

Abstract
DNA methylation is implicated in mammalian brain development and plasticity underlying learning and memory. We report the genome-wide composition, patterning, cell specificity, and dynamics of DNA methylation at single-base resolution in human and mouse frontal cortex throughout their lifespan. Widespread methylome reconfiguration occurs during fetal to young adult development, coincident with synaptogenesis. During this period, highly conserved non-CG methylation (mCH) accumulates in neurons, but not glia, to become the dominant form of methylation in the human neuronal genome. Moreover, we found an mCH signature that identifies genes escaping X-chromosome inactivation. Last, whole-genome single-base resolution 5-hydroxymethylcytosine (hmC) maps revealed that hmC marks fetal brain cell genomes at putative regulatory regions that are CG-demethylated and activated in the adult brain and that CG demethylation at these hmC-poised loci depends on Tet2 activity.
Dynamic epigenetic changes have been observed during brain development, maturation, and learning (1–6). DNA methylation (mC) is a stable covalent modification that persists in postmitotic cells throughout their lifetime, defining their cellular identity. However, the methylation status at each of the ~1 billion cytosines in the genome is potentially an information-rich and flexible substrate for epigenetic modification that can be altered by cellular activity (7, 8). Changes in DNA methylation were implicated in learning and memory (9, 10), as well as in age-related cognitive decline (11). Mice with a postnatal deletion of DNA methyltransferases Dnmt1 and Dnmt3a in forebrain excitatory neurons, or with a global deletion of methyl-CpG-binding protein 2 (MeCP2), show abnormal long-term neural plasticity and cognitive deficits (2, 12).
DNA methylation composition and dynamics in the mammalian brain are highly distinct. A modification of mC catalyzed by the Tet family of mC hydroxylase proteins, 5-hydroxymethylcytosine (hmC), accumulates in the adult brain (13–15) along with its more highly oxidized derivatives 5-formylcytosine and 5-carboxylcytosine. These modifications of mC were implicated as intermediates in an active DNA demethylation pathway (16–19). In addition, methylation in the non-CG context (mCH, where H = A, C, or T) is also present in the adult mouse and human brains (20, 21) but is rare or absent in other differentiated cell types (22, 23). Little is known about cell type–specific patterning of DNA methylation and its dynamics during mammalian brain development. Here, we provide integrated empirical data and analysis of DNA methylation at single-base resolution, across entire genomes, with cell-type and developmental specificity. These results extend our knowledge of the unique role of DNA methylation in brain development and function and offer a new framework for testing the role of the epigenome in healthy function and in pathological disruptions of neural circuits.
Accumulation of Non-CG DNA Methylation During Brain Development
To identify the composition and dynamics of transcription and methylation during mammalian brain development, we performed transcriptome profiling (mRNA-Seq) and whole-genome bisulfite sequencing [MethylC-Seq (24)] to comprehensively identify sites of cytosine DNA methylation (mC and hmC) and mRNA abundance at single-base resolution throughout the genomes of mouse and human frontal cortex (table S1). DNA methylation in embryonic stem (ES) cells occurs in both the CG (mCG) and non-CG (mCH) contexts, but mCH is largely lost upon cell differentiation (22, 23, 25, 26). We found that although mCH levels are negligible in fetal cortex, abundant mCH occurs in adult frontal cortex (Fig. 1A). mCH has previously been identified throughout the genome of the adult mouse brain (20) and at several hundred genomic positions in the human adult brain (21). Supporting previous studies, we found that mammalian brain mCH is typically depleted in expressed genes, with genic mCH level inversely proportional to the abundance of the associated transcript (Fig. 1, A and B) (20). This pattern is the opposite of that observed in ES cells (22) and suggests that genic mCH in the brain may inhibit transcription. The absence of mCH in fetal brain suggests that this signature for gene repression is added to the genome at a later developmental stage.
Fig. 1. Methylcytosine in mammalian frontal cortex is developmentally dynamic and abundant in CG and CH contexts.

(A) Browser representation of mC and mRNA transcript abundance in human and mouse frontal cortex and human ES cells. Chr2, chromosome 2. (B) mCH/CH within gene bodies exhibits opposite correlation with gene expression in ES cells (ESC) and brain. Contours show data point density, and red line shows smoothed mCH/CH as a function of mRNA. a.u., arbitrary units. (C) Synaptic density (for mouse, per 100 μm2; for human, per 100 μm3) and mC level in CG and CH contexts through development in mouse and human frontal cortex. †Synaptic density quantitation from De Felipe et al. (27) and Huttenlocher and Dabholkar (28). (D) DNA methyltransferase mRNA and protein abundance (mean ± SEM) in mouse frontal cortex through development. FPKM, fragments per kilobase of exon per million fragments mapped. (E) Fraction of cytosine base calls with each modification in fetal and adult mouse frontal cortex. (F) Cortex mC level in CG and CH contexts throughout mouse and human chromosome 12 in 100-kb bins smoothed with ~1-Mb resolution. CEN, centrosome. (G) Transcript abundance, chromatin accessibility [8-week mouse cortex ChIP input and DNaseI hypersensitivity (HS) normalized read density], and mC levels in 5-kb bins at the mouse immunoglobulin VH locus. (H) Density (z) plot of 10-week mouse frontal cortex mCH level (x) versus 8-week mouse cortex ChIP-input normalized read density (y) for all 10-kb bins of the mouse genome.
We performed MethylC-Seq on mouse and human frontal cortex during early postnatal, juvenile, adolescent, and adult stages (Fig. 1C). CH methylation level, defined as the fraction of all base calls at CH genome reference positions that were methylated (denoted mCH/CH), accumulates in mouse and human brain during early postnatal development to a maximum of 1.3 to 1.5% genome-wide at the end of adolescence before diminishing slightly during aging. mCH increases most rapidly during the primary phase of synaptogenesis in the developing postnatal brain, from 2 to 4 weeks in mouse (27) and in the first 2 years in humans (28), followed by slower accumulation of mCH during later adolescence. mCH accumulation initially parallels the increase in synapse density within human middle frontal gyrus (synaptogenesis lasts from birth to 5 years), but it subsequently continues to increase during the period of adolescent synaptic pruning, which in humans occurs between 5 and 16 years of age (Fig. 1C). Notably, the accumulation of mCH in mice from 1 to 4 weeks after birth coincides with a transient increase in abundance of the de novo methyltransferase Dnmt3a mRNA and protein (Fig. 1D). Analysis of the context of mCH sites showed that it is mainly present in the CA context (fig. S1, A to F), as previously reported for mCH (20, 22, 23, 26).
Overall, genomes in the frontal cortex are highly methylated. Whereas CG partially methylated domains (PMDs) account for about a third of the genome of various differentiated human cells (22, 25), human brain genomes have negligible CG PMDs, resembling pluripotent cell methylomes (25) (fig. S1, G and H). Given the high spatial concordance of CG PMDs and nuclear lamina-associated domains reported previously (29), the paucity of CG PMDs in these brain methylomes could indicate that lamina-associated domains are altered or much less frequent in the brain.
The adult mammalian brain contains the highest levels of hmC that have been observed (15), accounting for about 40% of methylated CG sites in cerebellar Purkinje cells (30). hmC accumulates during early postnatal brain development in mice (31, 32), becoming enriched in highly expressed genes (33). Given the evidence that hmC can be an intermediate in an active DNA demethylation pathway (16, 17), high-resolution analysis of the genomic distribution of hmC is needed to understand its role in the control of DNA methylation dynamics through brain development. Standard bisulfite-sequencing data does not distinguish between methylated and hydroxymethylated sites, so methylcytosines identified by MethylC-Seq analysis represent the sum of these two contributions. Therefore, we used Tet-assisted bisulfite sequencing [TAB-Seq (34)], a base-resolution technique that distinguishes hmC from C and mC genome-wide, to profile hmC in mouse fetal and adult frontal cortex (Fig. 1A). Integration of the genome-wide profiles of mC and hmC enabled a detailed breakdown of the methylated subset of the genome at these distinct developmental stages (Fig. 1E). hmC constitutes 0.20% of total cytosine base calls in fetal cortex and increases to 0.87% in adult cortex. This modification appears to be restricted to the CG context, as also observed in human and mouse ES cells (34); after correction for false detection, we estimated that 0.017% of cytosine base calls were hmCH genome-wide (99% confidence interval: 0 to 0.059%), and significant hmCH was detected at few individual sites (fig. S2, A and B). The overwhelming presence of hmC in the CG context (99.98%) in mouse adult and fetal frontal cortex is consistent with recent findings in human ES cells, where 99.89% of hmC is in the CG context (34). hmC was present at many highly methylated CG sites (fig. S2C). Therefore, although only a small fraction of all cytosines throughout the genome are methylated (mCG = 2.9%, mCH = 1.3%, hmC = 0.87%), mCH and hmC constitute major, and nonoverlapping, components of the methylated fraction of the genome in adult frontal cortex (mCG = 57.2%, mCH = 25.6%, hmC = 17.2%). These data suggest that the steady-state population of hmC in the adult brain is not an intermediate stage in the demethylation of mCH. However, these steady-state measurements do not preclude the possibility that hmCH could be rapidly turned over after conversion from mCH, leading to negligible detected hmCH despite Tet-mediated demethylation at CH sites.
Protection of Inaccessible Genomic Regions from de Novo Methylation
mCH accumulates in parallel across most of the genome (Fig. 1F). However, we found numerous (36 in human, 34 in mouse) noncentromeric, megabase-sized regions that do not accumulate mCH. These regions, which we termed mCH deserts, are enriched for large gene clusters that encode proteins involved in immunity and receptors required for sensory neuron function (table S2). One mCH desert spans the immunoglobulin VH locus, which encodes variable domains of the immunoglobulin heavy chain that rearrange in B lymphocytes. The VH locus is transcriptionally quiescent in the frontal cortex of 10-week-old mice, and the chromatin state is highly inaccessible, as inferred from deoxyribonuclease I (DNaseI) hypersensitivity profiling (35) and chromatin immunoprecipitation (ChIP) input sequence read density data (36, 37) (Fig. 1G). In contrast, mCG is not depleted in mCH deserts.
Genome-wide detection of hmC by cytosine 5-methylenesulphonate immunoprecipitation (CMS-IP) (38, 39) revealed that hmC is also strongly depleted in the VH locus. mCH deserts are observed at other loci in the genome, including olfactory receptor gene clusters that form heterochromatic aggregates required for monoallelic receptor expression in olfactory sensory neurons (40, 41). Genome-wide comparison of mCH/CH with chromatin accessibility, as inferred from ChIP input read density (36, 37), for all 10-kb windows of the mouse genome revealed two discrete groups of genomic regions (Fig. 1H). Low-accessibility regions tend to contain minimal mCH, whereas more-accessible regions of the genome show a proportional relationship between genome accessibility and mCH levels. Thus, although mCG is unaffected in these regions, lower chromatin accessibility appears to be highly inhibitory to deposition of mCH and hmC, potentially via inaccessibility to de novo methyltransferases and Tet mC hydroxylases. Furthermore, this indicates that accumulation of mCH and hmC during mammalian brain development occurs via processes that are at least partly independent from methylation at CG dinucleotides.
Cell Type–Specific DNA Methylation Patterns in Neurons and Glia
The diversity of neuronal and glial cells in the frontal cortex raises the question of which features of DNA methylation are found in specific cell types. We isolated populations of nuclei by fluorescence-activated cell sorting that were highly enriched for neurons (NeuN+) or glia (NeuN−) from human and mouse adult frontal cortex tissue. An additional glial population was isolated from mice expressing enhanced green fluorescent protein (eGFP) under the S100b promoter. MethylC-Seq revealed differences in the composition and patterning of mCG and mCH in neurons and glia (Fig. 2A). Whereas differential mCG between neurons and glia was restricted to localized regions, neurons were globally enriched for mCH compared with glia. Indeed, we discovered that the level of mCH in glia is similar to that of fetal and early postnatal cortical tissue, whereas adult neurons have the greatest frequency of mCH that has been observed in mammalian cells. This indicates that the rapid developmental increase in mammalian brain mCH that coincides with the period of synaptogenesis is primarily due to mCH accumulation in neurons. Furthermore, our data show that in human neurons mCH is the dominant form of methylation in the genome: It is more abundant than mCG and occurs in 5% of CH and 10% of CA sites (Fig. 2B and fig. S1, A, B, and H). Of the total methylated fraction of adult human neuronal genomes, mCH accounts for ~53%, whereas mCG constitutes ~47%.
Fig. 2. mCH is positionally conserved and is the dominant form of DNA methylation in human neurons.
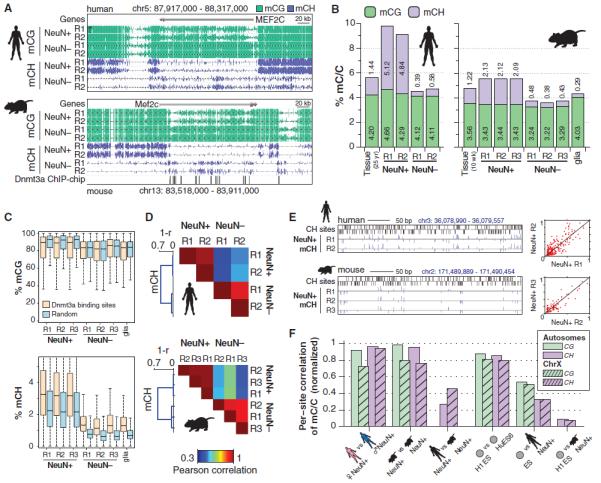
(A) Browser representation of mCG and mCH in NeuN+ and NeuN− cells. Human NeuN+/NeuN− samples: R1, 53-year-old female; R2, 55-year-old male. Mouse NeuN+/NeuN− samples: R1, 7-week males; R2, 6-week females; R3, 12-month females (not shown). (B) Percentage of methylated base calls in each sequence context throughout the genome. (C) Box and whisker plot of mCG and mCH level in neurons and glia at genomic regions bound by Dnmt3a versus a random set. Whiskers indicate 1.5 times the interquartile range. (D) mCH correlation between NeuN+ and NeuN− cells in mouse and human, measured in 10-kb bins. (E) Browser representation of mCH sites in neurons. Scatter plots (right) show consistent mCH/CH at all single sites in a 20-kb window overlapping the example region (left). (F) Correlation analysis of methylation state at single sites between neurons and ES cells in human and mouse. Correlation values are normalized by a simulation (62).
Although sparse in glia, mCH enrichment occurs within genes that are CH-hypomethylated in neurons, such as Mef2c (Fig. 2A), a transcriptional activator that plays critical roles in learning and memory, neuronal differentiation (42), synaptic plasticity (43), and regulation of synapse number and function (44). Genome-wide surveys identified 174 mouse genes in which glia were hypermethylated relative to neurons in the CH context (table S3). Unbiased gene ontology analysis revealed that these glial hyper-mCH genes are highly enriched for roles in neuronal and synaptic development and function (table S3). These genes also overlapped significantly with a set of 461 genes expressed at higher levels in neurons than in astrocytes (13-fold higher overlap than chance, P < 10−30, Fisher exact test, FET) (45) and 233 developmentally up-regulated genes (7.5-fold, P < 10−7, FET). These genes show hypomethylation of CG and CH in neurons and hypermethylation of CH in glia (fig. S3A), consistent with a potential role of mCH in transcriptional repression of neuronal genes in the glial genome. Furthermore, genes associated with oligodendrocyte or epithelial function accumulate mCH through development (fig. S3B), with oligodendrocyte up-regulated genes showing intragenic mCH hypermethylation in neurons and hypomethylation in glia, whereas epithelial genes display mCH hypermethylation in both neuronal and glial populations. Consistent with CH methylation requiring Dnmt3a, glial hyper-mCH genes frequently intersect areas of the genome bound by Dnmt3a in mouse postnatal neural stem cells (46). Dnmt3a-binding regions are greatly enriched for mCH, particularly in glia, whereas mCG is not enriched in Dnmt3a-binding regions in glia and is depleted in neurons (Fig. 2C). Thus, there is an association with Dnmt3a binding sites specific to mCH and not mCG, suggesting partial independence between these two marks.
mCH Position Is Highly Conserved
We examined whether the position of DNA methylation is stochastic or precisely controlled at different genomic scales. The level of mCH in 10-kb windows throughout the genome was highly reproducible between independent samples of the same cell type, with lower, but substantial, correlation between cell types (Fig. 2D). Closer inspection revealed consistency between the methylation level at individual mCH sites in neurons from different individuals in both mice and humans (Fig. 2E). At single-base resolution (fig. S4), perfect correlation between individuals would not be observed even if the true methylation level were identical at each site because of the stochastic effect of a finite number of sequenced reads. To correct for this, we normalized the observed correlation by that of simulated data sets with the same coverage per site as each of our experimental samples but with identical methylation levels (Fig. 2F and fig. S4). To assess statistical significance, we used a permutation test, which compared the data correlation with the correlation after randomly shuffling the relative positions of CH sites in each sample (fig. S4). This revealed that autosomal CG and CH sites have nearly identical methylation levels in neuronal populations isolated from different individuals of the same species. Observed differences could be explained by stochastic sampling rather than true individual variation. Unexpectedly, normalized per-site correlation is higher for mCH than mCG between neuronal populations isolated from the frontal cortices of different human individuals, and mouse neuronal mCG and mCH per-site correlations are equivalent. Per-site correlation between two human ES cell lines (H1 and HUES6) is also high (>0.8) for both mCG and mCH.
The high interindividual correlation of mCH at the kilobase and single-site scales indicates that methylation of CH positions, particularly in mammalian neurons, is a highly controlled process. It is not consistent with a stochastic event that takes place at any available CH position in a particular genomic region that accumulates mCH. Comparison of mCH between human and mouse neurons at conserved exonic CH positions revealed a low but significant interspecies correlation (Fig. 2F; P < 0.005, shuffle test), possibly indicating conservation of the cellular processes that precisely target or restrict mCH at these positions. Last, per-site mCG and mCH correlation between human ES cells and neurons is significantly lower, likely because of differences in the processes governing methylation of particular genomic features in the distinct cell types, for example, enrichment and depletion of mCH in highly transcribed genes in ES cells and neurons, respectively (Fig. 1, A and B).
The precise conservation of mCH position may be partly caused by the physical configuration of DNA within nucleosomes. Consistent with this, neuronal mCH patterns contain robust periodic components at the scale of nucleosome spacing [~170 base pairs (bp), fig. S5A] and the DNA helix coil length (~10.5 bp, fig. S5B). Such periodic components may arise from sequence-dependent constraints on mCH position, which would be the same in every neuronal cell. Alternatively, epigenetic heterogeneity within the population of NeuN+ nuclei in our sample may lead to stronger correlation for CH sites located on the same physical chromosome, compared with the correlation between the same locations on chromosomes from different cells. To test this, we measured the cross-correlation within individual reads, revealing a contribution of within-chromosome correlation to the periodic methylation pattern (fig. S5C).
Gender-Specific DNA Methylation Patterns on the X Chromosome
Interindividual correlation of mCG and mCH on chromosome X (ChrX) is frequently lower than on autosomes (Fig. 2F), prompting a closer analysis of ChrX mC patterns. ChrX mCG and mCH levels were generally lower in females compared with males, presumably because of the effect of ChrX inactivation (fig. S5, F and G) (47, 48). However, a subset of genes in both humans and mice have significantly greater intragenic mCH levels in females compared with males (Fig. 3A). Inspection of these genes revealed that most were previously found to escape inactivation in human females (X-escapees), displaying biallelic expression (49) and a reduction in promoter mCG hypermethylation, a DNA methylation signature of inactivated alleles (50). Quantification of human gender differences in neuronal DNA methylation for ChrX genes previously characterized as showing biallelic expression (49) revealed that females have reduced promoter mCG and a large increase in intragenic mCH but not intragenic mCG (Fig. 3, B and C, and fig. S5, D and E). The sequence composition of mCH is very similar in the whole genome, within autosomal gene bodies, and within X-chromosome inactivated and escapee gene bodies (fig. S5H). Analysis of gender-specific methylation in additional human cell types revealed that female promoter mCG hypomethylation is observed at X-escapee genes in glia and human embryonic stem cells (fig. S6). Intragenic mCH hypermethylation of X-escapees was also observed in female glia, albeit to a lesser extent than in neurons, but was not present in ES cells. Thus, X-escapee mCH hypermethylation may be a feature that is specific to neural cell types. Although both promoter CG hypomethylation and intragenic CH hypermethylation provide significant information for discriminating X-escapees [Fig. 3D, discriminability index (area under the curve, AUC) = 0.75 and 0.78, respectively], combining both mCG and mCH measurements boosts discriminability (AUC = 0.88). By using this intragenic mCH hypermethylation signature, we identified seven new putative X-escapee genes (table S4). On the basis of these data, we hypothesize that intragenic CH hypermethylation in neurons may play a compensatory role in genes that fail to acquire repressive CG hypermethylation in the promoter region, restoring equal gene expression between male and female cells (51).
Fig. 3. mCH is enriched in genes that escape X inactivation.
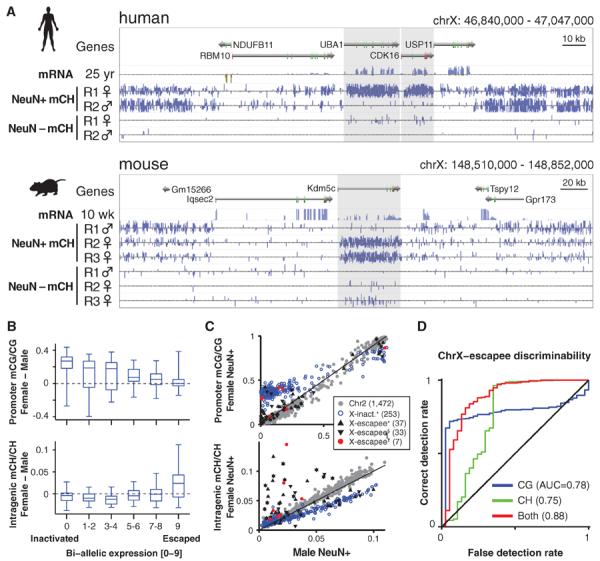
(A) Browser representation showing mCH-hypermethylated female human and mouse genes that escape X inactivation (shaded genes). (B) Box and whisker plots of gender differences in promoter mCG and intragenic mCH in inactivated and escapee genes on human chrX. (C) Scatter plot of gender differences in mCG and mCH in human chrX genes. Reported X inactivated and escapee genes: *Carrel and Willard (49); §Sharp et al. (50); †predicted escapee genes, and autosomal (Chr2) genes are indicated. (D) Discriminability analysis of genes that escape female X inactivation using mC data, showing correct versus false detection rate mapped for all possible mC/C thresholds.
Distinct Genic DNA Methylation States Demarcate Functionally Relevant Gene Clusters
DNA methylation within promoter regions and in gene bodies is implicated in regulation of gene expression (22, 52), suggesting that the precisely conserved, cell type–specific DNA methylation patterns may be related to specific neuronal and glial cellular processes. We therefore used an unbiased approach to classify patterns of mCG and mCH within each annotated gene body and in flanking regions extending 100 kb up- or downstream. After normalizing the methylation pattern around each autosomal gene by the local baseline mCG or mCH level in each adult neuronal or glial sample, we combined these features into a large data matrix containing 4200 individual DNA methylation measurements for each gene [seven samples, two contexts (CG and CH), 300 1-kb bins within and around each gene]. Using principal component (PC) analysis, we extracted five methylation features (PCs) that together account for 46% of the total data set variance (fig. S7A). Gene sets with specific neuronal or astrocytic expression, as well as ChrX genes, segregate within PC space (fig. S6B). We then used k-means clustering to classify all genes into 15 clusters on the basis of their mCG and mCH patterns (Fig. 4A and fig. S8). Several dominant patterns of DNA methylation and transcript abundance and dynamics between developmental and cellular states are evident. A cluster of genes that progressively loses gene-body mCG and mCH through development contains constitutively highly expressed genes that are strongly enriched for neuronal function and depleted for astrocyte-specific roles (Fig. 4A, box 1). These genes show intragenic mCG enrichment in glia and depletion in neurons (box 2), indicating that glial gene body mCG resembles that of the neural precursor cells that predominate the fetal brain. This indicates that the loss of mCG in brain tissue during development is due to CG hypomethylation in mature neurons. These constitutively highly expressed genes enriched for neuronal function also show extensive intragenic mCH hypomethylation in neurons in contrast to glia (box 3), and they are enriched for hmCG (box 4) as previously described (32, 33). Genes that are not as highly transcribed, but that are associated with neuronal function and are developmentally up-regulated, also show intragenic mCG and mCH hypomethylation in neurons but not glia (box 5). For these gene sets, mCG and mCH enrichment or depletion is precisely localized to transcribed regions, suggesting that this modification of genic mC is tightly coupled to transcription. Notably, the bodies of constitutively high genes that are not enriched for neuronal function (box 6) do not show marked fetal/glial mCG enrichment or neuronal mCH depletion, indicating that this differential methylation is specific for genes enriched for neuronal function and not simply an association with particular levels of transcriptional activity. Genes associated with astrocyte function show an opposite pattern to genes associated with neuronal function: a progressive increase in intragenic mCG and mCH in frontal cortex tissue over development, neuronal mCG and mCH hypermethylation, and glial mCG and mCH hypomethylation (Fig. 4A, boxes 7 to 9). Last, genes with constitutively low expression do not show developmental or cell type–specific DNA methylation patterns (box 10), demonstrating that dynamic DNA methylation in genes is highly associated with differential transcriptional activity in mammalian brain development and neural cell specialization.
Fig. 4. Celltype–specific and developmental differences in mC between mouse neurons and glia.

(A) Heat-map representation of 25,260 mouse genes organized in gene sets identified by k-means clustering using normalized genic mCG and mCH levels in adult developmental and NeuN+ and NeuN− samples. Left-hand plot shows mRNA abundance. (B) Enrichment or depletion of each cluster for developmental and cell-type specific gene sets. n.s., not significant (FET, FDR < 0.05). (C) mCG and mCH throughout gene body and flanking 100 kb for indicated gene sets. Transcript abundance (mRNA-Seq FPKM) over mouse development is shown for the same gene sets. Color scales are as in (A).
Each of the gene clusters identified in our unbiased analysis was significantly enriched or depleted for cell type–specific function [neuronal or astrocytic genes (45)] or particular expression patterns (constitutively high or low expression, developmentally up- or down-regulated) (Fig. 4B). Profiling the median mCG and mCH of genes within each of these categories allows direct comparison of developmental and cell type–specific DNA methylation in mouse (Fig. 4C) and human (fig. S3C). This analysis recapitulates many of the conclusions of the unbiased clustering (Fig. 4A).
The inverse relationship observed between genic mCH level and transcriptional activity is consistent with a model whereby intragenic accumulation of mCH impedes transcriptional activity. Alternatively, the process of transcription could interfere with mCH de novo methylation or induce active mCH demethylation, although these are not consistent with the DNMT3A-dependent intragenic mCH in human embryonic stem cells that is positively correlated with gene expression (22, 23). Overall, glial mCG and mCH patterns closely resemble those of the fetal and the early postnatal brain, indicating that DNA methylation in early mammalian brain developmental stages may be a default state that largely persists through to maturity in glial cells, whereas neuronal differentiation and maturation involve extensive reconfiguration of the DNA methylome that is highly associated with cell type–specific changes in transcriptional activity.
hmCG Is Enriched Within Active Genomic Regions in Fetal and Adult Mouse Brain
Our base-resolution analysis of hmC using TAB-Seq revealed that intragenic and global hmCG levels are largely equivalent between chromosomes, whereas hmCG/CG is 22% lower on the male ChrX, consistent with previous reports from enrichment based detection of hmC (32, 33) (Fig. 5A). Analysis of hmCG levels in different genomic regions revealed that, although adult hmCG/CG is similar across transcriptional end sites and intragenic, DNaseI-hypersensitive (DHS), and enhancer regions, the fetal frontal cortex shows a relative enrichment of hmCG in DHS regions and enhancers, in particular enhancer regions that are unique to the fetal developmental stage (Fig. 5B). The inverse pattern can be observed for adult mCG levels, which are lower in DHS regions and enhancers (fig. S9, A and B), suggesting that regions of relatively high hmCG levels in the fetal brain show relatively low mCG levels in the adult brain. Analysis of intragenic hmCG enrichment relative to flanking genomic regions, for cell type–specific or developmentally dynamic gene sets (Fig. 5C), showed that neuronal and astrocyte gene bodies that are highly enriched with hmCG in adult are also highly enriched at the fetal stage.
Fig. 5. hmCG is enriched within active genomic regions in fetal and adult mouse brain.
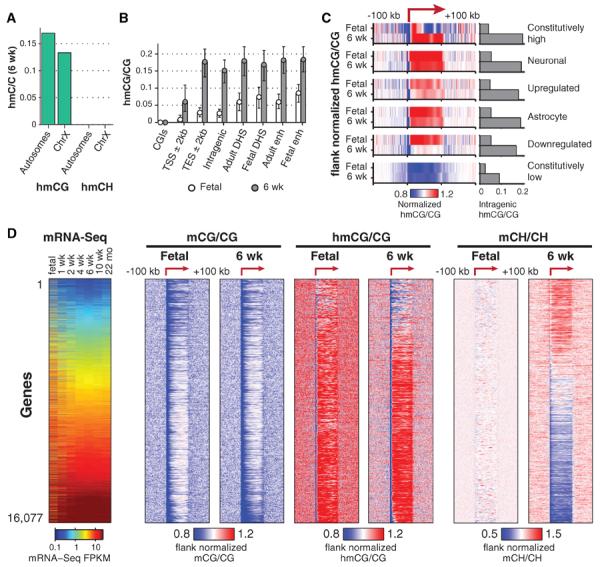
(A) hmCG level in 6-week mouse frontal cortex for autosomes and ChrX. (B) Median hmCG level within genomic features (error bars 32nd to 68th percentile). enh, enhancer. (C) Median normalized hmCG throughout gene body and flanking 100 kb for indicated gene sets. Bars show absolute hmCG/CG levels within gene bodies for each class. (D) mC and hmC throughout gene body and flanking 100 kb for each. Transcript abundance (mRNA-Seq FPKM) during mouse development is also shown (left).
Thus, despite lower absolute levels of intragenic hmCG in the fetal stage, the adult patterns of hmCG enrichment at these cell type–specific genes are already forming in utero. Constitutively lowly expressed genes show intragenic depletion of hmCG, in contrast to constitutively highly transcribed genes, which show localized enrichment of hmCG throughout part or all of the gene body. Developmentally down-regulated genes show enrichment of hmCG in the fetal frontal cortex but not in adults, indicating that reduced transcription is accompanied by a loss of hmCG enrichment. Overall, transcriptional activity is associated with intragenic hmCG enrichment, as reported (33), with in utero establishment of adult hmCG patterns for cell type–specific genes and loss of hmC enrichment associated with developmentally coupled transcriptional down-regulation.
Measurement of mC and hmC in all genes in fetal and adult mouse frontal cortex indicated that both mCG and hmCG are depleted at promoters and in gene bodies of lowly expressed genes, whereas hmCG is enriched throughout the gene bodies of more highly transcribed genes (Fig. 5D). The most highly expressed genes in the adult frontal cortex show intragenic mCG hypomethylation (Figs. 4 and 5D) but still retain high intragenic hmCG. Ranking all genes by transcript abundance, it is evident that the highest mean intragenic hmCG levels, which occur in the most highly transcribed genes, correspond to hmCG/CG ~ 0.25 and mCG/CG ~ 0.5 (fig. S10D). Frontal cortex development is accompanied by increased enrichment of hmCG at intragenic regions that are already hyper-hydroxymethylated at the fetal stage (Fig. 5D and fig. S10), demonstrating that adult patterns of genic hmC are already evident in the immature fetal brain.
CG Differentially Methylated Regions Enriched in Regulatory Regions
Because differences in genic mCG were observed over development and between neuronal and glial cell populations (Fig. 4), we scanned the human and mouse methylomes to comprehensively identify CG differentially methylated regions (CG-DMRs) throughout the genome. CG-DMRs were identified between fetal and adult frontal cortex, neurons and glia, and combined into four sets: neuronal and glial hyper- and hypo-methylated CG-DMRs. In total, 267,799 human and 142,835 mouse CG-DMRs were identified (median lengths: for mouse, 473 bp; human, 533 bp), revealing several predominant dynamics in mCG during brain development and cellular specialization (Fig. 6A and fig. S11). Neuronal CG-DMRs are the most numerous in both mice and humans, because of the very distinct mCG patterns that emerge during neuronal differentiation and maturation. At these sites, CG methylation in adult neurons is distinct compared with those in glial and/or fetal and early postnatal development frontal cortex tissue samples. Neuronal hypermethylated CG-DMRs also show mCH hypermethylation (fig. S11). In mouse, mCG/CG within neuronal hypomethylated CG-DMRs declines to a stable level by 1 week after birth. In contrast, neuronal hypermethylated CG-DMRs do not begin to change until 1 week after birth, after which they accumulate mCG until 2 to 4 weeks of age. These data indicate that increases in neuronal mCG occur during synaptogenesis after most decreases in neuronal mCG have already occurred. Furthermore, we found that hydroxymethylation in the adult cortex is highest in CG-DMRs that show neuronal hypermethylation and is depleted from CG-DMRs that display neuronal hypomethylation (Fig. 6A). This suggests that hmCG may be most abundant in neurons, rather than glial cells, in the frontal cortex.
Fig. 6. Developmental and cell type–specific differential mCG.

(A) Heat map of absolute mCG level in CG-DMRs identified between neurons and glia and over development in mouse (left) and human (right). (B) Fraction of all CG-DMRs located in distinct genomic features in mouse. (C) Enrichment or depletion of distinct cell type–specific and developmental CG-DMR sets within genomic features from (B). (D and E) Intersection of developmentally dynamic CG-DMRs and (D) DNaseI hypersensitive sites or (E) enhancers in mouse brain in thousands. (F) Browser representation of mouse developmentally dynamic CG-DMRs and quantification of local enrichment of chromatin modifications, genome accessibility, and mC. (G) TAB-Seq reads showing fetal-specific hmCG in the Fetal>Adult CG-DMR in mouse. (H) Proportion of mouse developmental CG-DMRs where mCG/CG is significantly increased or decreased in Tet2 knockout mice. (I) Distribution of mCG level difference between wild-type (WT) and Tet2 mutantat mouse CG-DMRs. Significantly different DMRs are indicated by coloration.
Analysis of the genomic features in which CG-DMRs are located revealed that although half are found within gene bodies, they are not common within promoters and transcriptional start and end regions. Instead, they are disproportionately located at DHS regions and enhancers unique to fetal or adult brain (Fig. 6B). Closer inspection of the enrichment and depletion of these CG-DMRs revealed that fetal enhancers and DHS sites unique to the fetal brain are enriched for hypermethylation in adult brain but not in the fetal brain (Fig. 6C). In contrast, adult enhancers and unique adult DHS sites are highly associated with CG hypermethylation in fetal stages but are not associated with hypermethylated CG-DMRs in the adult brain and in neurons. Thus, developmentally dynamic enhancers and DHS sites in frontal cortex have dynamic CG methylation that is depleted where chromatin accessibility and regulatory element activity increase, consistent with a range of human cell lines (53).
To characterize gene functions associated with the CG-DMRs, we analyzed the association between proximal genes (transcriptional start site within 5 kb of the DMR) and cell type–specific or developmentally dynamic gene sets (fig. S12A). We observed an inverse relationship between methylation and gene function. Genes associated with neuronal function and up-regulation during development are enriched for promoter hypermethylation in glia and hypomethylation in neurons, whereas genes down-regulated during brain development and those related to astrocyte function are enriched for promoter hypermethylation in neurons and hypomethylation in glia. Genes that are constitutively expressed at either high or low levels are not associated with promoter/transcription start site CG-DMRs, indicating that dynamic CG methylation is highly associated with changes in transcriptional activity and cell type–specific transcriptional regulation.
Because the majority of all developmentally dynamic CG-DMRs are associated with DHS sites, we examined the directional relationships between dynamic mCG and DNA accessibility states over development (Fig. 6D). Notably, DHS sites unique to fetal frontal cortex overlap with 28% of CG-DMRs that gain methylation through development (Adult>Fetal). However, these sites only overlap 7.3% of CG-DMRs that lose mCG during development (Fetal>Adult). Similarly, DHS sites unique to adult frontal cortex rarely overlap Adult>Fetal CG-DMRs. A similar analysis of developmentally dynamic enhancers active in only one of the developmental stages (37) (Fig. 6E) showed that enhancer activation is associated with mCG hypomethylation of the enhancer, whereas enhancer inactivation is associated with enhancer mCG hypermethylation.
This inverse relationship between genome accessibility and mCG level at putative functional regions of the genome suggests that nuclear factors that bind the region and increase accessibility may cause localized reduction in mCG, as previously reported for a small number of DNA binding proteins (54). Alternatively, mCG hypermethylation may cause reduced genome accessibility by direct inhibition of DNA-protein interactions or induction of chromatin compaction, with loss of mCG enabling increased chromatin accessibility and genome interaction with DNA binding factors.
Discrete regions that show increased or decreased CG methylation through development are associated with specific local chromatin modifications. We found that CG-methylated regions of the fetal frontal cortex that become hypomethylated in the adult (Fig. 6F, Fetal>Adult) gain localized histone modifications characteristic of active enhancers (H3K4me1 and H3K27ac) and increased DNaseI hypersensitivity in the adult. In addition, these regions have reduced accumulation of mCH (Fig. 6F), consistent with an overall decrease in mC linked to increased genome accessibility and enhancer activity. In contrast, genomic regions that gain mCG during development (Adult>Fetal) lose localized enrichment of H3K4me1, H3K27ac, and DNaseI hypersensitivity and show increased mCH. These changes indicate inactivation of these genomic regions through brain development and suggest that this inactivation is associated with increased local mCG.
A Hydroxymethylation Signature of Developmentally Activated Regions
Adult>Fetal CG-DMRs show broad low-level hmCG enrichment flanking the CG-DMR and a localized depletion of hmC at the center in both fetal and adult genomes (Fig. 6F), and the absolute abundance of hmC is several-fold lower in fetal compared with adult frontal cortex (Fig. 1E). This suggests that although Adult>Fetal CG-DMRs gain mCG through development, they tend to be refractory to conversion to hmC, potentially because of lower accessibility to the Tet hydroxylases. In contrast, Fetal>Adult CG-DMRs have a local enrichment of both mCG and hmCG in the fetal cortex that becomes a local depletion in the adult. Two enrichment-based genome-wide hmC profiling techniques, CMS-IP (38) and biotin-glucosyl tagging (31), confirmed the localized enrichment of hmC at Fetal>Adult CG-DMRs (fig. S12B). The localized enrichment of hmC at these inaccessible and quiescent genomic regions, which lose mCG and hmCG later in development, indicates that they may be premodified with hmCG in the fetal stage to create a dormant state that is poised for subsequent demethylation and activation at a later developmental stage. Closer inspection of base-resolution hmC data revealed that 4% of the hmCG bases that have significantly higher hmC levels in fetal compared with adult [false discovery rate (FDR) 0.05] directly overlap with Fetal>Adult CG-DMRs, far exceeding the number expected by chance (0.5%). This indicates that despite lower global levels of hmC in the fetal brain, developmentally demethylated CG-DMRs are enriched for hmCG bases that are more highly hydroxymethylated in fetal than in adult brain (Fig. 6G). The localized enrichment of mCG at these CG-DMRs in the fetal cortex indicates that CG-demethylation has not yet taken place.
If fetal hmCG is poised at dormant genomic regions in order to facilitate active DNA demethylation at later developmental stages, then the Tet hydroxylase enzymes that catalyze conversion of mC to hmC should be necessary for mCG hypomethylation in the adult frontal cortex at these regions. To test this, we performed MethylC-Seq of genomic DNA from frontal cortex tissue of adult Tet2−/− mice. Adult>Fetal CG-DMRs, which gain mCG through development, are largely unaffected in Tet2−/− compared with wild-type adult mice (Fig. 6, H and I; 3.6% hypermethylated, FET, FDR 0.05). By contrast, a substantial fraction of Fetal>Adult CG-DMRs are hypermethylated in Tet2−/− (19.7%) versus wild type. The mutant shows a small but significant increase in mCG at Fetal>Adult CG-DMRs (Fig. 6I and fig. S12C) (Tet2−/−: 7.9% ± 4.6%, P < 10−11, Wilcoxon signed rank test). The partial effect of the mutation on CG methylation is not unexpected given that all three Tet genes are expressed in the brain (fig. S9C) and may exhibit some functional redundancy. Additionally, a genome-wide search identified 14,340 CG-DMRs hypermethylated in Tet2−/− relative to wild type (6 weeks, 10 weeks, and 22 months), >fourfold more numerous than hypomethylated CG-DMRs (3099). This further indicates a role for Tet2 in mediating mCG demethylation during brain development.
Discussion
The essential role of frontal cortex in behavior and cognition requires the coordinated interaction, via electrical and chemical signaling, of multiple neuronal cell types and a diverse population of glial cells. Individual brain cells have unique roles within circuits that are defined by their location and pattern of connections as well as by their molecular identity. The development and maturation of the brain's physical structure and the refinement of the molecular identities of neurons and glial cells occur in parallel in a finely orchestrated process that starts early during the embryonic period and continues, in humans, well into the third decade of life (55, 56). An early postnatal burst of synaptogenesis is followed by activity-dependent pruning of excess synapses during adolescence (28, 57, 58). This process forms the basis for experience-dependent plasticity and learning in children and young adults (59), and its disruption leads to behavioral alterations and neuropsychiatric disorders (60). During this period, profound transcriptional changes lead to the appearance of adult electrophysiological characteristics in neocortical neurons.
Our study suggests a key role of DNA methylation in brain development and function. First, CH methylation accumulates significantly in neurons through early childhood and adolescence, becoming the dominant form of DNA methylation in mature human neurons. This shows that the period of synaptogenesis, during which the neural circuit matures, is accompanied by a parallel process of large-scale reconfiguration of the neuronal epigenome. Indeed, central nervous system deletion of Dnmt3a during late gestation induces motor deficits, and animals die prematurely (61). However, mice with a postnatal deletion restricted to the pyramidal cell population (complete recombination around 1 month old) do not show overt behavioral or transcriptional alterations (2). Our data suggest that expression of Dnmt3a specifically around the second postnatal week may be critical for establishing a normal brain DNA methylation profile and allowing healthy brain development.
Second, the precise positioning of mCG and mCH marks, which are conserved between individuals and across humans and mice, is consistent with a functional role. Whether this is the case, or whether the conserved patterns are instead a reflection of conserved nucleosome position or chromatin structure, requires further investigation. Third, the relationship between DNA methylation patterns and the function of neuron- or astrocyte-specific gene sets suggests a role for DNA methylation in distinguishing these two broad classes of cortical cells. DNA methylation could therefore play a key role in sculpting more-specific cellular identities. If this is the case, we expect that purified subpopulations will reveal high specificity of methylation at specific sites for particular cell types. Thus, the observation that most CH sites with nonzero methylation are methylated in ~20 to 25% of sampled cells (fig. S1H) could be explained by the heterogeneity of these brain circuits rather than by stochastic methylation within each cell. These conclusions obtained from our genome-wide, base-resolution, cell type–specific DNA methylomes for brain cells through key stages of development are the first steps toward unraveling the genetic program and experience-dependent epigenetic modifications leading to a fully differentiated nervous system.
Supplementary Material
Acknowledgments
We thank W. F. Loomis for critical reading of this manuscript; J. Chambers for technical assistance with animal breeding; D. Chambers for technical assistance with cell sorting; M. Lutz and T. Berggren for provision of the HUES6 cells; F. Yue, G. Hon, and B. Ren for providing mapped mouse ChIP-Seq data and assistance with TAB-Seq; and A. Nimmerjahn for providing the S100b-eGFP mice. Human brain tissue was obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, Maryland, the IDIBELL Biobank, which is part of the BrainNet Europe Bank funded by the European Commission (LSHM-CT-2004-503039). We thank the Stamatoyannopoulos laboratory (University of Washington) and the Mouse ENCODE Consortium for generating and providing access to the DNaseI hypersensitivity data sets (35). This work was supported by a National Institutes of Mental Health grant to M.M.B. and J.R.E. (MH094670); the Howard Hughes Medical Institute to T.J.S. and J.R.E.; the Gordon and Betty Moore Foundation (GMBF3034) to J.R.E.; NIH grant HG006827 to C.H.; NIH RO1 grants AI44432, HD065812, and CA151535; grant RM-01729 from the California Institute of Regenerative Medicine; and Translational Research grant TRP 6187-12 from the Leukemia and Lymphoma Society (to A.R.). R.L. was supported by an Australian Research Council Future Fellowship. Support was also provided by the Government of Western Australia through funding for the Western Australia CoE for Computational Systems Biology. E.A.M. was supported by the Center for Theoretical Biological Physics, University of California San Diego, and NIH (National Institute of Neurological Diseases and Stroke grant K99NS080911). W.A.P. was supported by a predoctoral graduate research fellowship from the NSF.
Footnotes
Analyzed data sets can be accessed at http://neomorph.salk.edu/brain_methylomes. Sequence data can be downloaded from National Center for Biotechnology Information GEO (GSE47966). Tet2 mutant mice are available under a material transfer agreement from the La Jolla Institute for Allergy and Immunology.
Supplementary Materials www.sciencemag.org/content/341/6146/1237905/suppl/DC1
References and Notes
- 1.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. doi: 10.1016/j.neuron.2008.10.012; pmid: 19109904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. doi: 10.1038/nn.2514; pmid: 20228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. doi: 10.1016/j.neuron.2007.02.022; pmid: 17359920. [DOI] [PubMed] [Google Scholar]
- 4.Numata S, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012;90:260–272. doi: 10.1016/j.ajhg.2011.12.020. doi: 10.1016/j.ajhg.2011.12.020; pmid: 22305529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suderman M, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. U.S.A. 2012;109(suppl. 2):17266–17272. doi: 10.1073/pnas.1121260109. doi: 10.1073/pnas.1121260109; pmid: 23045659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegmund KD, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE. 2007;2:e895. doi: 10.1371/journal.pone.0000895. doi: 10.1371/journal.pone.0000895; pmid: 17878930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinowich K, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. doi: 10.1126/science.1090842; pmid: 14593184. [DOI] [PubMed] [Google Scholar]
- 8.Guo JU, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. doi: 10.1038/nn.2900; pmid: 21874013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn. Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. doi: 10.1101/lm.026575.112; pmid: 23322554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levenson JM, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. doi: 10.1074/jbc.M511767200; pmid: 16606618. [DOI] [PubMed] [Google Scholar]
- 11.Oliveira AMM, Hemstedt TJ, Bading H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012;15:1111–1113. doi: 10.1038/nn.3151. doi: 10.1038/nn.3151; pmid: 22751036. [DOI] [PubMed] [Google Scholar]
- 12.Moretti P, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. doi: 10.1523/JNEUROSCI.2623-05.2006; pmid: 16399702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penn NW, Suwalski R, O'Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 1972;126:781–790. doi: 10.1042/bj1260781. pmid: 4538516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penn NW. Modification of brain deoxyribonucleic acid base content with maturation in normal and malnourished rats. Biochem. J. 1976;155:709–712. doi: 10.1042/bj1550709. pmid: 949331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. doi: 10.1371/journal.pone.0015367; pmid: 21203455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo JU, Su Y, Zhong C, Ming G-L, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. doi: 10.1016/j.cell.2011.03.022; pmid: 21496894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He Y-F, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. doi: 10.1126/science.1210944; pmid: 21817016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song C-X, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. doi: 10.1016/j.cell.2013.04.001; pmid: 23602153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen L, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. doi: 10.1016/j.cell.2013.04.002; pmid: 23602152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie W, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. doi: 10.1016/j.cell.2011.12.035; pmid: 22341451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varley KE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–567. doi: 10.1101/gr.147942.112. doi: 10.1101/gr.147942.112; pmid: 23325432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. doi: 10.1038/nature08514; pmid: 19829295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziller MJ, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. doi: 10.1371/journal.pgen.1002389; pmid: 22174693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. doi: 10.1016/j.cell.2008.03.029; pmid: 18423832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. doi: 10.1038/nature09798; pmid: 21289626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsahoye BH, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. doi: 10.1073/pnas.97.10.5237; pmid: 10805783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Felipe J, Marco P, Fairén A, Jones EG. Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb. Cortex. 1997;7:619–634. doi: 10.1093/cercor/7.7.619. doi: 10.1093/cercor/7.7.619; pmid: 9373018. [DOI] [PubMed] [Google Scholar]
- 28.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. doi: 10.1002/(SICI)1096-9861(19971020)387:2<167::AID-CNE1>3.0.CO;2-Z; pmid: 9336221. [DOI] [PubMed] [Google Scholar]
- 29.Berman BP, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012;44:40–46. doi: 10.1038/ng.969. doi: 10.1038/ng.969; pmid: 22120008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. 10.1126/science.1169786. doi: 10.1126/science. 1169786; pmid: 19372393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song C-X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. doi: 10.1038/nbt.1732; pmid: 21151123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. doi: 10.1038/nn.2959; pmid: 22037496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. doi: 10.1016/j.cell.2012.11.022; pmid: 23260135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. doi: 10.1016/j.cell.2012.04.027; pmid: 22608086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamatoyannopoulos JA, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418. doi: 10.1186/gb-2012-13-8-418. doi: 10.1186/gb-2012-13-8-418; pmid: 22889292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auerbach RK, et al. Mapping accessible chromatin regions using Sono-Seq. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14926–14931. doi: 10.1073/pnas.0905443106. doi: 10.1073/pnas.0905443106; pmid: 19706456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. doi: 10.1038/nature11243; pmid: 22763441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. doi: 10.1038/nature10102; pmid: 21552279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Pastor WA, Zepeda-Martínez JA, Rao A. The anti-CMS technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat. Protoc. 2012;7:1897–1908. doi: 10.1038/nprot.2012.103. doi: 10.1038/nprot.2012.103; pmid: 23018193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magklara A, et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011;145:555–570. doi: 10.1016/j.cell.2011.03.040. doi: 10.1016/j.cell.2011.03.040; pmid: 21529909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clowney EJ, et al. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell. 2012;151:724–737. doi: 10.1016/j.cell.2012.09.043. doi: 10.1016/j.cell.2012.09.043; pmid: 23141535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, et al. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9397–9402. doi: 10.1073/pnas.0802876105. doi: 10.1073/pnas.0802876105; pmid: 18599437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akhtar MW, et al. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS ONE. 2012;7:e34863. doi: 10.1371/journal.pone.0034863. doi: 10.1371/journal.pone.0034863; pmid: 22496871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbosa AC, et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9391–9396. doi: 10.1073/pnas.0802679105. doi: 10.1073/pnas.0802679105; pmid: 18599438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. doi: 10.1523/JNEUROSCI.4178-07.2008; pmid: 18171944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu H, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. doi: 10.1126/science.1190485; pmid: 20651149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: Evidence for X inactivation by DNA methylation. Science. 1981;211:393–396. doi: 10.1126/science.6164095. doi: 10.1126/science.6164095; pmid: 6164095. [DOI] [PubMed] [Google Scholar]
- 48.Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet. 2002;36:233–278. doi: 10.1146/annurev.genet.36.042902.092433. doi: 10.1146/annurev.genet.36.042902.092433; pmid: 12429693. [DOI] [PubMed] [Google Scholar]
- 49.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. doi: 10.1038/nature03479; pmid: 15772666. [DOI] [PubMed] [Google Scholar]
- 50.Sharp AJ, et al. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21:1592–1600. doi: 10.1101/gr.112680.110. doi: 10.1101/gr.112680.110; pmid: 21862626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnston CM, et al. Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet. 2008;4:e9. doi: 10.1371/journal.pgen.0040009. doi: 10.1371/journal.pgen.0040009; pmid: 18208332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. doi: 10.1038/nature09165; pmid: 20613842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. doi: 10.1038/nature11232; pmid: 22955617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. pmid: 22170606. [DOI] [PubMed] [Google Scholar]
- 55.Petanjek Z, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. doi: 10.1073/pnas.1105108108 pmid: 21788513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolb B, et al. Experience and the developing prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 2012;109(suppl. 2):17186–17193. doi: 10.1073/pnas.1121251109. doi: 10.1073/pnas.1121251109; pmid: 23045653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chugani HT. A critical period of brain development: Studies of cerebral glucose utilization with PET. Prev. Med. 1998;27:184–188. doi: 10.1006/pmed.1998.0274. doi: 10.1006/pmed.1998.0274; pmid: 9578992. [DOI] [PubMed] [Google Scholar]
- 58.Levitt P. Structural and functional maturation of the developing primate brain. J. Pediatr. 2003;143(suppl):S35–S45. doi: 10.1067/s0022-3476(03)00400-1. doi: 10.1067/S0022-3476(03)00400-1; pmid: 14597912. [DOI] [PubMed] [Google Scholar]
- 59.Johnston MV. Plasticity in the developing brain: implications for rehabilitation. Dev. Disabil. Res. Rev. 2009;15:94–101. doi: 10.1002/ddrr.64. doi: 10.1002/ddrr.64; pmid: 19489084. [DOI] [PubMed] [Google Scholar]
- 60.Uhlhaas PJ, Singer W. The development of neural synchrony and large-scale cortical networks during adolescence: Relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophr. Bull. 2011;37:514–523. doi: 10.1093/schbul/sbr034. doi: 10.1093/schbul/sbr034; pmid: 21505118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007;236:1663–1676. doi: 10.1002/dvdy.21176. doi: 10.1002/dvdy.21176; pmid: 17477386. [DOI] [PubMed] [Google Scholar]
- 62.Materials and methods are available as supplementary materials on Science Online.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.