-term hypoxemia in sheep. Am J Physiol. 1989;256:R1340–7. doi: 10.1152/ajpregu.1989.256.6.R1340. [DOI] [PubMed] [Google Scholar]
- [75].Cross KW, Dawes GS, Mott JC. Anoxia, oxygen consumption and cardiac output in new-born lambs and adult sheep. J Physiol. 1959;146:316–43. doi: 10.1113/jphysiol.1959.sp006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Longo LD. Respiratory gas exchange in the placenta. In: Fishman AP, Farhi LE, Tenney SM, editors. Handbook of physiology, Section 3: The Respiratory System. Vol IV. Gas exchange. American Physiological Society; Washington, DC: 1987. pp. 351–401. [Google Scholar]
- [77].Hill EP, Power GG, Longo LD. Mathematical simulation of pulmonary O2 and CO2 exchange. Am J Physiol. 1973;224:904–17. doi: 10.1152/ajplegacy.1973.224.4.904. [DOI] [PubMed] [Google Scholar]
- [78].Tomimatsu T, Pena JP, Longo LD. Fetal hypercapnia and cerebral tissue oxygenation: studies in near-term sheep. Pediatr Res. 2006;60:711–6. doi: 10.1203/01.pdr.0000246308.37154.ce. [DOI] [PubMed] [Google Scholar]
- [79].Menke J, Michel E, Rabe H, et al. Simultaneous influence of blood pressure, PCO2, and PO2 on cerebral blood flow velocity in preterm infants of less than 33 weeks' gestation. Pediatr Res. 1993;34:173–7. doi: 10.1203/00006450-199308000-00014. [DOI] [PubMed] [Google Scholar]
- [80].Panerai RB, Kelsall AW, Rennie JM, Evans DH. Analysis of cerebral blood flow autoregulation in neonates. IEEE Trans Biomed Eng. 1996;43:779–88. doi: 10.1109/10.508541. [DOI] [PubMed] [Google Scholar]
- [81].Toft PB, Leth H, Lou HC, Pryds O, Peitersen B, Henriksen O. Local vascular CO2 reactivity in the infant rat brain assessed by functional MRI. Pediatr Radiol. 1995;25:420–4. doi: 10.1007/BF02019053. [DOI] [PubMed] [Google Scholar]
- [82].Johnston MV, Trescher WH, Taylor GA. Hypoxic and ischemic central nervous system disorders in infants and children. Adv Pediatr. 1995;42:1–45. [PubMed] [Google Scholar]
- [83].Longo LD, Packianathan S. Hypoxia-ischaemia and the developing brain: Hypotheses regarding the pathophysiology of fetal-neonatal brain damage. Br J Obstet Gynaecol. 1997;104:652–62. doi: 10.1111/j.1471-0528.1997.tb11974.x. [DOI] [PubMed] [Google Scholar]
- [84].Palmer C. Hypoxic-ischemic encephalopathy. Therapeutic approaches against microvascular injury, and role of neutrophils, PAF, and free radicals. Clin Perinatol. 1995;22:481–517. [PubMed] [Google Scholar]
- [85].Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- [86].Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- [87].Harper SL, Bohlen HG, Rubin MH. Arterial and microvascular contributions to cerebral cortical autoregulation in rats. Am J Physiol. 1984;246:H17–24. doi: 10.1152/ajpheart.1984.246.1.H17. [DOI] [PubMed] [Google Scholar]
- [88].Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL., Jr Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol. 1978;234:H371–83. doi: 10.1152/ajpheart.1978.234.4.H371. [DOI] [PubMed] [Google Scholar]
- [89].Johansson B. Myogenic tone and reactivity: definitions based on muscle physiology. J Hypertens. 1989;7(Suppl 4):S5–S8. [PubMed] [Google Scholar]
- [90].Meininger GA, Davis MJ. Cellular mechanisms involved in the vascular myogenic response. Am J Physiol. 1992;263:H647–59. doi: 10.1152/ajpheart.1992.263.3.H647. [DOI] [PubMed] [Google Scholar]
- [91].Osol G. Mechanotransduction by vascular smooth muscle. J Vasc Res. 1995;32:275–92. doi: 10.1159/000159102. [DOI] [PubMed] [Google Scholar]
- [92].Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol. 2002;283:H2260–7. doi: 10.1152/ajpheart.00634.2002. [DOI] [PubMed] [Google Scholar]
- [93].Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39:183–238. doi: 10.1152/physrev.1959.39.2.183. [DOI] [PubMed] [Google Scholar]
- [94].Golding EM, Robertson CS, Bryan RM., Jr Comparison of the myogenic response in rat cerebral arteries of different calibers. Brain Res. 1998;785:293–8. doi: 10.1016/s0006-8993(97)01419-4. [DOI] [PubMed] [Google Scholar]
- [95].Helou S, Koehler RC, Gleason CA, Jones MD, Jr, Traystman RJ. Cerebrovascular autoregulation during fetal development in sheep. Am J Physiol. 1994;266:H1069–74. doi: 10.1152/ajpheart.1994.266.3.H1069. [DOI] [PubMed] [Google Scholar]
- [96].Müller T, Löhle M, Schubert H, et al. Developmental changes in cerebral autoregulatory capacity in the fetal sheep parietal cortex. J Physiol. 2002;539:957–67. doi: 10.1113/jphysiol.2001.012590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Szymonowicz W, Walker AM, Yu VYH, Stewart ML, Cannata J, Cussen L. Regional cerebral blood flow after hemorrhagic hypotension in the preterm, near-term, and newborn lamb. Pediatr Res. 1990;28:361–6. doi: 10.1203/00006450-199010000-00012. [DOI] [PubMed] [Google Scholar]
- [98].Tweed WA, Cote J, Pash M, Lou H. Arterial oxygenation determines autoregulation of cerebral blood flow in the fetal lamb. Pediatr Res. 1983;17:246–9. doi: 10.1203/00006450-198304000-00002. [DOI] [PubMed] [Google Scholar]
- [99].Papile LA, Rudolph AM, Heymann MA. Autoregulation of cerebral blood flow in the preterm fetal lamb. Pediatr Res. 1985;19:159–61. doi: 10.1203/00006450-198502000-00001. [DOI] [PubMed] [Google Scholar]
- [100].Verma PK, Panerai RB, Rennie JM, Evands DH. Grading of cerebral autoregulation in preterm and term neonates. Pediatr Neurol. 2000;23:236–42. doi: 10.1016/s0887-8994(00)00184-3. [DOI] [PubMed] [Google Scholar]
- [101].Gleason CA, Robinson R, Harris AP, Mayock DE, Traystman RJ. Cerebrovascular effects of intravenous dopamine infusions in fetal sheep. J Appl Physiol. 2002;92:717–24. doi: 10.1152/japplphysiol.00600.2001. [DOI] [PubMed] [Google Scholar]
- [102].Johnson GN, Palahniuk RJ, Twee WA, Jones MV, Wade JG. Regional cerebral blood flow changes during severe fetal asphyxia produced by slow partial umbilical cord compression. Am J Obstet Gynecol. 1979;135:48–52. [PubMed] [Google Scholar]
- [103].Lou HC, Lassen NA, Friis-Hansen B. Impaired autoregulation of cerebral blood flow in the distressed newborn infant. J Pediatr. 1979;94:118–21. doi: 10.1016/s0022-3476(79)80373-x. [DOI] [PubMed] [Google Scholar]
- [104].Milligan DW. Failure of autoregulation and intraventricular haemorrhage in preterm infants. Lancet. 1980;1:896–8. doi: 10.1016/s0140-6736(80)90836-3. [DOI] [PubMed] [Google Scholar]
- [105].Sandoval RJ, Injeti ER, Williams JM, Georthoffer WT, Pearce WJ. Myogenic contractility is more dependent on myofilament calcium sensitization in term fetal than adult ovine cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;293:H548–56. doi: 10.1152/ajpheart.00134.2007. [DOI] [PubMed] [Google Scholar]
- [106].Geary GG, Buchholz JN, Pearce WJ. Maturation depresses mouse cerebrovascular tone through endothelium-dependent mechanisms. Am J Physiol Regul Integr Comp Physiol. 2003;284:R734–41. doi: 10.1152/ajpregu.00510.2002. [DOI] [PubMed] [Google Scholar]
- [107].Geary GG, Osol GJ, Longo LD. Development affects in vitro vascular tone and calcium sensitivity in ovine cerebral arteries. J Physiol. 2004;558:883–96. doi: 10.1113/jphysiol.2003.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Bevan JA, Dodge J, Walters CL, Wellman T, Bevan RD. As human pial arteries (internal diameter 200–1000 microm) get smaller, their wall thickness and capacity to develop tension relative to their diameter increase. Life Sci. 1999;65:1153–61. doi: 10.1016/s0024-3205(99)00349-5. [DOI] [PubMed] [Google Scholar]
- [109].Brekke JF, Gokina NI, Osol G. Vascular smooth muscle cell stress as a determinant of cerebral artery myogenic tone. Am J Physiol Heart Circ Physiol. 2002;283:H2210–6. doi: 10.1152/ajpheart.00633.2002. [DOI] [PubMed] [Google Scholar]
- [110].Padget DH. The development of the cranial arteries in the human embryo. Contr Embryol. 1948;12:205–61. [Google Scholar]
- [111].Raybaud C. Development of the arterial supply to the brain tissue. In: Lasjaunias PL, Berenstein A, editors. Surgical neuroangiography. vol 3. Springer Verlag; New York: 1990. pp. 1–13. [Google Scholar]
- [112].Raybaud C. Normal and abnormal embryology and development of the intracranial vascular system. Neurosurg Clin N Am. 2010;21:399–426. doi: 10.1016/j.nec.2010.03.011. [DOI] [PubMed] [Google Scholar]
- [113].Streeter GL. The developmental alterations in the vascular system of the brain of the human embryo. Contr Embryol Carneg Instn. 1918;8:5–38. [Google Scholar]
- [114].Kathuria S, Gregg L, Chen J, Gandhi D. Normal cerebral arterial development and variations. Semin Ultrasound CT MRI. 2011;32:242–51. doi: 10.1053/j.sult.2011.02.002. [DOI] [PubMed] [Google Scholar]
- [115].Pfeifer RA. Die angioarchitektonik der grosshirnrinde. Verlag von Julius Springer; Berlin: 1928. [Google Scholar]
- [116].Jussila L, Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev. 2002;82:673–700. doi: 10.1152/physrev.00005.2002. [DOI] [PubMed] [Google Scholar]
- [117].Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- [118].Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- [119].Deaton RA, Gan Q, Owens GK. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296:H1027–37. doi: 10.1152/ajpheart.01230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kawai-Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292:C59–69. doi: 10.1152/ajpcell.00394.2006. [DOI] [PubMed] [Google Scholar]
- [121].McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428–41. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- [122].Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- [123].Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–78. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- [124].Chang L, Noseda M, Higginson M, et al. Differentiation of vascular smooth muscle cells from local precursors during embryonic and adult arteriogenesis requires Notch signaling. Proc Natl Acad Sci USA. 2012;109:6993–8. doi: 10.1073/pnas.1118512109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Herbert SP, Stainier DYR. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–64. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Patel-Hett S, D'Amore PA. Signal transduction in vasculogenesis and development angiogenesis. Int J Dev Biol. 2011;55:353–63. doi: 10.1387/ijdb.103213sp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Scher MS. Developmental origins of cerebrovascular disease I: Prenatal cerebrovascular development – classic findings in the context of advances in genetic and fetal surveillance evaluations. J Child Neurol. 2012;27:121–31. doi: 10.1177/0883073811417714. [DOI] [PubMed] [Google Scholar]
- [128].Takashima S, Tanaka K. Development of cerebrovascular architecture and its relationship to periventricular leukomalacia. Arch Neurol. 1978;35:11–16. doi: 10.1001/archneur.1978.00500250015003. [DOI] [PubMed] [Google Scholar]
- [129].Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–57. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- [130].Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–80. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- [131].Silpanisong J, Pearce WJ. Vasotrophic regulation of age-dependent vascular remodeling. Curr Vasc Pharmacol. 2013;(This issue) doi: 10.2174/1570161111311050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–65. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- [133].Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- [134].Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–80. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- [135].Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines Cancer Res. 1986;46:5629–32. [PubMed] [Google Scholar]
- [136].Connolly DT, Heuvelman DM, Nelson R, et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- [138].Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–39. [PMC free article] [PubMed] [Google Scholar]
- [139].Freitas-Andrade M, Carmeliet P, Stanimirovic DB, Moreno M. VEGFR-2-mediated increased proliferation and survival in response to oxygen and glucose deprivation in PIGF knockout astrocytes. J Neurochem. 2008;107:756–67. doi: 10.1111/j.1471-4159.2008.05660.x. [DOI] [PubMed] [Google Scholar]
- [140].Marko SB, Damon DH. VEGF promotes vascular sympathetic innervation. Am J Physiol Heart Circ Physiol. 2008;294:H2646–52. doi: 10.1152/ajpheart.00291.2008. [DOI] [PubMed] [Google Scholar]
- [141].Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–60. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- [142].Yang W, de Bono DP. A new role for vascular endothelial growth factor and fibroblast growth factors: increasing endothelial resistance to oxidative stress. FEBS Lett. 1997;403:139–42. doi: 10.1016/s0014-5793(96)01486-x. [DOI] [PubMed] [Google Scholar]
- [143].Osada-Oka M, Ikeda T, Imaoka S, Akiba S, Sato T. VEGF-enhanced proliferation under hypoxia by an autocrine mechanism in human vascular smooth muscle cells. J Atheroscler Thromb. 2008;15:26–33. doi: 10.5551/jat.e533. [DOI] [PubMed] [Google Scholar]
- [144].Krum JM, Mani N, Rosenstein JM. Roles of the endogenous VEGF receptors flt-1 and flk-1 in astroglial and vascular remodeling after brain injury. Exp Neurol. 2008;212:108–17. doi: 10.1016/j.expneurol.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Butler SM, Abrassart JM, Hubbell MC, et al. Contributions of VEGF to age-dependent transmural gradients in contractile protein expression in ovine carotid arteries. Am J Physiol Cell Physiol. 2011;301:C653–66. doi: 10.1152/ajpcell.00413.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–9. doi: 10.1161/STROKEAHA.109.564229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Elliott CF, Pearce WJ. Effects of maturation on cell water, protein, and DNA content in ovine cerebral arteries. J Appl Physiol. 1995;79:831–7. doi: 10.1152/jappl.1995.79.3.831. [DOI] [PubMed] [Google Scholar]
- [148].Fredriksson L, Nilsson I, Su EJ, et al. Platelet-derived growth factor C deficiency in C57BL/6 mice leads to abnormal cerebral vascularization, loss of neuroependymal integrity, and ventricular abnormalities. Am J Pathol. 2012;180:1136–44. doi: 10.1016/j.ajpath.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Lee WH, Wang GM, Seaman LB, Vannucci SJ. Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hypoxia-ischemia. J Cereb Blood Flow and Metab. 1996;16:227–36. doi: 10.1097/00004647-199603000-00007. [DOI] [PubMed] [Google Scholar]
- [150].Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- [151].Scarisbrick IA, Jones EG, Isackson PJ. Coexpression of mRNAs for NGF, BDNF, and NT-3 in the cardiovascular system of the pre-and postnatal rat. J Neurosci. 1993;13:875–93. doi: 10.1523/JNEUROSCI.13-03-00875.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci USA. 2008;105:7582–7. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Santhanam AVR, Smith LA, Katusic ZS. Brain-derived neurotrophic factor stimulates production of prostacyclin in cerebral arteries. Stroke. 2010;41:350–6. doi: 10.1161/STROKEAHA.109.564492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Charpie JR, Schreur KD, Papadopoulos SM, Webb RC. Endothelium dependency of contractile activity differs in infant and adult vertebral arteries. J Clin Invest. 1994;93:1339–43. doi: 10.1172/JCI117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Willis AP, Leffler CW. Endothelial NO and prostanoid involvement in newborn and juvenile pig pial arteriolar vasomotor responses. Am J Physiol Heart Circ Physiol. 2001;281:H2366–77. doi: 10.1152/ajpheart.2001.281.6.H2366. [DOI] [PubMed] [Google Scholar]
- [156].Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575–99. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- [157].Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003;116:1157–73. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- [158].Ingber DE. Tensegrity II. How structural networks influence cellular information processing networks. J Cell Sci. 2003;116:1397–408. doi: 10.1242/jcs.00360. [DOI] [PubMed] [Google Scholar]
- [159].Pollard TD. The cytoskeleton, cellular motility, and the reductionist agenda. Nature. 2003;422:741–5. doi: 10.1038/nature01598. [DOI] [PubMed] [Google Scholar]
- [160].Stein O, Eisenberg S, Stein Y. Aging of aortic smooth muscle cells in rats and rabbits. A morphologic and biochemical study. Lab Invest. 1969;21:386–97. [PubMed] [Google Scholar]
- [161].Small IV. Structure-function relationships in smooth muscle: the missing links. Bioessays. 1995;17:785–92. doi: 10.1002/bies.950170908. [DOI] [PubMed] [Google Scholar]
- [162].Small IV, Gimona M. The cytoskeleton of the vertebrate smooth muscle cell. Acta Physiol Scand. 1998;164:341–8. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- [163].Bradbury MW. The blood-brain barrier. Transport across the cerebral endothelium. Circ Res. 1985;57:213–22. doi: 10.1161/01.res.57.2.213. [DOI] [PubMed] [Google Scholar]
- [164].Harik SI, Kalaria RN. The blood-brain barrier: a selective overview. In: Ginsberg MD, Dietrich WD, editors. Cerebrovascular Diseases. Raven; New York: 1989. pp. 407–10. [Google Scholar]
- [165].Stonestreet BS, Patlak CS, Pettigrew KD, Reilly CB, Cserr HF. Ontogeny of blood-brain barrier function in ovine fetuses, lambs, and adults. Am J Physiol. 1996;271:R1594–601. doi: 10.1152/ajpregu.1996.271.6.R1594. [DOI] [PubMed] [Google Scholar]
- [166].Stonestreet BS, Sadowska GB, McKnight AJ, Patlak C, Petersson KH. Exogenous and endogenous corticosteroids modulate blood-brain barrier development in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2000;279:R468–77. doi: 10.1152/ajpregu.2000.279.2.R468. [DOI] [PubMed] [Google Scholar]
- [167].Stonestreet BS, Petersson KH, Sadowska GB, Pettigrew KD, Patlak CS. Antenatal steroids decrease blood-brain barrier permeability in the ovine fetus. Am J Physiol. 1999;276:R283–9. doi: 10.1152/ajpregu.1999.276.2.R283. [DOI] [PubMed] [Google Scholar]
- [168].Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17:894–904. doi: 10.1097/00004647-199708000-00008. [DOI] [PubMed] [Google Scholar]
- [169].Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–9. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- [170].Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–62. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].McCarty JH. Cell biology of the neurovascular unit: implications for drug delivery across the blood-brain barrier. Assay Drug Dev Technol. 2005;3:89–95. doi: 10.1089/adt.2005.3.89. [DOI] [PubMed] [Google Scholar]
- [172].Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- [173].Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–31. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- [174].McCarty JH. Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol. 2009;16:209–14. doi: 10.1097/MOH.0b013e32832a07eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Xu HL, Mao L, Ye S, Paisansathan C, Vetri F, Pelligrino DA. Astrocytes are a key conduit for upstream signaling of vasodilation during cerebral cortical neuronal activation in vivo. Am J Physiol Heart Circ Physiol. 2008;294:H622–32. doi: 10.1152/ajpheart.00530.2007. [DOI] [PubMed] [Google Scholar]
- [176].Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- [177].Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–9. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL. Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol. 2011;300:H440–7. doi: 10.1152/ajpheart.00722.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol. 2011;301:H1–11. doi: 10.1152/ajpheart.00230.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–76. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- [181].Ruhrberg C, Gerhardt H, Golding M, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–98. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Ward NL, LaManna JC. The neurovascular unit and its growth factors: coordinated response in the vascular and nervous systems. Neurol Res. 2004;26:870–883. doi: 10.1179/016164104X3798. [DOI] [PubMed] [Google Scholar]
- [183].Mammoto A, Mammoto T, Ingber DE. Rho signaling and mechanical control of vascular development. Curr Opin Hematol. 2008;15:228–34. doi: 10.1097/MOH.0b013e3282fa7445. [DOI] [PubMed] [Google Scholar]
- [184].Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–17. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–9. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- [186].Jessell TM, Sanes JR. Development. The decade of the developing brain. Curr Opin Neurobiol. 2000;10:599–611. doi: 10.1016/s0959-4388(00)00136-7. [DOI] [PubMed] [Google Scholar]
- [187].Rakic P. Cell migration and neuronal ectopias in the brain. Birth Defects Orig Artic Ser. 1975;11:95–129. [PubMed] [Google Scholar]
- [188].Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–94. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- [189].Tavazoie M, Van der Veken L, Silva-Vargas V, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–88. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kerever A, Schnack J, Vellinga D, et al. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells. 2007;25:2146–57. doi: 10.1634/stemcells.2007-0082. [DOI] [PubMed] [Google Scholar]
- [191].Braun A, Xu H, Hu F, et al. Paucity of pericytes in germinal matrix vasculature of premature infants. J Neurosci. 2007;27:12012–24. doi: 10.1523/JNEUROSCI.3281-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–7. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- [194].Domoki F, Perciaccante JV, Shimizu K, Puskar , Busija DW, Bari F. N-methyl-D-aspartate-induced vasodilation is mediated by endothelium-independent nitric oxide release in piglets. Am J Physiol Heart Circ Physiol. 2002;282:H1404–9. doi: 10.1152/ajpheart.00523.2001. [DOI] [PubMed] [Google Scholar]
- [195].Ayata C, Moskowitz MA. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-D-aspartate superfusion. Am J Physiol Heart Circ Physiol. 2006;290:H1837–41. doi: 10.1152/ajpheart.01102.2005. [DOI] [PubMed] [Google Scholar]
- [196].Busija DW, Bari F, Domoki F, Louis T. Mechanisms involved in the cerebrovascular dilator effects of N-methyl-D-aspartate in cerebral cortex. Brain Res Rev. 2007;56:89–100. doi: 10.1016/j.brainresrev.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Fiumana E, Parfenova H, Jaggar JH, Leffler CW. Carbon monoxide mediates vasodilator effects of glutamate in isolated pressurized cerebral arterioles of newborn pigs. Am J Physiol Heart Circ Physiol. 2003;284:H1073–9. doi: 10.1152/ajpheart.00881.2002. [DOI] [PubMed] [Google Scholar]
- [198].Leffler CW, Parfenova H, Fedinec AL, Basuroy S, Tcheranova D. Contributions of astrocytes and CO to pial arteriolar dilation to glutamate in newborn pigs. Am J Physiol Heart Circ Physiol. 2006;291:H2897–904. doi: 10.1152/ajpheart.00722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol. 2006;100:1065–76. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Zonta M, Angulo MC, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- [201].Iliff JJ, D'Ambrosio R, Ngai AC, Winn HR. Adenosine receptors mediate glutamate-evoked arteriolar dilation in the rat cerebral cortex. Am J Physiol Heart Circ Physiol. 2003;284:H1631–7. doi: 10.1152/ajpheart.00909.2002. [DOI] [PubMed] [Google Scholar]
- [202].Xi Q, Umstot E, Zhao G, Narayanan D, Leffler CW, Jaggar JH. Glutamate regulates Ca2+ signals in smooth muscle cells of newborn piglet brain slice arterioles through astrocyte- and heme oxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2010;298:H562–9. doi: 10.1152/ajpheart.00823.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Xi Q, Tcheranova D, Basuroy S, Parfenova H, Jaggar JH, Leffler CW. Glutamate-induced calcium signals stimulate CO production in piglet astrocytes. Am J Physiol Heart Circ Physiol. 2011; 301:H428–33. doi: 10.1152/ajpheart.01277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Edvinsson L, Owman C, Sjoberg NO. Autonomic nerves, mast cells, and amine receptors in human brain vessels. A histochemical and pharmacological study. Brain Res. 1976;115:377–93. doi: 10.1016/0006-8993(76)90356-5. [DOI] [PubMed] [Google Scholar]
- [205].Kobayashi S, Tsukahara S, Sugita K, Matsuo K, Nagata T. Histochemical studies on the postnatal development of autonomic nerves in mice cerebral arteries. Histochemistry. 1981;73:15–20. doi: 10.1007/BF00493128. [DOI] [PubMed] [Google Scholar]
- [206].Lou HC, Edvinsson L, MacKenzie ET. The concept of coupling blood flow to brain function: revision required? Ann Neurol. 1987;22:289–97. doi: 10.1002/ana.410220302. [DOI] [PubMed] [Google Scholar]
- [207].Sokoloff L. Local cerebral energy metabolism: its relationships to local functional activity and blood flow. In: Purves MS, Elliott K, editors. Cerebral vascular smooth muscle and its control. Elsevier; Amsterdam: 1978. pp. 171–97. [DOI] [PubMed] [Google Scholar]
- [208].Gotoh J, Kuang TY, Nakao Y, et al. Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am J Physiol Heart Circ Physiol. 2001;280:H821–9. doi: 10.1152/ajpheart.2001.280.2.H821. [DOI] [PubMed] [Google Scholar]
- [209].Buckley NM, Gootman PM, Yellin EL, Brazeau P. Age-related cardiovascular effects of catecholamines in anesthetized piglets. Circ Res. 1979;45:282–92. doi: 10.1161/01.res.45.2.282. [DOI] [PubMed] [Google Scholar]
- [210].Dunn JA, Lorch V, Sinha SN. Responses of small intrapulmonary arteries to vasoactive compounds in the fetal and neonatal lamb: norepinephrine, epinephrine, serotonin, and potassium chloride. Pediatr Res. 1989;25:360–3. doi: 10.1203/00006450-198904000-00009. [DOI] [PubMed] [Google Scholar]
- [211].Heymann MA, Iwamoto HS, Rudolph AM. Factors affecting changes in the neonatal systemic circulation. Annu Rev Physiol. 1981;43:371–83. doi: 10.1146/annurev.ph.43.030181.002103. [DOI] [PubMed] [Google Scholar]
- [212].Gauthier P, Nadeau RA, de Champlain J. The development of sympathetic innervation and the functional state of the cardiovascular system in newborn dogs. Can J Physiol Pharmacol. 1975;53:763–76. doi: 10.1139/y75-106. [DOI] [PubMed] [Google Scholar]
- [213].Hayashi S, Park MK, Kuehl TJ. Higher sensitivity of cerebral arteries isolated from premature and newborn baboons to adrenergic and cholinergic stimulation. Life Sci. 1984;35:253–60. doi: 10.1016/0024-3205(84)90108-5. [DOI] [PubMed] [Google Scholar]
- [214].MacKenzie ET, Scatton B. Cerebral circulatory and metabolic effects of perivascular neurotransmitters. CRC Crit Rev Clin Neurobiol. 1987;2:357–419. [PubMed] [Google Scholar]
- [215].Wagerle LC, Kurth CD, Roth RA. Sympathetic reactivity of cerebral arteries in developing fetal lamb and adult sheep. Am J Physiol. 1990;258:H1432–8. doi: 10.1152/ajpheart.1990.258.5.H1432. [DOI] [PubMed] [Google Scholar]
- [216].Toda N, Okamura T, Miyazaki M. Age-dependent changes in the response of isolated beagle coronary arteries to transmural electrical stimulation and catecholamines. J Pharmacol Exp Ther. 1986;238:319–26. [PubMed] [Google Scholar]
- [217].Robillard JE, Nakamura KT, Wilkin MK, McWeeny OJ, DiBona GF. Ontogeny of renal hemodynamic response to renal nerve stimulation in sheep. Am J Physiol. 1987;252:F605–12. doi: 10.1152/ajprenal.1987.252.4.F605. [DOI] [PubMed] [Google Scholar]
- [218].Wagerle LC, Moliken W, Russo P. Nitric oxide and beta-adrenergic mechanisms modify contractile responses to norepinephrine in ovine fetal and newborn cerebral arteries. Pediatr Res. 1995;38:237–42. doi: 10.1203/00006450-199508000-00017. [DOI] [PubMed] [Google Scholar]
- [219].Toda N, Shimizu I. Neuroeffector function in mesenteric arteries isolated from beagles of different ages. J Pharmacol Exp Ther. 1987;240:223–7. [PubMed] [Google Scholar]
- [220].Gitler MS, Piccio MM, Robillard JE, Jose PA. Characterization of renal alpha-adrenoceptor subtypes in sheep during development. Am J Physiol. 1991;260:R407–12. doi: 10.1152/ajpregu.1991.260.2.R407. [DOI] [PubMed] [Google Scholar]
- [221].Soltis EE, Newman PS. Ontogeny of vascular smooth muscle responsiveness in the postweaning rat. Dev Pharmacol Ther. 1992;18:44–54. [PubMed] [Google Scholar]
- [222].Monin P, Feillet F, Hascoet JM, Vert P. Effect of sympathetic nervous system on cerebral blood flow in the newborn piglet. Biol Neonate. 1990;58:192–9. doi: 10.1159/000243268. [DOI] [PubMed] [Google Scholar]
- [223].Duckles SP. Functional activity of the noradrenergic innervation of large cerebral arteries. Br J Pharmacol. 1980;69:193–9. doi: 10.1111/j.1476-5381.1980.tb07890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Tuor UI, Grewal D. Autoregulation of cerebral blood flow: influence of local brain development and postnatal age. Am J Physiol. 1994;267:H2220–8. doi: 10.1152/ajpheart.1994.267.6.H2220. [DOI] [PubMed] [Google Scholar]
- [225].Buchholz J, Edwards-Teunissen K, Duckles SP. Impact of development and chronic hypoxia on NE release from adrenergic nerves in sheep arteries. Am J Physiol. 1999;276:R799–808. doi: 10.1152/ajpregu.1999.276.3.R799. [DOI] [PubMed] [Google Scholar]
- [226].Buchholz J, Duckles SP. Chronic hypoxia alters prejunctional alpha(2)-receptor function in vascular adrenergic nerves of adult and fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;281:R926–34. doi: 10.1152/ajpregu.2001.281.3.R926. [DOI] [PubMed] [Google Scholar]
- [227].Kawamura K, Sakata N, Takebayashi S. Neuropeptide Y- and vasoactive intestinal polypeptide-containing nerve fibers in the human cerebral arteries: characteristics of distribution. Angiology. 1991;42:35–43. doi: 10.1177/000331979104200106. [DOI] [PubMed] [Google Scholar]
- [228].Tsai SH, Tew JM, Shipley MT. Cerebral arterial innervation II. Development of calcitonin-gene-related peptide and norepinephrine in the rat. J Comp Neurol. 1989;279:1–12. doi: 10.1002/cne.902790102. [DOI] [PubMed] [Google Scholar]
- [229].Tsai SH, Tew JM, Shipley MT. Development of cerebral arterial innervation: synchronous development of neuropeptide Y (NPY)-and vasoactive intestinal polypeptide (VIP)-containing fibers and some observations on growth cones. Brain Res Dev Brain Res. 1992;69:77–83. doi: 10.1016/0165-3806(92)90124-f. [DOI] [PubMed] [Google Scholar]
- [230].Nozaki K, Moskowitz MA, Maynard KI, et al. Possible origins and distribution of immunoreactive nitric oxide synthase-containing nerve fibers in cerebral arteries. J Cereb Blood Flow Metab. 1993;13:70–9. doi: 10.1038/jcbfm.1993.9. [DOI] [PubMed] [Google Scholar]
- [231].Gabella G. Structural apparatus for force transmission in smooth muscles. Physiol Rev. 1984;64:455–77. doi: 10.1152/physrev.1984.64.2.455. [DOI] [PubMed] [Google Scholar]
- [232].Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- [233].Janmey PA. The cytoskeleton and cell signaling. Component localization and mechanical coupling. Physiol Rev. 1998;78:763–81. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- [234].Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- [235].Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. 2007;9:1110–21. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- [236].Zhao Y, Xiao H, Long W, Pearce WJ, Longo LD. Expression of several cytoskeletal proteins in ovine cerebral arteries: developmental and functional considerations. J Physiol. 2004;558:623–32. doi: 10.1113/jphysiol.2004.064220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562–73. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- [238].Howard J, Hyman AA. Dynamics and mechanics of the microtubule plus end. Nature. 2003;422:753–8. doi: 10.1038/nature01600. [DOI] [PubMed] [Google Scholar]
- [239].Degani S. Evaluation of fetal cerebrovascular circulation and brain development: the role of ultrasound and Doppler. Semin Perinatol. 2009;33:259–69. doi: 10.1053/j.semperi.2009.04.004. [DOI] [PubMed] [Google Scholar]
- [240].Horowitz A, Menice CB, LaPorte R, Morgan KG. Mechanisms of smooth muscle contractions. Physiol Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- [241].Saunders NR, Habgood MD, Dziegielewska KM. Barrier mechanisms in the brain. II. Immature brain. Clin Exp Pharmacol Physiol. 1999;26:85–91. doi: 10.1046/j.1440-1681.1999.02987.x. [DOI] [PubMed] [Google Scholar]
- [242].Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- [243].Owens G. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- [244].Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–71. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Longo LD, Hull AD, Long DM, Pearce WJ. Cerebrovascular adaptations to high-altitude hypoxemia in fetal and adult sheep. Am J Physiol. 1993;264:R65–72. doi: 10.1152/ajpregu.1993.264.1.R65. [DOI] [PubMed] [Google Scholar]
- [246].Williams JM, Pearce WJ. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. J Appl Physiol. 2006;100:225–32. doi: 10.1152/japplphysiol.00221.2005. [DOI] [PubMed] [Google Scholar]
- [247].Williams JM, Hull AD, Pearce WJ. Maturational modulation of endothelium-dependent vasodilatation in ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2005;288:R149–57. doi: 10.1152/ajpregu.00427.2004. [DOI] [PubMed] [Google Scholar]
- [248].White CR, Hamade MW, Siami K, et al. Maturation enhances fluid shear-induced activation of eNOS in perfused ovine carotid arteries. Am J Physiol Heart Circ Physiol. 2005;289:H2220–7. doi: 10.1152/ajpheart.01013.2004. [DOI] [PubMed] [Google Scholar]
- [249].Parfenova H, Massie V, Leffler CW. Developmental changes in endothelium-derived vasorelaxant factors in cerebral circulation. Am J Physiol Heart Circ Physiol. 2000;278:H780–8. doi: 10.1152/ajpheart.2000.278.3.H780. [DOI] [PubMed] [Google Scholar]
- [250].White CR, Hao X, Pearce WJ. Maturational differences in soluble guanylate cyclase activity in ovine carotid and cerebral arteries. Pediatr Res. 2000;47:369–75. doi: 10.1203/00006450-200003000-00014. [DOI] [PubMed] [Google Scholar]
- [251].Williams JM, White CR, Chang MM, Injeti ER, Zhang L, Pearce WJ. Chronic hypoxic decreases in soluble guanylate cyclase protein and enzyme activity are age dependent in fetal and adult ovine carotid arteries. J Appl Physiol. 2006;100:1857–66. doi: 10.1152/japplphysiol.00662.2005. [DOI] [PubMed] [Google Scholar]
- [252].Pearce WJ, Williams JM, White CR, Lincoln TM. Effects of chronic hypoxia on soluble guanylate cyclase activity in fetal and adult ovine cerebral arteries. J Appl Physiol. 2009;107:192–9. doi: 10.1152/japplphysiol.00233.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Pearce WJ, Hull AD, Long DM, White CR. Effects of maturation on cyclic GMP-dependent vasodilation in ovine basilar and carotid arteries. Pediatr Res. 1994;36:25–33. doi: 10.1203/00006450-199407001-00005. [DOI] [PubMed] [Google Scholar]
- [254].White CR, Pearce WJ. Effects of maturation on cyclic GMP metabolism in ovine carotid arteries. Pediatr Res. 1996;39:25–31. doi: 10.1203/00006450-199601000-00004. [DOI] [PubMed] [Google Scholar]
- [255].Nauli SM, White CR, Hull AD, Pearce WJ. Maturation alters cyclic nucleotide and relaxation responses to nitric oxide donors in ovine cerebral arteries. Biol Neonate. 2003;83:123–35. doi: 10.1159/000067959. [DOI] [PubMed] [Google Scholar]
- [256].Hunter CJ, Blood AB, White CR, Pearce WJ, Power GG. Role of nitric oxide in hypoxic cerebral vasodilatation in the ovine fetus. J Physiol. 2003;549:625–33. doi: 10.1113/jphysiol.2002.038034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Lee SJ, Hatran DP, Tomimatsu T, Peña JP, McAuley G, Longo LD. Fetal cerebral blood flow, electrocorticographic activity, and oxygenation: responses to acute hypoxia. J Physiol. 2009;587:2033–47. doi: 10.1113/jphysiol.2009.166983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Longo LD. Carbon monoxide: effects on oxygenation of the fetus in utero. Science. 1976;194:523–5. doi: 10.1126/science.973133. [DOI] [PubMed] [Google Scholar]
- [259].Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–12. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- [260].Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- [261].Faraci FM. Role of endothelium-derived relaxing factor in cerebral circulation: large arteries vs. microcirculation. Am J Physiol. 1991;261:H1038–42. doi: 10.1152/ajpheart.1991.261.4.H1038. [DOI] [PubMed] [Google Scholar]
- [262].Hudetz AG, Shen H, Kampine JP. Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am J Physiol. 1998;274:H982–9. doi: 10.1152/ajpheart.1998.274.3.H982. [DOI] [PubMed] [Google Scholar]
- [263].McCrabb GJ, Harding R. Role of nitric oxide in the regulation of cerebral blood flow in the ovine foetus. Clin Exp Pharmacol Physiol. 1996;23:855–60. doi: 10.1111/j.1440-1681.1996.tb01133.x. [DOI] [PubMed] [Google Scholar]
- [264].Watkins LD. Nitric oxide and cerebral blood flow: an update. Cerebrovasc Brain Metab Rev. 1995;7:324–37. [PubMed] [Google Scholar]
- [265].Adachi K, Takahashi S, Melzer P, et al. Increases in local cerebral blood flow associated with somatosensory activation are not mediated by NO. Am J Physiol. 1994;267:H2155–62. doi: 10.1152/ajpheart.1994.267.6.H2155. [DOI] [PubMed] [Google Scholar]
- [266].Mbaku EM, Zhang L, Pearce WJ, Duckles SP, Buchholz J. Chronic hypoxia alters the function of NOS nerves in cerebral arteries of near-term fetal and adult sheep. J Appl Physiol. 2003;94:724–32. doi: 10.1152/japplphysiol.00771.2002. [DOI] [PubMed] [Google Scholar]
- [267].Buchanan JE, Phillis JW. The role of nitric oxide in the regulation of cerebral blood flow. Brain Res. 1993;610:248–55. doi: 10.1016/0006-8993(93)91408-k. [DOI] [PubMed] [Google Scholar]
- [268].Pelligrino DA, Koenig HM, Albrecht RF. Nitric oxide synthesis and regional cerebral blood flow responses to hypercapnia and hypoxia in the rat. J Cereb Blood Flow Metab. 1993;13:80–7. doi: 10.1038/jcbfm.1993.10. [DOI] [PubMed] [Google Scholar]
- [269].Armstead WM. Role of activation of calcium-sensitive K+ channels in NO- and hypoxia-induced pial artery vasodilation. Am J Physiol. 1997;272:H1785–90. doi: 10.1152/ajpheart.1997.272.4.H1785. [DOI] [PubMed] [Google Scholar]
- [270].Reid JM, Davies AG, Ashcroft FM, Paterson DJ. Effect of LNMMA, cromakalim, and glibenclamide on cerebral blood flow in hypercapnia and hypoxia. Am J Physiol. 1995;269:H916–22. doi: 10.1152/ajpheart.1995.269.3.H916. [DOI] [PubMed] [Google Scholar]
- [271].Pelligrino DA, Wang Q, Koenig HM, Albrecht RF. Role of nitric oxide, adenosine, N-methyl-D-aspartate receptors, and neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res. 1995;704:61–70. doi: 10.1016/0006-8993(95)01105-6. [DOI] [PubMed] [Google Scholar]
- [272].Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activation of ATP-sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab. 1997;17:100–8. doi: 10.1097/00004647-199701000-00013. [DOI] [PubMed] [Google Scholar]
- [273].Lindauer U, Megow D, Matsuda H, Dirnagl U. Nitric oxide: a modulator, but not a mediator, of neurovascular coupling in rat somatosensory cortex. Am J Physiol. 1999;277:H799–811. doi: 10.1152/ajpheart.1999.277.2.H799. [DOI] [PubMed] [Google Scholar]
- [274].Van Mil AH, Spilt A, Van Buchem MA, et al. Nitric oxide mediates hypoxia-induced cerebral vasodilation in humans. J Appl Physiol. 2002;92:962–6. doi: 10.1152/japplphysiol.00616.2001. [DOI] [PubMed] [Google Scholar]
- [275].Nauli SM, Zhang L, Pearce WJ. Maturation depresses cGMP-mediated decreases in [Ca2+]i and Ca2+ sensitivity in ovine cranial arteries. Am J Physiol Heart Circ Physiol. 2001;280:H1019–28. doi: 10.1152/ajpheart.2001.280.3.H1019. [DOI] [PubMed] [Google Scholar]
- [276].Green LR, Bennet L, Hanson MA. The role of nitric oxide synthesis in cardiovascular responses to acute hypoxia in the late gestation sheep fetus. J Physiol. 1996;497:271–7. doi: 10.1113/jphysiol.1996.sp021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Hunter CJ, Bennet L, Power GG, et al. Key neuroprotective role for endogenous adenosine A1 receptor activation during asphyxia in the fetal sheep. Stroke. 2003;34:2240–5. doi: 10.1161/01.STR.0000083623.77327.CE. [DOI] [PubMed] [Google Scholar]
- [278].Ioroi T, Yonetani M, Nakamura H. Effects of hypoxia and reoxygenation on nitric oxide production and cerebral blood flow in developing rat striatum. Pediatr Res. 1998;43:733–7. doi: 10.1203/00006450-199806000-00004. [DOI] [PubMed] [Google Scholar]
- [279].van Bel F, Sola A, Roman C, Rudolph AM. Role of nitric oxide in the regulation of the cerebral circulation in the lamb fetus during normoxemia and hypoxemia. Biol Neonate. 1995;68:200–10. doi: 10.1159/000244238. [DOI] [PubMed] [Google Scholar]
- [280].Rebich S, Devine JO, Armstead WM. Role of nitric oxide and cAMP in β-adrenoceptor-induced pial artery vasodilation. Am J Physiol. 1995;268:H1071–6. doi: 10.1152/ajpheart.1995.268.3.H1071. [DOI] [PubMed] [Google Scholar]
- [281].Armstead WM. cGMP and cAMP in prostaglandin-induced pial artery dilation and increased CSF opioid concentration. Am J Physiol. 1996;271:H166–72. doi: 10.1152/ajpheart.1996.271.1.H166. [DOI] [PubMed] [Google Scholar]
- [282].Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol Heart Circ Physiol. 2005;289:H1442–7. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Parfenova H, Parfenov VN, Shlopov BV, et al. Dynamics of nuclear localization sites for COX-2 in vascular endothelial cells. Am J Physiol Cell Physiol. 2001;281:C166–78. doi: 10.1152/ajpcell.2001.281.1.C166. [DOI] [PubMed] [Google Scholar]
- [284].Parfenova H, Levine V, Gunther WM, Pourcyrous M, Leffler CW. COX-1 and COX-2 contributions to basal and IL-1β-stimulated prostanoid synthesis in human neonatal cerebral microvascular endothelial cells. Pediatr Res. 2002;52:342–8. doi: 10.1203/00006450-200209000-00006. [DOI] [PubMed] [Google Scholar]
- [285].Leffler CW, Busija DW. Prostanoids and pial arteriolar diameter in hypotensive newborn pigs. Am J Physiol. 1987;252:H687–91. doi: 10.1152/ajpheart.1987.252.4.H687. [DOI] [PubMed] [Google Scholar]
- [286].Leffler CW, Parfenova H. Cerebral arteriolar dilation to hypoxia: role of prostanoids. Am J Physiol. 1997;272:H418–24. doi: 10.1152/ajpheart.1997.272.1.H418. [DOI] [PubMed] [Google Scholar]
- [287].Willis AP, Leffler CW. NO and prostanoids: age dependence of hypercapnic- and histamine-induced dilations of pig pial arterioles. Am J Physiol. 1999;277:H299–307. doi: 10.1152/ajpheart.1999.277.1.H299. [DOI] [PubMed] [Google Scholar]
- [288].Armstead WM. Relationship between opioids and prostaglandins in hypoxia-induced vasodilation of pial arteries in the newborn pig. Proc Soc Exp Biol Med. 1996;212:135–41. doi: 10.3181/00379727-212-44000. [DOI] [PubMed] [Google Scholar]
- [289].Armstead WM, Leffler CW. Neurohumoral regulation of the cerebral circulation. Proc Soc Exp Biol Med. 1992;199:149–57. doi: 10.3181/00379727-199-43340b. [DOI] [PubMed] [Google Scholar]
- [290].Armstead WM, Zuckerman SL, Shibata M, Parfenova H, Leffler CW. Different pial arteriolar responses to acetylcholine in the newborn and juvenile pig. J Cereb Blood Flow Metab. 1994;14:1088–95. doi: 10.1038/jcbfm.1994.142. [DOI] [PubMed] [Google Scholar]
- [291].Leffler CW, Armstead WM, Shibata M. Role of eicosanoids in cerebral hemodynamics. In: Phyllis JW, editor. The regulation of cerebral blood flow. CRC; Boca Raton, FL: 1993. pp. 297–313. [Google Scholar]
- [292].Parfenova H, Shibata M, Zuckerman S, Leffler CW. CO2 and cerebral circulation in newborn pigs: cyclic nucleotides and prostanoids in vascular regulation. Am J Physiol. 1994;266:H1494–501. doi: 10.1152/ajpheart.1994.266.4.H1494. [DOI] [PubMed] [Google Scholar]
- [293].Nishida N, Blood AB, Hunter CJ, et al. Role of prostanoids in the regulation of cerebral blood flow during normoxia and hypoxia in the fetal sheep. Pediatr Res. 2006;60:524–9. doi: 10.1203/01.pdr.0000242268.99726.53. [DOI] [PubMed] [Google Scholar]
- [294].Zuckerman SL, Armstead WM, Hsu P, Shibata M, Leffler CW. Age dependence of cerebrovascular response mechanisms in domestic pigs. Am J Physiol. 1996;271:H535–40. doi: 10.1152/ajpheart.1996.271.2.H535. [DOI] [PubMed] [Google Scholar]
- [295].Faraci FM. Role of nitric oxide in regulation of basilar artery tone in vivo. Am J Physiol. 1990;259:H1216–21. doi: 10.1152/ajpheart.1990.259.4.H1216. [DOI] [PubMed] [Google Scholar]
- [296].Armstead WM, Mirro R, Leffler CW, Busija DW. Acetylcholine produces cerebrovascular constriction through activation of muscarinic-1 receptors in the newborn pig. J Pharmacol Exp Ther. 1988;247:926–33. [PubMed] [Google Scholar]
- [297].Wagerle LC, Busija DW. Cholinergic mechanisms in the cerebral circulation of the newborn piglet: effect of inhibitors of arachidonic acid metabolism. Circ Res. 1989;64:1030–6. doi: 10.1161/01.res.64.5.1030. [DOI] [PubMed] [Google Scholar]
- [298].Armstead WM, Mirro R, Busija DW, Leffler CW. Permissive role of prostanoids in acetylcholine-induced cerebral vasoconstriction. J Pharmacol Exp Ther. 1989;251:1012–9. [PubMed] [Google Scholar]
- [299].Zhang Y, Leffler CW. Compensatory role of NO in cerebral circulation of piglets chronically treated with indomethacin. Am J Physiol Regul Integr Comp Physiol. 2002;282:R400–10. doi: 10.1152/ajpregu.00256.2001. [DOI] [PubMed] [Google Scholar]
- [300].Berne RM, Rubio R. Adenine nucleotide metabolism in the heart. Circ Res. 1974;35(Suppl 3):109–20. [PubMed] [Google Scholar]
- [301].Phillis JW, Walter GA, Simpson RE. Brain adenosine and transmitter amino acid release from the ischemic rat cerebral cortex: effects of the adenosine deaminase inhibitor deoxycoformycin. J Neurochem. 1991;56:644–50. doi: 10.1111/j.1471-4159.1991.tb08198.x. [DOI] [PubMed] [Google Scholar]
- [302].Van Wylen DG, Park TS, Rubio R, Berne RM. The effect of local infusion of adenosine and adenosine analogues on local cerebral blood flow. J Cereb Blood Flow Metab. 1989;9:556–62. doi: 10.1038/jcbfm.1989.79. [DOI] [PubMed] [Google Scholar]
- [303].Winn HR, Rubio GR, Berne RM. The role of adenosine in the regulation of cerebral blood flow. J Cereb Blood Flow Metab. 1981;1:239–44. doi: 10.1038/jcbfm.1981.29. [DOI] [PubMed] [Google Scholar]
- [304].Hoffman WE, Albrecht RF, Miletich DJ. The role of adenosine in CBF increases during hypoxia in young vs aged rats. Stroke. 1984;15:124–9. doi: 10.1161/01.str.15.1.124. [DOI] [PubMed] [Google Scholar]
- [305].Morii S, Ngai AC, Ko KR, Winn HR. Role of adenosine in regulation of cerebral blood flow: effects of theophylline during normoxia and hypoxia. Am J Physiol. 1987;253:H165–75. doi: 10.1152/ajpheart.1987.253.1.H165. [DOI] [PubMed] [Google Scholar]
- [306].Kurth CD, Wagerle LC. Cerebrovascular reactivity to adenosine analogues in 0.6–0.7 gestation and near-term fetal sheep. Am J Physiol. 1992;262:H1338–42. doi: 10.1152/ajpheart.1992.262.5.H1338. [DOI] [PubMed] [Google Scholar]
- [307].Newman JP, Peebles DM, Hanson MA. Adenosine produces changes in cerebral hemodynamics and metabolism as assessed by near-infrared spectroscopy in late-gestation fetal sheep in utero. Pediatr Res. 2001;50:217–21. doi: 10.1203/00006450-200108000-00009. [DOI] [PubMed] [Google Scholar]
- [308].Blood AB, Hunter CJ, Power GG. The role of adenosine in regulation of cerebral blood flow during hypoxia in the near-term fetal sheep. J Physiol. 2002;543:1015–23. doi: 10.1113/jphysiol.2002.023077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Blood AB, Hunter CJ, Power GG. Adenosine mediates decreased cerebral metabolic rate and increased cerebral blood flow during acute moderate hypoxia in the near-term fetal sheep. J Physiol. 2003;553:935–45. doi: 10.1113/jphysiol.2003.047928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Booth LC, Tummers L, Jensen EC, et al. Differential effects of the adenosine A1 receptor agonist adenosine amine congener on renal, femoral and carotid vascular conductance in preterm fetal sheep. Clin Exp Pharmacol Physiol. 2008;35:1316–20. doi: 10.1111/j.1440-1681.2008.05013.x. [DOI] [PubMed] [Google Scholar]
- [311].Leffler CW, Fedinec AL, Parfenova H, Jaggar JH. Permissive contributions of NO and prostacyclin in CO-induced cerebrovascular dilation in piglets. Am J Physiol Heart Circ Physiol. 2005;289:H432–8. doi: 10.1152/ajpheart.01195.2004. [DOI] [PubMed] [Google Scholar]
- [312].Marks GS, Brien JF, Nakatsu K. What role does the heme—heme oxygenase—carbon monoxide system play in vasoregulation? Am J Physiol Regul Integr Comp Physiol. 2003;285:R522–3. doi: 10.1152/ajpregu.00317.2003. [DOI] [PubMed] [Google Scholar]
- [313].Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–4. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- [314].Wang R. Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor. Can J Physiol Pharmacol. 1998;76:1–15. doi: 10.1139/cjpp-76-1-1. [DOI] [PubMed] [Google Scholar]
- [315].Wang R, Wang Z, Wu L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol. 1997;121:927–34. doi: 10.1038/sj.bjp.0701222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Durante W, Schafer AI. Carbon monoxide and vascular cell function. Int J Mol Med. 1998;2:255–62. doi: 10.3892/ijmm.2.3.255. [DOI] [PubMed] [Google Scholar]
- [317].Christodoulides N, Durante W, Kroll MH, Schafer AI. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclasestimulatory carbon monoxide. Circulation. 1995;91:2306–9. doi: 10.1161/01.cir.91.9.2306. [DOI] [PubMed] [Google Scholar]
- [318].Koneru P, Leffler CW. Role of cGMP in carbon monoxide-induced cerebral vasodilation in piglets. Am J Physiol Heart Circ Physiol. 2004;286:H304–9. doi: 10.1152/ajpheart.00810.2003. [DOI] [PubMed] [Google Scholar]
- [319].Wang R, Wu L, Wang Z. The direct effect of carbon monoxide on KCa channels in vascular smooth muscle cells. Pflügers Arch. 1997;434:285–91. doi: 10.1007/s004240050398. [DOI] [PubMed] [Google Scholar]
- [320].Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, E S, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–7. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- [321].Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activate KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of α-subunits. Am J Physiol Heart Circ Physiol. 2004;286:H610–8. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- [322].Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- [323].Kanu A, Gilpin D, Fedinec AL, Leffler CW. Cyclooxygenase products stimulate carbon monoxide production by piglet cerebral microvessels. Exp Biol Med. 2006;231:181–5. doi: 10.1177/153537020623100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Kanu A, Leffler CW. Roles of glia limitans astrocytes and carbon monoxide in adenosine diphosphate-induced pial arteriolar dilation in newborn pigs. Stroke. 2009;40:930–5. doi: 10.1161/STROKEAHA.108.533786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Kanu A, Leffler CW. Arachidonic acid- and prostaglandin E2-induced cerebral vasodilation is mediated by carbon monoxide, independent of reactive oxygen species in piglets. Am J Physiol Heart Circ Physiol. 2011;301:H2482–7. doi: 10.1152/ajpheart.00628.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Li A, Xi Q, Umstot ES, et al. Astrocyte-derived CO is a diffusible messenger that mediates glutamate-induced cerebral arteriolar dilation by activating smooth muscle cell KCa channels. Circ Res. 2008;102:234–41. doi: 10.1161/CIRCRESAHA.107.164145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Wu L, Cao K, Lu Y, Wang R. Different mechanisms underlying the stimulation of KCa channels by nitric oxide and carbon monoxide. J Clin Invest. 2002;110:691–700. doi: 10.1172/JCI15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Hartsfield CL. Cross talk between carbon monoxide and nitric oxide. Antioxid Redox Signal. 2002;4:301–7. doi: 10.1089/152308602753666352. [DOI] [PubMed] [Google Scholar]
- [329].Holt DC, Fedinec AL, Vaughn AN, Leffler CW. Age and species dependence of pial arteriolar responses to topical carbon monoxide in vivo. Exp Biol Med. 2007;232:1465–9. doi: 10.3181/0705-BC-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Longo LD. The biological effects of carbon monoxide on the pregnant woman, fetus, and newborn infant. Am J Obstet Gynecol. 1977;129:69–103. doi: 10.1016/0002-9378(77)90824-9. [DOI] [PubMed] [Google Scholar]
- [331].Kinobe RT, Vlahakis JZ, Soong JM, et al. Heme oxygenase activity in fetal and adult sheep is not altered by acclimatization to high altitude hypoxia. Can J Physiol Pharmacol. 2006;84:893–901. doi: 10.1139/y06-034. [DOI] [PubMed] [Google Scholar]
- [332].Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- [333].Robinson JS, Fedinec AL, Leffler CW. Role of carbon monoxide in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol Heart Circ Physiol. 2003;282:H2371–6. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- [334].Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. Circ Res. 1993;72:476–80. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- [335].Armstead WM. Opioids and nitric oxide contribute to hypoxia-induced pial artery vasodilation. Am J Physiol. 1995;268:H226–32. doi: 10.1152/ajpheart.1995.268.1.H226. [DOI] [PubMed] [Google Scholar]
- [336].Armstead WM. Role of opioids in the physiologic and pathophysiologic control of the cerebral circulation. Proc Soc Exp Biol Med. 1997;212:210–21. doi: 10.3181/00379727-214-44089. [DOI] [PubMed] [Google Scholar]
- [337].Armstead WM, Mirro R, Busija DW, Leffler CW. Opioids in cerebrospinal fluid in hypotensive newborn pigs. Circ Res. 1991;68:922–9. doi: 10.1161/01.res.68.4.922. [DOI] [PubMed] [Google Scholar]
- [338].Armstead WM, Mirro R, Shibata M, Leffler CW. Prostanoids modulate opioid-induced increases in CSF vasopressin concentration. Am J Physiol. 1992;263:H1670–4. doi: 10.1152/ajpheart.1992.263.6.H1670. [DOI] [PubMed] [Google Scholar]
- [339].Armstead WM. Hypotension dilates pial arteries by KATP and KCa channel activation. Brain Res. 1999;816:158–64. doi: 10.1016/s0006-8993(98)01146-9. [DOI] [PubMed] [Google Scholar]
- [340].Armstead WM. Relationship between NOC/oFQ and cerebral hemodynamics following hypoxia/ischemia in piglets. Am J Physiol Heart Circ Physiol. 2000;278:H477–83. doi: 10.1152/ajpheart.2000.278.2.H477. [DOI] [PubMed] [Google Scholar]
- [341].Ben-Haim G, Armstead WM. Role of cAMP and K+ channel-dependent mechanisms in piglet hypoxic/ischemic impaired nociception/orphanin FQ-induced cerebrovasodilation. Brain Res. 2000;884:51–8. doi: 10.1016/s0006-8993(00)02882-1. [DOI] [PubMed] [Google Scholar]
- [342].Sanders KM. Signal Transduction in Smooth Muscle. Invited review: Mechanisms of calcium handling in smooth muscles. J Appl Physiol. 2001;91:1438–49. doi: 10.1152/jappl.2001.91.3.1438. [DOI] [PubMed] [Google Scholar]
- [343].Putney JW, Ribiero CM. Signaling pathways between the plasma membrane and endoplasmic reticulum calcium stores. Cell Mol Life Sci. 2000;57:1272–86. doi: 10.1007/PL00000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Long W, Zhang L, Longo LD. Fetal and adult cerebral artery KATP and KCa channel responses to long-term hypoxia. J Appl Physiol. 2002;92:1692–701. doi: 10.1152/japplphysiol.01110.2001. [DOI] [PubMed] [Google Scholar]
- [345].Lin MT, Hessinger DA, Pearce WJ, Longo LD. Developmental differences in Ca2+-activated K+ channel activity in ovine basilar artery. Am J Physiol Heart Circ Physiol. 2003;285:H701–9. doi: 10.1152/ajpheart.00138.2003. [DOI] [PubMed] [Google Scholar]
- [346].Akopov SE, Zhang L, Pearce WJ. Developmental changes in the calcium sensitivity of rabbit cranial arteries. Biol Neonate. 1998;74:60–71. doi: 10.1159/000014011. [DOI] [PubMed] [Google Scholar]
- [347].Nauli SM, Williams JM, Gerthoffer WT, Pearce WJ. Chronic hypoxia modulates relations among calcium, myosin light chain phosphorylation, and force differently in fetal and adult ovine basilar arteries. J Appl Physiol. 2005;99:120–7. doi: 10.1152/japplphysiol.01131.2004. [DOI] [PubMed] [Google Scholar]
- [348].Zurcher SD, Pearce WJ. Maturation modulates serotonin- and potassium-induced calcium-45 uptake in ovine carotid and cerebral arteries. Pediatr Res. 1995;38:493–500. doi: 10.1203/00006450-199510000-00004. [DOI] [PubMed] [Google Scholar]
- [349].Lin MT, Longo LD, Pearce WJ, Hessinger DA. Ca2+-activated K+ channel-associated phosphatase and kinase activities during development. Am J Physiol Heart Circ Physiol. 2005;289:H414–25. doi: 10.1152/ajpheart.01079.2004. [DOI] [PubMed] [Google Scholar]
- [350].Atkinson SA. Calcium and phosphorus needs of premature infants. Nutrition. 1994;10:66–8. [PubMed] [Google Scholar]
- [351].Koo WW, Warren L. Calcium and bone health in infants. Neonatal Netw. 2003;22:23–37. doi: 10.1891/0730-0832.22.5.23. [DOI] [PubMed] [Google Scholar]
- [352].Specker B. Nutrition influences bone development from infancy through toddler years. J Nutr. 2004;134:691S–695S. doi: 10.1093/jn/134.3.691S. [DOI] [PubMed] [Google Scholar]
- [353].Long W, Zhao Y, Zhang L, Longo LD. Cerebral artery sarcoplasmic reticulum Ca2+ stores and contractility: changes with development. Am J Physiol Regul Integr Comp Physiol. 2000;279:R860–73. doi: 10.1152/ajpregu.2000.279.3.R860. [DOI] [PubMed] [Google Scholar]
- [354].Nauli SM, Williams JM, Akopov SE, Zhang L, Pearce WJ. Developmental changes in ryanodine- and IP3-sensitive Ca2+ pools in ovine basilar artery. Am J Physiol Cell Physiol. 2001;281:C1785–96. doi: 10.1152/ajpcell.2001.281.6.C1785. [DOI] [PubMed] [Google Scholar]
- [355].Akopov SE, Zhang L, Pearce WJ. Regulation of Ca2+ sensitization by PKC and RHO proteins in ovine cerebral arteries: effects of artery size and age. Am J Physiol. 1998;275:H930–9. doi: 10.1152/ajpheart.1998.275.3.H930. [DOI] [PubMed] [Google Scholar]
- [356].Akopov SE, Zhang L, Pearce WJ. Intracranial-extracranial differences in the Ca2+ sensitivity of rabbit arteries. Proc Soc Exp Biol Med. 1997;214:76–82. doi: 10.3181/00379727-214-44072. [DOI] [PubMed] [Google Scholar]
- [357].Akopov SE, Zhang L, Pearce WJ. Physiological variations in ovine cerebrovascular calcium sensitivity. Am J Physiol. 1997;272:H2271–81. doi: 10.1152/ajpheart.1997.272.5.H2271. [DOI] [PubMed] [Google Scholar]
- [358].Lompre AM. Sarcoplasmic reticulum in vascular cells in hypertension and during proliferation. Clin Exp Pharmacol Physiol. 1999;26:553–7. doi: 10.1046/j.1440-1681.1999.03079.x. [DOI] [PubMed] [Google Scholar]
- [359].Vallot O, Combettes L, Jourdon P, et al. Intracellular Ca2+ handling in vascular smooth muscle cells is affected by proliferation. Arterioscler Thromb Vasc Biol. 2000;20:1225–35. doi: 10.1161/01.atv.20.5.1225. [DOI] [PubMed] [Google Scholar]
- [360].Ray LB, Gough NR, Adler EM. Calcium signaling: a meeting of the minds. Sci STKE. 2004;2004:eg8. [Google Scholar]
- [361].McCalden TA, Bevan JA. Sources of activator calcium in rabbit basilar artery. Am J Physiol. 1981;241:H129–33. doi: 10.1152/ajpheart.1981.241.2.H129. [DOI] [PubMed] [Google Scholar]
- [362].Grunstein MM, Rosenberg SM, Schramm CM, Pawlowski NA. Mechanisms of action of endothelin 1 in maturing rabbit airway smooth muscle. Am J Physiol. 1991;260:L434–43. doi: 10.1152/ajplung.1991.260.6.L434. [DOI] [PubMed] [Google Scholar]
- [363].Hillemeier AC, Bitar KN, Biancani P. Developmental characteristics of the kitten antrum. Gastroenterology. 1991;101:339–43. doi: 10.1016/0016-5085(91)90009-a. [DOI] [PubMed] [Google Scholar]
- [364].Zderic SA, Sillen U, Liu GH, et al. Developmental aspects of bladder contractile function: evidence for an intracellular calcium pool. J Urol. 1993;150:623–5. doi: 10.1016/s0022-5347(17)35564-7. [DOI] [PubMed] [Google Scholar]
- [365].Chin TK, Friedman WF, Klitzner TS. Developmental changes in cardiac myocyte calcium regulation. Circ Res. 1990;67:574–9. doi: 10.1161/01.res.67.3.574. [DOI] [PubMed] [Google Scholar]
- [366].Kojima M, Sperelakis N, Sada H. Ontogenesis of transmembrane signaling systems for control of cardiac Ca2+ channels. J Dev Physiol. 1990;1:181–219. [PubMed] [Google Scholar]
- [367].Xiong Z, Sperelakis N. Regulation of L-type calcium channels of vascular smooth muscle cells. J Moll Cell Cardiol. 1995;27:75–91. doi: 10.1016/s0022-2828(08)80009-0. [DOI] [PubMed] [Google Scholar]
- [368].Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–27. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Shelly DA, He S, Moseley A, et al. Na(+) pump alpha 2-isoform specifically couples to contractility in vascular smooth muscle: evidence from gene-targeted neonatal mice. Am J Physiol Cell Physiol. 2004;286:C813–20. doi: 10.1152/ajpcell.00389.2003. [DOI] [PubMed] [Google Scholar]
- [370].Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann N Y Acad Sci. 1997;834:524–36. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- [371].Mir MA, Morgan K, Lewis M, et al. Problems and pitfalls in the isolation of an endogenous Na+, K+-ATPase inhibitor. Hypertension. 1987;10:I57–60. doi: 10.1161/01.hyp.10.5_pt_2.i57. [DOI] [PubMed] [Google Scholar]
- [372].Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- [373].Knot HG, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Knot HJ, Zimmermann PA, Nelson MT. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J Physiol. 1996;492:419–30. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Long W, Zhang L, Longo LD. Cerebral artery KATP- and KCa-channel activity and contractility: changes with development. Am J Physiol Regul Integr Comp Physiol. 2000;279:R2004–14. doi: 10.1152/ajpregu.2000.279.6.R2004. [DOI] [PubMed] [Google Scholar]
- [376].Teng GQ, Nauli SM, Brayden JE, Pearce WJ. Maturation alters the contribution of potassium channels to resting and 5HT-induced tone in small cerebral arteries of the sheep. Brain Res Dev Brain Res. 2002;133:81–91. doi: 10.1016/s0165-3806(01)00304-2. [DOI] [PubMed] [Google Scholar]
- [377].Wellman GC, Cartin L, Eckman DM, et al. Membrane depolarization, elevated Ca2+ entry, and gene expression in cerebral arteries of hypertensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H2559–67. doi: 10.1152/ajpheart.2001.281.6.H2559. [DOI] [PubMed] [Google Scholar]
- [378].Gollasch M, Wellman GC, Knot HJ, et al. Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res. 1998;83:1104–14. doi: 10.1161/01.res.83.11.1104. [DOI] [PubMed] [Google Scholar]
- [379].Pérez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–38. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci. 2001;22:505–12. doi: 10.1016/s0165-6147(00)01775-2. [DOI] [PubMed] [Google Scholar]
- [381].Lin MT, Hessinger DA, Pearce WJ, Longo LD. Modulation of BK channel calcium affinity by differential phosphorylation in developing ovine basilar artery myocytes. Am J Physiol Heart Circ Physiol. 2006;291:H732–40. doi: 10.1152/ajpheart.01357.2005. [DOI] [PubMed] [Google Scholar]
- [382].Pearce WJ, Elliott SR. Maturation enhances the sensitivity of ovine cerebral arteries to the ATP-sensitive potassium channel activator lemakalim. Pediatr Res. 1994;35:729–32. doi: 10.1203/00006450-199406000-00021. [DOI] [PubMed] [Google Scholar]
- [383].Lefkowitz RJ. Historical review: a brief history and personal retro-spective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–22. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- [384].Sealfon SC. Teaching resources. G-protein-coupled receptors. Sci STKE. 2005;2005:tr11. doi: 10.1126/stke.2792005tr11. [DOI] [PubMed] [Google Scholar]
- [385].Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- [386].Luttrell LM, Roudabush FL, Choy EW, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–54. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Bevan RD. Trophic effects of peripheral adrenergic nerves on vascular structure. Hypertension. 1984;6:III19–26. doi: 10.1161/01.hyp.6.6_pt_2.iii19. [DOI] [PubMed] [Google Scholar]
- [388].Edvinsson L, MacKenzie ET. Amine mechanisms in the cerebral circulation. Pharmacol Rev. 1976;28:275–348. [PubMed] [Google Scholar]
- [389].Kenakin TP. Theoretical and practical problems with the assessment of intrinsic efficacy of agonists: efficacy of reputed beta-1 selective adrenoceptor agonists for beta-2 adrenoceptors. J Pharmacol Exp Ther. 1982;223:416–23. [PubMed] [Google Scholar]
- [390].McCalden TA. Sympathetic control of the cerebral circulation. J Auton Pharmacol. 1981;1:421–31. doi: 10.1111/j.1474-8673.1981.tb00082.x. [DOI] [PubMed] [Google Scholar]
- [391].Wagerle LC, Hefferna TM, Sacks LM, Delivoria-Papadopoulos M. Sympathetic effect on cerebral blood flow regulation in hypoxic newborn lambs. Am J Physiol. 1983;245:H487–94. doi: 10.1152/ajpheart.1983.245.3.H487. [DOI] [PubMed] [Google Scholar]
- [392].Asada Y, Lee JF. α2-Adrenoreceptors mediate NE constriction of porcine pial veins. Am J Physiol. 1992;263:H1907–10. doi: 10.1152/ajpheart.1992.263.6.H1907. [DOI] [PubMed] [Google Scholar]
- [393].Elliott SR, Pearce WJ. Effects of maturation on α-adrenergic receptor affinity and occupancy in small cerebral arteries. Am J Physiol. 1994;267:H757–63. doi: 10.1152/ajpheart.1994.267.2.H757. [DOI] [PubMed] [Google Scholar]
- [394].Wagerle LC, Busija DW. Effect of thromboxane A2/endoperoxide antagonist SQ29548 on the contractile response to acetylcholine in newborn piglet cerebral arteries. Circ Res. 1990;66:824–31. doi: 10.1161/01.res.66.3.824. [DOI] [PubMed] [Google Scholar]
- [395].Ruffolo RR, Jr, Nichols AJ, Stadel JM, Hieble JP. Pharmacologic and therapeutic applications of α2-adrenoceptor subtypes. Annu Rev Pharmacol Toxicol. 1993;32:243–79. doi: 10.1146/annurev.pa.33.040193.001331. [DOI] [PubMed] [Google Scholar]
- [396].Bishai J, Penninga L, Nijland R, et al. Pre- and postjunctional α2-adrenergic receptors in fetal and adult ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1654–62. doi: 10.1152/ajpregu.00475.2001. [DOI] [PubMed] [Google Scholar]
- [397].Van Riper DA, Bevan JA. Selective variation of agonist and neurally mediated vasoconstriction with rabbit middle cerebral artery branch order. J Pharmacol Exp Ther. 1991;257:879–86. [PubMed] [Google Scholar]
- [398].Su C, Bevan JA, Assali NS, Brinkman CR., III Regional variation of lamb blood vessel responsiveness to vasoactive agents during fetal development. Circ Res. 1977;41:844–8. doi: 10.1161/01.res.41.6.844. [DOI] [PubMed] [Google Scholar]
- [399].Matz RL, Alvarez de Sotomayor M, Schott C. Andriantsitohaina R. Preservation of vascular contraction during ageing: dual effect on calcium handling and sensitization. Br J Pharmacol. 2003;138:745–50. doi: 10.1038/sj.bjp.0705104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Toda N. Alpha adrenergic receptor subtypes in human, monkey and dog cerebral arteries. J Pharmacol Exp Ther. 1983;226:861–8. [PubMed] [Google Scholar]
- [401].Busija DW, Leffler CW. Exogenous norepinephrine constricts cerebral arterioles via α2-adrenoreceptors in newborn pigs. J Cereb Blood Flow Metab. 1987;7:184–8. doi: 10.1038/jcbfm.1987.42. [DOI] [PubMed] [Google Scholar]
- [402].Wagerle LC, Delivoria-Papadopoulos M. α-adrenergic receptor subtypes in the cerebral circulation of newborn piglets. Am J Physiol. 1987;252:R1092–8. doi: 10.1152/ajpregu.1987.252.6.R1092. [DOI] [PubMed] [Google Scholar]
- [403].Bevan RD, Tsuru H, Bevan JA. Cerebral artery mass in the rabbit is reduced by chronic sympathetic denervation. Stroke. 1983;14:393–6. doi: 10.1161/01.str.14.3.393. [DOI] [PubMed] [Google Scholar]
- [404].Langer SZ, Hicks PE. Alpha-adrenoceptors subtypes in blood vessels: physiology and pharmacology. J Cardiovasc Pharmacol. 1984;6(Suppl 4):S547–58. doi: 10.1097/00005344-198406004-00001. [DOI] [PubMed] [Google Scholar]
- [405].Leech CJ, Faber JE. Different α-adrenoceptor subtypes mediate constriction of arterioles and venules. Am J Physiol. 1996;270:H710–22. doi: 10.1152/ajpheart.1996.270.2.H710. [DOI] [PubMed] [Google Scholar]
- [406].Kurth CD, Wagerle LC, Delivoria-Papadopoulos M. Sympathetic regulation of cerebral blood flow during seizures in newborn lambs. Am J Physiol. 1988;255:H563–8. doi: 10.1152/ajpheart.1988.255.3.H563. [DOI] [PubMed] [Google Scholar]
- [407].Longo LD, Pearce WJ. High altitude hypoxic-induced modulation of noradrenergic-mediated responses in fetal and adult cerebral arteries. Comp Biochem Physiol. 1998;119A:683–94. doi: 10.1016/s1095-6433(98)01006-x. [DOI] [PubMed] [Google Scholar]
- [408].Ueno N, Zhao Y, Zhang L, Longo LD. High altitude-induced changes in alpha1-adrenergic receptors and Ins(1,4,5)P3 responses in cerebral arteries. Am J Physiol. 1997;272:R669–74. doi: 10.1152/ajpregu.1997.272.2.R669. [DOI] [PubMed] [Google Scholar]
- [409].Tsukahara T, Taniguchi T, Fujiwara M, Honda H, Nichikawa M. Alteration in α-adrenergic receptors in human cerebral arteries after subarachnoid hemorrhage. Stroke. 1985;16:53–8. doi: 10.1161/01.str.16.1.53. [DOI] [PubMed] [Google Scholar]
- [410].Usui H, Fujiwara M, Tsukahara T, Taniguchi T, Kurahashi K. Differences in contractile responses to electrical stimulation and α-adrenergic binding sites in isolated cerebral arteries of humans, cows, dogs, and monkeys. J Cardiovasc Pharmacol. 1985;7(Suppl 3):S47–52. doi: 10.1097/00005344-198500073-00006. [DOI] [PubMed] [Google Scholar]
- [411].Edvinsson L, MacKenzie ET, Robert JP, Skärby T, Young AR. Cerebrovascular responses to haemorrhagic hypotension in anaesthetized cats. Effects of α-adrenoceptor antagonists. Acta Physiol Scand. 1985;123:317–23. doi: 10.1111/j.1748-1716.1985.tb07594.x. [DOI] [PubMed] [Google Scholar]
- [412].McPherson RW, Koehler RC, Traystman RJ. Hypoxia, α2-adrenergic, and nitric oxide-dependent interactions on canine cerebral blood flow. Am J Physiol. 1994;266:H476–82. doi: 10.1152/ajpheart.1994.266.2.H476. [DOI] [PubMed] [Google Scholar]
- [413].Bylund DB. Subtypes of α1- and α2-adrenergic receptors. FASEB J. 1992;6:832–9. doi: 10.1096/fasebj.6.3.1346768. [DOI] [PubMed] [Google Scholar]
- [414].Guimãraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319–56. [PubMed] [Google Scholar]
- [415].Aleixandre A, Pintado A, Puerro M. α2-Adrenoceptor-mediated contractions in the rabbit aorta treated with BAY K 8644. Eur J Pharmacol. 1992;221:129–34. doi: 10.1016/0014-2999(92)90781-x. [DOI] [PubMed] [Google Scholar]
- [416].Tsai SH, Buchholz J, Duckles SP. Postjunctional α2-adrenoceptors in blood vessels: effect of age. Eur J Pharmacol. 1993;237:311–6. doi: 10.1016/0014-2999(93)90283-n. [DOI] [PubMed] [Google Scholar]
- [417].Xiao X, Rand MJ. α2-Adrenoceptor agonists enhance vasoconstrictor responses to α1-adrenoceptor agonists in the rat tail artery by increasing the influx of Ca2+ Br J Pharmacol. 1989;98:1032–8. doi: 10.1111/j.1476-5381.1989.tb14635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [418].Szabo C, Emilsson K, Hardebo JE, Hystedt S, Owman C. Uptake and release of serotonin in rat cerebrovascular nerves after subarachnoid hemorrhage. Stroke. 1992;23:54–61. doi: 10.1161/01.str.23.1.54. [DOI] [PubMed] [Google Scholar]
- [419].Bonvento G, MacKenzie ET, Edvinsson L. Serotonergic innervation of the cerebral vasculature: relevance to migraine and ischaemia. Brain Res Brain Res Rev. 1991;16:257–63. doi: 10.1016/0165-0173(91)90009-w. [DOI] [PubMed] [Google Scholar]
- [420].Hoyer D, Clarke DE, Fozard JR, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- [421].Hoyer D, Martin GR. Classification and nomenclature of 5-HT receptors: a comment on current issues. Behav Brain Res. 1996;73:263–8. doi: 10.1016/0166-4328(96)00109-x. [DOI] [PubMed] [Google Scholar]
- [422].Ullmer C, Schmuck K, Kalkman HO, Lubbert H. Expression of serotonin receptor mRNAs in blood vessels. FEBS Lett. 1995;370:215–21. doi: 10.1016/0014-5793(95)00828-w. [DOI] [PubMed] [Google Scholar]
- [423].Teng GQ, Williams J, Zhang L, Purdy R, Pearce WJ. Effects of maturation, artery size, and chronic hypoxia on 5-HT receptor type in ovine cranial arteries. Am J Physiol. 1998;275:R742–53. doi: 10.1152/ajpregu.1998.275.3.R742. [DOI] [PubMed] [Google Scholar]
- [424].Akopov SE, Zhang L, Pearce WJ. Maturation alters the contractile role of calcium in ovine basilar arteries. Pediatr Res. 1998;44:154–60. doi: 10.1203/00006450-199808000-00003. [DOI] [PubMed] [Google Scholar]
- [425].Hardebo JE, Suzuki N, Ekblad E, Owman C. Vasoactive intestinal polypeptide and acetylcholine coexist with neuropeptide Y, dopa-mine-beta-hydroxylase, tyrosine hydroxylase, substance P or calcitonin gene-related peptide in neuronal subpopulations in cranial parasympathetic ganglia of rat. Cell Tissue Res. 1992;267:291–300. doi: 10.1007/BF00302967. [DOI] [PubMed] [Google Scholar]
- [426].Kimura T, Yu JG, Edvinsson L, Lee TJ. Cholinergic, nitric oxidergic innervation in cerebral arteries of the cat. Brain Res. 1997;773:117–24. doi: 10.1016/s0006-8993(97)00889-5. [DOI] [PubMed] [Google Scholar]
- [427].Suzuki N, Hardebo JE, Kährstrom J, Owman C. Neuropeptide Y co-exists with vasoactive intestinal polypeptide and acetylcholine in parasympathetic cerebrovascular nerves originating in the sphenopalatine, otic, and internal carotid ganglia of the rat. Neuroscience. 1990;36:507–19. doi: 10.1016/0306-4522(90)90001-7. [DOI] [PubMed] [Google Scholar]
- [428].Chorobski J, Penfield W. Cerebral vasodilator nerves and their pathway from the medulla oblongata with observations on the pial and intracerebral vascular plexus. Arch Neur Psych. 1932;28:1257–89. [Google Scholar]
- [429].Brayden JE, Bevan JA. Acetylcholine and vasoactive intestinal polypeptide in the cerebral circulation: Histochemical and biochemical indices of innervation. In: Owman C, Hardebo JE, editors. Neural regulation of brain circulation. Elsevier Science Publishing Co.; Amsterdam: 1986. pp. 371–81. [Google Scholar]
- [430].Duckles SP. Evidence for a functional cholinergic innervation of cerebral arteries. J Pharmacol Exp Ther. 1981;217:544–8. [PubMed] [Google Scholar]
- [431].Edvinsson L, MacKenzie ET, McCulloch J, Uddman R. Perivascular innervation and receptor mechanisms in cerebrovascular bed. In: Wood JH, editor. Cerebral blood flow: Physiologic and clinical aspects. McGraw-Hill; New York: 1987. pp. 145–72. [Google Scholar]
- [432].Saito A, Wu JY, Lee TJ. Evidence for the presence of cholinergic nerves in cerebral arteries: an immunohistochemical demonstration of choline acetyltransferase. J Cereb Blood Flow Metab. 1985;5:327–34. doi: 10.1038/jcbfm.1985.42. [DOI] [PubMed] [Google Scholar]
- [433].Sato A, Sato Y, Uchida S. Regulation of cerebral cortical blood flow by the basal forebrain cholinergic fibers and aging. Auton Neurosci. 2002;96:13–9. doi: 10.1016/s1566-0702(01)00367-8. [DOI] [PubMed] [Google Scholar]
- [434].Sato A, Sato Y, Uchida S. Activation of the intracerebral cholinergic nerve fibers originating in the basal forebrain increases regional cerebral blood flow in the rat's cortex and hippocampus. Neurosci Lett. 2004;361:90–3. doi: 10.1016/j.neulet.2004.01.004. [DOI] [PubMed] [Google Scholar]
- [435].Hardeo JE, Lindvall O, Nilsson B. On the possible influence of adrenergic and cholinergic mechanisms in normo- and hypercapnia. In: Heistad D, Marcus ML, editors. Cerebral blood flow: effects of nerves and neurotransmitter. Elsevier; New York: 1982. pp. 377–83. [Google Scholar]
- [436].Suzuki N, Hardebo JE. The cerebrovascular parasympathetic innervation. Cerebrovasc Brain Metab Rev. 1993;5:33–46. [PubMed] [Google Scholar]
- [437].Wagerle LC, Kurth CD, Busija DW. Cholinergic reactivity of cerebral arteries in the developing fetal and newborn lamb. J Dev Physiol. 1992;17:51–4. [PubMed] [Google Scholar]
- [438].Docherty CC, Kalmar-Nagy J, Engelen M, Nathanielsz PW. Development of fetal vascular responses to endothelin-1 and acetylcholine in the sheep. Am J Physiol Regul Integr Comp Physiol. 2001;280:R554–62. doi: 10.1152/ajpregu.2001.280.2.R554. [DOI] [PubMed] [Google Scholar]
- [439].Zhao Y, Long W, Zhang L, Longo LD. Extracellular signal-regulated kinases and contractile responses in ovine in adult and fetal cerebral arteries. J Physiol. 2003;551:691–703. doi: 10.1113/jphysiol.2003.046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [440].Zhao Y, Zhang L, Longo LD. PKC-induced ERK1/2 interactions and downstream effectors in ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2005;289:R164–71. doi: 10.1152/ajpregu.00847.2004. [DOI] [PubMed] [Google Scholar]
- [441].Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–25. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- [442].Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–6. doi: 10.1038/372231a0. Corrigenda. Nature 1994; 372: 812. [DOI] [PubMed] [Google Scholar]
- [443].Jaggar JH, Wellman GC, Heppner TJ, et al. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand. 1998;164:577–87. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- [444].Himpens B, Kitazawa T, Somlyo AP. Agonist-dependent modulation of Ca2+ sensitivity in rabbit pulmonary artery smooth muscle. Plfügers Arch. 1990;417:21–8. doi: 10.1007/BF00370764. [DOI] [PubMed] [Google Scholar]
- [445].Karaki H, Ozaki H, Hori M, et al. Calcium movements, distribution, and functions in smooth muscle. Pharmacol Rev. 1997;49:157–230. [PubMed] [Google Scholar]
- [446].Pabelick CM, Sieck GC, Prakash YS. Significance of spatial and temporal heterogeneity of calcium transients in smooth muscle. J Appl Physiol. 2001;91:488–96. doi: 10.1152/jappl.2001.91.1.488. [DOI] [PubMed] [Google Scholar]
- [447].Wilson SM, Mason HS, Ng LC, et al. Role of basal extracellular Ca2+ entry during 5-HT-induced vasoconstriction of canine pulmonary arteries. Br J Pharmacol. 2005;144:252–64. doi: 10.1038/sj.bjp.0706077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [448].Bruce L, Nixon GF. Increased sensitization of the myofilaments in rat neonatal portal vein: a potential mechanism. Exp Physiol. 1997;82:985–93. doi: 10.1113/expphysiol.1997.sp004084. [DOI] [PubMed] [Google Scholar]
- [449].Zhou J, Zhao Y, Nijland R, Zhang L, Longo LD. Ins(1,4,5)P3 receptors in cerebral arteries: Changes with development and high altitude hypoxia. Am J Physiol. 1997;272:R1954–59. doi: 10.1152/ajpregu.1997.272.6.R1954. [DOI] [PubMed] [Google Scholar]
- [450].Injeti ER, Sandoval RJ, Williams JM, Smolensky AV, Ford LE, Pearce WJ. Maximal stimulation-induced in situ myosin light chain kinase activity is upregulated in fetal compared with adult ovine carotid arteries. Am J Physiol Heart Circ Physiol. 2008;295:H2289–98. doi: 10.1152/ajpheart.00606.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [451].Sandoval RJ, Injeti ER, Gerthoffer WT, Pearce WJ. Postnatal maturation modulates relationships among cytosolic Ca2+, myosin light chain phosphorylation, and contractile tone in ovine cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;293:H2183–92. doi: 10.1152/ajpheart.00647.2007. [DOI] [PubMed] [Google Scholar]
- [452].Somlyo AV. Ultrastructure of vascular smooth muscle. In: Bohr DF, Somlyo AP, Sparks HV Jr, editors. Handbook of physiology. Section 2: The cardiovascular system, Volume II. Vascular smooth muscle. American Physiological Society; Bethesda, MD: 1980. pp. 33–67. [Google Scholar]
- [453].Chadwick CC, Saito A, Fleischer S. Isolation and characterization of the inositol trisphosphate receptor from smooth muscle. Proc Natl Acad Sci USA. 1990;87:2132–6. doi: 10.1073/pnas.87.6.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [454].Schramm CM, Chuang ST, Grunstein MM. Maturation of inositol 1,4,5-trisphosphate receptor binding in rabbit tracheal smooth muscle. Am J Physiol. 1992;263:L501–5. doi: 10.1152/ajplung.1992.263.4.L501. [DOI] [PubMed] [Google Scholar]
- [455].Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508:211–21. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [456].Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–30. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- [457].Laporte R, Laher I. Sarcoplasmic reticulum-sarcolemma interactions and vascular smooth muscle tone. J Vasc Res. 1997;34:325–43. doi: 10.1159/000159242. [DOI] [PubMed] [Google Scholar]
- [458].Nixon GF, Mignery GA, Somlyo AV. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultra-structural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J Muscle Res Cell Motil. 1994;15:682–700. doi: 10.1007/BF00121075. [DOI] [PubMed] [Google Scholar]
- [459].Tribe RM, Borin ML, Blaustein MP. Functionally and spatially distinct Ca2+ stores are revealed in cultured vascular smooth muscle cells. Proc Natl Acad Sci USA. 1994;91:5908–12. doi: 10.1073/pnas.91.13.5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [460].Stauderman KA, McKinney RA, Murawsky MM. The role of caffeine-sensitive Ca 2+ stores in agonist- and inositol 1,4,5-trisphosphate-induced Ca2+ release from bovine adrenal chromaffin cells. Biochem J. 1991;278:643–50. doi: 10.1042/bj2780643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [461].Somlyo AV, Bond M, Somlyo AP, Scarpa A. Inositol trisphosphate-induced calcium release and contraction in vascular smooth muscle. Proc Natl Acad Sci USA. 1985;82:5231–5. doi: 10.1073/pnas.82.15.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [462].Iino M. Calcium release mechanisms in smooth muscle. Jpn J Pharmacol. 1990;54:345–54. doi: 10.1254/jjp.54.345. [DOI] [PubMed] [Google Scholar]
- [463].Lesh RE, Nixon GF, Fleischer S, Airey JA, Somlyo AP, Somlyo AV. Localization of ryanodine receptors in smooth muscle. Circ Res. 1998;82:175–85. doi: 10.1161/01.res.82.2.175. [DOI] [PubMed] [Google Scholar]
- [464].Bian JH, Ghosh TK, Wang JC, Gill DL. Identification of intracellular calcium pools. Selective modification by thapsigargin. J Biol Chem. 1991;266:8801–6. [PubMed] [Google Scholar]
- [465].Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–23. [PubMed] [Google Scholar]
- [466].Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–8. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- [467].Brandt L, Andersson KE, Edvinsson L, Ljunggren B. Effect of extracellular calcium and of calcium antagonists on the contractile responses of isolated human pial and mesenteric arteries. J Cereb Blood Flow Metab. 1981;1:339–47. doi: 10.1038/jcbfm.1981.37. [DOI] [PubMed] [Google Scholar]
- [468].Nakanishi T, Gu H, Abe K, Momma K. Developmental changes in the contractile system of the mesenteric small artery of rabbit. Pediatr Res. 1997;41:65–71. doi: 10.1203/00006450-199701000-00010. [DOI] [PubMed] [Google Scholar]
- [469].Tasker PN, Michelangeli F, Nixon GF. Expression and distribution of the type 1 and type 3 inositol 1,4,5-trisphosphate receptor in developing vascular smooth muscle. Circ Res. 1999;84:536–42. doi: 10.1161/01.res.84.5.536. [DOI] [PubMed] [Google Scholar]
- [470].Le Jemtel TH, Lambert F, Levitsky DO, et al. Age-related changes in sarcoplasmic reticulum Ca2+-ATPase and α-smooth muscle actin gene expression in aortas of normotensive and spontaneously hypertensive rats. Circ Res. 1993;72:341–8. doi: 10.1161/01.res.72.2.341. [DOI] [PubMed] [Google Scholar]
- [471].Mahony L, Jones LR. Developmental changes in cardiac sarcoplasmic reticulum in sheep. J Biol Chem. 1986;261:15257–65. [PubMed] [Google Scholar]
- [472].Lincoln TM, Cornwell TL. Towards an understanding of the mechanism of action of cyclic AMP and cyclic GMP in smooth muscle relaxation. Blood Vessels. 1991;28:129–37. doi: 10.1159/000158852. [DOI] [PubMed] [Google Scholar]
- [473].Murad F, Waldman S, Molina C, Bennett B, Leitman D. Regulation and role of guanylate cyclase-cyclic GMP in vascular relaxation. Prog Clin Biol Res. 1987;249:65–76. [PubMed] [Google Scholar]
- [474].Zellers TM, Vanhoutte PM. Endothelium-dependent relaxations of piglet pulmonary arteries augment with maturation. Pediatr Res. 1991;30:176–80. doi: 10.1203/00006450-199108000-00011. [DOI] [PubMed] [Google Scholar]
- [475].Rand MJ. Nitrergic transmission: nitric oxide as a mediator of non-adrenergic, non-cholinergic neuro-effector transmission. Clin Exp Pharmacol Physiol. 1992;19:147–69. doi: 10.1111/j.1440-1681.1992.tb00433.x. [DOI] [PubMed] [Google Scholar]
- [476].Iwamoto J, Yoshinaga M, Yang SP, Krasney E, Krasney J. Methylene blue inhibits hypoxic cerebral vasodilation in awake sheep. J Appl Physiol. 1992;73:2226–32. doi: 10.1152/jappl.1992.73.6.2226. [DOI] [PubMed] [Google Scholar]
- [477].Twort CH, van Breemen C. Cyclic guanosine monophosphate-enhanced sequestration of Ca2+ by sarcoplasmic reticulum in vascular smooth muscle. Circ Res. 1988;62:961–4. doi: 10.1161/01.res.62.5.961. [DOI] [PubMed] [Google Scholar]
- [478].Chen XL, Rembold CM. Nitroglycerin relaxes rat tail artery primarily by lowering Ca2+sensitivity and partially by repolarization. Am J Physiol. 1996;271:H962–8. doi: 10.1152/ajpheart.1996.271.3.H962. [DOI] [PubMed] [Google Scholar]
- [479].Colyer J. Phosphorylation states of phospholamban. Ann N Y Acad Sci. 1998;853:79–91. doi: 10.1111/j.1749-6632.1998.tb08258.x. [DOI] [PubMed] [Google Scholar]
- [480].Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113:1671–6. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- [481].Kawada T, Toyosato A, Islam MO, Yoshida Y, Imai S. cGMP-kinase mediates cGMP- and cAMP-induced Ca2+ desensitization of skinned rat artery. Eur J Pharmacol. 1997;323:75–82. doi: 10.1016/s0014-2999(97)00028-9. [DOI] [PubMed] [Google Scholar]
- [482].Pfitzer G, Ruegg JC, Flockerzi V, Hofmann F. cGMP-dependent protein kinase decreases calcium sensitivity of skinned cardiac fibers. FEBS Lett. 1982;149:171–5. doi: 10.1016/0014-5793(82)81095-8. [DOI] [PubMed] [Google Scholar]
- [483].Rokolya A, Ahn HY, Moreland S, van Breemen C, Moreland RS. A hypothesis for the mechanism of receptor and G-protein-dependent enhancement of vascular smooth muscle myofilament Ca2+ sensitivity. Can J Physiol Pharmacol. 1994;72:1420–6. doi: 10.1139/y94-205. [DOI] [PubMed] [Google Scholar]
- [484].Savineau JP, Marthan R. Modulation of the calcium sensitivity of the smooth muscle contractile apparatus: molecular mechanisms, pharmacological and pathophysiological implications. Fundam Clin Pharmacol. 1997;11:289–99. doi: 10.1111/j.1472-8206.1997.tb00841.x. [DOI] [PubMed] [Google Scholar]
- [485].George MJ, Shibata EF. Regulation of calcium-activated potassium channels by S-nitrosothiol compounds and cyclic guanosine mono-phosphate in rabbit coronary artery myocytes. J Invest Med. 1995;43:451–8. [PubMed] [Google Scholar]
- [486].Zhou XB, Arntz C, Kamm S, et al. A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J Biol Chem. 2001;276:43239–45. doi: 10.1074/jbc.M104202200. [DOI] [PubMed] [Google Scholar]
- [487].Wu X, Somlyo AV, Somlyo AP. Cyclic GMP-dependent stimulation reverses G-protein-coupled inhibition of smooth muscle myosin light chain phosphate. Biochem Biophys Res Commun. 1996;220:658–63. doi: 10.1006/bbrc.1996.0460. [DOI] [PubMed] [Google Scholar]
- [488].Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol. 1991;260:L280–5. doi: 10.1152/ajplung.1991.260.4.L280. [DOI] [PubMed] [Google Scholar]
- [489].Butt E, Notle C, Schulz S, et al. Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochem Pharmacol. 1992;43:2591–600. doi: 10.1016/0006-2952(92)90148-c. [DOI] [PubMed] [Google Scholar]
- [490].Kumar R, Joyner RW, Komalavilas P, Lincoln TM. Analysis of expression of cGMP-dependent protein kinase in rabbit heart cells. J Pharmacol Exp Ther. 1999;291:967–75. [PubMed] [Google Scholar]
- [491].Moreland RS, Cilea J, Moreland S. Calcium dependent regulation of vascular smooth muscle contraction. Adv Exp Med Biol. 1991;308:81–94. doi: 10.1007/978-1-4684-6015-5_7. [DOI] [PubMed] [Google Scholar]
- [492].Somlyo AV, Somlyo AP. Intracellular signaling in vascular smooth muscle. Adv Exp Med Biol. 1993;346:31–8. doi: 10.1007/978-1-4615-2946-0_4. [DOI] [PubMed] [Google Scholar]
- [493].Bradley AB, Morgan KG. Alterations in cytoplasmic calcium sensitivity during porcine coronary artery contractions as detected by aequorin. J Physiol. 1987;385:437–48. doi: 10.1113/jphysiol.1987.sp016500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [494].Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by rho and rho-associated kinase. Science. 1996;473:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- [495].Seager JM, Murphy TV, Garland CJ. Importance of inositol (1,4,5)-trisphosphate, intracellular Ca2+ release and myofilament Ca2+ sensitization in 5-hydroxytryptamine-evoked contraction of rabbit mesenteric artery. Br J Pharmacol. 1994;111:525–32. doi: 10.1111/j.1476-5381.1994.tb14769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [496].Hartshorne DJ, Ito M, Erdödi F. Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem. 2004;279:37211–4. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- [497].Carpenter CL. Actin cytoskeleton and cell signaling. Crit Care Med. 2000;28:N94–9. doi: 10.1097/00003246-200004001-00011. [DOI] [PubMed] [Google Scholar]
- [498].Charles SM, Zhang L, Longo LD, Buchholz JN, Pearce WJ. Postnatal maturation attenuates pressure-evoked myogenic tone and stretch-induced increases in Ca2+ in rat cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2007;293:R737–44. doi: 10.1152/ajpregu.00869.2006. [DOI] [PubMed] [Google Scholar]
- [499].Dunn WR, Wellman GC, Bevan JA. Enhanced resistance artery sensitivity to agonists under isobaric compared with isometric conditions. Am J Physiol. 1994;266:H147–55. doi: 10.1152/ajpheart.1994.266.1.H147. [DOI] [PubMed] [Google Scholar]
- [500].VanBavel E, Mulvany MJ. Role of wall tension in the vasoconstrictor response of cannulated rat mesenteric small arteries. J Physiol. 1994;477:103–15. doi: 10.1113/jphysiol.1994.sp020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [501].Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–83. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- [502].Nishizuka Y. The family of protein kinase C for signal transduction. JAMA. 1989;262:1826–33. [PubMed] [Google Scholar]
- [503].Horowitz A, Clément-Chomienne O, Walsh MP, Morgan KG. ε-Isoenzyme of protein kinase C induces a Ca2+-independent contraction in vascular smooth muscle. Am J Physiol Cell Physiol. 1996;271:C589–94. doi: 10.1152/ajpcell.1996.271.2.C589. [DOI] [PubMed] [Google Scholar]
- [504].Singer HA. Protein kinase C. In: Bárány M, editor. Biochemistry of smooth muscle contraction. Academic Press; San Diego, CA: 1996. pp. 155–65. [Google Scholar]
- [505].Walsh MP, Horowitz A, Clement-Chomienne O, Andrea JE, Allen BG, Morgan KG. Protein kinase C mediation of Ca2+-independent contractions of vascular smooth muscle. Biochem Cell Biol. 1996;74:485–502. doi: 10.1139/o96-053. [DOI] [PubMed] [Google Scholar]
- [506].Haller H, Smallwood JI, Rasmussen H. Protein kinase C translocation in intact vascular smooth muscle strips. Biochem J. 1990;270:375–81. doi: 10.1042/bj2700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [507].Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca2+-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104:265–86. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [508].Bazan E, Campbell AK, Rapoport RM. Effects of protein kinase C activation on norepinephrine-induced phosphatidylinositide hydrolysis in intact rat aorta. Eur J Pharmacol. 1993;245:173–7. doi: 10.1016/0922-4106(93)90125-s. [DOI] [PubMed] [Google Scholar]
- [509].Minneman KP. α1-Adrenergic receptor subtypes, inositol phosphates, and sources of cell Ca2+ Pharmacol Rev. 1988;40:87–119. [PubMed] [Google Scholar]
- [510].Danthuluri NR, Deth RC. Phorbol ester-induced contraction of arterial smooth muscle and inhibition of α-adrenergic response. Biochem Biophys Res Commun. 1984;125:1103–9. doi: 10.1016/0006-291x(84)91397-4. [DOI] [PubMed] [Google Scholar]
- [511].Osol G, Laher I, Cipolla M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ Res. 1991;68:359–67. doi: 10.1161/01.res.68.2.359. [DOI] [PubMed] [Google Scholar]
- [512].Rasmussen H, Forder J, Kojima I, Scriabine A. TPA-induced contraction of isolated rabbit vascular smooth muscle. Biochem Biophys Res Commun. 1984;122:776–84. doi: 10.1016/s0006-291x(84)80101-1. [DOI] [PubMed] [Google Scholar]
- [513].Nishimura J, Moreland S, Morland RS, van Breemen C. Regulation of the Ca2+ force relationship in permeabilized arterial smooth muscle. Adv Exp Med Biol. 1991;304:111–127. doi: 10.1007/978-1-4684-6003-2_11. [DOI] [PubMed] [Google Scholar]
- [514].Khalil RA, Morgan KG. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. Am J Physiol. 1993;265:C406–11. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- [515].Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–92. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [516].Boulton T, Cobb M. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2:357–71. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [517].Adam LP, Franklin MT, Raff GJ, Hathaway DR. Activation of mitogen-activated protein kinase in porcine carotid arteries. Circ Res. 1995;76:183–90. doi: 10.1161/01.res.76.2.183. [DOI] [PubMed] [Google Scholar]
- [518].Cobb MH, Robbins DJ, Boulton TG. ERKs, extracellular signal-regulated MAP-2 kinases. Curr Opin Cell Biol. 1991;3:1025–32. doi: 10.1016/0955-0674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- [519].Dessy C, Kim I, Sougnes CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoreceptor stimulation. Am J Physiol Heart Circ Physiol. 1998;275:C1081–6. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- [520].Alessandrini A, Crews CM, Erikson RL. Phorbol ester stimulates a protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product. Proc Natl Acad Sci USA. 1992;89:8200–4. doi: 10.1073/pnas.89.17.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [521].Childs TJ, Watson MH, Sanghera JS, Campbell DL, Pelech SL, Mak AS. Phosphorylation of smooth muscle caldesmon by mitogen-activated protein (MAP) kinase and expression of MAP kinases in differentiated smooth muscle. J Biol Chem. 1992;267:22853–9. [PubMed] [Google Scholar]
- [522].Sugden PH, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal. 1997;9:337–51. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- [523].Epstein AM, Throckmorton D, Brophy CM. Mitogen-activated protein kinase activation: an alternate signaling pathway for sustained vascular smooth muscle contraction. J Vasc Surg. 1997;26:327–32. doi: 10.1016/s0741-5214(97)70196-4. [DOI] [PubMed] [Google Scholar]
- [524].Katoch SS, Morland RS. Agonist and membrane depolarization induced activation of MAP kinase in the swine carotid artery. Am J Physiol. 1995;269:H222–9. doi: 10.1152/ajpheart.1995.269.1.H222. [DOI] [PubMed] [Google Scholar]
- [525].Lagaud GJL, Lam E, Lui A, van Breemen C, Laher I. Nonspecific inhibition of myogenic tone by PD98059, a MEK1 inhibitor, in rat middle cerebral arteries. Biochem Biophys Res Comm. 1999;257:523–7. doi: 10.1006/bbrc.1999.0350. [DOI] [PubMed] [Google Scholar]
- [526].Ratz PH. Regulation of ERK phosphorylation in differentiated arterial muscle of rabbits. Am J Physiol Heart Circ Physiol. 2001;281:H114–23. doi: 10.1152/ajpheart.2001.281.1.H114. [DOI] [PubMed] [Google Scholar]
- [527].Touyz RM, He G, Deng L-Y, Schiffrin EL. Role of extracellular signal-regulated kinases in angiotensin II-stimulated contraction of smooth muscle cells from human resistance arteries. Circulation. 1999;99:392–9. doi: 10.1161/01.cir.99.3.392. [DOI] [PubMed] [Google Scholar]
- [528].Watts SW. Serotonin activates the mitogen-activated protein kinase pathway in vascular smooth muscle: use of the mitogen-activated protein kinase kinase inhibitor PD-098059. J Pharmacol Exp Ther. 1996;279:1541–50. [PubMed] [Google Scholar]
- [529].Xiao D, Zhang L. ERK MAP kinases regulate smooth muscle contraction in ovine uterine artery: effect of pregnancy. Am J Physiol Heart Circ Physiol. 2002;282:H292–300. doi: 10.1152/ajpheart.2002.282.1.H292. [DOI] [PubMed] [Google Scholar]
- [530].Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164:623–35. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- [531].Jiang MJ, Morgan KG. Intracellular calcium levels in phorbol ester-induced contractions of vascular muscle. Am J Physiol. 1987;253:H1365–71. doi: 10.1152/ajpheart.1987.253.6.H1365. [DOI] [PubMed] [Google Scholar]
- [532].Walsh MP, Andrea JE, Allen BG, Clement-Chomienne O, Collins EM, Morgan KG. Smooth muscle protein kinase C. Can J Physiol Pharmacol. 1994;72:1392–9. doi: 10.1139/y94-201. [DOI] [PubMed] [Google Scholar]
- [533].Rembold CM, Murphy RA. [Ca2+]-dependent myosin phosphorylation in phorbol diester stimulated smooth muscle contraction. Am J Physiol. 1988;255:C719–23. doi: 10.1152/ajpcell.1988.255.6.C719. [DOI] [PubMed] [Google Scholar]
- [534].Gokina NI, Osol G. Temperature and protein kinase C modulate myofilament Ca2+ sensitivity in pressurized rat cerebral arteries. Am J Physiol. 1998;274:H1920–7. doi: 10.1152/ajpheart.1998.274.6.H1920. [DOI] [PubMed] [Google Scholar]
- [535].Löhn M, Kämpf D, Gui-Xuan C, Haller H, Luft FC, Gollasch M. Regulation of arterial tone by smooth muscle myosin type II. Am J Physiol Cell Physiol. 2002;283:C1383–9. doi: 10.1152/ajpcell.01369.2000. [DOI] [PubMed] [Google Scholar]
- [536].Takahashi E, Berk BC. MAP kinases and vascular smooth muscle function. Acta Physiol Scand. 1998;164:611–21. doi: 10.1111/j.1365-201x.1998.tb10705.x. [DOI] [PubMed] [Google Scholar]
- [537].Gorenne I, Su X, Moreland RS. Inhibition of p42 and p44 MAP kinase does not alter smooth muscle contraction in swine carotid artery. Am J Physiol. 1998;275:H131–8. doi: 10.1152/ajpheart.1998.275.1.H131. [DOI] [PubMed] [Google Scholar]
- [538].Nixon GF, Iizuka K, Haystead CM, Haystead TA, Somlyo AP, Somlyo AV. Phosphorylation of caldesmon by mitogen-activated protein kinase with no effect on Ca2+ sensitivity in rabbit smooth muscle. J Physiol. 1995;48:283–9. doi: 10.1113/jphysiol.1995.sp020879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [539].Philip S, Armstead WM. Differential role of PTK, ERK and p38 MAPK in superoxide impairment of NMDA cerebrovasodilation. Brain Res. 2003;979:98–103. doi: 10.1016/s0006-8993(03)02879-8. [DOI] [PubMed] [Google Scholar]
- [540].Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- [541].Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rhokinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522:177–85. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [542].McDaniel NL, Rembold CM, Murphy RA. Cyclic nucleotide dependent relaxation in vascular smooth muscle. Can J Physiol Pharmacol. 1994;72:1380–5. doi: 10.1139/y94-199. [DOI] [PubMed] [Google Scholar]
- [543].Murphy RA, Walker JS. Inhibitory mechanisms for cross-bridge cycling: the nitric oxide-cGMP signal transduction pathway in smooth muscle relaxation. Acta Physiol Scand. 1998;164:373–80. doi: 10.1046/j.1365-201X.1998.00434.x. [DOI] [PubMed] [Google Scholar]
- [544].Crichton CA, Templeton AG, Smith GL. Effect of altered bathing pH on calcium activated force in alpha toxin permeabilised rat portal vein and human umbilical artery. Cardiovasc Res. 1994;28:1378–84. doi: 10.1093/cvr/28.9.1378. [DOI] [PubMed] [Google Scholar]
- [545].Li Y, Je HD, Malek S, Morgan KG. ERK1/2-mediated phosphorylation of myometrial caldesmon during pregnancy and labor. Am J Physiol Regul Integr Comp Physiol. 2003;284:R192–9. doi: 10.1152/ajpregu.00290.2002. [DOI] [PubMed] [Google Scholar]
- [546].Xiao D, Zhang L. Calcium homeostasis and contraction of the uterine artery: effect of pregnancy and chronic hypoxia. Biol Re-prod. 2004;70:1171–7. doi: 10.1095/biolreprod.103.024943. [DOI] [PubMed] [Google Scholar]
- [547].Ishitani T, Hattori Y, Sakuraya F, et al. Effects of Ca2+ sensitizers on contraction, [Ca2+]i transient and myofilament Ca2+ sensitivity in diabetic rat myocardium: potential usefulness as inotropic agents. J Pharmacol Exp Ther. 2001;298:613–22. [PubMed] [Google Scholar]
- [548].Xavier FE, Davel AP, Rossoni LV, Vassallo DV. Time-dependent hyperreactivity to phenylephrine in aorta from untreated diabetic rats: role of prostanoids and calcium mobilization. Vascul Pharmacol. 2003;40:67–76. doi: 10.1016/s1537-1891(02)00315-4. [DOI] [PubMed] [Google Scholar]
- [549].Shaw LM, Ohanian J, Heagerty AM. Calcium sensitivity and agonist-induced calcium sensitization in small arteries of young and adult spontaneously hypertensive rats. Hypertension. 1997;30:442–8. doi: 10.1161/01.hyp.30.3.442. [DOI] [PubMed] [Google Scholar]
- [550].Ungvari Z, Koller A. Endothelin and prostaglandin H2/thromboxane A2 enhance myogenic constriction in hypertension by increasing Ca2+ sensitivity of arteriolar smooth muscle. Hypertension. 2000;36:856–61. doi: 10.1161/01.hyp.36.5.856. [DOI] [PubMed] [Google Scholar]
- [551].Charles SM, Zhang L, Cipolla MJ, Buchholz JN, Pearce WJ. Roles of cytosolic Ca2+ concentration and myofilament Ca2+ sensitization in age-dependent cerebrovascular myogenic tone. Am J Physiol Heart Circ Physiol. 2010;299:H1034–44. doi: 10.1152/ajpheart.00214.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [552].LeClair R, Lindner V. The role of collagen triple helix repeat containing 1 in injured arteries, collagen expression, and transforming growth factor beta signaling. Trends Cardiovasc Med. 2007;17:202–5. doi: 10.1016/j.tcm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- [553].Tang L, Dai DL, Su M, Martinka M, Li G, Zhou Y. Aberrant expression of collagen triple helix repeat containing 1 in human solid cancers. Clin Cancer Res. 2006;12:3716–22. doi: 10.1158/1078-0432.CCR-06-0030. [DOI] [PubMed] [Google Scholar]
- [554].Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–73. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- [555].Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006;12:6869–75. doi: 10.1158/1078-0432.CCR-06-1405. [DOI] [PubMed] [Google Scholar]
- [556].McClellan WJ, Dai Y, Abad-Zapatero C, et al. Discovery of potent and selective thienopyrimidine inhibitors of Aurora kinases. Bioorg Med Chem Lett. 2011;21:5620–4. doi: 10.1016/j.bmcl.2011.06.041. [DOI] [PubMed] [Google Scholar]
- [557].Skibbens RV, Hieter P. Kinetochores and the checkpoint mechanism that monitors for defects in the chromosome segregation machinery. Annu Rev Genet. 1998;32:307–37. doi: 10.1146/annurev.genet.32.1.307. [DOI] [PubMed] [Google Scholar]
- [558].van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Over-expression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188:83–100. doi: 10.1083/jcb.200906147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [559].Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–68. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- [560].Watanabe Y, Heistad DD. Targeting cerebral arteries for gene therapy. Exp Physiol. 2005;90:327–31. doi: 10.1113/expphysiol.2004.028498. [DOI] [PubMed] [Google Scholar]
- [561].Villar J, Smeriglio V, Martorell R, Brown CH, Klein RE. Heterogeneous growth and mental development of intrauterine growth-retarded infants during the first 3 years of life. Pediatrics. 1984;74:783–91. [PubMed] [Google Scholar]
- [562].Rurak DW. Plasma vasopressin levels during hypoxaemia and the cardiovascular effects of exogenous vasopressin in foetal and adult sheep. J Physiol. 1978;277:341–57. doi: 10.1113/jphysiol.1978.sp012275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [563].Cohen WR, Piasecki GJ, Cohn HE, Young JB, Jackson BT. Adrenal secretion of catecholamines during hypoxemia in fetal lambs. Endocrinology. 1984;114:383–90. doi: 10.1210/endo-114-2-383. [DOI] [PubMed] [Google Scholar]
- [564].Jensen A, Roman C, Rudolph AM. Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. J Dev Physiol. 1991;15:309–23. [PubMed] [Google Scholar]
- [565].Peeters LL, Sheldon RE, Jones MD, Jr, Makowski EL, Meschia G. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol. 1979;135:637–46. doi: 10.1016/s0002-9378(16)32989-1. [DOI] [PubMed] [Google Scholar]
- [566].Richardson B, Korkola S, Asano H, Challis J, Polk D, Fraser M. Regional blood flow and the endocrine response to sustained hypoxemia in the preterm ovine fetus. Pediatr Res. 1996;40:337–43. doi: 10.1203/00006450-199608000-00024. [DOI] [PubMed] [Google Scholar]
- [567].Koos BJ, Chau A, Ogunyemi D. Adenosine mediates metabolic and cardiovascular responses to hypoxia in fetal sheep. J Physiol. 1995;488:761–6. doi: 10.1113/jphysiol.1995.sp021007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [568].Iwamoto HS, Kaufman T, Keil LC, Rudolph AM. Responses to acute hypoxemia in fetal sheep at 0.6–0.7 gestation. Am J Physiol. 1989;256:H613–20. doi: 10.1152/ajpheart.1989.256.3.H613. [DOI] [PubMed] [Google Scholar]
- [569].Mulder AL, van Golde JC, Prinzen FW, Blanco CE. Cardiac output distribution in response to hypoxia in the chick embryo in the second half of the incubation time. J Physiol. 1998;508:281–7. doi: 10.1111/j.1469-7793.1998.281br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [570].Giussani DA, Riquelme RA, Moraga FA, et al. Chemoreflex and endocrine components of cardiovascular responses to acute hypoxemia in the llama fetus. Am J Physiol. 1996;271:R73–83. doi: 10.1152/ajpregu.1996.271.1.R73. [DOI] [PubMed] [Google Scholar]
- [571].Giussani DA, Riquelme RA, Sanhueza EM, Hanson MA, Blanco CE, Llanos AJ. Adrenergic and vasopressinergic contributions to the cardiovascular response to acute hypoxaemia in the llama fetus. J Physiol. 1999;515:233–41. doi: 10.1111/j.1469-7793.1999.233ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [572].Jackson BT, Piasecki GJ, Novy MJ. Fetal responses to altered maternal oxygenation in rhesus monkey. Am J Physiol. 1987;252:R94–101. doi: 10.1152/ajpregu.1987.252.1.R94. [DOI] [PubMed] [Google Scholar]
- [573].Fumia FD, Edelstone DI, Holzman IR. Blood flow and oxygen delivery to fetal organs as functions of fetal hematocrit. Am J Obstet Gynecol. 1984;150:274–82. doi: 10.1016/s0002-9378(84)90365-x. [DOI] [PubMed] [Google Scholar]
- [574].Fletcher AJW, Gardner DS, Edwards CM, Fowden AL, Giussani DA. Development of the ovine fetal cardiovascular defense to hypoxemia towards full term. Am J Physiol Heart Circ Physiol. 2006;291:H3023–34. doi: 10.1152/ajpheart.00504.2006. [DOI] [PubMed] [Google Scholar]
- [575].Giussani DA, Spencer JA, Hanson MA. Fetal cardiovascular reflex responses to hypoxaemia. Fetal Matern Med Rev. 1994;6:17–37. [Google Scholar]
- [576].Llanos AJ, Riquelme RA, Sanhueza EM, et al. The fetal llama versus the fetal sheep: different strategies to withstand hypoxia. High Alt Med Biol. 2003;4:193–202. doi: 10.1089/152702903322022794. [DOI] [PubMed] [Google Scholar]
- [577].Liggins GC. Premature delivery of foetal lambs infused with glucocorticoids. J Endocrinol. 1969;45:515–23. doi: 10.1677/joe.0.0450515. [DOI] [PubMed] [Google Scholar]
- [578].Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–25. [PubMed] [Google Scholar]
- [579].Unno N, Wong CH, Jenkins SL, et al. Blood pressure and heart rate in the ovine fetus: ontogenic changes and effects of fetal adrenalectomy. Am J Physiol. 1999;276:H248–56. doi: 10.1152/ajpheart.1999.276.1.H248. [DOI] [PubMed] [Google Scholar]
- [580].Derks JB, Giussani DA, Jenkins SL, et al. A comparative study of cardiovascular, endocrine and behavioural effects of betamethasone and dexamethasone administration to fetal sheep. J Physiol. 1997;499:217–26. doi: 10.1113/jphysiol.1997.sp021922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [581].Anwar MA, Schwab M, Poston L, Nathanielsz PW. Betamethasone-mediated vascular dysfunction and changes in hematological profile in the ovine fetus. Am J Physiol. 1999;276:H1137–43. doi: 10.1152/ajpheart.1999.276.4.H1137. [DOI] [PubMed] [Google Scholar]
- [582].Molnar J, Nijland MJ, Howe DC, Nathanielsz PW. Evidence for microvascular dysfunction after prenatal dexamethasone at 0.7, 0.75, and 0.8 gestation in sheep. Am J Physiol Regul Integr Comp Physiol. 2002;283:R561–7. doi: 10.1152/ajpregu.00031.2002. [DOI] [PubMed] [Google Scholar]
- [583].Schwab M, Roedel M, Anwar MA, et al. Effects of betamethasone administration to the fetal sheep in late gestation on fetal cerebral blood flow. J Physiol. 2000;528:619–32. doi: 10.1111/j.1469-7793.2000.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [584].Molnar J, Howe DC, Nijland MJ, Nathanielsz PW. Prenatal dexamethasone leads to both endothelial dysfunction and vasodilatory compensation in sheep. J Physiol. 2003;547:61–6. doi: 10.1113/jphysiol.2002.032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [585].Fletcher AJW, McGarrigle HHG, Edwards CMB, Fowden AL, Giussani DA. Effects of low dose dexamethasone treatment on basal cardiovascular and endocrine function in fetal sheep during late gestation. J Physiol. 2002;545:649–60. doi: 10.1113/jphysiol.2001.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [586].Löhle M, Müller T, Wicher C, et al. Betamethasone effects on fetal sheep cerebral blood flow are not dependent on maturation of cerebrovascular system and pituitary-adrenal axis. J Physiol. 2005;564:575–88. doi: 10.1113/jphysiol.2004.077537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [587].Miller SL, Supramaniam VG, Jenkin G, Walker DW, Wallace EM. Cardiovascular responses to maternal betamethasone administration in the intrauterine growth-restricted ovine fetus. Am J Obstet Gynecol. 2009;201:613.e1–8. doi: 10.1016/j.ajog.2009.07.028. [DOI] [PubMed] [Google Scholar]
- [588].Long JB, Holaday JW. Blood-brain barrier: endogenous modulation by adrenal-cortical function. Science. 1985;227:1580–3. doi: 10.1126/science.3975627. [DOI] [PubMed] [Google Scholar]
- [589].Ment LR, Oh W, Ehrenkranz RA, Philip AG, Duncan CC, Makuch RW. Antenatal steroids, delivery mode, and intraventricular hemorrhage in preterm infants. Am J Obstet Gynecol. 1995;172:795–800. doi: 10.1016/0002-9378(95)90001-2. [DOI] [PubMed] [Google Scholar]
- [590].Shankaran S, Bauer CR, Bain R, Wright LL, Zachary J. Relationship between antenatal steroid administration and grades III and IV intracranial hemorrhage in low birth weight infants. The NICHD Neonatal Research Network. Am J Obstet Gynecol. 1995;173:305–12. doi: 10.1016/0002-9378(95)90219-8. [DOI] [PubMed] [Google Scholar]
- [591].Giussani DA, Gardner DS, Cox DT, Fletcher AJW. Purinergic contribution to circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;280:R678–85. doi: 10.1152/ajpregu.2001.280.3.R678. [DOI] [PubMed] [Google Scholar]
- [592].Jellyman JK, Gardner DS, McGarrigle HH, Fowden AL, Giussani DA. Antenatal glucocorticoid therapy increases glucose delivery to cerebral circulations during acute hypoxemia in fetal sheep during late gestation. Am J Obstet Gynecol. 2009;201:82.e1–8. doi: 10.1016/j.ajog.2009.01.012. [DOI] [PubMed] [Google Scholar]
- [593].Gudmundsson S, Marsál K. Blood velocity waveforms in the fetal aorta and umbilical artery as predictors of fetal outcome: a comparison. Am J Perinatol. 1991;8:1–6. doi: 10.1055/s-2007-999326. [DOI] [PubMed] [Google Scholar]
- [594].Wladimiroff JW, vd Wijngaard JA, Degani S, Noordam MJ, van Eyck J, Tonge HM. Cerebral and umbilical arterial blood flow velocity waveforms in normal and growth-retarded pregnancies. Obstet Gynecol. 1987;69:705–9. [PubMed] [Google Scholar]
- [595].Mari G, Deter Rl. Middle cerebral artery flow velocity waveforms in normal and small-for-gestational-age fetuses. Am J Obstet Gynecol. 1992;166:1262–70. doi: 10.1016/s0002-9378(11)90620-6. [DOI] [PubMed] [Google Scholar]
- [596].Wladimiroff JW, Tonge HM, Stewart PA. Doppler ultrasound assessment of cerebral blood flow in the human fetus. Br J Obstet Gynaecol. 1986;93:471–5. [PubMed] [Google Scholar]
- [597].Woo JS, Liang ST, Lo RL, Chan FY. Middle cerebral artery Doppler flow velocity waveforms. Obstet Gynecol. 1987;70:613–6. [PubMed] [Google Scholar]
- [598].Dubiel M, Gudmundsson S, Gunnarsson G, Marsál K. Middle cerebral artery velocimetry as a predictor of hypoxemia in fetuses with increased resistance to blood flow in the umbilical artery. Early Hum Dev. 1997;47:177–84. doi: 10.1016/s0378-3782(96)01777-x. [DOI] [PubMed] [Google Scholar]
- [599].Figueras F, Cruz-Martinez R, Sanz-Cortes M, et al. Neurobehavioral outcomes in preterm, growth-restricted infants with and without prenatal advanced signs of brain-sparing. Ultrasound Obstet Gynecol. 2011;38:288–94. doi: 10.1002/uog.9041. [DOI] [PubMed] [Google Scholar]
- [600].Baschat AA. Integrated fetal testing in growth restriction: combining multivessel Doppler and biophysical parameters. Ultrasound Obstet Gynecol. 2003;21:1–8. doi: 10.1002/uog.21. [DOI] [PubMed] [Google Scholar]
- [601].Baschat AA. Neurodevelopment following fetal growth restriction and its relationship with antepartum parameters of placental dys-function. Ultrasound Obstet Gynecol. 2011;37:501–14. doi: 10.1002/uog.9008. [DOI] [PubMed] [Google Scholar]
- [602].Bhide A. Fetal growth restriction and developmental delay: current understanding and future possibilities. Ultrasound Obstet Gynecol. 2011;38:243–5. doi: 10.1002/uog.10055. [DOI] [PubMed] [Google Scholar]
- [603].Baumbach GL, Heistad DD. Effects of sympathetic stimulation and changes in arterial pressure on segmental resistance of cerebral vessels in rabbits and cats. Circ Res. 1983;52:527–33. doi: 10.1161/01.res.52.5.527. [DOI] [PubMed] [Google Scholar]