

Abstract
The melanocortin 1 receptor (MC1R) is overexpressed in most melanoma metastases, making it a promising target for imaging of melanomas. In this study, the expression of MC1R in a large fraction of patients with melanoma was confirmed using mRNA and tissue microarray. Here, we have characterized the in vivo tumor and tissue distribution and pharmacokinetics (PK) of uptake and clearance of a MC1R specific peptidomimetic ligand conjugated to a near-infrared fluorescent dye. We propose an interdisciplinary framework to bridge the different time and space scales of ligand-tumor-host interactions: intravital fluorescence microscopy to quantify probe internalization at the cellular level, a xenograft tumor model for whole body pharmacokinetics, and a computational pharmacokinetic model for integration and interpretation of experimental data. Administration of the probe into mice bearing tumors with high and low MC1R expression demonstrated normalized image intensities that correlated with expression levels (p<0.05). The biodistribution study showed high kidney uptake as early as 30 min post-injection. The PK computational model predicted the presence of receptors in the kidneys with a lower affinity, but at higher numbers than in the tumors. As the mouse kidney is known to express the MC5R, this hypothesis was confirmed by both co-injection of a ligand with higher MC5R affinity compared to MC1R and by injection of lower probe concentrations (e.g. 1 nmol/kg), both leading to decreased kidney accumulation of the MC1R ligand. In addition, through this interdisciplinary approach we could predict the rates of ligand accumulation and clearance into and from organs and tumors, and the amount of injected ligand required to have maximum specific retention in tumors. These predictions have potential to aid in the translation of a targeted agent from lab to the clinic. In conclusion, the characterized MC1R-specific probe has excellent potential for in vivo detection of melanoma metastases. The process of cell-surface marker validation, targeted imaging probe development, and in vitro, in vivo and in silico characterization described in this study can be generally applied to preclinical development of targeted agents.
Keywords: malignant melanoma, Melanocortin 1 receptor, targeted imaging agent, pharmacokinetics and biodistribution
INTRODUCTION
Melanoma is the most common cause of death from cutaneous malignancies and the fastest increasing cancer in the US.1, 2 Assessment of metastatic spread to regional lymph nodes that drain the tumor site as well as detection of distant metastasis are important for staging, prognosis and to determine the course of therapy.3, 4 About 16% of patients develop metastases to regional lymph nodes, lung, liver, and brain.5, 6 Surgery remains the mainstay of melanoma therapy at all sites.5, 7
Sentinel lymph node biopsy (SLNB) is the current gold standard for evaluating regional lymph node involvement 4. This method is associated with morbidities 7, and 80% of patients are negative for lymph node metastasis and therefore have undergone a potentially avoidable procedure. Also, indocyanine green (ICG) near-infrared fluorescent (NIRF) dye has been used intraoperatively for SLNB.9, 10 The radiocolloid and dyes are not tumor-specific, hence, cannot directly detect lymph node involvement.
Standard imaging techniques have been used for detection of distant metastasis, including CT, MRI and 18F-FDG PET.11, 12 However, all of these techniques have limitations [reviewed by 8, 9]. 18F-FDG PET lacks sufficient sensitivity to detect micrometastasis, especially for the assessment of SLNs.14–16 In addition, this tracer cannot discriminate between malignancy and inflammation, including nonneoplastic postoperative changes.17, 18 Also, some normal tissues can accumulate 18F-FDG, including bowel, bladder, muscle and brown fat, contributing to background signal.10 Specificity could be improved through the use of a molecular imaging probe targeting a cell-surface receptor that is highly and densely expressed in melanoma metastases relative to surrounding tissues.
Therefore, melanoma-specific molecular imaging probes are needed for the noninvasive and sensitive detection of regional lymph node involvement for staging of melanoma. In addition, they can used to follow therapy response. Such probes could also be used for the intraoperative margin assessment and detection of regional lymph node involvement during surgery 20, 21 and there is increasing interest in the use of novel targeted therapies for melanoma, which is notoriously resistant to most systemic therapies.11 For example, by attachment to a targeted agent with a cleavable linker, cytotoxins may be targeted to tumor cells for receptor-mediated endocytosis and intracellular release 12, increasing the effective local concentration at the metastasis, while maintaining tolerable systemic levels of cytotoxin, i.e., increasing the therapeutic window.
Melanoma progression is associated with altered expression of cell surface proteins. It has been estimated that over 80% of melanomas express high levels of the MC1R. 13 MC1R is a member of a family of five G protein-coupled melanocortin receptors (MC1R – MC5R) 14, which bind melanocyte-stimulating hormone (MSH) and related ligands. Because of the high expression of MC1R in melanoma, MSH has been investigated as a ligand for selective imaging and therapeutic agents, and a number of ligands have been developed.15–17 As off-target binding inside the melanocortin receptor family is undesirable given the presence mRNA coding for MC5R in human and mouse kidney.18, 19 Since the kidney is involved in agent clearance, MC1R selective ligands are needed. The other two MSH avid melanocortin receptor isoforms (MC3R and MC4R) are primarily expressed in the brain and CNS, which is protected by the blood brain barrier.20–22
In this study, we evaluated the expression of MC1R through RNA expression microarray and immunohistochemistry (IHC) analysis in melanoma patient samples and cell lines. While MC1R is not a novel target, this represents the most extensive study on its distribution in melanoma to date. In addition, a high affinity MC1R selective ligand with lower affinity for MC5R has been described, and we have recently modified this ligand with moieties to facilitate attachments.23 More recently, we have conjugated near-infrared fluorescent (NIRF) dyes to this ligand and demonstrated in vivo specificity for tumors with MC1R expression.24
In order to characterize the biodistribution and the tumor-host-ligand interactions for these novel NIRF probes, we used an interdisciplinary approach: first, we used intravital confocal fluorescence microscopy of a dorsal skin-fold window-chamber mouse xenograft tumor model to demonstrate probe diffusion from blood vessels into tissue and uptake into tumor cells; then, in a higher time and spatial scale, in vivo time-resolved fluorescence imaging was used to characterize the heterogeneity of tumor uptake, biodistribution and pharmacokinetics of the probe; and finally, a novel compartmental mathematical model was applied to interpret the experimental data and to estimate the pharmacokinetic parameters for the whole animal. These studies demonstrate that our probe has excellent potential for in vivo detection of melanoma metastasis, and that the combination of quantitative imaging with PK mathematical modeling can be used to better characterize the behavior of targeted agents in vivo, simplify the interpretation of an otherwise complex system of multi-organ interactions, and shorten the gap between pre-clinical and clinical studies.
EXPERIMENTAL SECTION
Cell culture
A375 human malignant melanoma cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (Life Technologies), 100 units/mL penicillin and 100 μg/mL streptomycin in 5% CO2 at 37°C. The cell line was obtained from American Type Culture Collection (ATCC), expanded for two passages, and cryopreserved. A375 cells engineered to express MC1R (A375/MC1R) were cultured in media containing 300 μg/ml of G418 for selection.24 All experiments were performed with cells of passage number less than 25. Cells were authenticated as negative for mycoplasma by testing at the ATCC and were monitored by microscopy and confirmed to maintain morphological traits over subsequent passages.
DNA microarray analysis
RNA extracts of melanoma cell lines were analyzed using the Affymetrix U133A array platform. Data generated from these arrays have been published previously 36 and have been deposited in the NCBI’s Gene Expression Omnibus (GEO). Data are accessible using GEO Series accession GSE4845. For patients, affymetrix expression data for MC1R in patient tissue samples were compiled from publicly available datasets and the analysis was done as described before.37
Immunohistochemistry (IHC) of melanoma tissue microarray (TMA)
A TMA was constructed at the Moffitt Tissue Core containing patient tissue samples of formalin-fixed and paraffin-embedded (FFPE) cutaneous specimens, including normal skin, a range of different types of nevi, primary melanomas and melanoma metastases (Table 1). The same method was previously reported for construction of a Ewing sarcoma TMA at the Moffitt Tissue Core Facility 38, except the melanoma TMA has only one sample per case due to the large number of cases. No identifiable human subject information was associated with the melanoma TMA. Rabbit MC1R polyclonal antibody (GTX70735, GeneTex), 1:200 dilution, was used for staining with diaminobenzidine (DAB). The slides were scanned and scored as described before.25
Table 1.
IHC scoring of MC1R expression in patient tissue samples.
| Tissue type | n | Pathology score | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 6 | 9 | % = 4 | ||
| normal skin | 19 | 0 | 1 | 5 | 13 | 0 | 0 | 0 | 0 |
| compound nevi | 9 | 0 | 0 | 0 | 6 | 0 | 3 | 0 | 33 |
| junctional nevi | 5 | 0 | 0 | 0 | 4 | 0 | 1 | 0 | 20 |
| intradermal nevi | 7 | 0 | 0 | 0 | 5 | 0 | 2 | 0 | 29 |
| Clark’s, atypical, dysplastic nevi | 33 | 0 | 1 | 5 | 22 | 0 | 5 | 0 | 15 |
| Primary cutaneous melanoma (in situ) | 11 | 0 | 0 | 0 | 9 | 0 | 2 | 0 | 18 |
| Primary cutaneous melanoma (0.1–0.75 mm) | 12 | 0 | 2 | 1 | 5 | 0 | 4 | 0 | 33 |
| Primary cutaneous melanoma (0.75–1mm) | 13 | 0 | 0 | 3 | 7 | 0 | 3 | 0 | 23 |
| Primary cutaneous melanoma (1–2 mm) | 13 | 0 | 0 | 3 | 4 | 0 | 6 | 0 | 46 |
| Primary cutaneous melanoma (2–4 mm) | 12 | 0 | 0 | 1 | 3 | 1 | 6 | 1 | 67 |
| Primary cutaneous melanoma (>4 mm) | 14 | 0 | 1 | 1 | 1 | 0 | 8 | 3 | 79 |
| Primary mucosal melanoma | 11 | 0 | 1 | 1 | 3 | 0 | 4 | 2 | 55 |
| Melanoma in regional lymph nodes | 35 | 1 | 3 | 10 | 12 | 0 | 9 | 0 | 26 |
| Melanoma distant metastasis, M1 | 9 | 0 | 1 | 0 | 4 | 0 | 3 | 1 | 44 |
| Melanoma distant metastasis, M2 | 9 | 0 | 0 | 1 | 2 | 0 | 5 | 1 | 67 |
| Melanoma distant metastasis, M3 | 10 | 0 | 1 | 0 | 5 | 0 | 4 | 0 | 40 |
Intravital imaging of the dorsal skin-fold window chamber tumor xenograft model
A previously described 26 dorsal skin-fold window chamber tumor xenograft model was used to study the pharmacokinetics of extravasation, tumor penetration, and tumor cell binding and uptake of the MC1R specific ligand conjugated to Cy5 NIRF dye (MC1RL-Cy5), except that A375/MC1R or A375 parental melanoma cells were used to form a tumor and the cells were mixed with GFP-expressing microvessel fragments from rat.27
Seven days after implantation of microvessels and tumor cells, mice were intravenously injected with 100 μl of 5% 10,000 MW cascade blue dextran (Invitrogen) in sterile H2O to verify microvessel patency. Then, 5 nmol/kg of the MC1RL-Cy5 probe was injected into the tail vein. Confocal images of probe uptake into the melanoma tumor cells were continuously acquired prior to, during and after injection of probe using the Olympus FV1000 (MPE) Multiphoton Laser Scanning Microscope (Lisa Muma Weitz Advanced Microscopy and Cell Imaging facility at USF) using 4X U-Plan Apo 0.16NA and 25X XL Plan N 1.05 NA water immersion lenses. The presence of MC1RL-Cy5 was measured by excitation with a 635 nm wavelength IR laser and the emitted light was detected using a 655–755 nm filter.
Fluorescence image intensities were quantified using Image Pro Plus v6.2 (Media Cybernetics, Bethesda, MD). The histogram count/size selection tool was used to analyze groups of 3–4 cells in triplicate for 0 and 24 hour time points following ligand injection. A threshold was set (R 10–255; G 0–255; B 0–255) to quantify the pixels positive for presence of the MC1RL-Cy5 probe. The Cy5 (R) channel intensity area and average intensity values were evaluated to determine the mean, standard deviation and P-value by single tailed, paired Student’s T-test.
In vivo time-resolved fluorescence imaging and time-domain based fluorescence tomography
Female nu/nu mice 6–8 weeks old (Harlan Sprague Dawley, Inc., Minneapolis, IN) were injected subcutaneously with 1 × 106 MC1R expressing A375 cells in the right flank and parental cells in the left flank. Tumor volume was determined with calipers using the formula: volume = (length × width2)/2. Once tumors reached 500–800 mm3, 1–30 nmol/kg of the MC1R specific ligand conjugated to IRDye800CW from Li-Cor (MC1RL-800) in 100 μL sterile saline was injected into the tail vein. In vivo fluorescence lifetime images were acquired using the Optix-MX3 (Advanced Research Technologies, Inc. a subsidiary of SoftScan Healthcare Group, Saint-Laurent, Canada). Animals were positioned on a heating pad and anesthetized using isoflurane (flow 2–2.5 l/min). Images were acquired using a scan resolution of 1.5 mm and a 790 nm pulsed laser diode with 40 MHz frequency and 12 ns time window. Images were analyzed using Optix-MX3 Optiview Software (version 3.01). The mean tumor fluorescence signal was determined prior to injection then subtracted from mean normalized intensity values of post-injection ROIs and tomographic image reconstructions were generated using the time-domain information from fluorescence lifetime scans.
Ex vivo studies
Tumors and kidneys were excised and a ~1 mm center section was cut from each tumor while maintaining registration with the in vivo position of the tumor as best as possible. The center section was imaged ex vivo using both the Optix-MX3 and IVIS 200 (Caliper LifeSciences, Hopkinton, MA) imaging systems. For IVIS 200 acquisitions, the standard ICG excitation and emission filter set was used for imaging. After imaging, the crude center sections were fixed in formalin and embedded in paraffin for histology and fine sectioning. Formalin-fixed sections (5 μm) were IHC stained with MC1R primary antibody as described above for TMAs.
Biodistribution studies
Mice were imaged and euthanized at 2–72 h post-injection (3 nmol/kg) time-points. Tumors, kidneys and liver were excised, rinsed with PBS, blotted dry, and then imaged ex vivo with the Optix-MX3 as described above. Images were analyzed as described above.
Mathematical modeling
In this work, we developed a new computational multi-compartmental model to represent the accumulation and clearance of the ligand in the high MC1R-expressing tumor, kidney, bloodstream, and low MC1R-expressing tissue. This model was based on concepts introduced by Gabrielsson et al.28 We assumed conservation of mass of ligand, which would only be excreted through the kidneys. The volumes of kidneys, high MC1R-expressing tumor, blood, and mouse obtained from ex vivo experiments were used in the computational model to determine the size of each compartment.
The computational model of the tumor is composed of two compartments: the first represents the specific binding to the MC1R receptor, while the second represents non-specific tumor uptake. The specific binding compartment of the tumor is described by three constants: Kti, Kto and Tmax which are the rates of probe accumulation, clearance and the maximum concentration at saturation, respectively.
The non-MC1R specific tissue of the mouse (muscle, liver, etc.) was represented by one unique compartment, which accumulates and clears the ligand at rates Kni and Kno, respectively. These two constants were also used to describe the non-specific uptake compartments of the tumor and the kidneys.
The accumulation of ligand in the kidneys suggested that there might be some interaction between the ligand and kidney cells. To test this hypothesis, we included in the model a melanocortin-receptor specific binding compartment in the kidney, represented by the constants Kki, Kko and Kmax, meaning the rate of accumulation of ligand in the kidney, the rate of clearance of the ligand back to the bloodstream, and the maximum concentration of ligand at saturation, respectively. Both the specific and nonspecific compartments of the kidney were modeled to filter the ligand into the bladder at a rate, Kb.
The model was simulated by numerically solving a system of six different equations (Supplementary equation 1) with time steps of 0.005 h (18 s). We implemented a Genetic Algorithm (G.A.) using the Java library JGAP (http://jgap.sourceforge.net/) to fit the model parameters to the experimental measurements ex vivo and in vivo. Briefly, this algorithm creates a random group of possible fit solutions (500), and for a number of generations (1,000) it evaluates the goodness of fit of the different solutions, attributing the highest scores to those with the best fit (shortest distance between the simulated curves and actual measurements). The best solutions increase their frequency in the pool of possible solutions, while the lowest scores are removed. Spontaneous “mutations” of these parameter values gradually change these solutions in order to add diversity to the population, and increase the chances of finding the best possible fit. The final product of this genetic algorithm is the set of pharmacokinetic parameters (Table 2) which, when incorporated into the mathematical model, generates ligand distribution curves which best fit the ex vivo and in vivo measurements.
Table 2.
Parameter value estimates by the multi-compartmental pharmacokinetic computational model derived from ex vivo experiments.
| Parameter* | Mean±s.d. of 10 simulations |
|---|---|
| Kti (NU**.h)−1 | 2.7+1.4 |
| Kto (h)−1 | 26+12 |
| Kki (NU.h)−1 | 1E-2+6.8E-3 |
| Kb (h)−1 | 7.8E-2+1.4E-2 |
| VB (cm3) | 1.4 † |
| VT (cm3) | 0.8 cm3† |
| VK (cm3) | 0.9 cm3† |
| VN (cm3) | 25 cm3† |
| Kni (NU.h)−1 | 1.8+0.2 |
| Kno (h)−1 | 2.3+0.6 |
| TMAX (NU) | 130+9 |
| KMAX (NU) | 2.7E3+2.3E3 |
| Kko (h)−1 | 0.96+0.14 |
Each parameter is defined in the materials and methods section.
Denotes parameters whose values were obtained from weighing the mouse organs ex vivo, and were fixed in the beginnig of the simulation, thus no standard deviation exists. VB is the volume of blood, VT is the volume of the MC1R high expressing tumor, VK is the combined volume of both kidneys, and VN is the total volume of the mouse minus kidneys, blood and MC1R+ tumor.
NU stands for Normalized Unit of fluorescence measured, which is proportional to ligand concentration.
Statistics
Data are represented as mean ± s.d. All statistical analyses were performed with GraphPad Prism version 5.01. Unpaired Student’s t-test was used to determine the statistical significance of differences between two independent groups of variables. For all tests, a P ≤ 0.05 was considered significant.
RESULTS
MC1R expression in patient tissue samples
It has been estimated that 80% of malignant melanomas express high levels of MC1R.13 For further confirmation and to characterize mRNA expression in patient tissue samples, we analyzed publicly available DNA microarray data sets, which showed that MC1R mRNA expression was highly and generally expressed in a large fraction of melanomas (Figure 1A, note log scale). In contrast, MC1R expression was not elevated in other skin cancers, normal skin and organs involved in toxicity and drug clearance, i.e., heart, lung, spleen, liver and kidney.
Figure 1.
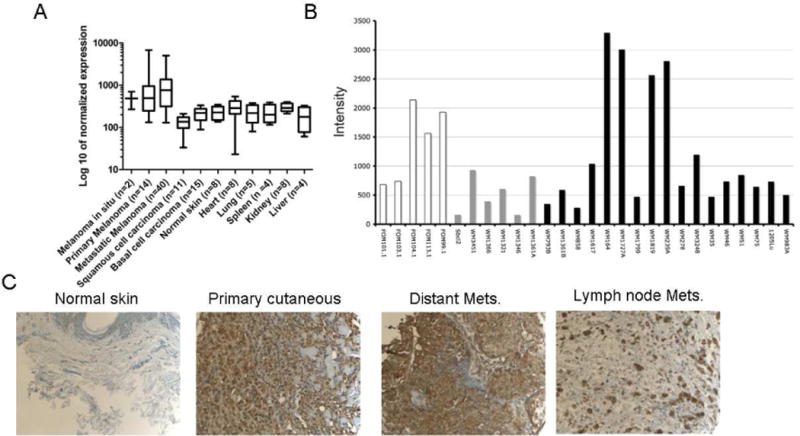
A) DNA microarray expression profile of MC1R in melanoma, other skin cancers and normal tissues. Data are represented as mean ± SD. Note the log10 scale. B) DNA microarray of primary human melanocytes (white), melanoma cell lines with the NRAS mutation (gray) and melanoma cell lines with the BRAF mutation (black). C) Representative IHC staining of MC1R in normal skin with a pathology score of 3 and different types of melanoma with pathology score of ≥4.
To determine MC1R protein expression in patient samples, IHC was performed on a melanoma tissue microarray containing 267 samples. Figure 1C shows representative staining in normal skin relative to staining in a primary cutaneous melanoma, distant metastasis and lymph node metastasis. None of the normal skin samples (n = 19) had staining with a pathology score of ≥4, i.e., homogeneous moderate to high staining (Table 1). Benign lesions (n = 65) had percentages of ≥4 staining ranging from 15–33%. A relationship between primary cutaneous melanoma lesion size and pathology score was observed, with smaller lesions ranging from melanoma in situ to lesions 1 mm in thickness scoring 18% to 33% ≥4, and lesions from 1 mm to >4 mm in thickness scoring 46–79% ≥4. Primary mucosal melanomas (n = 11) and melanoma distant metastases had scores ranging from 40% to 67% ≥4.
MC1R expression in melanoma cell lines
MC1R expression was examined on a previously published mRNA expression microarray of a panel of melanoma cell lines (Figure 1B) 29, 30 Moderate to high expression was observed in all primary melanocytes, and NRAS- mutant melanoma cell lines had low to moderate expression. In contrast, expression in BRAF-mutant melanoma cell lines was highly heterogeneous, with some lines exhibiting very low expression (WM 858, 793, 1799, 35) and some with extremely high expression (WM 164, 1727A, 1819, 239A). Notably, 3 of the 4 high expressing lines were metastatic: WM164, WM239A and WM1727A.
In vivo tumor cell uptake studies
We have recently demonstrated in vivo tumor selectivity and in vitro tumor cell uptake of NIRF dye labeled probes with high affinity and selectivity for MC1R. 34, 35 A375 melanoma cells transfected to stably overexpress MC1R with receptor number of 75,000 per cell were used as positive cells with high expression 24 and parental A375 cells with endogenous expression were used as low-expressing cells.44 The high expressing A375/MC1R cells were used herein to form dorsal-window chamber (DWC) xenografts for intravital confocal fluorescence microscopy to demonstrate tumor cell uptake in vivo.
Since standard microscopy (metal halide) excitation light sources are inefficient at longer wavelengths and the commercially available fluorescence microscope detectors are generally inefficient at 800 nm wavelengths, we used the previously reported Cy5 dye conjugate (MC1RL-Cy5) 24 for use in intravital fluorescence microscopy studies. The MC1RL-Cy5 probe has high affinity for MC1R, with a Ki of 0.3 ± 0.05 nM.24 The binding affinity is comparable to that of unlabeled ligand, 0.24 nM Ki.23
To study the distribution of the probe into tumors and tumor cells by intravital fluorescence microscopy, xenograft tumors were implanted under the glass cover of dorsal skin-fold window chambers (Figure 2A). The tumors were constructed using A375/MC1R cells mixed with GFP rat microvessels in type I collagen. Following a 5–7 day period of tumor growth, microvessel patency was verified by i.v. injection of blue dextran and regions of the tumor with patent GFP vessels were chosen for study (Figure 2B). Extravasation, tumor cell binding and uptake of the probe were observed by continuous confocal microscope acquisitions after i.v. injection of 5 nmol/kg MC1RL-Cy5 probe. Figure 2C shows the whole tumor surrounded by GFP microvessels at 24 h post-injection of probe (red) using low magnification (4X). Different post-injection timepoints are shown in Figure 2D using higher magnification (25X). Immediately following injection, the probe was observed to extravasate and penetrate into the tumor. Probe fluorescence was observed at the periphery of cells as early as 5 min after injection. At 5 min to 2 h, probe fluorescence increased on the tumor cell surfaces, with some apparent cell uptake. At 24 h post-injection, the tumor cells had fully internalized the probe and unbound circulating probe had mostly cleared from circulation within the tumor and significant fluorescence intensity was retained, p=0.025, relative to pre-injection images (Figure 2E).
Figure 2.
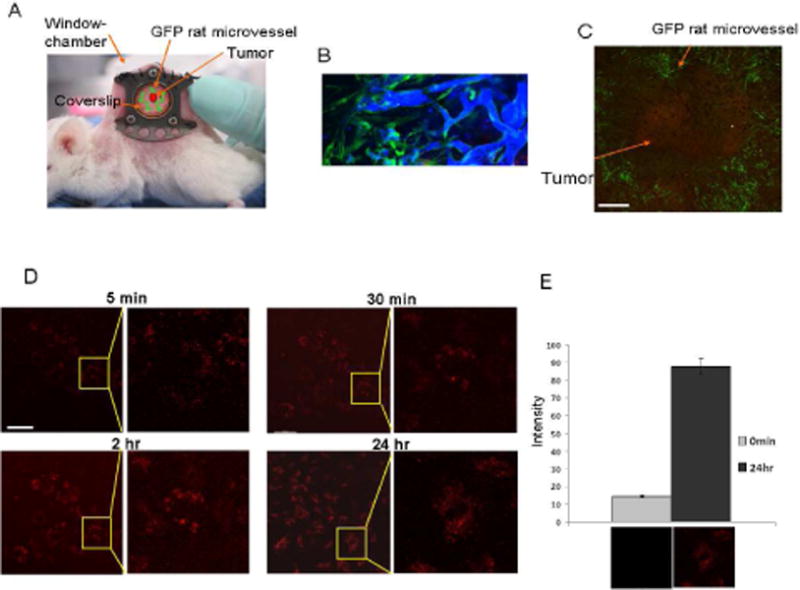
In vivo uptake studies of the MC1RL-Cy5 probe using the dorsal skin-fold window chamber xenograft tumor model for intravital confocal imaging. A) Mouse bearing a dorsal skin-fold window-chamber. B) Verification of GFP rat microvessel (green) patency following intravenous injection of Blue Dextran using confocal fluorescence microscopy. C) Intravital confocal microscope image of probe (red) labeling tumor cells 24 h after administration and green microvessels using 4X magnification. Bar = 1000μm. D) At 25X magnification (left panel for each time-point), probe fluorescence was detected on the cell surface by 5 min after i.v. injection of the probe, and was detected inside the cells by 24 h after injection. A close-up image (right panel for each time-point) is shown to better illustrate cell binding and uptake. Bar = 100μm. E) Quantification of the probe-related fluorescence signal before injection and at 24 hr after injection (P=0.025). Representative images are underneath each graph.
Heterogeneity of probe uptake into tumor
To investigate the heterogeneity of probe uptake in vivo, bilateral subcutaneous high and low MC1R expressing xenograft tumors (A375/MC1R engineered and A375 endogenous) were established.24 After tumor growth to approximately 500–800 mm3, the previously reported IRDye800CW conjugate (MC1RL-800) 24 was injected intravenously and fluorescence accumulation was monitored over time. The MC1RL-800 probe has high affinity for MC1R, with a 0.4 ± 0.1 nM Ki24 which is comparable to that of unlabeled ligand, 0.24 nM Ki.23 At 2h post-injection, A375 tumors with low MC1R expression had significantly lower normalized fluorescence signal compared to A375/MC1R tumors (P≤0.05, n=3) (Figure 3A). In vivo tomographic slices were generated based on the fluorescence time-domain shift showing that probe distribution is heterogeneous in different slices of the tumor, where it has the highest accumulation ~2 mm depth from the top of the ~5 mm thick tumor (Figure 3B).
Figure 3.
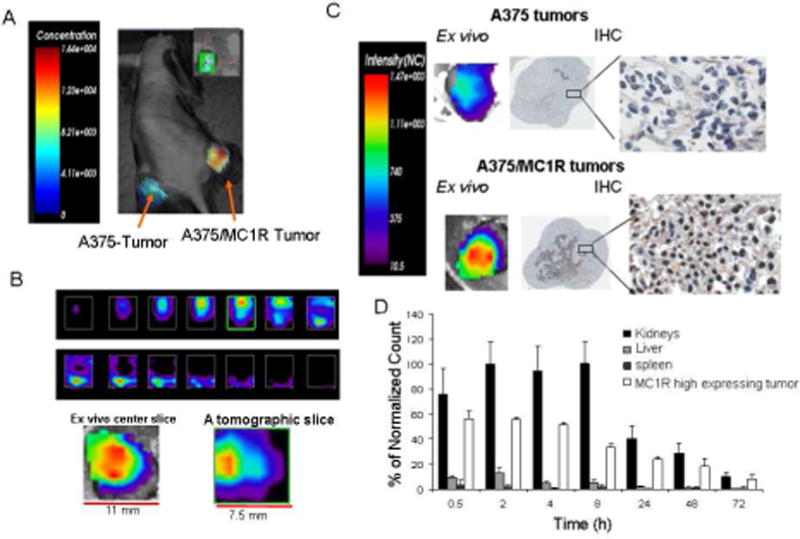
In vivo and ex vivo uptake of MC1RL-800 into tumor. A) Representative time-domain based fluorescence tomography image of MC1RL-800 accumulation in mouse xenograft tumors 2 hours after intravenous injection of 5 nmol/kg probe. A375 cells that constitutively express low levels of MC1R were used to form the low-expressing tumor (left flank) and A375/MC1R cells were used to form the high expressing tumor (right flank). Only the tumor areas were scanned. B) Top: In vivo tomographic slices (0.5 mm thickness) through the high-expression tumor from top to bottom (ordered from left to right). Below: Ex vivo surface radiance image of a center section from the high-expressing tumor (left) compared to the center slice from the in vivo tomographic image of the same tumor. An effort was made to maintain registration of the ex vivo section with the in vivo image slice. All analyses were performed using the Optix-MX3 Optiview Software (version 3.01). C) Ex vivo images of center sections from low- and high-expressing tumors with corresponding IHC staining of MC1R. D) Biodistribution of MC1RL-800 probe determined by quantification of ex vivo fluorescence values from the tumor, kidneys and liver at different time-points post-injection of 3 nmol/kg probe. No signal was detected in the heart, lung, brain and other organs (not shown). The values were normalized as percentage of the highest signal.
Ex vivo images of the corresponding center sections of the high- and low-MC1R expressing tumors confirmed the in vivo results. IHC staining confirmed the high and low MC1R expression in the two tumor types, and areas with the highest IHC staining corresponded to areas with the highest fluorescence signal (Figure 3C).
Biodistribution studies
For biodistribution studies, mice bearing high expressing (A375/MC1R) tumors were injected with probe (n=3), and tissue distribution of fluorescence signal was determined after removing tumors and organs from 30 min to 72 h post-administration (Figure 3D). At 30 min post-injection, probe was retained at relatively high levels in the MC1R high expressing tumor. At early time points, probe accumulation in the kidneys was significantly higher than in the tumors, e.g. at 2 h after injection, kidney signal was 2 fold higher (P<0.001). However, the difference in accumulation was no longer significant by 72 h after injection, and the probe was fully cleared from both tumors and kidneys by 96 h after injection. MC1RL-800 did not accumulate in the liver and no signal was detected in the other organs, such as spleen, heart, brain, etc.
Estimation of whole animal pharmacokinetics parameters
A compartmental mathematical model was used to estimate the pharmacokinetics of probe uptake and clearance. The model includes tumor, kidney and whole mouse compartment volumes, and assumes mass conservation of the probe (Figure 4A). This model was used to account for the interference of tumor probe release in the uptake and clearance dynamics of blood and kidneys. The fit of ex vivo data for the 3 nmol/kg dosage in Figure 4B (left) shows a statistically significant correlation between experimental and simulated dynamics in two of the three measurable compartments: tumor (slope=0.9298, R2=0.9163), kidneys (slope=0.8371, R2=0.9370) and negative tissue (slope=0.3371, R2=0.7040), (Supplemental Figure S1). The poor correlation between computer simulations and actual measurements in the negative tissue was mainly due to the fluorescence levels measured being closer to background.
Figure 4.
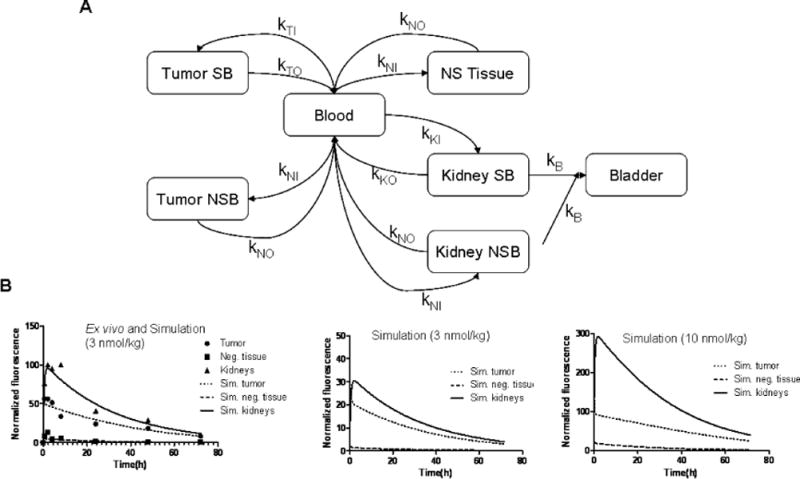
Mathematical modeling of MC1RL-800 uptake in the body. A) Graphical representation of the multi-compartment model used in this study. The targeted probe injected into the blood stream diffuses to all tissues in the mouse (NS Tissue), preferentially accumulating in the positive tumor in a specific and non-specific manner (Tumor SB and Tumor NSB). Targeted probe also accumulates in the kidney in a specific and non-specific way (Kidney SB and Kidney NSB), and is filtered into the bladder. B) Multi-compartmental model simulations of the pharmacokinetic distribution of probe over time at a range of dosages. On the left, the ex vivo imaging data for an injection of 3 nmol/kg were used to determine the model parameters with the best fit, the simulation (lines) overlay the data (points). In the middle and right are simulations of lower 1 nmol/kg and higher (10 nmol/kg) injected doses, respectively.
The parameters obtained from the model (Table 2 and Supplementary Figure S1) help clarify the dynamics of this system: the rate of accumulation of probe is faster in the high expressing tumor compared to the kidneys in the specific binding compartments (Table 2) and the tumor has higher affinity for the ligand (Kd=8.7±0.5) than the kidneys (Kd=40±17) (Supplementary Fig. S1), which explains why the model predicts that the probe reaches peak concentration in the tumor before reaching peak in the kidneys (30 min for the tumor, 2 h for the kidneys). Therefore, the PK computational model predicted the presence of receptors in the kidneys with a lower affinity [this prediction was confirmed by an experiment that will be explained below, Figure 5C].
Figure 5.
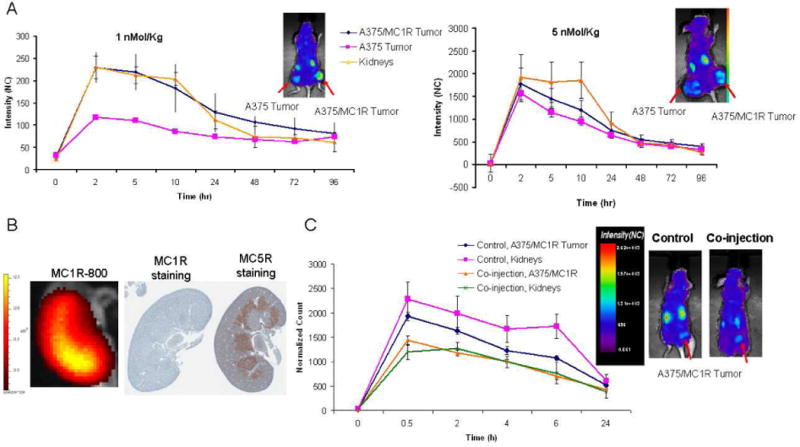
In vivo pharmacokinetics study of MC1RL-800. A) Plots of normalized fluorescence values for high- and low-expressing tumors and kidneys over a longitudinal time-course following administration of low (1 nmol/kg) and high (5 nmol/kg) doses of probe. Insets show representative normalized fluorescence intensity map overlays on visible light images of mice bearing bilateral flank xenograft tumors, 2 hours post intravenous injection of the probe that were acquired using the Optix-MX-3 instrument. B) A representative ex vivo fluorescence image (left) of kidney 2 hr after probe injection acquired using the IVIS 200 instrument and corresponding IHC stained sections of MC1R (center) and MC5R (right). C) Co-injection of 1 nmol/kg of MC5R specific ligand and 5 nmol/kg of MC1RL-800 (right image) to reduce kidney uptake of the probe. Inset shows representative normalized fluorescence intensity map overlays, 30 min post intravenous injection of the probe (control, left image) and probe plus MC5R ligand co-injection (right image). Images were acquired using the Optix-MX-3 instrument. Data represent mean ± s.d. NC: Normalized Counts.
The actual clearance of probe observed in the tumor and kidneys is the combination of the dynamics between negative tissue, tumor, kidneys and blood. In the early moments after injection, most of the probe is present in the blood. At this point, the accumulation of probe in the tumor, kidneys and negative tissue is determined by tissue-specific kinetic properties. However, once equilibrium is reached, the only outlet for this system is the rate of transport of the probe from the kidney into the bladder, Kb, which should lead to a clearance with a half-life of ~9 h. The actual 20 h half-life observed in the kidneys is due to further uptake of probe from the MC1R expressing tumors and negative tissue, due to clearance through the blood, which in turn determines the clearance of these tissues (22 h for the tumor and 16 h for negative tissue).
Effect of probe concentration on uptake and clearance
To determine whether the amount of the MC1RL-800 probe injected had an effect on kidney uptake and retention, different amounts of the probe were injected i.v. and the pharmacokinetics of uptake and clearance monitored (Supplementary Figure S2). As expected, the lower dose (1 nmol/kg) provided greater discrimination and more rapid renal clearance compared to the higher dose (5 nmol/kg) (Figure 5A). Therefore, the renal uptake can be reduced by injection of lower probe concentrations (e.g. 1 nmol/kg) which has higher affinity against MC1R, compared to MC5R (Ki values of the ligand against MC1R and MC5R are 0.24 nM and 46 nM, correspondingly).23 MC5R is expressed in kidney mouse (see below for details). Furthermore, with either dose, the measurable tumor and kidney half-lives were approximately 20 h for the kidneys and 22 h for the tumors. Following specific uptake of probe into cells, the fluorescent dye may be quenched in the acidic lysosomal environment. Hence, this half-life may represent a combination of agent clearance into the blood and acid quenching of dye following cellular internalization. This half-life is also dependent on the clearance of the ligand through the kidneys, into the bladder, which in the computational model was denominated Kb (Table 2). We plan to further characterize the rate of tumor clearance in future studies using Eu-labeled ligands, which cannot be acid-quenched.
The compartmental math model simulations also confirm the behavior observed in vivo when the probe is injected at increasingly higher concentrations: at low concentrations (Figure 4B, middle) the tumor and kidneys show similar probe levels, but at higher probe injection concentrations (Figure 4B, right), the kidney becomes increasingly brighter while the signal measured in the positive tumor and negative control tissue in the mice become closer (Figure 4B and Supplementary Figure S2).
Effect of differential marker isoform affinities and expression levels on probe uptake and clearance
As it was mentioned earlier, the computational model predicted that the ligand has lower affinity for receptors in the kidneys compared to the receptors in tumors. As both human and mouse kidneys express MC5R 29, 30 and have little or no expression of MC1R, we hypothesized that the different affinity in kidneys and tumor is occurring via off-target binding of the probe to MC5R. IHC staining of a mouse kidney section shows strong expression of MC5R in the medulla (Figure 5B, right panel), and ex vivo fluorescence imaging of the corresponding kidney shows probe retention in the area of the medulla (Figure 5B, left panel). To test the hypothesis, 1 nmol/kg of an MC5R-selective ligand, H-Tyr-Val-Nle-Gly-His-DNal(2′)-Arg-DPhe-Asp-Arg-Phe-Gly-NH2, 23 was injected. This agent has Ki values of 5.6 and 0.71 nM for MC1R and MC5R, respectively. Co-injection of the MC5R-specific compound along with 5 nmol/kg of MC1RL-800 significantly reduced renal uptake, i.e. 36% decrease at 2 h after injection (p<0.05), compared to control animals (Figure 5C), and the A375/MC1R tumors also had significantly lower signal following blocking (28% decrease at 2 h). Thus, it appears that the renal uptake of MC1RL-800 is due to off-target binding, which can be pharmacologically blocked.
In agreement, the computational pharmacokinetic model estimated the Kd (probe concentration needed for 50% saturation of target receptors) in the tumor (MC1R) and kidneys (MC 5R) to be ~8.7 and ~40 units of normalized fluorescence, respectively, indicating that the tumor affinity for the probe is 5-fold higher. On the other hand, it also suggested a higher number of receptors in the kidney (6-fold difference between Kmax and Tmax, the maximum concentrations for specific binding of probe in the kidneys and tumor, respectively, Table 2), which explains why images of higher concentration probe injections show kidneys that are brighter than the positive tumor. The tumor specific binding compartment saturates at a normalized probe concentration of ~160, while the kidneys’ specific binding compartment saturates at approximately 1,000. This explains why at higher probe concentrations, 10 nmol/kg or more, the peak of fluorescence of the kidneys is significantly higher than in the tumors. At higher doses (>30 nmol/kg), nonspecific uptake in all organs increases significantly and obfuscates the measurement of specific binding in the tumor, and thus, such high concentrations should be avoided.
DISCUSSION
It has been previously reported that up to 80% of melanoma cell lines express high levels of MC1R.13 Our cell line data suggest that this may be an optimistic estimate, as only 67% of the cell lines had comparable mRNA expression levels compared to primary human melanocytes and only 19% had expression higher than any primary melanocyte. Regardless, these data were obtained using human cell lines, rather than patient tissue samples. It is not uncommon for protein expression in cultured cell lines to differ from that of tumors.31
Previous reports of MC1R expression in melanoma patient tissue samples have been as high as 95%.46, 47 However, these studies have taken a “present/not present” binary approach to data scoring. In this publication, we have sought to rigorously quantify MC1R expression in patient tumor and nonneoplastic samples with the assistance of a dermatopathologist and showed that MC1R protein expression is moderate to high in 20–80% of primary melanoma tumors and 25–70% of melanoma metastases, depending on the classification (Table 1). The nonneoplastic “normal” skin samples had relatively low or sparse expression of MC1R (100% had a pathologist score less than 4). Our findings, combined with previous reports, suggest that MC1R may be useful as a marker for specific targeting of melanoma in a large subset of patients.
We recently described a high affinity and selective MC1R ligand 4-phenylbutyryl-His-DPhe-Arg-Trp-Gly-Lys(hex-5-ynoyl)-NH223, that was designed for attachment via a C-terminal lysine. Ki values for this ligand against MC1R and MC5R were 0.24 nM and 46 nM, respectively. The attachment and lysine insertion did not significantly alter ligand binding affinity or specificity. We have also described the attachment of this ligand to a NIRF dye with an excitation wavelength of 773 nm and emission wavelength of 792 nm (IRDye800CW Maleimide, LI-COR) to make 4-phenylbutyryl-His-DPhe-Arg-Trp-Gly-Lys(Mpr-IRDye800CW)-NH2 (MC1RL-800) and confirmed the specificity of the probe by blocking experiment.24 Since longer wavelengths have lower tissue absorption and autofluorescence, this conjugate was used for in vivo fluorescence imaging studies. The conjugate retained high binding affinities comparable to the unmodified ligand.24 The use of small peptidomimetic ligands as carriers for the delivery of imaging or therapeutic moieties to diseased tissues offers several advantages, such as high biostability, easy synthesis and modification, faster blood clearance, high affinity and specificity, and low toxicity and immunogenicity.32
In this study, tumor cell uptake was evaluated as an important factor for the optimal utility of targeted imaging and therapeutic agents using intravital confocal fluorescence microscopy and showed the rapid extravasation and penetration of probe into the tumor, binding to the tumor cell surface at early time-points, and the complete cellular internalization and extracellular clearance from the tumor by 24 h after injection.
Endogenous MC1R expression ranges from several hundred to around 10,000 receptors per cell in different human cell lines.13 Our in vivo results indicate that the MC1RL-800 imaging probe is specifically retained in MC1R expressing tumors, and normalized fluorescence counts distinguished A375 (400 ± 93 receptors/cell) from A375/MC1R tumors with engineered expression (75,000 receptors/cell). Although the difference in fluorescence values between high and low MC1R expressing tumors was not proportional to receptor numbers estimated by in vitro assays, it has been observed that protein expression in cell culture can be different when compared to expression in xenograft tumors made using the same cells.24 Therefore, in vivo expression levels can be estimated by scaling. We have also reported the specific in vitro uptake of the MC1RL-Cy5 probe into B16F10 cells and in vivo retention of the MC1RL-800 probe in B16F10 tumors.24 B16F10 cells and tumors are melanotic, and endogenously express MC1R at high levels (~22,000 ± 4200 MC1R/cell).
The in vivo biodistribution of MC1RL-800 probe indicates high tumor and kidney uptake as early as 15 min after injection. The kidney uptake was significantly decreased by co-injection of probe with an unlabeled ligand that has higher affinity for MC5R compared to MC1R, or by injection of lower probe concentrations (e.g. 1 nmol/kg). These observations are in agreement with the in vitro binding data, confirming higher affinity of probe to MC1R compared to MC5R, and IHC data confirming MC5R expression in mouse kidney. When using non-toxic versions of the probe, e.g., imaging agents, kidney accumulation should not be a serious problem, as complete clearance from the kidneys was observed within a few days (~96 h) after injection. However, the ligand may still have potential for tumor-specific delivery of cytotoxic agents through careful dose determination and use of MC5R specific blocking agents, or by regional administration in the form of isolated limb perfusion or infusion, which keeps the kidneys out of drug circulation and is used to treat in-transit melanoma metastasis.33
The intra-tumor heterogeneity of the image reconstructions was confirmed by comparing the center slice from the in vivo images with the surface fluorescence measurement from the center section of the tumor ex vivo (Figure 3B). While not in perfect registration, the two images were similar in distribution of signal. Future studies will explore the possibility of using in vivo tomographic images for quantification of probe retention for longitudinal biodistribution studies, potentially eliminating the need to sacrifice multiple animals at a series of time points.
Our multi-compartmental mathematical model correctly fit the empirical data of the dynamic uptake and clearance of probe from tumor, kidney and melanocortin-receptor negative tissue, generating rate constants and maximum probe concentrations in the tumor and kidneys. The parameter values generated were independent of the administered probe concentration (dosage). By using the computational model, we were able to estimate the pharmacokinetic parameters of different tissues using measurements obtained from a complex in vivo system, and use these parameters to understand the interactions between ligand and tumor, and ligand and host. This information will be invaluable for optimizing dosing and time points in future research with clinical trials.
The multi-compartmental mathematical model presented in this work allows for a better understanding of the behavior of a targeted imaging probe in the mouse body, and quantitatively answers important questions such as: (1) how much probe should be injected in animal models and in future patients to maximize the tumor signal while minimizing non-specific uptake in negative tissue, (2) what is the accumulation rate of a targeted probe in tumors, (3) what is the rate a targeted probe can clear from tumor and organs, and (4) what are the ideal time points for imaging. Therefore, the mathematical model developed in this study has potential to aid in the translation of a targeted drug or imaging agent from lab to the clinic.
In conclusion, the imaging probe developed in this study has potential for the intraoperative detection of regional lymph node involvement, and for margin detection in real-time during surgical removal of primary and metastatic melanoma lesions, potentially improving patient outcomes. In addition, the MC1R-specific ligand could be used to develop a PET or SPECT tracer for the noninvasive identification of regional lymph node metastasis and distant metastasis. The process reported herein that includes characterization of marker expression in patient samples, development of a marker-specific molecular imaging probe, and in vitro, in vivo and in silico characterization of probe tumor specificity, pharmacokinetics and biodistribution is a framework that can be generally applied to pre-clinical development of targeted imaging and therapeutic agents.
Supplementary Material
Acknowledgments
The authors wish to acknowledge the Molecular Genomics, Tissue, Analytic Microscopy, and Small Animal Modeling and Imaging Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute; and the Comparative Medicine and Lisa Muma Weitz advanced Microscopy and Cell Imaging facilities at the University of South Florida for their technical support. We also thank LI-COR Biosciences for providing IRDye800CW. This research was founded by NIH/NCI R01CA097360.
Abbreviations
- MC1R
Melanocortin 1 receptor
Footnotes
Supporting Information Available
Scatterplot of experimental and simulations of model-predicted accumulation of ligand for positive tumor, negative tissue and kidneys, pharmacokinetic profiles of different probe concentrations and multi-compartmental whole body pharmacokinetic equations. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Thompson JF, Uren RF. Lymphatic mapping in management of patients with primary cutaneous melanoma. Lancet Oncol. 2005;6:877–85. doi: 10.1016/S1470-2045(05)70423-X. [DOI] [PubMed] [Google Scholar]
- 3.Leung AM, Hari DM, Morton DL. Surgery for distant melanoma metastasis. Cancer J. 2012;18:176–84. doi: 10.1097/PPO.0b013e31824bc981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong SL, Balch CM, Hurley P, Agarwala SS, Akhurst TJ, Cochran A, Cormier JN, Gorman M, Kim TY, McMasters KM, Noyes RD, Schuchter LM, Valsecchi ME, Weaver DL, Lyman GH. Sentinel Lymph Node Biopsy for Melanoma: American Society of Clinical Oncology and Society of Surgical Oncology Joint Clinical Practice Guideline. J Clin Oncol. 2012 doi: 10.1200/JCO.2011.40.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caudle AS, Ross MI. Metastasectomy for stage IV melanoma: for whom how much? Surg Oncol Clin N Am. 2011;20:133–44. doi: 10.1016/j.soc.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Kalady MF, White RR, Johnson JL, Tyler DS, Seigler HF. Thin melanomas: predictive lethal characteristics from a 30-year clinical experience. Ann Surg. 2003;238:528–35. doi: 10.1097/01.sla.0000090446.63327.40. discussion 535–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cigna E, Gradilone A, Ribuffo D, Gazzaniga P, Fino P, Sorvillo V, Scuderi N. Morbidity of selective lymph node biopsy for melanoma: meta-analysis of complications. Tumori. 2012;98:94–8. doi: 10.1177/030089161209800113. [DOI] [PubMed] [Google Scholar]
- 8.Frangioni JV. New technologies for human cancer imaging. J Clin Oncol. 2008;26:4012–21. doi: 10.1200/JCO.2007.14.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho Shon IA, Chung DK, Saw RP, Thompson JF. Imaging in cutaneous melanoma. Nucl Med Commun. 2008;29:847–76. doi: 10.1097/MNM.0b013e32830439fb. [DOI] [PubMed] [Google Scholar]
- 10.Wechalekar K, Sharma B, Cook G. PET/CT in oncology–a major advance. Clin Radiol. 2005;60:1143–55. doi: 10.1016/j.crad.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Monzon JG, Dancey J. Targeted agents for the treatment of metastatic melanoma. Onco Targets Ther. 2012;5:31–46. doi: 10.2147/OTT.S21259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 13.Siegrist W, Solca F, Stutz S, Giuffre L, Carrel S, Girard J, Eberle AN. Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells. Cancer Res. 1989;49:6352–8. [PubMed] [Google Scholar]
- 14.Yang Y. Structure, function and regulation of the melanocortin receptors. Eur J Pharmacol. 2011;660:125–30. doi: 10.1016/j.ejphar.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai M, Varga EV, Stankova M, Mayorov A, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. Cell signaling trafficking of human melanocortin receptors in real time using two-photon fluorescence confocal laser microscopy: differentiation of agonists antagonists. Chem Biol Drug Des. 2006;68:183–93. doi: 10.1111/j.1747-0285.2006.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koikov LN, Ebetino FH, Solinsky MG, Cross-Doersen D, Knittel JJ. Sub-nanomolar hMC1R agonists by end-capping of the melanocortin tetrapeptide His-D-Phe-Arg-Trp-NH(2) Bioorg Med Chem Lett. 2003;13:2647–50. doi: 10.1016/s0960-894x(03)00552-3. [DOI] [PubMed] [Google Scholar]
- 17.Mayorov AV, Han SY, Cai M, Hammer MR, Trivedi D, Hruby VJ. Effects of macrocycle size rigidity on melanocortin receptor-1-5 selectivity in cyclic lactam alpha-melanocyte-stimulating hormone analogs. Chem Biol Drug Des. 2006;67:329–35. doi: 10.1111/j.1747-0285.2006.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chhajlani V. Distribution of cDNA for melanocortin receptor subtypes in human tissues. Biochem Mol Biol Int. 1996;38:73–80. [PubMed] [Google Scholar]
- 19.Labbe O, Desarnaud F, Eggerickx D, Vassart G, Parmentier M. Molecular cloning of a mouse melanocortin 5 receptor gene widely expressed in peripheral tissues. Biochemistry. 1994;33:4543–9. doi: 10.1021/bi00181a015. [DOI] [PubMed] [Google Scholar]
- 20.Hruby VJ, Cai M, Cain JP, Mayorov AV, Dedek MM, Trivedi D. Design synthesis biological evaluation of ligands selective for the melanocortin-3 receptor. Curr Top Med Chem. 2007;7:1107–19. doi: 10.2174/156802607780906645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, Balthasar N, Williams KW, Elmquist JK. Melanocortin 4 receptors reciprocally regulate sympathetic parasympathetic preganglionic neurons. Cell. 2013;152:612–9. doi: 10.1016/j.cell.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biebermann H, Kuhnen P, Kleinau G, Krude H. The neuroendocrine circuitry controlled by POMC, MSH, and AGRP. Handb Exp Pharmacol. 2012:47–75. doi: 10.1007/978-3-642-24716-3_3. [DOI] [PubMed] [Google Scholar]
- 23.Barkey NM, Tafreshi NK, Josan JS, De Silva CR, Sill KN, Hruby VJ, Gillies RJ, Morse DL, Vagner J. Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. J Med Chem. 2011;54:8078–84. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tafreshi NK, Huang X, Moberg VE, Barkey NM, Sondak VK, Tian H, Morse DL, Vagner J. Synthesis characterization of a melanoma-targeted fluorescence imaging probe by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Bioconjug Chem. 2012;23:2451–9. doi: 10.1021/bc300549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tafreshi NK, Bui MM, Bishop K, Lloyd MC, Enkemann SA, Lopez AS, Abrahams D, Carter BW, Vagner J, Grobmyer SR, Gillies RJ, Morse DL. Noninvasive detection of breast cancer lymph node metastasis using carbonic anhydrases IX XII targeted imaging probes. Clin Cancer Res. 2012;18:207–19. doi: 10.1158/1078-0432.CCR-11-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 2006;66:5216–23. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 27.Kirkpatrick ND, Andreou S, Hoying JB, Utzinger U. Live imaging of collagen remodeling during angiogenesis. Am J Physiol Heart Circ Physiol. 2007;292:H3198–206. doi: 10.1152/ajpheart.01234.2006. [DOI] [PubMed] [Google Scholar]
- 28.Gabrielsson JL, Weiner DL. Methodology for pharmacokinetic/pharmacodynamic data analysis. Pharm Sci Technolo Today. 1999;2:244–252. doi: 10.1016/s1461-5347(99)00162-5. [DOI] [PubMed] [Google Scholar]
- 29.Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M, Schadendorf D, Dummer R. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006;19:290–302. doi: 10.1111/j.1600-0749.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- 30.Smalley KS, Contractor R, Nguyen TK, Xiao M, Edwards R, Muthusamy V, King AJ, Flaherty KT, Bosenberg M, Herlyn M, Nathanson KL. Identification of a novel subgroup of melanomas with KIT/cyclin-dependent kinase-4 overexpression. Cancer Res. 2008;68:5743–52. doi: 10.1158/0008-5472.CAN-08-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stein WD, Litman T, Fojo T, Bates SE. A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: comparing solid tumors with cell lines comparing solid tumors from different tissue origins. Cancer Res. 2004;64:2805–16. doi: 10.1158/0008-5472.can-03-3383. [DOI] [PubMed] [Google Scholar]
- 32.Bullok KE, Gammon ST, Violini S, Prantner AM, Villalobos VM, Sharma V, Piwnica-Worms D. Permeation peptide conjugates for in vivo molecular imaging applications. Mol Imaging. 2006;5:1–15. [PubMed] [Google Scholar]
- 33.Creech O, Jr, Krementz ET, Ryan RF, Winblad JN. Chemotherapy of cancer: regional perfusion utilizing an extracorporeal circuit. Ann Surg. 1958;148:616–32. doi: 10.1097/00000658-195810000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.