

Abstract
Attention-deficit/hyperactivity disorder (ADHD) is the most commonly diagnosed behavioral disorder in childhood and likely represents an extreme of normal behavior. ADHD significantly impacts learning in school-age children and leads to impaired functioning throughout the life span. There is strong evidence for a genetic etiology of the disorder, although putative alleles, principally in dopamine-related pathways suggested by candidate-gene studies, have very small effect sizes. We use affected-sib-pair analysis in 203 families to localize the first major susceptibility locus for ADHD to a 12-cM region on chromosome 16p13 (maximum LOD score 4.2; P=.000005), building upon an earlier genomewide scan of this disorder. The region overlaps that highlighted in three genome scans for autism, a disorder in which inattention and hyperactivity are common, and physically maps to a 7-Mb region on 16p13. These findings suggest that variations in a gene on 16p13 may contribute to common deficits found in both ADHD and autism.
Attention-deficit/hyperactivity disorder (ADHD [MIM 143465]) is the most commonly diagnosed behavioral disorder in childhood and likely represents an extreme of normal behavior (Levy et al. 1997; Brown et al. 2001). ADHD significantly impacts learning in school-age children and leads to impaired functioning throughout the life span (Cantwell 1996). There is strong evidence for a genetic etiology of the disorder (Smalley 1997), although putative alleles, principally in dopamine-related pathways suggested by candidate-gene studies, have very small effect sizes (Faraone et al. 2001; Swanson et al. 2001). In the first genomewide scan for ADHD, three chromosomal regions, 10q26, 12q23, and 16p13, yielded multipoint maximum LOD scores >1.5, but no region exceeded the criteria for significant or suggestive linkage (Fisher et al. 2002).
The sample used in the present study consists of 277 affected sib pairs (ASPs) in 203 families, including 126 ASPs (104 families) from the previous genomewide scan of ADHD (Fisher et al. 2002). ADHD was diagnosed using DSM-IV criteria, with 95% of affected individuals meeting full criteria and 5% of affected individuals falling one symptom short but meeting age-at-onset and impairment criteria (American Psychiatric Association 1994). This definition of ADHD corresponds to the “broad” classification used in our previous genomewide scan (Fisher et al. 2002). All families with ASP had at least one affected member meeting full criteria (for a description of the measures and diagnostic methods, see Smalley et al. 2000). The affected members (322 male and 118 female) in the ASPs had a mean IQ of 106, a mean age of 11 years, and were largely white (80%).
Individuals were genotyped for 11 microsatellites and 5 SNPs spanning ∼25 cM on chromosome 16p, analyzed using standard procedures (Hirschhorn et al. 2000; Fisher et al. 2002). Both parents were genotyped in 185 (91%) of the families, and only one parent was genotyped in 18 (9%) of the families. Because of our interest in the 16p region, resulting both from the initial genome scan results (Fisher et al. 2002) and from our work in autism and tuberous sclerosis complex (Smalley et al. 1998; International Molecular Genetic Study of Autism Consortium [IMGSAC] 1998, 2001; Khare et al. 2001), we are developing SNPs in the region. We identified five SNPs from larger sets available on chromosome 16, because they were located in the 25-cM region, genotyped well, and were relatively polymorphic (i.e., allelic variants had frequencies >10%). The five SNPs fell within a narrow region (1 Mb) and were used in linkage analyses by setting the distances between them according to physical maps.
The microsatellite order and the SNP placement among them were determined using the Marshfield genetic database and physical databases (Center for Medical Genetics Web site), the Human Genome Project–Santa Cruz (HGP-sc) (International Human Genome Sequence Consortium 2001; UCSC Human Genome Project Working Draft Web site), and Celera (CEL) (Venter et al. 2001; Celera Web site), as well as genetic mapping in our own data set. Because this region has inconsistencies across the genetic and physical databases available, the following procedure was used to determine the map order and distances for linkage analyses. The Marshfield map was used to determine microsatellite order, and, for markers genetically positioned at a single point, physical databases were used to order them. When inconsistencies were identified across different physical databases (e.g., HGP-sc August and December 2001 builds and CEL), orders that were consistent in at least two of the physical builds were selected. Genetic mapping was performed using the ASPEX program package (available from the ASPEX Linkage Analysis Package FTP site) and the “do_shuffle” command, which computes a multipoint LOD score for a marker against a fixed map of markers, moving the location of the unknown marker from the distal end through each position along the map. This method was used to check the order of the microsatellites (e.g., D16S764) in our data when discrepancies were noted between different physical databases and the Marshfield map. The SNPs were positioned on the basis of physical data (HGP-sc and CEL) and genetic mapping of the two most informative SNPs (rs1127293 and rs1107143) in the current sample against the microsatellite panel.
Single and multipoint maximum LOD scores (MLSs) were computed under the possible triangle method, under the assumption of no dominance variance, through use of ASPEX. Transmission/disequilibrium testing (TDT) of the SNPs and ADHD was performed using ASPEX, which allows for multiple siblings to be included in a test of association in the presence of linkage, by permuting parental alleles while fixing the identity-by-descent (IBD) status of the siblings.
As shown in figure 1, significant linkage (MLS=4.2) was observed for ADHD and the markers on 16p, with the maximum obtained near marker D16S3114. The 1-LOD support interval falls between markers D16S519 and D16S405, within estimated distances of ∼12 cM in our data set and ∼7 Mb on the basis of physical maps (HGP-sc and CEL) of the region between these two markers. The significant linkage in the extended sample was a function of both the increased density of markers and the additional ASPs. In the initial genome scan, the linkage finding for markers on chromosome 16 and ADHD (∼10-cM density) was a multipoint MLS of 1.5 near marker D16S3075 (Fisher et al. 2002). The markers in the present analyses (∼2-cM density) yielded a multipoint MLS of 3.1 (P=.00008) at marker D16S3114 in the same set of ASPs, despite similar levels of marker heterozygosity (i.e., the average information content of the first set was 79%, and that of the second set was 81%). The additional set of 153 ASPs independently yielded a multipoint MLS of 1.9 (P=.0015).
Figure 1.

MLSs for ADHD and markers on chromosome 16p. The relative marker locations, estimated in the current data set, are shown by diamonds. The SNPs are shown as a single point, with the average MLS indicated in the figure at that position. The SNP distances were based on physical distances (HGP-sc, August 2001 build) and were ordered as follows (with intermarker distances given, in Mb): rs153783–.005–rs1125972–.02–rs1065838–.05–rs1127293–.005–rs1107143.
As indicated in table 1, three genomewide scans for autism have highlighted the region on 16p13, with maximum linkage peaks located within the 1-LOD support interval and spanning contigs positioned 13–20 Mb from the telomere of 16p (HGP-sc and CEL). It should be noted, however, that only two of the three scans in autism (IMGSAC 2001; Liu et al. 2001) met criteria for suggestive linkage and none met criteria for significant linkage (Lander and Kruglyak 1995).
Table 1.
Single-Point IBD Proportions, Multipoint Sharing Proportions, and MLS Values (Single-Point and Multipoint) for ADHD and Markers on Chromosome 16p
|
Distance(cM) |
Single-Point |
Multipoint |
Linkage Peak from Study byb |
|||||||||
| Marker | Marshfield | PresentStudyc | No. ofInformativeMeioses | % IBD | MLS | % Sharing | MLS | Pd | LinkagePeak and1-LODSupportIntervala | IMGSAC(2001) | Philippe et al.(1999) | Liu et al.(2001) |
| D16S404 | 18.07 | 0 | 250 | 53.2 | .27 | 56.5 | 1.23 | .0086 | ||||
| D16S519 | 20.77 | 4.9 | 240 | 54.2 | .39 | 57.8 | 2.03 | .0011 | ||||
| D16S3114 | 23.28 | 4.5 | 261 | 62.5 | 3.63 | 60.4 | 4.22 | .0000052 | *** | |||
| D16S3075 | 23.28 | 2.8 | 268 | 53.0 | .23 | 59.7 | 3.75 | .000016 | *** | |||
| D16S3047 | 23.28 | .5 | 223 | 61.4 | 2.80 | 59.9 | 3.84 | .000013 | ||||
| D16S3102 | 24.53 | *** | ||||||||||
| D16S2619 | 28.3 | *** | ||||||||||
| rs153783 | 2.9 | 38 | 63.2 | .64 | 59.0 | 3.28 | .000051 | |||||
| rs1125972 | .005 | 38 | 39.5 | .00 | 59.1 | 3.29 | .000050 | |||||
| rs1065838 | .02 | 94 | 57.4 | .52 | 59.1 | 3.35 | .000043 | |||||
| rs1127293 | .05 | 123 | 62.6 | 1.68 | 59.5 | 3.81 | .000014 | |||||
| rs1107143 | .005 | 110 | 60.9 | 1.06 | 59.0 | 3.25 | .000055 | |||||
| D16S3060 | 28.3 | 2.9 | 276 | 58.7 | 2.29 | 60.1 | 3.79 | .000015 | ||||
| D16S405 | 28.3 | 1.6 | 232 | 55.2 | .55 | 59.0 | 3.01 | .000098 | ||||
| D16S3079 | 28.3 | 1.8 | 198 | 56.1 | .70 | 58.9 | 2.80 | .00017 | ||||
| D16S764 | 29.97 | 3.0 | 205 | 54.1 | .35 | 58.0 | 2.15 | .00083 | ||||
| D16S499 | 33.14 | 4.1 | 237 | 58.6 | 1.53 | 56.9 | 1.52 | .0041 | ||||
| D16S3046 | 40.65 | 7.5 | 296 | 53.7 | .39 | 56.7 | 1.43 | .0051 | ||||
Support interval is demarcated by a box; position of linkage peak is indicated by asterisks.
Positions of linkage peaks are indicated by asterisks. The maximum LOD scores for the autism scans were as follows: IMGSAC (2001), MLS = 2.9; Philippe et al. (1999), MLS = 0.74; Liu et al. (2001), MLS = 2.19.
Intermarker distances are estimated from the data set; inter-SNP distances are based on physical distances (HGP-sc, CEL).
The P values for the multipoint MLS values are found by multiplying 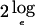 and determining significance from χ2 tables, taking into account the one-sided nature of the linkage test, as described elsewhere (Kruglyak and Lander 1995).
and determining significance from χ2 tables, taking into account the one-sided nature of the linkage test, as described elsewhere (Kruglyak and Lander 1995).
As shown in table 1, single-point MLS values were fairly consistent with the multipoint analyses, with three markers having an MLS >2.0 and seven markers having MLS values of .5–2. We tested each SNP for association with ADHD through use of the TDT, but no associations were observed (all P values >.05; data not shown).
There are 159 unigene clusters that map to the 7-Mb interval highlighted by the current scan; of these, 35 are known genes, but none are hypothesized candidates for ADHD (HGP-sc, August 2001 build). There are two possible candidates, reflected by brain-expressed mRNAs and a potential role in neurobehavioral disorders: SNN, the gene that encodes stannin, which interacts with neural toxins and exposure to which produces behavioral changes; and Nude1, a gene that encodes a Lis1-interacting protein. Hemizygous mutations in Lis1 result in a severe brain malformation, lissencephaly. Two additional candidates map within 2 Mb of the 1-LOD support interval: GRIN2A, which encodes an N-methyl-D-aspartate glutamate receptor subunit proposed to play a role in long-term potentiation; and DOC2A, which encodes a C2-like domain–containing protein implicated in neurotransmission. It is interesting to note that a gene coding for tuberous sclerosis complex (TSC2) is located ∼5 Mb telomeric from this region and that TSC is an autosomal dominant disorder in which high rates of autism and ADHD have been reported (Smalley 1998). Genotype-phenotype studies of large deletions on 16p in patients with TSC may provide additional data to identify a risk gene in ADHD and autism.
The multipoint MLS of 4.2 exceeds that recommended for significant linkage (i.e., 3.93) for the specific analytic method used in the present study (Nyholt 2000). Although neither set of ASPs independently reached significance for linkage, increased IBD sharing is evident in the first and second sets of ASPs, reflected by multipoint MLS values of 3.1 (P=.00008) and 1.9 (P=.0015), respectively. Although the MLS obtained in the second sample exceeds the recommended significance level for a replication (P=.01; Lander and Kruglyak 1995), it cannot be thought of as a true replication, because the original sample alone did not yield significant linkage; the second set of ASPs provides significant evidence for linkage of ADHD at the .05 level, when a conservative Bonferonni correction is used for 15 markers in the interval under analysis (Lander and Kruglyak 1995). The estimated effect size for the putative gene, based on the multipoint sharing proportions of 58%–60% in the extended sample, is equivalent to sibling recurrence risk ratio (λs) values of 1.4–1.6 (Risch et al. 1990a, 1990b), suggesting that such a locus could account for as much as 28%–32% of the observed λs, which has been estimated as ∼5 in ADHD (Smalley 1997).
The question of whether the region highlighted on 16p13 contains a risk gene for ADHD, autism, or both disorders requires additional investigation. Independent replication of the linkage finding on 16p13 for ADHD is necessary for confirmation of linkage. Further studies are needed to determine the significance of linkage between autism and markers in this region. If linkages are replicated, then the phenotypic overlap of ADHD and autism may shed light on a pleiotropic risk allele, if present. Clinically, ADHD and autism are quite distinct, and, although inattention and hyperactivity are common among children with autism, a diagnosis of ADHD is precluded under DSM-IV criteria if symptoms are better accounted for by autism or pervasive developmental disorder (American Psychiatric Association 1994). Despite the clear clinical boundaries, there are certain behavioral, cognitive, and neurobiological deficits that suggest some degree of phenotypic overlap. First, maladaptive social functioning (a clinical criterion of autism) is common in ADHD, and, although a majority of such deficits are thought to arise from core features of ADHD (e.g., impulsivity), other deficits, such as failing to monitor and react to social interactions and misinterpretation of social cues (Greene et al. 1996), may be similar in type to those seen in autism. Second, executive function (EF) deficits, processes involved in the planning and implementation of goal-oriented strategies, are noted in both ADHD and autism, although underlying mechanisms, including inhibition, set-shifting, attention, and working memory, may differ (Sergeant et al. 2002). For example, Ozonoff and Jensen (1999) found deficits in flexibility and planning in autism, whereas inhibition deficits accounted for the EF deficits in ADHD; however, additional work in this area is greatly needed (Sergeant et al. 2002). Third, there is a significant subgroup of children with autism and symptoms of ADHD who respond to stimulant medication (Handen et al. 2000), suggesting that common neurobiological mediators may be present in a subset of cases. Further identification of phenotypic overlap in ADHD and autism at the behavioral, neurocognitive, and brain levels is warranted, particularly in light of the linkage findings. The current data provide the first evidence, to our knowledge, of significant linkage of this common neurobehavioral disorder, using a genomewide approach and anonymous polymorphic markers.
Acknowledgments
This work was supported by National Institute of Mental Health grants MH58277 (to S.L.S.), MH01969 (to J.J.M.), and MH01805 (to J.T.M.); by UC LSI grant L9808 (to S.F.N.); and by the Wellcome Trust (support to A.P.M.). A.P.M. is a Wellcome Trust Principal Research Fellow. S.E.F. is a Royal Society Research Fellow. Thanks to all the families who participated in this research; to Elva Rios, Tae Kim, Laura Combs, Leah Pressman, Ph.D., and Deborah Lynn, M.D., for their help in data collection; to May Yang, for analytic support; to Helen Hotta, for administrative support; to Lori Crawford, for molecular support; and to Dr. Dennis Cantwell, who inspired our work on ADHD.
Electronic-Database Information
The accession number and URLs for data presented herein are as follows:
- ASPEX Linkage Analysis Package, ftp://lahmed.stanford.edu/pub/aspex/index.html
- Celera, http://www.celera.com/
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ADHD [MIM 143465])
- UCSC Human Genome Project Working Draft, http://genome.cse.ucsc.edu/ (for August and December 2001 builds)
References
- American Psychiatric Association (1994) DSM IV: diagnostic and statistical manual of mental disorders. American Psychiatric Association, Washington, DC [Google Scholar]
- Brown RT, Freeman WS, Perrin JM, Stein MT, Amler RW, Feldman HM, Pierce K, Wolraich ML (2001) Prevalence and assessment of attention-deficit/hyperactivity disorder in primary care settings. Pediatrics 107:E43 [DOI] [PubMed] [Google Scholar]
- Cantwell DP (1996) Attention deficit disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 35:978–987 [DOI] [PubMed] [Google Scholar]
- Faraone SV, Doyle AE, Mick E, Biederman J (2001) Meta-analysis of the association between the 7-repeat allele of the dopamine D(4) receptor gene and attention deficit hyperactivity disorder. Am J Psychiatry 158:1052–1057 [DOI] [PubMed] [Google Scholar]
- Fisher SE, Francks C, McCracken JT, McGough JJ, Marlow AJ, MacPhie IL, Newbury DF, Crawford LR, Palmer CG, Woodward JA, Del'Homme M, Cantwell DP, Nelson SF, Monaco AP, Smalley SL (2002) A genomewide scan for loci involved in attention deficit/hyperactivity disorder. Am J Hum Genet 70:1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RW, Biederman J, Faraone SV, Ouellette CA, Penn C, Griffin SM (1996) Toward a new psychometric definition of social disability in children with attention-deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 35: 571–578 [DOI] [PubMed] [Google Scholar]
- Handen BL, Johnson CR, Lubetsky M (2000) Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord 30:245–255 [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Sklar P, Lindblad-Toh K, Lim YM, Ruiz-Gutierrez M, Bolk S, Langhorst B, Schaffner S, Winchester E, Lander ES (2000) SBE-TAGS: an array-based method for efficient single-nucleotide polymorphism genotyping. Proc Natl Acad Sci USA 97:12164–12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequence Consortium (2001) Initial sequence and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (1998) A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet 7:571–578 [DOI] [PubMed] [Google Scholar]
- ——— (2001) A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet 69:570–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare L, Strizheva GD, Bailey JN, Au KS, Northrup H, Smith M, Smalley SL, Henske EP (2001) A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. J Med Genet 38:347–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Levy F, Hay DA, McStephen M, Wood C, Waldman I (1997) Attention-deficit hyperactivity disorder: a category or a continuum? Genetic analysis of a large-scale twin study. J Am Acad Child Adolesc Psychiatry 36:737–744 [DOI] [PubMed] [Google Scholar]
- Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, Lord C, Iversen P, Hoh J, Ott J, Gilliam TC (2001) A genomewide screen for autism susceptibility loci. Am J Hum Genet 69:327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholt DR (2000) All LODs are not created equal. Am J Hum Genet 67:282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, Jensen J (1999) Brief report: specific executive function profiles in three neurodevelopmental disorders. J Autism Dev Disord 29:171–177 [DOI] [PubMed] [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, Penet C, Feingold J, Brice A, Leboyer M, Paris Autism Research International Sibpair Study (1999) Genome-wide scan for autism susceptibility genes. Hum Mol Genet 8:805–812 [DOI] [PubMed] [Google Scholar]
- Risch N (1990a) Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 46:222–228 [PMC free article] [PubMed] [Google Scholar]
- ——— (1990b) Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet 46:229–241 [PMC free article] [PubMed] [Google Scholar]
- Sergeant JA, Geurts H, Oosterlaan J (2002) How specific is a deficit of executive functioning for attention-deficit/hyperactivity disorder? Behav Brain Res 130:3–28 [DOI] [PubMed] [Google Scholar]
- Smalley SL (1997) Genetic influences in childhood-onset psychiatric disorders: autism and attention-deficit/hyperactivity disorder. Am J Hum Genet 60:1276–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1998) Autism and tuberous sclerosis. J Autism Dev Disord 28:407–414 [DOI] [PubMed] [Google Scholar]
- Smalley SL, McGough JJ, Del'Homme M, NewDelman J, Gordon E, Kim T, Liu A, McCracken JT (2000) Familial clustering of symptoms and disruptive behaviors in multiplex families with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 39:1135–1143 [DOI] [PubMed] [Google Scholar]
- Swanson J, Posner M, Fusella J, Wasdell M, Sommer T, Fan J (2001) Genes and attention deficit hyperactivity disorder. Curr Psychiatry Rep 3:92–100 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, et al (2001) The sequence of the human genome. Science 291:1304–1351 [DOI] [PubMed] [Google Scholar]