

Abstract
Infectious diseases are known as one of the most life-threatening disabilities worldwide. Approximately 13 million deaths related to infectious diseases are reported each year. The only way to combat infectious diseases is by chemotherapy using antimicrobial agents and antibiotics. However, due to uncontrolled and unnecessary use of antibiotics in particular, surviving bacteria have evolved resistance against several antibiotics. Emergence of multidrug resistance in bacteria over the past several decades has resulted in one of the most important clinical health problems in modern medicine. For instance, approximately 440,000 new cases of multidrug-resistant tuberculosis are reported every year leading to the deaths of 150,000 people worldwide. Management of multidrug resistance requires understanding its molecular basis and the evolution and dissemination of resistance; development of new antibiotic compounds in place of traditional antibiotics; and innovative strategies for extending the life of antibiotic molecules. Researchers have begun to develop new antimicrobials for overcoming this important problem. Recently, platensimycin – isolated from extracts of Streptomyces platensis – and its analog platencin have been defined as promising agents for fighting multidrug resistance. In vitro and in vivo studies have shown that these new antimicrobials have great potential to inhibit methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and penicillin-resistant Streptococcus pneumoniae by targeting type II fatty acid synthesis in bacteria. Showing strong efficacy without any observed in vivo toxicity increases the significance of these antimicrobial agents for their use in humans. However, at the present time, clinical trials are insufficient and require more research. The strong antibacterial efficacies of platensimycin and platencin may be established in clinical trials and their use in humans for coping with multidrug resistance may be allowed in the foreseeable future.
Keywords: drug resistance, antibiotics, bacterial infections, platensimycin, platencin
Introduction to managing drug resistance in the treatment of bacterial infections
Infectious diseases that are caused by bacteria, fungi, parasites, and viruses can arise from the invasion of a host cell by one of these pathogens. Infectious diseases are one of the most life-threatening groups of disabilities worldwide. According to World Health Organization data, approximately 13 million deaths related to infectious diseases have been reported worldwide. It is also known that infectious diseases cause 45% of all deaths in developing countries. At present, six groups of disabilities are considered the most lethal infectious diseases since they lead to 90% of total deaths related to infectious diseases: acute respiratory infections, acquired immunodeficiency syndrome, diarrheal diseases, tuberculosis (TB), malaria, and measles.1,2 Besides these illnesses, other tropical diseases, some sexually transmitted disabilities, and nosocomial infections can also threaten health of humans and cause death. The only way to combat infectious diseases is by chemotherapy using antimicrobial agents and antibiotics. Antimicrobial agents have been used for >70 years in the treatment of infectious diseases. These agents have great potential to reduce illnesses and treat affected patients. Since their first introduction, use of antibiotics has become widespread around the world because of their significant effectiveness on microbial agents.3 However, due to their uncontrolled and unnecessary use, bacteria have evolved resistance against several antibiotics.4 This situation has led to a reduction in effectiveness of conventional therapy (antibiotics). The development of resistance in bacteria is thought to be due to natural selection/adaptation. According to this mechanism, susceptible bacteria are inhibited and do not divide or die when exposed to an antimicrobial agent, while remaining resistant bacteria maintain viability. Resistant organisms multiply and spread to different geographic regions of the world. Moreover, resistant organisms can also transfer their resistant genes to susceptible individuals and make them more resistant to antimicrobial agents.5,6 Natural selection of resistant organisms occurs so rapidly in bacteria that some antibiotics have lost effectiveness in bacteria within a few months after its administration.7
Currently, antibiotic resistance of microbial agents has become a serious global concern. For instance, about 440,000 new cases of multidrug-resistant (MDR) TB are reported every year, leading to the deaths of 150,000 people worldwide.8 The prevalence of Staphylococcus aureus strains that are resistant to methicillin has reached high levels worldwide.9 The number of methicillin-resistant S. aureus (MRSA) strains has increased at a rate of 40% in the last 10 years.10 Since S. aureus is one of the most commonly seen causative agents of nosocomial infections, MRSA has recently become the most fatal bacteria in intensive care units of hospitals. Similarly, the rate of resistant Streptococcus pyogenes strains worldwide are 80% for erythromycin and 50% for penicillin, which are otherwise known as effective antibiotics.11 Another big problem related to drug resistance has emerged for malaria. It is known that 30% of all people worldwide are at risk of malarial infection, and every year 1.5 million people infected with malaria die worldwide.12 Moreover, this number is expected to increase in the future since Plasmodium spp. have evolved resistance against chloroquine, which had been the most effective drug used in treatment of this illness. All of these data reveal that drug resistance is one of the most important risks that threaten the health of the human population. In response, researchers have begun to develop new approaches to overcome this important problem. Among these approaches, combined use of conventional antibiotics composes one of the preliminary efforts. This application depends on enhancement of the mode of action mechanisms in addition to decreasing toxicity by using low dosages of different kinds of antibiotics with distinct effects on bacteria.13,14 Another approach is the use of plant derivatives, such as extracts and essential oils, and their applications combined with current antibiotics.15,16 On the other hand, some groups have focused on the molecular mechanisms of resistance and have targeted the prevention of the expression of resistance genes within bacteria.17 There are also efforts directed toward the synthesis of new antibiotics and antimicrobial agents. Researchers are developing new drugs with different mechanisms of action that are expected to replace current antibiotics. One of these new synthesized antibiotics is platensimycin, with its promising antibacterial efficacy. This review considers the threat of drug resistance in the world in detail and then introduces effective mechanisms, pharmacologic properties, efficacy, and clinical trials of platensimycin for its use in the clinical setting.
Drug resistance in significant pathogens
Antimicrobial resistance of pathogens is increasing yearly. Recently, vancomycin-resistant Enterococcus, MDR TB, MRSA, and amantadine/rimantadine-resistant and oseltamivir-resistant influenza virus have been a serious public health threat.18 Nearly every major pathogen has obtained resistance to a therapeutic agent. Resistant pathogens can lead to serious infections. They are managed with the few alternative drugs available for treatment. Streptococcus pneumoniae is an example of these criteria. In 2006, national surveys of invasive pneumococcal disease in the USA were 5,000 deaths and 41,400 cases. Meningitis caused 6.1% of cases, and bacteremia without focus caused 23.0%.19 The case/fatality ratio may be higher than 25% for specific high-risk groups with meningitis and bacteremia despite proper treatment.20 In 1994, drug-resistant S. pneumoniae became a nationwide notifiable disease in the USA.21 In 2006, 21.6% of the resistant isolates were nonsusceptible to erythromycin, 22.7% were nonsusceptible to trimethoprim/sulfamethoxazole, and 25.6% were nonsusceptible to penicillin.22 Chloroquine-resistant malaria is a worldwide problem of public health importance; 41.0% of the world’s population live in malaria-transmitted areas.23 Every year, 350–500 million cases of malaria occur, and nearly one million people die.24 Plasmodium falciparum resistance to chloroquine has been confirmed in Egypt, Central America west of the Panama Canal, Haiti, Dominican Republic, and other countries in the Middle East.25 In some regions, most parasites (90%) may be resistant to chloroquine.26 Contributors to the progression of parasitic resistance have been the limited number of attainable antimalarial drugs and defective dosing for malaria treatment.27 TB’s public health implications have become more significant as the pathogen has become more and more resistant to antimicrobial therapy. In 2006, nearly 14,000 TB cases were examined in the USA.28 The second most prevalently reported notifiable disease in the USA is TB. Gonorrhea is an additional example of a great antimicrobial resistance concern. In 2006, nearly 14% of isolates were tested resistant to fluoroquinolones, which was an increase from 2.2% in 2002.29
Resistance of Gram-positive bacteria
Pneumococci
S. pneumoniae is the causative agent of diseases, such as pneumonia, meningitis, otitis media, and bacteremia. Infection of S. pneumoniae mainly results in higher death rates among younger, older, and immunocompromised people.30 Resistance to penicillin in S. pneumoniae was first detected in 1967. Since then the penicillin-resistant strain of S. pneumoniae has become progressively prevalent worldwide.31 Variants of the Spain23F-1 or Spain6B-2 clones were the predominant penicillin nonsusceptible S. pneumoniae (PNSP) isolated from 1994–1997 and remained so, but Taiwan19F-14 and Taiwan serotype 6B clones were disseminated in Hong Kong in 1999 and 2000.32 The emergent clones are penicillin nonsusceptible. PNSP clones are generally resistant to tetracyclines, cotrimoxazole, chloramphenicol, and macrolides. Recent studies have indicated that the prevalence of PNSP between clinical isolates of S. pneumoniae is >40% in 16 of 60 countries observed, and <5% in only three of them.33 An S. pneumoniae isolate is measured to lack susceptibility when penicillin’s minimal inhibitory concentration (MIC) is >0.06 mg/L,34 and is related to a PNSP.
Enterococcus
Enterococci have stood out among Gram-positive bacteria because of their essential relevant resistance to multiple antimicrobials. In addition, they have the function to obtain or improve characteristics that render them resistant to even higher concentrations of several antibiotics.35 The revelation of vancomycin resistance among strains of Enterococcus in the late 1980s was a crushing development.35 Enterococci, especially Enterococcus faecalis, are one of the serious causative factors of infective endocarditis. Unluckily, vancomycin, penicillin, and other cell wall-active antimicrobials repress but do not destroy enterococci.36 High-level streptomycin resistance (MIC 2000 μg/mL) in enterococci can result from adenylylation of aminoglycosides. Aminoglycoside-inactivating enzymes make it inactive against the ribosomal target.37
High-level kanamycin resistance (MIC 2000 μg/mL) was already clearly typical among enterococci by the 1970s.38 Therefore, for gentamicin, an MIC >500 μg/mL indicates high-level resistance. In the USA, it was found that 3.6% of enterococci isolated in hospitals with >500 patients were resistant to vancomycin, but only 1.8% in hospitals with <200 patients. Enterococcal vancomycin resistance in intensive care unit patients increased from 0.4% to 13.6% between 1989 and 1993. Most of the reports from the USA only mentioned vancomycin-resistant enterococci, as vancomycin remains their only commercially available glycopeptide.39
MRSA
According to a USA study in 2007, 880,000 people are infected by MRSA every year and 2.4% of the patients stay in different hospital units annually. This type of infection also leads to six extra hospital days on average for patients. Therefore, MRSA infection results in $8 billion in total costs annually. Unfortunately, 5% of people with MRSA infection are dying and this number corresponds to 20,000–40,000 deaths per year.40 Methicillin-resistant strains of S. aureus have the ability to grow in medium that includes methylpenicillin and derivatives, including oxacillin, nafcillin, and methicillin. This resistance is induced by expression of a modified penicillin-binding protein, PBP2a.41 Methicillin-susceptible strains are repressed by oxacillin at concentrations of 4 μg/mL or methicillin at 8 μg/mL. MRSA grow in the presence of 16 μg/mL to >2,000 μg/mL of methicillin. Through the application of high antibiotic concentrations, S. aureus developed various resistance mechanisms against penicillin, including methicillin. Despite the fact that methicillin is resistant to hydrolysis by staphylococcal β-lactamase, S. aureus strains that produce large amounts of β-lactamase have been isolated.42 These hyperproducers of β-lactamase tend to resist methicillin through limited hydrolysis of the antibiotic, resulting in a phenotype that, with respect to methicillin, is intermediate between susceptible and resistant.42 There is an obvious need for more effective antibiotic therapy for MRSA infections. Reports describing treatment failure of vancomycin for MDR MRSA infections have raised concern for the emergence of strains of MRSA for which there will be no effective therapy.
Resistance of Gram-negative bacteria
Gram-negative bacteria are major causal agents of infections such as urinary tract infections, otitis media, sexually transmitted diseases, acute and chronic bronchitis, and community-acquired pneumonia. These diseases are frequently caused by Gram-negative organisms. Gramnegative microorganisms that are mostly seen in primary care units include Escherichia coli (urinary tract infection), Haemophilus influenzae (community-acquired pneumonia and bronchitis), Klebsiella pneumoniae (community-acquired pneumonia and urinary tract infection), Neisseria gonorrhoeae (gonorrhea), and Neisseria meningitidis (meningitis). With increasing antimicrobial use in society, a relative shortage of new antimicrobials with novel mechanisms of action and increases in outpatient therapy for many diseases have resistance issues in the ambulatory setting.
Recently, resistance to antimicrobial agents mostly used to treat Gram-negative pathogens has increased. Excessive use of antibiotics, uncontrolled infection, and plasmid-mediated resistance gene transfer all play important roles in this emerging threat. Reports of Gram-negative pathogens resistant to all commonly used antimicrobial agents have become progressively commonplace, both in the USA and abroad. One example of the genetic adaptability of these organisms, Pseudomonas aeruginosa often combines multiple mechanisms to result in broad-spectrum resistance.43 It has been described as a public health crisis, but only few novel antimicrobial agents are in development for the treatment of MDR Gram-negative pathogens.44 Efforts to preserve the antimicrobials are crucial to future success in treating Gram-negative infections.
Suitable diagnosis, liable antimicrobial use, and familiarity with local resistance patterns can help the clinician to treat patients. In the last few years, resistance to antimicrobial agents in the treatment of Gram-negative microorganisms has generally increased.
P. aeruginosa, Acinetobacter baumannii, and K. pneumoniae are among the most common bacteria, referred to as MDR bacteria. Excessive use of antimicrobials, careless infection control, and plasmid-mediated resistance gene transfer are playing important roles in this problem.43
Resistance to β-lactam antibiotics
Bacterial resistance to β-lactam antibiotics and β-lactamase inhibitors is an important problem that decreases the clinical efficacy of drugs that are the cornerstone of antibiotics throughout the world. Gram-negative pathogens frequently synthesize new β-lactamases in order to respond to the efficacy of these kinds of antibiotics. More than 340 β-lactamase enzymes have been identified to date.45 These drug inhibitory agents include class A SHV- and TEM-derived extended-spectrum β-lactamases (ESBLs). In addition to inhibitor-resistant enzymes, non-TEM, non-SHV class A ESBLs, and carbapenemases, class B metallo-β-lactamases and some of their new inhibitors, plasmid and chromosomally encoded class C enzymes, the OXA-type oxacillinases, ESBLs, and carbapenemases of class D are the other types of suppressing agents against β-lactam antibiotics.46
The most common plasmid-mediated β-lactamase is TEM-1, which has been reported in approximately 75%–80% of plasmid-mediated β-lactamase resistance cases.47–50 The TEM enzyme was originally isolated from a single strain of E. coli. It was also isolated from blood culture in 1965.51
The TEM-1 β-lactamase spread into the Enterobacteriaceae family within 10 years. A close relative of TEM-1, TEM-2, was recorded in P. aeruginosa in 1969. Furthermore, plasmids encoding the TEM-1 enzyme had spread in H. influenzae and N. gonorrhoeae in 1970.52 Another common plasmid mediated β-lactamase was found in K. pneumoniae and E. coli and named as SHV-1 (for sulfhydryl variable). The SHV-1 β-lactamase is chromosomally encoded in most of the isolates of K. pneumoniae, and it is usually plasmid-mediated in E. coli.53
One of the β-lactam agents, carbapenems, has an action against MDR Gram-negative bacteria. Imipenem, meropenem, and doripenem show high antibacterial activity against P. aeruginosa, but ertapenem does not show such efficacy.54 In recent years, some Gram-negative bacteria have started to produce β-lactamase enzymes which degrade β-lactam rings and deactivate the efficacy of β-lactam antibiotics. It was recently demonstrated that Acinetobacter, Pseudomonas, and members of the Enterobacteriaceae family exhibited strong resistance to β-lactam.55 B-lactamase production was also commonly seen in N. gonorrhoeae and this activity limited the usefulness of penicillin G for the treatment of N. gonorrhoeae.
Penicillin’s Gram-negative activity increases from penicillin to ampicillin and ticarcillin to piperacillin. Like penicillin, cephalosporins change their Gram-negative activity. First-generation agents like cefazolin act against pathogens such as E. coli and K. pneumoniae. Third-generation agents such as ceftazidime, cefotaxime, and ceftriaxone and fourth-generation agents such as cefepime show greater potency against these pathogen microorganisms.44
Resistance to fluoroquinolones
MDR quinolones in Enterobacteriaceae has been rapidly rising all around the world.56–60
Resistance to fluoroquinolones has been shown to be frequently associated with alterations in gyrA.61,62 The mutations are localized at the 59 end of the gene (nucleotides 199–318 in the E. coli gene sequence), the quinolone resistance-determining region, near Tyr122, which binds transiently cleaved DNA.63,64
Genetic characterization of gyrA mutations associated with fluoroquinolone resistance in E. coli has been well defined by DNA sequence analysis of resistant strains of both clinical isolates and mutants selected in vitro.64–67 These mutations are most commonly found in the Ser83 and Asp87 codons and in the corresponding codon positions of the gyrA genes from several other organisms.61,62,68,69
The first plasmid-mediated quinolone-resistant protein Qnr was identified from urine in a K. pneumoniae isolate in 1994. It began a novel era in resistance to quinolones. Quinolones insert into bacteria through porins or directly through the lipid cytoplasmic membrane and target DNA topoisomerases.60 Resistance to quinolones in Enterobacteriaceae most commonly occurs slowly as a result of mutation, usually accumulating in the genes encoding primarily DNA gyrase and also topoisomerase IV. Decreased permeability caused by changes in the amount of porins (in particular outer membrane protein F) or increased efflux in regulatory genes of chromosomally-encoded MDR by mutations may cause increments in quinolone resistance.56,70,71
Fluoroquinolones have become well-used agents in ambulatory settings for a wide range of infections such as urinary tract infections, pneumonia and other respiratory infections, and gastrointestinal infections.
Recently, resistance to fluoroquinolones in Gram-negative bacteria has been increasing. Changes in DNA gyrase and topoisomerase IV – enzymes of bacteria that are targeted by fluoroquinolone – leads to mutational resistance. Furthermore, the presence of multidrug efflux pumps is a significant contributor to fluoroquinolone resistance. Multidrug efflux pumps remove the drug before its antimicrobial action can be achieved, seen in P. aeruginosa in particular.
Worldwide, there are several reported rates of fluoroquinolone resistance in both community-acquired and nosocomial pathogens. Many factors, such as patient characteristics, local epidemiological factors, antibiotic policies, inadequate or excessive usage, lower standards of living in developing countries, and lack of information may be responsible for such divergent data.72
Multistep selection of resistant strains resulting from the association of different resistance mechanisms is responsible for the emergence of high-level resistance to fluoroquinolones. Thus, recognition of strains with low-level resistance is important in order to modify standard therapeutic regimens. As can be seen in vitro, selection of a resistant clone is more likely to occur with the use of less effective compounds. All resistant mutants selected with one compound display cross-resistance to the other compounds but at a variable MIC level. High-level resistance is threatening because such bacteria are already resistant to or have reduced susceptibility to new antibiotics. When resistant strains are first selected, they may spread in the community or in the hospital environment if they have epidemic potential (salmonellae and pneumococci) or if favorable conditions exist. Follow-up of fluoroquinolone resistance is compulsory and should be performed everywhere, including individual communities, so that infections can be better managed and the spread of resistant strains can be counteracted.73
Resistance to aminoglycosides
There are three mechanisms of resistance to aminoglycosides:
Reduced uptake or decreased cell permeability. Reduced drug uptake, mostly seen in Pseudomonas spp. and other nonfermenting Gram-negative bacilli, is due to membrane impermeabilization, but the underlying molecular mechanisms are unknown.74 Aerobic Gramnegative bacilli in general show a phenomenon of adaptive resistance.75,76
Altered ribosome binding sites. Mutations of aminogly-coside attachment may interfere with ribosomal binding. Resistance to streptomycin can occur by this mechanism if this agent binds to a single site on the 30S subunit of the ribosome. Resistance to other aminoglycosides by this mechanism is uncommon. Because they bind to multiple sites on both ribosomal subunits, high-level resistance cannot be selected by a single step.77
Enzymatic modifications. This is the most common type. About 50 different enzymes have been identified,78 and enzymatic modification leads to high-level resistance.77 Encoding for aminoglycoside-modifying enzymes are generally found on plasmids and transposons. Because of multiple genes, most enzyme-mediated resistance occurs in Gram-negative bacilli.79
There are three types of aminoglycoside-modifying enzymes:78
N-acetyltransferases, which catalyze acetyl coenzyme A (CoA)-dependent acetylation of an amino group.
O-adenyltransferase, which catalyze adenosine triphosphate-dependent adenylylation of a hydroxyl group.
O-phosphotransferases, which catalyze adenosine triphosphate-dependent phosphorylation of a hydroxyl group.
For the treatment of Gram-negative infections, currently used agents include gentamicin, tobramycin, and amikacin. Their spectrum of activity includes Enterobacteriaceae, Acinetobacter spp., and P. aeruginosa. Intrinsic aminoglycoside resistance happens mainly via drug exclusion from the cell.
Gram-negative organisms and resistance
E. coli
E. coli is the most common Gram-negative pathogen encountered in clinical practice. E. coli, as one of the normal human gastrointestinal flora, is the primary cause of urinary tract infections in the outpatient setting. Infections caused by E. coli include uncomplicated urinary tract infections to hospital-acquired pneumonia and sepsis. Thus, antimicrobial resistance in E. coli is of major importance in both the ambulatory and hospitalized patient.
Recently, resistance to fluoroquinolones among clinical strains of E. coli has been rising. In 2004, according to the National Nosocomial Infections Surveillance project, about 10% of E. coli strains isolated from hospitalized patients were resistant to fluoroquinolones.72,79 Mostly, this resistance is due to either efflux pumps or target-site modification.
K. pneumoniae
K. pneumoniae is in contrast to E. coli, a constant producer of SHV-1. While ampicillin and amoxicillin could be expected to penetrate the cell wall of K. pneumoniae, they have no defense to β-lactamases, preventing therapy with these agents. The addition of a β-lactamase inhibitor should repair the effect of the penicillin agent, provided no ESBLs or other resistance mechanisms are present. The MDR profile of ESBL-producing Gram-negative bacteria reveals that this organism has broad-spectrum resistance from penicillin to aztreonam. Fluoroquinolone resistance in K. pneumoniae is similar to E. coli and other Enterobacteriaceae. Resistance most commonly occurs as a mutation of DNA gyrase and topoisomerase IV.
P. aeruginosa
P. aeruginosa is naturally resistant to many agents, either through β-lactamase production or through the exclusion of antimicrobial agents from the cell.43 Mutational resistance has been increasing in frequency.
A. baumannii
Several studies show that carbapenem overuse affects the MDR profile of A. baumannii.80–82
Resistance of TB
TB is caused by Mycobacterium tuberculosis. While drug-resistant TB organisms are resistant to at least one first-line anti-TB drug, MDR TB is caused by isoniazid (INH) and rifampin (RIF) – the two most potent TB drugs. Both treating and curing drug-resistant TB is complicated. Inappropriate or inadequate management can threaten life.83
Drug resistance is diagnosed by drug-susceptibility testing in the laboratory. However, since these tests can take several weeks, treatment could be started with an empirical treatment regimen based on expert advice, depending on the probability that the person has drug-resistant TB disease. When testing results are known, the treatment regimen should be adjusted according to the results. Patients should be monitored closely throughout treatment. Directly observed therapy should always be used in the treatment of drug-resistant TB to ensure adherence.
The problem of resistance of TB is very old. The first two drugs used in formal clinical trials, streptomycin84,85 and para-aminosalicylic acid,86 were developed in the 1940s. In 1994, it was thought that 2.2 billion people were infected with M. tuberculosis87 and the organism caused 1.6 million deaths. Drug resistance can be natural (primary) or acquired. If natural, the patient is infected with a resistant organism even though the patient has never received therapy. Acquired resistance means that the patient’s initially sensitive organism becomes resistant after using antimicrobials.
In 2005, the overall prevalence of primary resistance to INH was 7.3%, the overall prevalence of acquired resistance to INH was 14.6%, and the overall prevalence of primary MDR TB was 1% in the USA.87 In 2006 in the USA, the prevalence of MDR TB was about 0.02% (17 cases). More than 80% of cases had no previous exposure to TB, and therefore considered to be primary MDR TB.88
Spontaneous mutations in the chromosome can lead to drug resistance in M. tuberculosis. Mutations associated with RIF and fluoroquinolone resistance occur at about 10−8, mutations leading to INH, streptomycin, ethambutol, kanamycin, or para-aminosalicylic acid resistance occur at a frequency of approximately 10−6, and mutations leading to ethionamide, capreomycin, cycloserine, or thiacetazone resistance occur at a frequency of about 10−8.89 These frequencies are important as resistance to a combination of antibiotics (eg, RIF with INH) would be expected to occur at a combined rate.90,91
In general, bacteria are resistant to both drugs. However, the possibility of cure using both drugs is high because INH would kill RIF-resistant bacteria and RIF would kill INH-resistant bacteria. RIF resistance would improve among the INH-resistant population at a frequency of 10−8. At first, the patient might improve while RIF kills the majority of the bacteria, but then strains resistant to both drugs would occur, and the patient would relapse. On the other hand, a clinician’s prescribing behavior may lead to drug resistance.
Treatment of drug-resistant TB
Standard short-course TB therapy is the best way to prevent MDR TB. Standard management is effective in patients with bacilli that are resistant to INH and/or streptomycin. Drug resistance should be suspected in a patient who presents these conditions:
Has had first therapy;
Is in an area where drug resistance is widespread;
Sputum smears are positive after 2 months of therapy;
Sputum smears are positive after 4 months of therapy;
The patient’s chest radiography is getting worse.
If the anti-TB drugs are insufficient, drug resistance can occur. Second-line agents must be employed when INH, RIF, pyrazinamide, or ethambutol cannot be used because of drug resistance.
Prevention of drug-resistant TB
Patients exposed to INH mono-resistant TB can be treated for 2 months with a combination of rifamycin and pyrazinamide.92,93 If the patient cannot tolerate pyrazinamide, a 4–6 month regimen of rifamycin alone is acceptable.
Patients exposed to RIF mono-resistant TB can be treated with an ordinary INH regimen.
The treatment and cure of MDR-TB cases is very difficult because there are no sufficient published data concerning the composition, duration, or effectiveness of treatment regimens for MDR-TB. Standard regimens that have been suggested include pyrazinamide and ethambutol or pyrazinamide and quinolone. These drugs are expected to treat active disease for 12 months in immunocompromised patients and ≥6 months in patients who are immunocompetent.92
Finally, the success of therapy should be measurable with drug-susceptible TB. Patients should heal clinically and cultures should become negative. In addition, smears and cultures should be observed every 2 months for the next 2 years.
The challenges of MDR strains of bacteria and current management
The discovery of antibiotics was one of the major advancements in public health during the 20th Century.94 During the early period of antibiotic usage, bacterial infections were considered defeated. Antibiotics provided prevention of potentially lethal infections.95 However, the emergence of MDR bacteria in the last few decades is resulting in one of the most important clinical health problems in modern medicine.96
Antibiotic resistance occurs by vertical transmission of the product of innate genetics and physiology of bacteria through species.96 Resistance can also be transferred horizontally by plasmids or by chromosomally located conjugative transposons across species and genera.97 This combinatorial genetic strategy has resulted in the accumulation of MDR phenotypes in many species of bacteria.96 However, the emergence of MDR could not be adequately explained on the basis of random, independent mutational events.98 Alteration or modification of the target of antibiotics, degradation of the antibiotic structure, and decreased permeability and energy-dependent (or active) efflux are some of the resistance action of bacteria.95 Efflux-mediated resistance is mostly drug specific. On the other hand, some of the efflux systems accommodate multiple drugs and thus contribute significantly to bacterial intrinsic and acquired MDR.95
MDR organisms, predominantly bacteria, are resistant to one or more classes of antimicrobial agents.99 More strains of pathogens have become antibiotic resistant, and some have become resistant to many antibiotics and chemotherapeutic agents.100 Even if the known MDR organisms were defined as resistant to at least three antibiotics belonging to three different families, these organisms are frequently resistant to most available antimicrobial agents.101 It has been shown by different researchers that some strains of E. coli, K. pneumoniae, and A. baumannii are resistant to all antimicrobial agents, and Stenotrophomonas maltophilia, Burkholderia cepacia, and Ralstonia pickettii are intrinsically resistant to the broadest spectrum antimicrobial agents.102 The increasing antimicrobial resistance of infectious agents is defined as one of the most important health threats worldwide.103
It was estimated that at least 25,000 patients die each year from an infection caused by MDR bacteria in the European Union.104 Antibiotic resistance is also a major public health issue in low and middle income countries.105 The potential for transfer of drug resistance to human bacteria and MDR has become a worldwide concern.98 Therefore, there is an urgent and critical need to develop new antibacterial agents with activity against MDR bacteria.104
Managing MDR requires understanding of the molecular basis, evolution, and dissemination of resistance; development of new antibiotic compounds in place of traditional antibiotics; and innovative strategies for extending the life of antibiotic molecules.96 One of the major problems facing the antibiotic drug discovery sector is the difficulty in identifying new druglike compounds with suitable antibacterial activity.96
Over the last 50 years, medicinal chemists have been highly successful in developing new antibiotics, both natural and synthetic, such as fourth-generation β-lactam and third-generation macrolides. However, significantly new approaches and strategies for new molecules are needed.106
Generally, antibiotics have been founded by testing natural products. Natural product antibiotic effects can be directly isolated from soil and marine microorganisms or antibiotic-producing organisms that can be modified genetically. Another approach is the diversion of natural metabolic pathways of antibiotic-producing organisms by introduction of substrate precursors into the fermentation system.107 However, different approaches should be developed for these products to improve their spectrum, pharmacokinetics, human safety, and ability to cope with resistance.
Recently, an attempt to develop antibiotics with an effect on bacteria, viruses, fungi, and protozoa was based on nanotechnology. Nanoemulsions pass through pores of skin and hair follicles to the infected area, without harming the dermal cells, and disrupt the microorganism and decrease the risk of resistance against antibiotics.108
The molecular basis of tolerance to antimicrobial agents should be examined in detail with gene knockouts, genomics, and proteomics. Combinations of antibacterial agents that prolong the life of these drugs can be used after a detailed performance study.109 Even if research and development of antibacterial agents against infectious agents starts in the near future, the issue of antibiotic resistance is likely to continue to increase.104 Side effects of newly developed antibiotics constitute another serious problem. Therefore, potential approaches should be to increase patient compliance, decrease the potential for resistance development, reduce drug costs, and increase efficacy, ultimately extending the life of these antimicrobial agents.109
Platensimycin and platencin
Bacterial infections are increasingly defying antibiotics that are heavily used in hospitals.110 For this reason, treatment costs and therapy failures are increasing with regard to the fight against infectious diseases. In particular, resistant Grampositive pathogens such as MSRA,111,112 vancomycin-resistant enterococci,113 and penicillin-resistant S. pneumoniae are posing a serious threat.114,115
There are many approaches to developing an effective antibacterial agent that target an essential constituent or pathway in the infection agents. However, some of these attempts have been undesirable due to inefficient pharmacokinetics, drug resistance, and toxicity. Moreover, when drug research costs are considered, the problem is increasingly difficult to solve. On the other hand, initiatives about an encouragement program for governments to develop antimicrobial agents have been published.116 As a result of these initiatives, considerable effort has been put into developing new techniques to improve antibiotics, for example the antisense ribonucleic acid (RNA) method has been developed for use in the discovery and improvement of antibiotics.117–121
In the antisense RNA method, drug targets are determined with the antisense RNA by decreasing expression levels of a certain protein which is thought to be effective in an essential pathway. Comparisons can then be made about the strain with the depleted protein. According to the test results, special drugs can be developed that target the protein of interest.120 To demonstrate accuracy of the method, FabF (a β-ketoacyl-acyl carrier protein [ACP] synthase II) and related other targets were investigated.122
It is expected that decreasing fabF transcript levels due to expression of antisense fabF transcript impairs the target proteins. When control organisms were compared to cells expressing fabF antisense RNA with zone of inhibition measurement, only FabF-inhibited cells indicated significant distinction from others.122 As a result, researchers focused on the inhibition of the FabF pathway. Eventually, in 2006, Wang et al screened platensimycin as an inhibitor of the FabF/H pathway.123–126
Platensimycin was identified as a metabolite of a filamentous bacterium Streptomyces platensis MA7327 strain found in soil samples from Spain and South Africa in 2006.122,124,126–129 Platensimycin is a bacteriostatic drug that does not exhibit cross-resistance to Gram-positive bacteria with MIC values of 0.1–0.32 mg/mL, therefore making it a potentially useful antibiotic candidate.130,131 Platensimycin was efficiently used in a murine model of a common S. aureus infection and no toxicity was observed in these animals. In addition, lack of toxicity in HeLa cells with platensimycin and lack of antifungal activity are clear signs for the promising marked selectivity of platensimycin.134,135 Furthermore, platensimycin shows strong efficacy without any observed toxicity in vivo and it is nontoxic to humans. Its unique target is bacterial fatty acid synthesis (FAS).
Platensimycin blocks FabF and stops bacterial FAS selectively.122,123,128,132 Different procedures (reversephase high-performance liquid chromatography, mass spectrometry, two-dimensional nuclear magnetic resonance, and X-ray crystallographic analysis) were utilized to acquire the structure of platensimycin. Among them, X-ray crystallographic analysis presented absolute stereochemistry of the molecule.124 Its molecular formula is C24H27NO7 and molecular weight is 441.47 g/mol. The structure of platensimycin consists of two distinct structural motifs that are joined by an amide bond.125 Platensimycin is formed by two structures. One of these is an aromatic unit which has a hydrophobic characteristic and the other one is a tetracyclic lipophilic unit.126,131,133 The aromatic and tetracyclic portions are joined by a flexible amide chain. The aromatic unit binds to FabF and penetrates into the catalytic site.134
After discovery and patenting of platensimycin, researchers focused on the synthesis of platensimycin125,135–138 and its analogs.123,139 These studies revealed that platensimycin, which had great potential to penetrate through bacterial cell membranes, demonstrated high antibacterial activity against Gram-positive bacterium including staphylococci and enterococci. Despite the promising efficacy of platensimycin, FASII is not a suitable antibiotic target for Gram-positive pathogens as exogenous fatty acids fully bypass inhibition of the FAS pathway in both in vivo and in vitro conditions.
Platensimycin is not effective against Gram-negative bacteria.123,131 This is due to the extrusion mechanism of Gramnegative bacteria. This mechanism pumps platensimycin out of the cell membrane in Gram-negative bacteria. It demonstrates that platensimycin can inhibit Gram-negative bacteria when this pump mechanism is disabled due to mutations. For example, E. coli is an extrusion-negative mutant bacterium and platensimycin is effective against these bacteria.123,140,141
The Merck team recently described their finding of platencin as an antibiotic with an analogous structure and profile to platensimycin.128,142 Platencin is actually an analog of platensimycin. It also demonstrates antibacterial activity against Gram-positive bacteria (25% of the MIC 0.5–32 mg/mL) through the inhibition of FAS. In vitro studies have shown that it is active against vancomycin-resistant enterococci, MRSA, and both linezolid- and macrolide-resistant pathogens.123,128,131
This antibiotic is an agent for another rising drug applicant due to its in vivo capability and lack of toxic side effects. Platencin is produced by the S. platensis strain MA7339. S. platensis was discovered in a soil sample collected from Spain. The similar whole-cell differential assay was used. The molecular formula of platencin is C24H25NO6 and its molecular weight is 425.47 g/mol, marginally smaller than platensimycin. It is comprised of a tetracyclic unit lacking the ether ring (thus different from the pentacyclic ketolide of platensimycin). The tetracyclic unit is connected to an aminobenzoic acid moiety by an amide bond.142 Platensimycin inhibits the elongation-condensing enzyme FabF. In contrast to platensimycin, platencin is a binary inhibitor of FabF and the condensing enzyme FabH. The fact that platencin fundamentally targets two enzymes in the FAS pathway may assist in reducing the potential for resistance advancement against the drug.
MIC rates of platencin against a group of organisms were nearly the same, but it showed lower activity in the existence of serum. It also demonstrated acceptable antibacterial activity in vivo. The most critical conflict was the enzyme inhibition profile of platencin. It determined a dual activity against FabF and FabH. Capability against FabF (half maximal inhibitory concentration [IC50] = 0.113 μM) was about six times less than platensimycin while inhibition of FabH (IC50 = 16.2 μM) was four times more intense. By themselves these IC50 rates are not remarkable, but in combination these two antibiotic candidates have a synergistic impact on suppressing bacterial cell growth.128,142
Jayasuriya et al used docking assays to demonstrate these potency differences. It is tempting to visualize the design that maximizes inhibition of both these enzymes to exploit the synergistic effect on antibacterial activity.142
Pharmacology of platensimycin and platencin
Platensimycin belongs to a unique structural class of natural antibiotics and has no cross-resistance to other classes of antibiotic-resistant bacteria. MIC values of platensimycin are 0.5 μg/mL against S. aureus (MRSA) and S. aureus (MRSA, macrolide), 1 μg/mL against S. aureus (MRSA, linezolid), 0.5 μg/mL against S. aureus (vancomycin-intermediate S. aureus, vancomycin), 0.1 μg/mL against Enterococcus faecium (vancomycin-resistant enterococci), and >64 μg/mL against E. coli (wild-type).123 Molecular weight of platensimycin is 441.47 g/mol (Figure 1). Platencin (molecular weight 425.2 g/mol) is another novel natural product that has similar structure and profile to platensimycin (Figure 2). MIC values of platencin against vancomycin-resistance E. faecium and efflux-negative E. coli are <0.06 μg/mL and 2 μg/mL, respectively. Platencin does not exhibit cross-resistance to MRSA.
Figure 1.
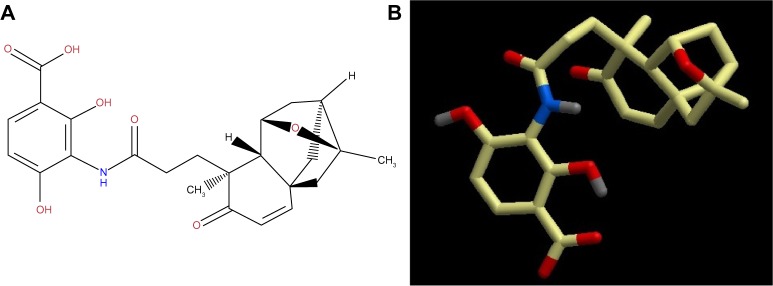
(A) two-dimensional and (B) three-dimensional molecular structure of platensimycin (molecular weight 441.47 g/mol).
Figure 2.
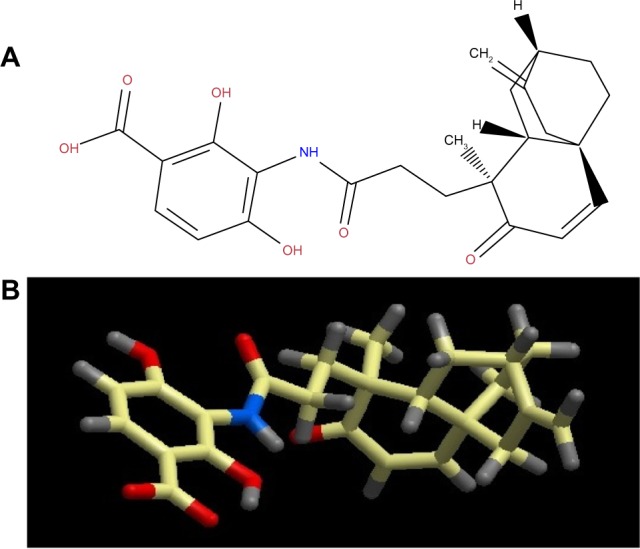
(A) two-dimensional and (B) three-dimensional molecular structure of platencin (molecular weight 425.2 g/mol).
Both platensimycin and platencin have different ways of killing Gram-positive pathogens, including MDR strains of staphylococci and enterococci. It is known that most antibiotics target one of four essential bacterial processes including cell wall biosynthesis, nucleic acid biosynthesis, protein synthesis, and folic acid biosynthesis.143 Platensimycin shows antibacterial activity by targeting FASII. FASII is present in prokaryotes, plants, fungi, and parasites, as well as in mitochondria. In animals, as well as yeast and some fungi, these same reactions occur in FASI. FASI is less efficient than FASII.144 The ACP is a key component in type II systems and carries the fatty acid intermediates from enzyme to enzyme through the cytosol. Usually, C16–C18 fatty acids are the principal end products of the pathway; in case of bacteria, the pathway is more complicated and consists of FASI and FASII. The first enzyme of the fatty acid biosynthesis is the acetyl-CoA carboxylase (AccABCD) that transfers acetyl-CoA to malonyl-CoA, which subsequently interacts with ACP by malonyl-CoA–ACP transacylase (FabD) to form malonyl-ACP. The original fatty acid cycle is initiated by the condensation of malonyl-ACP with acetyl-CoA by the β-ketoacyl-ACP synthase III (FabH) to form β-ketobutyryl-ACP and carbon dioxide. The following steps are repeated until the desired chain length of the fatty acid is achieved: the first is the nicotinamide adenine dinucleotide phosphate-dependent reduction of the keto group to the hydroxyl group to give a β-hydroxyacyl-ACP intermediate by the p-ketoacyl-ACP reductase (FabG). In the next step, water is removed by either a FabA or FabZ isoform of the β-hydroxyacyl-ACP dehydratase to give a trans-2-enoyl-ACP. The third step involves the reduction of the produced double bond by the nicotinamide adenine dinucleotide-dependent trans-2-enoyl-ACP reductase I (FabI) to form an acyl-ACP. The formed acyl-ACP is then condensed with a new malonyl-ACP by either β-ketoacyl-ACP synthase I or II (FabB and FabF) to start a subsequent cycle.145 So far, the only enzyme of this pathway successfully inhibited by antibiotics (INH and triclosan) in clinical use is FabI. Further inhibitors of enzymes in the fatty acid biosynthesis have been elucidated, among them two natural inhibitors of the condensing enzymes, thiolactomycin (FabH, FabB/F) and cerulenin.146,147 According to studies, new antibiotics must be developed for the inhibition of FASII. Fatty acids have to be synthesized for bacterial viability. If fatty acids are not synthesized in bacteria, bacteria will not build a cell wall. Platensimycin prohibits the pathway of FASII by specifically inhibiting β-ketoacyl-ACP synthase II (FabF).124 Platencin is less active than platensimycin against FabF. However, it is more active than platensimycin against FabH.130
Pharmacokinetics
Application of pharmacokinetic principles allows the relationship between dose and antibiotic concentration to be described. This is one important component of the relationship between dosage regimen and time course of effect. The existence of the second component is the concentration–effect relationship (pharmacodynamics). Therefore, by predicting the time course of antibiotic concentrations, one important step towards predicting the time course of antibiotic effect has been made. If the same dosage regimen of an antibiotic is given to different patients, there is true between-subject variability in the observed antibiotic concentrations. A reason for high between-subject variability may be that patients differ in their ability to absorb or eliminate an antibiotic. By including the variability of the pharmacokinetic parameters into a pharmacokinetic model, one can predict drug effects more precisely and optimize dosage regimens for the patient population.148 Platensimycin and platencin are promising antibiotics against important infection agents. However, their pharmacokinetic and safety profiles in humans are still not fully understood. In a mouse study, test animals were infected with S. aureus by intravenous injection with continuous infusion of platensimycin. As a result, the infection rate decreased approximately 40% in the kidney. Moreover, no toxic effect was detected when different concentrations of platensimycin were applied to the test animals. Platensimycin shows low toxicity in vitro in HeLa cells (IC50 >1000 μg/mL).123 According to all reported studies, platensimycin and platencin are promising antibiotics. Therefore, studies should be conducted on pharmacokinetics.
Analogs/derivatives of platensimycin and platencin
Many chemists, biochemists, and pharmacologists have tried to synthesize analogs/derivatives of platensimycin and platencin because of their strong antibacterial properties. One study has demonstrated that 7-phenylplatensimycin and 11-methyl-7-phenylplatensimycin are more potent analogs of platensimycin by carbonyl ylide cycloaddition strategy, and that they showed higher activity than platensimycin against MRSA and vancomycin-insensitive S. aureus.149 Barykina et al have synthesized nor-platencin, which is four to 16 times less active than platencin against several resistant strains of S. aureus, macrolide-resistant E. faecalis, and vancomycin-resistant E. faecium.150 In another study, Patra et al synthesized and characterized four new ferrocene-containing bioorganometallics inspired by the antibiotic platensimycin lead structure. 3-(4,4-diferrocenoylpentanamido)-2,4-dihydroxybenzoic acid; 3-(4,4-diferrocenoylbutanamido)-2,4-dihydroxybenzoic acid; 3-(4-[acetylferrocene] butanamide)-2,4-dihydroxybenzoic acid; and 3-(4-ferrocenoylbutanamido)-2,4-dihydroxybenzoic acid were found to be inactive against Gram-negative E. coli and P. aeruginosa. However, among all the new compounds tested, 3-(4-[acetylferrocenoyl]butanamido)-2,4-dihydroxybenzoic acid, bearing a ferrocenyl 1,3-diketone functionality, inhibits S. aureus growth selectively at an MIC value of 128 μg/mL.151 Wang et al have synthesized and tested biological activity of oxazinidinyl platensimycin, the methyl ester analog, and the simplified oxazinidinyl analog. Although its structure mimics that of platensimycin, oxazinidinyl platensimycin is two orders of magnitude less potent.152 Another study by Wang et al synthesized dialkylamino-2,4-dihydroxybenzoic acids as analogs of platensimycin and platencin.153
Patient-focused perspectives related to the use of platensimycin and platencin
Today, the bacterial resistance against existing antibiotics is well known and it has been a major issue for the last few decades.154 There are many chemicals that are lethal for bacteria, but they have the same effect on the host cells and hence cannot be used to cure bacterial infection. Thus, an attack on bacterial metabolic pathways selectively in the presence of a host is an important step in solving the problem. After a search for new antibiotics for fighting against MDR strains, two novel antibiotics, platensimycin and platencin, have been discovered. The advantage of these antibiotics is that they inhibit the bacterial fatty acid biosynthesis, which is essential for the survival of bacteria. Their structures are related with slightly different mechanisms of action, and they target different enzymes in the bacterial fatty acid biosynthesis.123,155
Platensimycin has strong, broad-spectrum Gram-positive antibacterial activity and is selective for inhibiting cellular lipid biosynthesis. Wang et al showed that the antibacterial effect is via the selective targeting of FabF and FabB in the synthetic pathway of fatty acids.123 The inhibitor with the most potential for the FabF/B enzymes is platensimycin, which shows in vivo efficacy with no observed toxicity.123 Both platensimycin and platencin show in vivo activity, but the usefulness of this target in Gram-positive organisms is doubtful due to the ability of exogenous unsaturated fatty acids to remediate FASII inhibition.156 It has been shown that platensimycin is active against M. tuberculosis through the inhibition of mycolic acid synthesis, and that it inhibits both KasA and KasB enzymes.157 It also provides a new lead for anti-TB drug design.156 Modifying either the aromatic portion or the ketolide domain of platensimycin and platencin has produced many analogs recently.158–164 However, none of them have better activity than platensimycin and platencin.155 Thus, to improve their poor pharmacokinetic properties, several congeners of these products have been discovered from the fermentation of broth but, again, none of them have exhibited better activity.165–168 For a major breakthrough in antibiotic research, platensimycin and platencin may still provide many derivative compounds, which are waiting to be discovered.155
The preclinical trials of these new antibiotics are targeted to combat MRSA in a mouse model.123 It has been reported that platensimycin is also a potent and highly selective inhibitor for mammalian FAS for the treatment of diabetes and related metabolic disorders in animal models. In particular, platensimycin inhibits FAS, but not the sterol synthesis in rat primary hepatocytes. It also preferentially concentrates in liver when administered orally to mice and potently inhibits hepatic de novo lipogenesis, increases glucose oxidation, and reduces fatty acid oxidation. In chronic applications, the platensimycin led to a reduction in liver triglyceride levels and improved insulin sensitivity in db/+ mice fed a high-fructose diet. The ambient glucose levels in db/db mice were also reduced with use of platensimycin.169
Platensimycin is not ready for clinical use. The in vivo activity has been established in a mouse model, but to maintain efficacy, continuous delivery is necessary. The pharmacokinetic profile for platensimycin is poor, and needs improvement before clinical use.170
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.World Health Organization . The Global Burden of disease: 2004 Update. Geneva: World Health Organization; 2008. Available from: http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf. [Google Scholar]
- 2.World Health Organization . Global Health Risks: mortality and burden of disease attributable to selected major risks. Geneva: World Health Organization; 2009. Available from: http://www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf. [Google Scholar]
- 3.Zinner SH. Antibiotic use: present and future. New Microbiol. 2007;30(3):321–325. [PubMed] [Google Scholar]
- 4.Cunha BA. Antibiotic resistance. Med Clin North Am. 2000;84(6):1407–1429. doi: 10.1016/s0025-7125(05)70295-9. [DOI] [PubMed] [Google Scholar]
- 5.Maclean RC, Hall AR, Perron GG, Buckling A. The evolution of antibiotic resistance: insight into the roles of molecular mechanisms of resistance and treatment context. Discov Med. 2010;10(51):112–118. [PubMed] [Google Scholar]
- 6.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10(Suppl 12):S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 7.Bush K. Antibacterial drug discovery in the 21st century. Clin Microbiol Infect. 2004;10(Suppl 4):10–17. doi: 10.1111/j.1465-0691.2004.1005.x. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization 2011/2012 Tuberculosis Global Facts Geneva: World Health Organization; 2011Available from: http://www.who.int/tb/publications/2011/factsheet_tb_2011.pdfAccessed April 16, 2013 [Google Scholar]
- 9.Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob. 2006;5:2. doi: 10.1186/1476-0711-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borg MA, de Kraker M, Scicluna E, et al. Prevalence of methicillin-resistant Staphylococcus aureus (MRSA) in invasive isolates from southern and eastern Mediterranean countries. J Antimicrob Chemother. 2007;60(6):1310–1315. doi: 10.1093/jac/dkm365. [DOI] [PubMed] [Google Scholar]
- 11.Sibanda T, Okoh AI. The challenges of overcoming antibiotic resistance: plant extracts as potential sources of antimicrobial and resistance modifying agents. Afr J Biotechnol. 2007;6(25):2886–2896. [Google Scholar]
- 12.Garcia LS. Malaria. Clin Lab Med. 2010;30(1):93–129. doi: 10.1016/j.cll.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Wright GD. Q&A: antibiotic resistance: where does it come from and what can we do about it? BMC Biol. 2010;8:123. doi: 10.1186/1741-7007-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahal JJ. Novel antibiotic combinations against infections with almost completely resistant Pseudomonas aeruginosa and Acinetobacter species. Clin Infect Dis. 2006;43(Suppl 2):S95–S99. doi: 10.1086/504486. [DOI] [PubMed] [Google Scholar]
- 15.Stefanovic OD, Stanojevic DD, Comic LR. Synergistic antibacterial activity of Salvia officinalis and Cichorium intybus extracts and antibiotics. Acta Pol Pharm. 2012;69(3):457–463. [PubMed] [Google Scholar]
- 16.Kang HK, Kim HY, Cha JD. Synergistic effects between silibinin and antibiotics on methicillin-resistant Staphylococcus aureus isolated from clinical specimens. Biotechnol J. 2011;6(11):1397–1408. doi: 10.1002/biot.201000422. [DOI] [PubMed] [Google Scholar]
- 17.Tan YT, Tillett DJ, McKay IA. Molecular strategies for overcoming antibiotic resistance in bacteria. Mol Med Today. 2000;6(8):309–314. doi: 10.1016/s1357-4310(00)01739-1. Swedish. [DOI] [PubMed] [Google Scholar]
- 18.Deyde VM, Xu X, Bright RA, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196(2):249–257. doi: 10.1086/518936. French. [DOI] [PubMed] [Google Scholar]
- 19.Centers of Disease Control and Prevention Active Bacterial Core Surveillance (ABCs) Report: Emerging Infections Program Network, Streptococcus pneumoniae, 2007 Atlanta, GA: Centers for Disease Control and Prevention; 2008Available from: http://www.cdc.gov/abcs/reports-findings/survreports/spneu07.pdfAccessed April 16, 2013 [Google Scholar]
- 20.Laurichesse H, Grimaud O, Waight P, Johnson AP, George RC, Miller E. Pneumococcal bacteraemia and meningitis in England and Wales, 1993 to 1995. Commun Dis Public Health. 1998;1(1):22–27. [PubMed] [Google Scholar]
- 21.Council of State and Territorial Epidemiologists . National Surveillance for Drug-Resistant Streptococcus Pneumoniae (DRSP) Invasive Disease. CSTE Position Statement 1994–10. Atlanta, GA: Council of State and Territorial Epidemiologists; 1994. [Google Scholar]
- 22.Centers for Disease Control and Prevention Extensively drug resistant tuberculosis – United States, 1993–2006. Morb Mortal Wkly Rep. 2007;56:250–253. [PubMed] [Google Scholar]
- 23.World Health Organization World malaria situation in 1994. Part I. Population at risk. Wkly Epidemiol Rec. 1997;72(36):269–274. [PubMed] [Google Scholar]
- 24.Guinovart C, Navia MM, Tanner M, Alonso PL. Malaria: burden of disease. Curr Mol Med. 2006;6(2):137–140. doi: 10.2174/156652406776055131. [DOI] [PubMed] [Google Scholar]
- 25.Arguin PM, Kozarsky PE, Reed C, Centers for Disease Control and Prevention . CDC Health Information for International Travel 2008. St Louis, MO: Mosby; 2007. [Google Scholar]
- 26.Bjorkman A, Phillips-Howard PA. The epidemiology of drug-resistant malaria. Trans R Soc Trop Med Hyg. 1990;84(2):177–180. doi: 10.1016/0035-9203(90)90246-b. [DOI] [PubMed] [Google Scholar]
- 27.Krogstad DJ. Malaria as a reemerging disease. Epidemiol Rev. 1996;18(1):77–89. doi: 10.1093/oxfordjournals.epirev.a017918. [DOI] [PubMed] [Google Scholar]
- 28.Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993–2006. Clin Infect Dis. 2009;49(9):1350–1357. doi: 10.1086/605559. Swedish. [DOI] [PubMed] [Google Scholar]
- 29.Wang FD, Chen YY, Chen TL, Liu CY. Risk factors and mortality in patients with nosocomial Staphylococcus aureus bacteremia. Am J Infect Control. 2008;36(2):118–122. doi: 10.1016/j.ajic.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Feldman C, Klugman K. Pneumococcal infections. Curr Opin Infect Dis. 1997;10(2):109–115. [Google Scholar]
- 31.Dowson CG, Coffey TJ, Spratt BG. Origin and molecular epidemiology of penicillin-binding-protein-mediated resistance to β-lactam antibiotics. Trends Microbiol. 1994;2(10):361–366. doi: 10.1016/0966-842x(94)90612-2. [DOI] [PubMed] [Google Scholar]
- 32.Ip M, Lyon DJ, Yung RW, Tsang L, Cheng AF. Introduction of new clones of penicillin-nonsusceptible Streptococcus pneumoniae in Hong Kong. J Clin Microbiol. 2002;40(4):1522–1525. doi: 10.1128/JCM.40.4.1522-1525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doern GV, Pfaller MA, Kugler K, Freeman J, Jones RN. Prevalence of antimicrobial resistance among respiratory tract isolates of Streptococcus pneumoniae in North America: 1997 results from the SENTRY antimicrobial surveillance program. Clin Infect Dis. 1998;27(4):764–770. doi: 10.1086/514953. [DOI] [PubMed] [Google Scholar]
- 34.Hiramatsu K, Aritaka N, Hanaki H, et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet. 1997;350(9092):1670–1673. doi: 10.1016/S0140-6736(97)07324-8. Finnish. [DOI] [PubMed] [Google Scholar]
- 35.Eliopoulos GM. Antibiotic resistance in Enterococcus species: an update. Curr Clin Top Infect Dis. 1996;16:21–51. [PubMed] [Google Scholar]
- 36.Storch GA, Krogstad DJ. Antibiotic-induced lysis of enterococci. J Clin Invest. 1981;68(3):639–645. doi: 10.1172/JCI110298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krogstad DJ, Korfhagen TR, Moellering RC, Jr, Wennersten C, Swartz MN. Aminoglycoside-inactivating enzymes in clinical isolates of Streptococcus faecalis. An explanation for resistance to antibiotic synergism. J Clin Invest. 1978;62(2):480–486. doi: 10.1172/JCI109149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moellering RC, Jr, Korzeniowski OM, Sande MA, Wennersten CB. Species-specific resistance to antimicrobial synergism in Streptococcus faecium and Streptococcus faecalis. J Infect Dis. 1979;140(2):203–208. doi: 10.1093/infdis/140.2.203. Swedish. [DOI] [PubMed] [Google Scholar]
- 39.Centers for Disease Control and Prevention Nosocomial enterococci resistant to vancomycin – United States, 1989–1993. MMWR Morb Mortal Wkly Rep. 1993;42(30):597–599. [PubMed] [Google Scholar]
- 40.Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763–1771. doi: 10.1001/jama.298.15.1763. French. [DOI] [PubMed] [Google Scholar]
- 41.Hartman BJ, Tomasz A. Low-affinity penicillin-binding protein associated with β-lactam resistance in Staphylococcus aureus. J Bacteriol. 1984;158(2):513–516. doi: 10.1128/jb.158.2.513-516.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDougal LK, Thornsberry C. The role of β-lactamase in staphylococcal resistance to penicillinase-resistant penicillins and cephalosporins. J Clin Microbiol. 1986;23(5):832–839. doi: 10.1128/jcm.23.5.832-839.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis. 2002;34(5):634–640. doi: 10.1086/338782. [DOI] [PubMed] [Google Scholar]
- 44.Andes D, Craig WA. Cephalosporins. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2005. pp. 294–311. French. [Google Scholar]
- 45.Shah AA, Hasan F, Ahmed S, Hameed A. Characteristics, epidemiology and clinical importance of emerging strains of Gram-negative bacilli producing extended-spectrum β-lactamases. Res Microbiol. 2004;155(6):409–421. doi: 10.1016/j.resmic.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Helfand MS, Bonomo RA. B-lactamases: a survey of protein diversity. Curr Drug Targets Infect Disord. 2003;3(1):9–23. doi: 10.2174/1568005033342181. [DOI] [PubMed] [Google Scholar]
- 47.Matthew M. Plasmid-mediated β-lactamases of Gram-negative bacteria: properties and distribution. J Antimicrob Chemother. 1979;5(4):349–358. doi: 10.1093/jac/5.4.349. [DOI] [PubMed] [Google Scholar]
- 48.Perilli M, Segatore B, de Massis MR, et al. TEM-72, a new extended-spectrum β-lactamase detected in Proteus mirabilis and Morganella morganii in Italy. Antimicrob Agents Chemother. 2000;44(9):2537–2539. doi: 10.1128/aac.44.9.2537-2539.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenau A, Cattier B, Gousset N, Harriau P, Philippon A, Quentin R. Capnocytophaga ochracea: characterization of a plasmid-encoded extended-spectrum TEM-17 β-lactamase in the phylum Flavobacter-bacteroides. Antimicrob Agents Chemother. 2000;44(3):760–762. doi: 10.1128/aac.44.3.760-762.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy C, Foz A, Segura C, Tirado M, Foster C, Reig R. Plasmid-determined β-lactamases identified in a group of 204 ampicillin-resistant Enterobacteriaceae. J Antimicrob Chemother. 1983;12(5):507–510. doi: 10.1093/jac/12.5.507. [DOI] [PubMed] [Google Scholar]
- 51.Datta N, Kontomichalou P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature. 1965;208(5007):239–241. doi: 10.1038/208239a0. [DOI] [PubMed] [Google Scholar]
- 52.Sirot D, Sirot J, Labia R, et al. Transferable resistance to third-generation cephalosporins in clinical isolates of Klebsiella pneumoniae: identification of CTX-1, a novel β-lactamase. J Antimicrob Chemother. 1987;20(3):323–334. doi: 10.1093/jac/20.3.323. Finnish. [DOI] [PubMed] [Google Scholar]
- 53.De Champs C, Sauvant MP, Chanal C, et al. Prospective survey of colonization and infection caused by expanded-spectrum-β-lactamase-producing members of the family Enterobacteriaceae in an intensive care unit. J Clin Microbiol. 1989;27(12):2887–2890. doi: 10.1128/jcm.27.12.2887-2890.1989. French. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chambers HF. Other β-lactam antibiotics. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2005. pp. 311–318. [Google Scholar]
- 55.Livermore DM, Woodford N. The β-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 2006;4(9):413–420. doi: 10.1016/j.tim.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 56.Ruiz J. Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemother. 2003;51(5):1109–1117. doi: 10.1093/jac/dkg222. [DOI] [PubMed] [Google Scholar]
- 57.Burman LG. Apparent absence of transferable resistance to nalidixic acid in pathogenic Gram-negative bacteria. J Antimicrob Chemother. 1977;3(5):509–516. doi: 10.1093/jac/3.5.509. [DOI] [PubMed] [Google Scholar]
- 58.Munshi MH, Sack DA, Haider K, Ahmed ZU, Rahaman MM, Morshed MG. Plasmid-mediated resistance to nalidixic acid in Shigella dysenteriae type 1. Lancet. 1987;2(8556):419–421. doi: 10.1016/s0140-6736(87)90957-3. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Martinez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet. 1998;351(9105):797–799. doi: 10.1016/S0140-6736(97)07322-4. [DOI] [PubMed] [Google Scholar]
- 60.Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61(3):377–392. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deguchi T, Yasuda M, Nakano M, et al. Quinolone-resistant Neisseria gonorrhoeae: correlation of alterations in the gyrA subunit of DNA gyrase and the parC subunit of topoisomerase IV with antimicrobial susceptibility profiles. Antimicrob Agents Chemother. 1996;40(4):1020–1023. doi: 10.1128/aac.40.4.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Georgiou M, Munoz R, Roman F, et al. Ciprofloxacin-resistant Haemophilus influenzae strains possess mutations in analogous positions of gyrA and parC. Antimicrob Agents Chemother. 1996;40(7):1741–1744. doi: 10.1128/aac.40.7.1741. French. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horowitz DS, Wang JC. Mapping the active site tyrosine of Escherichia coli DNA gyrase. J Biol Chem. 1987;262(11):5339–5344. [PubMed] [Google Scholar]
- 64.Moniot-Ville N, Guibert J, Moreau N, Acar JF, Collatz E, Gutmann L. Mechanisms of quinolone resistance in a clinical isolate of Escherichia coli highly resistant to fluoroquinolones but susceptible to nalidixic acid. Antimicrob Agents Chemother. 1991;35(3):519–523. doi: 10.1128/aac.35.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conrad S, Oethinger M, Kaifel K, Klotz G, Marre R, Kern WV. GyrA mutations in high-level fluoroquinolone-resistant clinical isolates of Escherichia coli. J Antimicrob Chemother. 1996;38(3):443–455. doi: 10.1093/jac/38.3.443. [DOI] [PubMed] [Google Scholar]
- 66.Heisig P, Tschorny R. Characterization of fluoroquinolone-resistant mutants of Escherichia coli selected in vitro. Antimicrob Agents Chemother. 1994;38(6):1284–1291. doi: 10.1128/aac.38.6.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vila J, Ruiz J, Marco F, et al. Association between double mutation in gyrA gene of ciprofloxacin-resistant clinical isolates of Escherichia coli and MICs. Antimicrob Agents Chemother. 1994;38(10):2477–2479. doi: 10.1128/aac.38.10.2477. Portuguese. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tankovic J, Perichon B, Duval J, Courvalin P. Contribution of mutations in gyrA and parC genes to fluoroquinolone resistance of mutants of Streptococcus pneumoniae obtained in vivo and in vitro. Antimicrob Agents Chemother. 1996;40(11):2505–2510. doi: 10.1128/aac.40.11.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Huang WM, Taylor DE. Cloning and nucleotide sequence of the Campylobacter jejuni gyrA gene and characterization of quinolone resistance mutations. Antimicrob Agents Chemother. 1993;37(3):457–463. doi: 10.1128/aac.37.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hooper DC. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin Infect Dis. 2000;31(Suppl 2):S24–S28. doi: 10.1086/314056. [DOI] [PubMed] [Google Scholar]
- 71.Hawkey PM. Mechanisms of quinolone action and microbial response. J Antimicrob Chemother. 2003;51(Suppl 1):29–35. doi: 10.1093/jac/dkg207. [DOI] [PubMed] [Google Scholar]
- 72.Hooper DC. Mechanisms of fluoroquinolone resistance. Drug Resist Updat. 1999;2(1):38–55. doi: 10.1054/drup.1998.0068. [DOI] [PubMed] [Google Scholar]
- 73.Acar JF, Goldstein FW. Trends in bacterial resistance to fluoroquinolones. Clin Infect Dis. 1997;24(Suppl 1):S67–S73. doi: 10.1093/clinids/24.supplement_1.s67. [DOI] [PubMed] [Google Scholar]
- 74.Chambers HF, Sande MA. Antimicrobial agents: the aminoglycosides. In: Hardman JG, Limbird LE, editors. The Pharmacological Basis of Therapeutics. New York, NY: McGraw-Hill; 1995. pp. 1103–1121. [Google Scholar]
- 75.Karlowsky JA, Hoban DJ, Zelenitsky SA, Zhanel GG. Altered denA and anr gene expression in aminoglycoside adaptive resistance in Pseudomonas aeruginosa. J Antimicrob Chemother. 1997;40(3):371–376. doi: 10.1093/jac/40.3.371. [DOI] [PubMed] [Google Scholar]
- 76.Xiong YQ, Caillon J, Kergueris MF, et al. Adaptive resistance of Pseudomonas aeruginosa induced by aminoglycosides and killing kinetics in a rabbit endocarditis model. Antimicrob Agents Chemother. 1997;41(4):823–826. doi: 10.1128/aac.41.4.823. French. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kucers A, Crowe S, Grayson ML, Hoy J. The Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal, and Antiviral Drugs. Oxford: Butterworth-Heinemann; 1997. [Google Scholar]
- 78.Davies J, Wright GD. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997;5:234–240. doi: 10.1016/S0966-842X(97)01033-0. [DOI] [PubMed] [Google Scholar]
- 79.Gilbert D. Aminoglycosides. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2000. pp. 307–355. [Google Scholar]
- 80.National Nosocomial Infections Surveillance System National Nosocomial Infections Surveillance (NNIS) System Report, data summary from Jan 1992 through Jun 2004, issued Oct 2004. Am J Infect Control. 2004;32(8):470–185. doi: 10.1016/S0196655304005425. French. [DOI] [PubMed] [Google Scholar]
- 81.Rahal JJ, Urban C, Segal-Maurer S. Nosocomial antibiotic resistance in multiple Gram-negative species: experience at one hospital with squeezing the resistance balloon at multiple sites. Clin Infect Dis. 2002;34(4):499–503. doi: 10.1086/338639. [DOI] [PubMed] [Google Scholar]
- 82.Manikal VM, Landman D, Saurina G, Oydna E, Lal H, Quale J. Endemic carbapenem-resistant Acinetobacter species in Brooklyn, New York: citywide prevalence, interinstitutional spread, and relation to antibiotic usage. Clin Infect Dis. 2000;31(1):101–106. doi: 10.1086/313902. [DOI] [PubMed] [Google Scholar]
- 83.Blumberg HM, Burman WJ, Chaisson RE, et al. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167(4):603–662. doi: 10.1164/rccm.167.4.603. French. [DOI] [PubMed] [Google Scholar]
- 84.World Health Organization Anti-Tuberculosis Drug Resistance in the World Geneva: World Health Organization; 2008Available from: http://whqlibdoc.who.int/hq/2008/WHO_HTM_TB_2008.394_eng.pdfAccessed April 16, 2013 [Google Scholar]
- 85.Centers for Disease Control and Prevention Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs – worldwide, 2000–2004. MMWR Morb Mortal Wkly Rep. 2006;55(11):301–305. [PubMed] [Google Scholar]
- 86.Shimao T. Drug resistance in tuberculosis control. Tubercle. 1987;68(Suppl 2):5–18. doi: 10.1016/0041-3879(87)90014-6. [DOI] [PubMed] [Google Scholar]
- 87.World Health Organization TB: A Global Emergency. WHO Report on the TB Epidemic Geneva: World Health Organization; 1994Available from: http://whqlibdoc.who.int/hq/1994/WHO_TB_94.177.pdfAccessed April 16, 2013 [Google Scholar]
- 88.Schatz A, Bugle E, Waksman SA. Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Exp Biol Med. 1944;55(1):66–69. doi: 10.1097/01.blo.0000175887.98112.fe. Italian. [DOI] [PubMed] [Google Scholar]
- 89.Lehmann J. Para-aminosalicylic acid in the treatment of tuberculosis. Lancet. 1946;1(6384):15. doi: 10.1016/s0140-6736(46)91185-3. [DOI] [PubMed] [Google Scholar]
- 90.Bloch AB, Simone PM, McCray E, Castro KG. Preventing multidrug-resistant tuberculosis. JAMA. 1996;275(6):487–489. Italian. [PubMed] [Google Scholar]
- 91.Frieden TR, Sterling TR, Munsiff SS, Watt CJ, Dye C. Tuberculosis. Lancet. 2003;362(9387):887–899. doi: 10.1016/S0140-6736(03)14333-4. [DOI] [PubMed] [Google Scholar]
- 92.Centers for Disease Control and Prevention Prevention and treatment of tuberculosis among patients infected with human immunodeficiency virus: principles of therapy and revised recommendations. MMWR Recomm Rep. 1998;47(RR-20):1–58. [PubMed] [Google Scholar]
- 93.Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, Jul 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America (IDSA), Sep 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 Pt 2):S221–S247. doi: 10.1164/ajrccm.161.supplement_3.ats600. [DOI] [PubMed] [Google Scholar]
- 94.Bowler PG, Welsby S, Towers V, et al. Multidrug-resistant organisms, wounds and topical antimicrobial protection. Int Wound J. 2012;9(4):387–396. doi: 10.1111/j.1742-481X.2012.00991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar A, Schweizer HP. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv Drug Deliv Rev. 2005;57(10):1486–1513. doi: 10.1016/j.addr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 96.Wright GD, Sutherland AD. New strategies for combating multidrug-resistant bacteria. Trends Mol Med. 2007;13(6):260–267. doi: 10.1016/j.molmed.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 97.Neu HC. The crisis in antibiotic resistance. Science. 1992;257(5073):1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 98.Wax RG, Lewis K, Salyers AA, Taber H. Bacterial Resistance to Antimicrobials. Boca Raton, FL: CRC Press; 2008. [Google Scholar]
- 99.Boicheva M, Lazarova G, Rachkova K, Rukanova D, Teneva M, Dzeneva H. Bacterial pathogens with multidrug resistance. Etiological structure of infections in ICU, Umbal-Ead Stara Zagora. Trakia Journal of Sciences. 2010;8(Suppl 2):165–170. [Google Scholar]
- 100.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. Italian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Siegel JD, Rhinehart E, Jackson M, Chiarello L. Management of multidrug-resistant organisms in health care settings, 2006. Am J Infect Control. 2007;35(10 Suppl 2):S165–S193. doi: 10.1016/j.ajic.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 102.Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell. 2007;128(6):1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 103.Amato-Gauci A, Ammon A.The First European Communicable Disease Epidemiological Report Stockholm: European Centre for Disease Prevention and Control; 2007Available from: http://www.ecdc.europa.eu/en/publications/Publications/0706_SUR_First_%20Annual_Epidemiological_Report_2007.pdfAccessed April 16, 2013 [Google Scholar]
- 104.Freire-Moran L, Aronsson B, Manz C, et al. Critical shortage of new antibiotics in development against multidrug-resistant bacteria – time to react is now. Drug Resist Updat. 2011;14(2):118–124. doi: 10.1016/j.drup.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 105.Zaidi AK, Huskins WC, Thaver D, Bhutta ZA, Abbas Z, Goldmann DA. Hospital-acquired neonatal infections in developing countries. Lancet. 2005;365(9465):1175–1188. doi: 10.1016/S0140-6736(05)71881-X. German. [DOI] [PubMed] [Google Scholar]
- 106.Committee on New Directions in the Study of Antimicrobial Therapeutics . Treating Infectious Diseases in a Microbial World: Report of Two Workshops on Novel Antimicrobial Therapeutics. Washington, DC: National Academies Press; 2006. [PubMed] [Google Scholar]
- 107.Gootz TD. Discovery and development of new antimicrobial agents. Clin Microbiol Rev. 1990;3(1):13–31. doi: 10.1128/cmr.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Akyar I. Searching for Antibiotics Against Super Bug Bacteria. Acibadem University Health Sciences Journal. 2010;1(2):62–67. [Google Scholar]
- 109.Coates A, Hu Y, Bax R, Page C. The future challenges facing the development of new antimicrobial drugs. Nat Rev Drug Discov. 2002;1(11):895–910. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- 110.Pearson H. “Superbug” hurdles key drug barrier. Nature. 2002;418(6897):469. doi: 10.1038/418469b. [DOI] [PubMed] [Google Scholar]
- 111.Barrett JF. MRSA: status and prospects for therapy? An evaluation of key papers on the topic of MRSA and antibiotic resistance. Expert Opin Ther Targets. 2004;8(6):515–519. doi: 10.1517/14728222.8.6.515. [DOI] [PubMed] [Google Scholar]
- 112.Salgado CD, Farr BM, Calfee DP. Community-acquired methicillin-resistant Staphylococcus aureus: a meta-analysis of prevalence and risk factors. Clin Infect Dis. 2003;36(2):131–139. doi: 10.1086/345436. Portuguese. [DOI] [PubMed] [Google Scholar]
- 113.Deshpande LM, Fritsche TR, Moet GJ, Biedenbach DJ, Jones RN. Antimicrobial resistance and molecular epidemiology of vancomycin-resistant enterococci from North America and Europe: a report from the SENTRY antimicrobial surveillance program. Diagn Microbiol Infect Dis. 2007 Jun;58(2):163–170. doi: 10.1016/j.diagmicrobio.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 114.Gordon KA, Biedenbach DJ, Jones RN. Comparison of Streptococcus pneumoniae and Haemophilus influenzae susceptibilities from community-acquired respiratory tract infections and hospitalized patients with pneumonia: five-year results for the SENTRY Antimicrobial Surveillance Program. Diagn Microbiol Infect Dis. 2003;46(4):285–289. doi: 10.1016/s0732-8893(03)00087-7. [DOI] [PubMed] [Google Scholar]
- 115.Whitney CG, Farley MM, Hadler J, et al. Increasing prevalence of multidrug-resistant Streptococcus pneumoniae in the United States. N Engl J Med. 2000;343(26):1917–1924. doi: 10.1056/NEJM200012283432603. [DOI] [PubMed] [Google Scholar]
- 116.Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42(5):657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 117.Ji Y, Zhang B, Van SF, et al. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science. 2001;293(5538):2266–2269. doi: 10.1126/science.1063566. Dutch. [DOI] [PubMed] [Google Scholar]
- 118.Hutchison CA, Peterson SN, Gill SR, et al. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286(5447):2165–2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- 119.Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, Mekalanos JJ. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc Natl Acad Sci U S A. 2002;99(2):966–971. doi: 10.1073/pnas.012602299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Forsyth RA, Haselbeck RJ, Ohlsen KL, et al. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol. 2002;43(6):1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 121.Kobayashi K, Ehrlich S, Albertini A, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100(8):4678–4683. doi: 10.1073/pnas.0730515100. Italian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Young K, Jayasuriya H, Ondeyka JG, et al. Discovery of FabH/FabF inhibitors from natural products. Antimicrob Agents Chemother. 2006;50(2):519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang J, Soisson SM, Young K, et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441(7091):358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 124.Singh SB, Jayasuriya H, Ondeyka JG, et al. Isolation, structure, and absolute stereochemistry of platensimycin, a broad spectrum antibiotic discovered using an antisense differential sensitivity strategy. J Am Chem Soc. 2006;128(36):11916–11920. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]
- 125.Singh SB, Herath KB, Wang J, Tsou N, Ball RG. Chemistry of platensimycin. Tetrahedron Lett. 2007;48:5429–5433. [Google Scholar]
- 126.Herath KB, Attygalle AB, Singh SB. Biosynthetic studies of platensimycin. J Am Chem Soc. 2007;129(50):15422–15423. doi: 10.1021/ja0758943. [DOI] [PubMed] [Google Scholar]
- 127.Arnaud C. Antibiotic halts lipid synthesis. Chem Eng News. 2006;84:7. [Google Scholar]
- 128.Wang J, Kodali S, Lee SH, et al. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc Natl Acad Sci. 2007;104(18):7612–7616. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barton S. New antibiotic on the horizon? Nat Rev Drug Discov. 2006;5:539–539. [Google Scholar]
- 130.Singh SB, Young K. New antibiotic structures from fermentations. Expert Opin Ther Pat. 2010;20:1359–1371. doi: 10.1517/13543776.2010.509347. [DOI] [PubMed] [Google Scholar]
- 131.Habich D, von Nussbaum F. Platensimycin, a new antibiotic and “superbug challenger” from nature. Chem Med Chem. 2006;1(9):951–954. doi: 10.1002/cmdc.200600145. [DOI] [PubMed] [Google Scholar]
- 132.Nicolaou KC, Stepan AF, Lister T, et al. Design, synthesis, and biological evaluation of platensimycin analogues with varying degrees of molecular complexity. J Am Chem Soc. 2008;130(39):13110–13119. doi: 10.1021/ja8044376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Holzgrabe U, Dingermann T, Zundorf I. An old pathway to new antibiotics – platensimycin. Alte wege zu neuen antibiotika – platensimycin. Pharm Unserer Zeit. 2006;35(5):388–389. doi: 10.1002/pauz.200690104. German. [DOI] [PubMed] [Google Scholar]
- 134.Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. Inhibition of β-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J Biol Chem. 2001;276(9):6551–6559. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
- 135.Nicolaou KC, Li A, Edmonds DJ. Total synthesis of platensimycin. Angew Chem Int Ed Engl. 2006;45(42):7086–7090. doi: 10.1002/anie.200603892. [DOI] [PubMed] [Google Scholar]
- 136.Kaliappan KP, Ravikumar V. An expedient enantioselective strategy for the oxatetracyclic core of platensimycin. Org Lett. 2007;9(12):2417–2419. doi: 10.1021/ol070848t. [DOI] [PubMed] [Google Scholar]
- 137.Zou Y, Chen CH, Taylor CD, Foxman BM, Snider BB. Formal synthesis of (+/−)-platensimycin. Org Lett. 2007;9(9):1825–1828. doi: 10.1021/ol070563g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li P, Payette JN, Yamamoto H. Enantioselective route to platensimycin: an intramolecular Robinson annulation approach. J Am Chem Soc. 2007;129(31):9534–9535. doi: 10.1021/ja073547n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nicolaou KC, Lister T, Denton RM, Montero A, Edmonds DJ. Adamantaplatensimycin: a bioactive analogue of platensimycin. Angew Chem Int Ed Engl. 2007;46(25):4712–4714. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]
- 140.Potera C. Novel pentacyclic from soils blocks bacterial fatty acid synthesis. Microbe. 2006;1:350–351. [Google Scholar]
- 141.Manallack D, Crosby I, Khakham Y, Capuano B. Platensimycin: a promising antimicrobial targeting fatty acid synthesis. Curr Med Chem. 2008;15(7):705–710. doi: 10.2174/092986708783885255. [DOI] [PubMed] [Google Scholar]
- 142.Jayasuriya H, Herath KB, Zhang C, et al. Isolation and structure of platencin: a FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew Chem Int Ed Engl. 2007;46(25):4684–4688. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]
- 143.Pearson H. Antibiotic faces uncertain future. Nature. 2006;441(7091):260–261. doi: 10.1038/441260a. [DOI] [PubMed] [Google Scholar]
- 144.Chan DI, Vogel HJ. Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem J. 2010;430(1):1–19. doi: 10.1042/BJ20100462. [DOI] [PubMed] [Google Scholar]
- 145.Heath RJ, White SW, Rock CO. Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res. 2001;40(6):467–497. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 146.Campbell JW, Cronan JE., Jr Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu Rev Microbiol. 2001;55:305–332. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 147.Miyakawa S, Suzuki K, Noto T, Harada Y, Okazaki H. Thiolactomycin, a new antibiotic. IV Biological properties and chemotherapeutic activity in mice. J Antibiot (Tokyo) 1982;35(4):411–419. doi: 10.7164/antibiotics.35.411. [DOI] [PubMed] [Google Scholar]
- 148.Holford NH, Sheiner LB. Kinetics of pharmacologic response. Pharmacol Ther. 1982;16(2):143–166. doi: 10.1016/0163-7258(82)90051-1. [DOI] [PubMed] [Google Scholar]
- 149.Jang KP, Kim CH, Na SW, et al. 7-phenylplatensimycin and 11-methyl-7-phenylplatensimycin: more potent analogs of platensimycin. Bioorg Med Chem Lett. 2010;20(7):2156–2158. doi: 10.1016/j.bmcl.2010.02.037. [DOI] [PubMed] [Google Scholar]
- 150.Barykina OV, Rossi KL, Rybak MJ, Snider BB. Synthesis and antibacterial properties of (−)-nor-platencin. Org Lett. 2009;11(22):5334–5337. doi: 10.1021/ol902194q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Patra M, Gasser G, Wenzel M, Merz K, Bandow JE, Metzler-Nolte N. Synthesis and biological evaluation of ferrocene-containing bioorganometallics inspired by the antibiotic platensimycin lead structure. Organometallics. 2010;29:4312–4319. [Google Scholar]
- 152.Wang J, Lee V, Sintim HO. Efforts towards the identification of simpler platensimycin analogues – the total synthesis of oxazinidinyl platensimycin. Chemistry. 2009;15(12):2747–2750. doi: 10.1002/chem.200802568. [DOI] [PubMed] [Google Scholar]
- 153.Wang J, Sintim HO. Dialkylamino-2,4-dihydroxybenzoic acids as easily synthesized analogues of platensimycin and platencin with comparable antibacterial properties. Chemistry. 2011;17(12):3352–3357. doi: 10.1002/chem.201002410. [DOI] [PubMed] [Google Scholar]
- 154.Cohen R. Approaches to reduce antibiotic resistance in the community. Pediatr Infect Dis J. 2006;25(10):977–980. doi: 10.1097/01.inf.0000239271.10784.1e. [DOI] [PubMed] [Google Scholar]
- 155.Palanichamy K, Kaliappan KP. Discovery and syntheses of “superbug challengers” – platensimycin and platencin. Chem Asian J. 2010;5(4):668–703. doi: 10.1002/asia.200900423. [DOI] [PubMed] [Google Scholar]
- 156.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24(1):71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Brown AK, Taylor RC, Bhatt A, Futterer K, Besra GS. Platensimycin activity against mycobacterial β-ketoacyl-ACP synthases. PLoS One. 2009;4(7):e6306. doi: 10.1371/journal.pone.0006306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nicolaou KC, Tria GS, Edmonds DJ. Total synthesis of platencin. Angew Chem Int Ed Engl. 2008;47(9):1780–1783. doi: 10.1002/anie.200800066. [DOI] [PubMed] [Google Scholar]
- 159.Hayashida J, Rawal VH. Total synthesis of (+/–)-platencin. Angew Chem Int Ed Engl. 2008;47(23):4373–4376. doi: 10.1002/anie.200800756. [DOI] [PubMed] [Google Scholar]
- 160.Tiefenbacher K, Mulzer J. Synthesis of platensimycin. Synthese von platensimycin. Angew Chem. 2008;120(14):2582–2590. doi: 10.1002/anie.200705303. German. [DOI] [PubMed] [Google Scholar]
- 161.Yun SY, Zheng JC, Lee D. Concise synthesis of the tricyclic core of platencin. Angew Chem Int Ed Engl. 2008;47(33):6201–6203. doi: 10.1002/anie.200801587. [DOI] [PubMed] [Google Scholar]
- 162.Waalboer DCJ, Schaapman MC, van Delft FL, Rutjes FP. High-pressure entry into platencin. Angew Chem Int Ed Engl. 2008;47(35):6576–6578. doi: 10.1002/anie.200802912. [DOI] [PubMed] [Google Scholar]
- 163.Nicolaou KC, Toh QY, Chen DY. An expedient asymmetric synthesis of platencin. J Am Chem Soc. 2008;130(34):11292–11293. doi: 10.1021/ja804588r. [DOI] [PubMed] [Google Scholar]
- 164.Austin KA, Banwell MG, Willis AC. A formal total synthesis of platencin. Org Lett. 2008;10(20):4465–4468. doi: 10.1021/ol801647h. [DOI] [PubMed] [Google Scholar]
- 165.Jayasuriya H, Herath KB, Ondeyka JG, et al. Structure of homoplatensimide A: a potential key biosynthetic intermediate of platensimycin isolated from Streptomyces platensis. Tetrahedron Lett. 2008;49(22):3648–3651. [Google Scholar]
- 166.Herath KB, Zhang C, Jayasuriya H, et al. Structure and semisynthesis of platensimide A, produced by Streptomyces platensis. Org Lett. 2008;10(9):1699–1702. doi: 10.1021/ol800251v. [DOI] [PubMed] [Google Scholar]
- 167.Zhang C, Ondeyka J, Zink DL, Burgess B, Wang J, Singh SB. Isolation, structure and fatty acid synthesis inhibitory activities of platensimycin B1–B3 from Streptomyces platensis. Chem Commun (Camb) 2008;(40):5034–5036. doi: 10.1039/b810113b. [DOI] [PubMed] [Google Scholar]
- 168.Singh SB, Jayasuriya H, Herath KB, et al. Isolation, enzyme-bound structure, and activity of platensimycin A1 from Streptomyces platensis. Tetrahedron Lett. 2009;50(37):5182–5185. doi: 10.1016/j.bmcl.2009.06.061. [DOI] [PubMed] [Google Scholar]
- 169.Wu M, Singh SB, Wang J, et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc Natl Acad Sci U S A. 2011;108(13):5378–5383. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Skold O. Antibiotics and Antibiotic Resistance. Hoboken, NJ: John Wiley & Sons; 2011. [Google Scholar]