

Abstract
The potential role of conventional and regulatory T cells (Tregs) in protection from HIV-1 infection remains unclear. To address this question, we analyzed samples from 129 HIV-1-exposed seronegative individuals (HESN) from an HIV-1-serodiscordant couples cohort. To assess the presence of HIV-specific T cell responses and Treg function, we measured the proliferation of T cells in response to HIV-1 peptide pools in peripheral blood mononuclear cells (PBMCs) and PBMCs depleted of Tregs. We identified HIV-specific CD4+ and CD8+ T cell responses and, surprisingly, the overall CD4+ and CD8+ T cell response rate was not increased when Tregs were removed from cell preparations. Of the 20 individuals that had HIV-1-specific CD4+ T cell responses, only eight had Tregs that could suppress this proliferation. When compared with individuals whose Tregs could suppress HIV-1-specific CD4+ T cell proliferation, individuals with Tregs unable to suppress showed a trend toward increased T cell activation and Treg frequency and a significant increase in HIV-1-specific production of microphage inflammatory protein-1β (MIP-1β) by CD4+ T cells, autocrine production of which has been shown to be protective in terms of HIV-1 infection of CD4+ T cells.
Introduction
An important challenge related to the development of an HIV-1 vaccine is the lack of a validated experimental model for transmission. While investigations using nonhuman primate models lend considerable insight into possible immune correlates of protection against HIV-1, fundamental differences between simian immunodeficiency virus (SIV) and HIV-1, and between host immune responses in humans and other primates, preclude direct extrapolation of immunogenicity and correlates from monkey models to human infection. Rare individuals who remain seronegative despite repeated HIV-1 exposure (HIV-1-exposed seronegatives, HESN) represent an exquisitely relevant human model of HIV-1 protection through their potential ability to either resist or markedly control infection. A number of studies of immunologic factors potentially relevant to HIV-1 protection have been done among HESN populations including sex workers from areas of high HIV-1 prevalence, men who have sex with men, and HIV- 1-serodiscordant couples.1,2
The degree and persistence of immune protection in HESN may be dependent on continuous virus exposure; cellular immune responses against HIV-1 have been shown to fade in HESN infants born to HIV-1-infected mothers, HESN healthcare workers reporting a single accidental work exposure to HIV-1, and high-risk HESN women who adopt safer sexual practices, in whom late HIV-1 seroconversion has been seen as exposure and immunity have waned.3–5 Thus, HESN studies carried out using cohorts where the viral exposure varies over time may be less likely to yield data on immune correlates of protection against HIV-1. HIV-1-serodiscordant couples, given ongoing exposure, offer an opportunity to overcome this problem.
The reasons for possible resistance to HIV-1 infection in HESN subjects remain controversial and under investigation, although several nonmutually exclusive mechanisms have been proposed. First, there are known genetic factors at play, such as the CCR5Δ32 mutation that results in a CCR5 receptor unable to bind HIV-1 and therefore prevention of infection in homozygous individuals.6 Second, there could be an antigen-specific immune response generated upon HIV-1 exposure that is sufficient to prevent infection. The first immunologic evidence of such HIV-1 resistance was seen in the uninfected members of HIV-1-serodiscordant partnerships, who specifically mounted CD8+ T cell responses to HIV-1 envelope and core proteins.7 Similar HIV-1-specific T cell responses have since been found in a variety of HESN populations, including sex workers and infants born to HIV-1-infected women.8,9 Compared to unexposed individuals, a number of specific phenotypic and functional characteristics of CD8+ T cells have been identified in HESN, including a higher percentage of central memory T cells and a decreased percentage of effector memory cells,10–12 an increased percentage of activated CD8+ T cells,11,12 increased antiviral cytolytic function,13 and increased cellular proliferative capacity.14 In support of this last finding, it is notable that in HIV-1-infected persons the ability of HIV-1-specific CD8+ T cells to proliferate upon antigen exposure is the only functional attribute that inversely correlates with viral loads.15,16
Additionally, a distinctive chemokine pattern has been reported in HESN. In a cohort of commercial sex workers, an increased level of microphage inflammatory protein-1α (MIP-1α) and microphage inflammatory protein-1β (MIP-1β) in HESN versus control groups was detected.17 Similarly, in a serodiscordant couples cohort, in vitro secretion of beta chemokines has been shown to be higher in exposed seronegative individuals, both at baseline and upon stimulation with p24.18 Furthermore, beta chemokines have been shown to protect CD4+ T cells from HIV-1 infection by binding to CCR5, the coreceptor for HIV-1 entry.19 Another mechanism proposed to account for HIV-1 resistance in HESN is an overall CD4+ T cell quiescence, as defined by decreased expression of activation markers,20 cytokine secretion,21 and gene expression profiling22 in HESN commercial sex workers. This reduced T cell activation has been proposed to be at least partially due to an increased percentage of regulatory T cells (Tregs).20
Tregs, a subset of CD4+ T cells, have demonstrated roles in regulating the immune system under homeostatic conditions as well as during infection. Tregs can suppress proliferation and function of several immune cell types, including Th1, Th2, and Th17 CD4+ T cells and CD8+ T cells. Moreover, they can modulate the migration of immune cells to the site of infection.23 Specifically, in a mouse model, it has been demonstrated that in the absence of Tregs the expression of CCL5/RANTES, a beta chemokine that binds to CCR5, was decreased in vaginal tissues following herpes simplex virus (HSV) infection, demonstrating that Tregs control the chemokine gradient in response to a viral exposure.24
The role of Tregs specifically in HIV-1 infection remains unclear, though the recent finding of HIV-1-Gag-specific Tregs using class II tetramer staining25 suggests that they indeed participate in anti-HIV immunity. Given their role in suppressing immunity, they could be beneficial by dampening generalized immune activation, thereby reducing the pool of HIV-1-susceptible cells; alternately, they could be detrimental, by reducing the HIV-1-specific immune response. In support of the latter idea, in one study among HESN neonates, depletion of Tregs revealed strong HIV-1-specific CD8+ and CD4+ immune responses.26
In our study, we addressed the role of Tregs in protection from HIV-1 infection by characterizing the Treg and conventional T cell phenotypes in HESN belonging to a serodiscordant couples cohort. By using the highly specific CFSE assay to detect HIV-specific proliferation, we observed that a group of HESN subjects whose Tregs were unable to suppress the HIV-1-driven T cell proliferation were characterized by an increase in T cell activation and increased CD4+ T cell secretion of MIP-1β in response to HIV-1 stimulation.
Materials and Methods
Study participants
Between August 2007 and July 2010, 400 HIV-1-serodiscordant couples from Kampala, Uganda were enrolled in an observational study of immune correlates of HIV-1 protection. Participants were ≥18 years of age and sexually active. HIV-1-seropositive partners had no history of AIDS-defining conditions and were not using antiretroviral therapy. The study protocol was approved by institutional review boards at the University of Washington and the AIDS Research Committee of the Uganda National Council of Science and Technology; all participants provided written informed consent.
For the present study, a subset of HIV-1-uninfected partners was tested, and peripheral blood mononuclear cell (PBMC) samples obtained at the time of study enrollment were used. The number of analyzed samples varied between assays due to variable cell recovery and viability.
Reagents
For the in vitro stimulation, global potential T cell epitope peptides27 for Gag, Pol, Env, and Vpu, each including the 40 most frequent 15-mers among all sequences, were synthesized by BioSynthesis (Lewisville, TX) and used at 2 μg/ml per peptide.
For the characterization of Tregs, anti-CD3 (ECD, clone UCHT1) was from Beckman Coulter (Brea, CA); CD4 (QDot 605, clone S3.5) was from Invitrogen (Grand Island, NY); and CD8 (Ax700, clone RPA-T8), CD25 (PerCP Cy5.5, clone M-A251), CD127 (V450, clone HIL-7R-M21), and FoxP3 (Ax488, clone 236A/E7) were from BD (San Jose, CA).
For the T cell analysis CD14 (QDot605, clone TüK4) was from Invitrogen; CD3 (Pac blue, clone UCHT1), CD4 (APCCy7, clone RPA-T4), Ki67 (FITC, clone B56), and Bcl-2 (PE, clone Bcl-2/100) were from BD.
For intracellular cytokine staining, Golgi Stop, anti-CD3 (ECD, clone UCHT1), CD4 (Ax700, clone RPA-T8), CD8 (PerCP Cy5.5, RPA-T8), MIP-1β (PE, clone D21-1351), interferon (IFN)-γ (V450, clone B27), and tumor necrosis factor (TNF)-α (FITC, clone 6401.1111) were from BD. Phorbol 12-myristate 13-acetate (PMA), ionomycin, dimethyl sulfoxide (DMSO), propidium iodide (PI), and Brefeldin A were from Sigma-Aldrich (St. Louis, MO).
For cell sorting, cells were stained with anti-CD4 (APC Cy7, clone RPA-T4), CD8 (Ax700, clone RPA-T8), CD127 (Ax647, clone HIL-7R-M21), and CD25 (PECy7, clone M-A251), all from BD. Anti-CD3 (purified, clone HIT3a) and CD28 (purified, clone CD28.2) for the stimulation were from BD Biosciences.
Aqua live-dead cell staining kit and CellTrace-CFSE proliferation kit were from Invitrogen.
Treg frequency and T cell activation detection
PBMCs were thawed according to standard procedures28 and two different flow cytometry staining panels were used on each sample to measure Treg and T cell frequency and phenotype. The Treg panel included a live-dead discrimination dye and antibodies recognizing CD3, CD4, CD8, CD25, CD127, and FoxP3. The T cell panel included a live-dead discrimination dye and antibodies recognizing CD3, CD14, CD4, CD8, Ki67, and Bcl-2. For each sample, 500,000 cells were incubated with the antibody mix for the extracellular markers in the dark for 20 min and then fixed with Fix Perm buffer (eBioscience) for the Treg staining and with the permeabilization kit (BD) for the T cell staining. The intracellular staining was then performed and, after incubating for 30 min in the dark, samples were washed and analyzed on an LSRII system (BD). FACS data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Proliferation assay
The proliferation assay we used was modified from the one previously reported.29 PBMCs were rested overnight and stained with anti-CD4, CD8, CD25, and CD127 as described above. Before sorting, PI was added to exclude dead cells. Each sample was divided into two and the Treg fraction, defined as PI negative, CD4+, CD127dim, and CD25high, was depleted from one. The other aliquot was used to sort live total PBMCs. Sorting was performed with a FACS ARIA II (BD). Once sorted, the samples were CFSE labeled per the manufacturer's directions. Briefly, cells were washed with phosphate-buffered saline (PBS) and incubated in the dark for 8 min with 1 μM CFSE, and 10 ml of fetal bovine serum (FBS) was added to quench the reaction. After incubating for 5 min on ice and washing twice, cells were plated at 100,000/well and incubated for 5 days in the presence of one of the peptide pools (2 mg/ml), or anti-CD3/CD28 (1 μg/ml and 0.5 μg/ml, respectively) as a positive control, or with peptide diluent (0.8% DMSO) as a negative control. To detect the fraction of proliferating cells, the samples were stained with anti-CD3, CD4, CD8, CD25, CD127, and FoxP3, together with the live-dead dye, and assessed by flow cytometry as described above.
To determine the response rate, we identified the HIV-1-specific responses as those in which the percentage of CFSE low cells in peptide-stimulated samples was greater than 3× that of DMSO-stimulated cells. The response rate was calculated as the percentage of positive responses out of the total number of analyzed samples.
Intracellular cytokine staining
PBMCs were rested overnight and plated at 500,000 cells/well with 100 ml heat-inactivated autologous serum and stimulated with either 2 mg/ml HIV-1 peptide pool (Env, Gag, Vpu, Tat, Pol, or Nef), DMSO alone (0.8%) as a negative control, or PMA (1 mg/ml)/ionomycin (1 mM) as a positive control, all in the presence of Brefeldin A (1 mg/ml) and Golgi Stop. After incubating the samples at 37°C for 5 h, the cells were stained with the live-dead stain and anti-CD3, CD4, and CD8, then permeabilized and stained with anti-IFN-γ, TNF-α, and MIP-1ß.
Gene expression microarrays
Sample preparation and array processing were performed by the Genomics Shared Resource at the Fred Hutchinson Cancer Research Center. Briefly, total RNA was extracted by an RNeasy Plus Microkit (Qiagen, Germantown, MD), following the manufacturer's instructions. RNA integrity was checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA) and quantified using a Qubit 2.0 Fluorometer (Life Technologies, Grand Island, NY). RNA samples that were determined to be of high quality were converted to cDNA using the Ovation PicoSL WTA System V2 (NuGEN Technologies, San Carlos, CA) and labeled for subsequent microarray analysis using the Encore BiotinIL Module (NuGEN Technologies). Biotin-labeled cRNAs were processed on HumanHT-12 v4 Expression BeadChips (Illumina, San Diego, CA).
The complete data set was assessed for quality and quantile normalized using the Bioconductor package lumi.30 The dataset was initially filtered by flagging probes that were below a signal “noise floor,” which was calculated as the 75th percentile of the negative control probe signals within each array. We subsequently filtered the dataset by employing a variance filter using the “shorth” function of the Bioconductor package genefilter. Differential gene expression between the increased and decreased proliferation states was determined using the Bioconductor package limma,31 and a false discovery rate (FDR) method was used to correct for multiple testing.32 Significant differential gene expression was defined as |log2 (ratio) |≥0.585 (±1.5-fold) with the FDR set to 10%. Finding no expression changes using the above methods, we also employed SAM,33 using the Bioconductor package siggenes, which confirmed our earlier findings. Data were deposited in the GEO repository (access number GSE38778).
Statistical analyses
The McNemar test was used to compare the frequency of individuals with any proliferative response in PBMCs vs. in PBMCs-Tregs. The magnitude of proliferative responses was estimated as the ratio of the proportion of cells proliferating in response to a peptide pool vs. the proportion proliferating to DMSO. The Wilcoxon signed rank test was used to compare the magnitude of responses in PBMCs vs. PBMCs-Tregs. Spearman's rho was used to estimate correlations between immune parameters. Statistical analyses were performed using SAS 9.2.
Results
Demographic and clinical characteristics of the study group
PBMC samples were obtained from 129 HIV-1-uninfected members of Ugandan HIV-1-serodiscordant heterosexual couples. All of the selected couples were married or living together and the median partnership duration was 6 years. The median viral load of the partner was 2,565, ranging from undetectable to 108,170, and 16.3% reported unprotected sex during the month before the sample was obtained. Demographic and clinical characteristics of the subjects are reported in Table 1.
Table 1.
Participant Characteristics
| Characteristic | Number (%) or median (IQR) |
|---|---|
| Demographic characteristics, HIV-1-negative partner | |
| Female | 63 (48.8) |
| Age | 32 (26, 40) |
| Couple characteristics | |
| Married or living with partner | 129 (100) |
| Partnership duration, years | 5.9 (2.4, 13.0) |
| Number of children | 2 (0, 3) |
| Any unprotected sex acts, prior month | 21 (16.3) |
| Clinical characteristics, HIV-1-negative partner | |
| Sexually transmitted infection | 7 (5.4) |
| Circumcised (men only) | 29 (43.9) |
| Hormonal contraceptive use (women only) | 10 (15.9) |
| Clinical characteristics, HIV-1-infected partner | |
| CD4 count | 406 (274, 660) |
| Viral load | 2,565 (undetectable—108,170) |
HIV-1 exposure results in virus-specific T cell responses that can be increased or decreased by Treg depletion
To determine whether HIV-1 exposure resulted in HIV-1-specific CD4+ and CD8+ T cell responses, we performed a CFSE-based proliferation assay in response to four HIV-1 peptide pools (Gag, Pol, Env, and Vpu). Overall, we detected a virus-specific CD4+ T cell response in 12.8% of the analyzed participants and a CD8+ T cell response rate of 6.4% among our tested HESN individuals (Fig. 1A). All samples showed proliferation when stimulated with anti-CD3 and anti-CD28 (data not shown).
FIG. 1.

HIV-driven T cell proliferation in HIV-exposed seronegatives. (A) The response rates for total peripheral blood mononuclear cells (PBMCs) (white bars) and PBMCs-Tregs (black bars) for CD4+ and CD8+ T cells were calculated as the percentage of positive proliferative responses out of the total number of analyzed samples. The McNemar test was used to compare the frequency of individuals with any proliferative response in PBMCs vs. in PBMCs-Tregs: for CD4+ T cells, p=0.37 and for CD8+ T cells, p=0.74. (B) A FACS-based sorting scheme was used for the T cell proliferation assay. The PBMCs from each sample were stained with propidium iodide (PI) for dead cell exclusion and with anti-CD4, CD8, CD127, and CD25, and divided in half. The PI-negative fraction was sorted from one aliquot, and considered the “PBMCs.” To prepare the “PBMCs-Treg” fraction, the second aliquot was also sorted based on PI negativity. Next, the CD4-negative cells were sorted and combined with the CD4+CD127+CD25– (T conv) cells to obtain the PBMCs-Tregs.
In addition to testing the T cell proliferation in response to HIV peptides, the CFSE assay was also used to evaluate the Treg suppressive capacity. To perform this assay, two fractions were sorted from each sample prior to labeling cells with CFSE and in vitro culture (Fig. 1B). One included all the live PBMCs, identified by their exclusion of PI, and the other, termed “PBMCs-Tregs,” included all the CD4− cells plus the CD4+ CD127+ CD25lo cells (conventional T cells), all selected by PI exclusion as well. The proliferation of the two fractions in response to the same peptide pools, as well as to a polyclonal stimulus (anti-CD3 and anti-CD28), was measured by CFSE dilution and compared. This assay allows an indirect measurement of Treg function with a limited number of cells.
Surprisingly, when the PBMCs were depleted of Tregs, the CD8+ T cell proliferation response rate decreased from 6.4% to 5.3%, and the response rate for CD4+ T cells dropped from 12.8% to 8.5% (Fig. 1A). Thus, the data suggest that the role of Tregs in regulating the HIV-1-specific T cell response in HESN is multifaceted, as Tregs do not appear consistently capable of suppressing HIV-1-driven T cell responses.
When we looked in detail at the different subjects, we saw that among the 24 CD4+ T cell proliferative responses to one of the tested peptide pools, eight increased when Tregs were depleted (Fig. 2A), while the remaining 16 decreased (Fig. 2B). There were 12 detectable CD8+ T cell responses, six of which decreased upon Treg depletion whereas six increased (Fig. 2C and D).
FIG. 2.

Treg depletion can result in an increased or decreased CD4+ and CD8+ T cell proliferation in different subjects. The ratios between the percentage of cells proliferating in response to a peptide pool and to DMSO are shown for samples where the Treg depletion caused an increased (A, C) and decreased (B, D) CD4+ and CD8+ T cell proliferation, respectively. Each data pair represents the response to one peptide pool. N=number of responses. The p value was calculated by the Wilcoxon signed rank test.
To identify any potential intrinsic differences between Tregs that could and could not suppress HIV-1-specific CD4+ T cell proliferation in HESN, the Treg expression profiles in the two groups were analyzed by microarray. No significant differences in the expression profile were detected between the two groups of Tregs when using two different standard methods for filtering and assessing the microarray data (data not shown).
In total, these results show that HIV-1-specific CD4+ and CD8+ T cell responses are present in selected individuals. Furthermore, depletion of Tregs can lead to an increased or decreased T cell proliferation in response to HIV-1 peptides.
Lack of Treg suppressive capacity results in an increased CD4+ T cell activation and MIP-1β secretion
We next took all individuals with an HIV-1-specific CD4+ T cell proliferative response and grouped them based on whether their Tregs suppressed or enhanced this response. We then evaluated Treg frequency, T cell activation, and cytokine secretion in response to HIV-1 peptides (Env, Gag, Vpu, Tat, Pol, or Nef) in these two groups. Subjects whose Tregs were unable to suppress HIV-1-specific CD4+ T cell responses showed a trend toward an increased frequency of Tregs (CD3+ CD4+ CD127dim CD25high and FoxP3+) relative to total CD4+ T cells (p=0.07) as well as elevated CD4+ T cell activation as measured by the expression of Ki67 and downmodulation of Bcl-2 (p=0.07) (Fig. 3A and B) in comparison to subjects whose Tregs were suppressing the CD4+ T cell proliferation. These two variables, Treg frequency and T cell activation, directly correlated (ρ=0.76, p=0.0001 by Spearman test, Fig. 3C). Interestingly, the same direct correlation between Treg frequency and CD4+ and CD8+ T cell activation remained significant when we analyzed the data from all the subjects (data not shown).
FIG. 3.
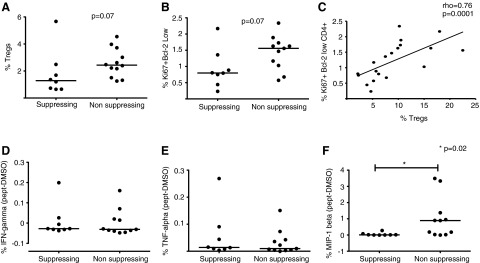
Lack of Treg suppressive capacity results in increased CD4+ T cell activation and microphage inflammatory protein-1β (MIP-1β) secretion. (A) Frequencies of Tregs and (B) activated CD4+ T cells are compared between the groups where the Tregs could or could not suppress CD4+ T cell proliferation. (C) Linear correlation between Treg frequency and CD4+ T cell activation (p=0.0008 by Spearman test). (D) Interferon (IFN)-γ, (E) tumor necrosis factor (TNF)-α, and (F) MIP-1β secretion by CD4+ T cells in response to stimulation by HIV-1 peptide pools are compared between the two groups. After background subtraction, the values in the graphs were calculated as the average between the responses upon stimulation with different peptide pools. The bars represent the mean between the samples. p=0.02, Wilcoxon rank sum test.
We next evaluated cytokine production in response to stimulation with HIV-1 peptide pools by intracellular cytokine staining. IFN-γ and TNF-α production by CD4+ T cells was comparable between the two groups (Fig. 3D and E). However, the magnitude of CD4+ T cells producing MIP-1β was significantly higher in subjects whose Tregs were unable to suppress HIV-1-driven CD4+ T cell proliferation (p=0.02, Fig. 3F).
In sum, these results suggest that the group of subjects whose Tregs lack suppressive capacity toward HIV-1 specific CD4+ T cell responses is characterized by an overall increase in CD4+ T cell activation and Treg frequency and has an increased HIV-1-driven secretion of MIP-1β from CD4+ T cells.
Discussion
In this study, we addressed the role of Tregs in protection from HIV-1 infection in HIV-1-discordant couples. We identified virus-specific CD4+ and CD8+ T cell responses and, surprisingly, the overall CD4+ T cell response rate was not increased when Tregs were removed from cell preparations. Of the 20 individuals that had HIV-1-specific CD4+ T cell responses, only eight had Tregs that could suppress this proliferation. When compared with individuals whose Tregs could suppress HIV-1-specific CD4+ T cell proliferation, individuals with Tregs unable to suppress showed a trend toward increased T cell activation and Treg frequency and a significant increase in HIV-1-specific production of MIP-1β by CD4+ T cells, autocrine production of which has been shown to be protective in terms of HIV-1 infection of CD4+ T cells.
HIV-1-exposed seronegative individuals represent an opportunity to better understand the natural protective immunity to HIV-1 and to improve our understanding of what it will take to design a successful HIV-1 vaccine. Multiple mechanisms underlying this “resistance” to infection have been proposed, ranging from genetic characteristics6 to innate and adaptive immunity. In terms of innate immunity, HIV-1 protection has been correlated with an increased function of NK cells34 as well as higher concentrations of secreted factors, such as the beta chemokines MIP-1α, MIP-1β, and RANTES.35,36 As for adaptive immunity, HIV-1-specific CD4+ and CD8+ T cell responses have been detected in HESN in a number of different studies.37–43
These proposed mechanisms of resistance are not mutually exclusive, and moreover, it is possible that they differ between subjects as well as between cohorts. Furthermore, although these individuals are at high risk of exposure, the rate of viral transmission per coital act is low, estimated around 0.0007 when the HIV-1-infected partner is virally suppressed.44 It is thus challenging to discriminate between the lack of seroconversion due to a lack of sufficient viral exposure or due to a “special” protective immune setting. Finally, there remains the dilemma of how to define exposed seronegative subjects; in the literature, there is a lack of consensus on the duration and intensity of exposure and other clinical characteristics required to be considered “HIV-1 exposed.” In our cohort, all subject were married or living with an HIV+ partner for an average of 5 years, increasing the possibility of a low but constant exposure to HIV.
We focused on the potential role of adaptive immunity in protection from HIV-1, given the disagreement in the literature regarding the role of T cells in protection from HIV-1 in HESN, although this discordance can be at least in part explained by the lack of a common method of analyzing the responses as well as by the intrinsic differences between cohorts and subjects studied. In a study from Restrepo et al., a very high percentage of subjects belonging to a discordant couple cohort showed HIV-1-specific responses.45 Another study conducted using HIV-1-serodiscordant couples showed that a higher percentage of responses was detected by cytokine secretion than by a thymidine incorporation method,46 while in another study no responses were reported.47 Taking this into consideration, we used a CFSE-based proliferation assay, which allowed a specific identification of HIV-1-driven proliferative responses as well as the discrimination between CD4+ and CD8+ T cell responses.
To the best of our knowledge, our study is the first in HESN to assess both T cell responses and the Treg functional status in HESN subjects. We identified HIV-specific T cell responses for both CD4+ and CD8+ T cells. The high specificity of the assay, with the very low level of proliferation in unstimulated samples, together with our stringent positivity call, supports our findings. Our results are not in disagreement with those reported by Restrepo et al.45 since the response evaluated was different: proliferation or cytokine secretion.
When we evaluated the effect of Tregs on the HIV-specific responses, we unexpectedly identified two different subsets of subjects: in one, the Tregs were able to suppress the CD4+ T cell proliferation, whereas in the other they were not. Treg instability has been addressed before in animal models, where it has been shown that Tregs can be reprogrammed to exert functions other than suppression. For instance, in a vaccination model, it was demonstrated that the adjuvant CpG caused a reprogramming of Tregs into Th1 cells essential for the priming of CD8+ T cells.48 Additionally, Tregs have the potential to be reprogrammed into Th17-like cells in inflammatory conditions, driven by interleukin (IL)-6.49 Thus, a situation of chronic inflammation can induce the loss of suppressive function that we observed in Tregs. We believe that in our study, for the first time, we observed the loss of the suppressive activity of human Tregs. We cannot exclude that this phenomenon was due to reasons other than HIV exposure, such as the presence of inflammation due to other pathogenic events or chronic conditions such as autoimmunity in the subjects studied.
In our study, although we did not have the ability to look at Tregs cytokine secretion due to our limited number of cells, we saw that in the samples where Treg suppressive activity was altered, CD4+ T cells were able to secrete more MIP-1β in response to HIV peptides as compared to the group characterized by suppressive Tregs. Interestingly, it was previously shown that MIP-1β has a protective activity against HIV-1 infection.50 Moreover, the group characterized by the lack of Treg suppressive activity had a trend toward higher CD4+ T cell activation and Treg frequency, and these two phenotypes were directly correlated, which could be explained by a compensatory increase of Treg frequency in response to higher T cell activation. This phenomenon has been previously described in different infection models.51,52
Future analyses aimed at examining subjects longitudinally may provide additional insights.53 Moreover, a more detailed study of cytokine secretion and characterization of Tregs in subjects in which they have lost the ability to suppress HIV responses would allow a better understanding of the mechanism identified in our study.
In conclusion, our results support the hypothesis that there are some HESN subjects with an intrinsic immunity to HIV-1, which could be a result of a low but constant HIV-1 exposure. The unique immune response we identified for a subset of these subjects is a lack of HIV-1-specific suppressive activity of Tregs toward CD4+ T cells, possibly resulting in an increased CD4+ T cell activation and secretion of the protective molecule MIP-1β in response to HIV-1. Although we cannot exclude the presence of other mechanisms of resistance in these subjects, these findings could further our search for immune correlates of protection from HIV-1 as well as aid in the design of new HIV-1 vaccines. Additionally, it is possible that reprogramming or blockade of Tregs could be therapeutically beneficial in protection from HIV-1 infection.
Acknowledgments
We wish to acknowledge Stephen Voght for critical reading of the manuscript; Stephen DeRosa, Nicole Frahm, and Helen Horton for helpful discussion; and Ryan Basom, Crissa Bennett, and Neil Shafer for technical assistance. We thank the James B. Pendleton Charitable Trust for their generous equipment donation. We are grateful to the research staff and study participants in Kampala who made this study possible.
Funding for this study was provided by NIAID/NIH 3 R01 AI047086-10S1 (to M.J.M. and J.M.L.), by the Bill and Melinda Gates foundation 41185, and by NIAID/NIH 1R01AI096968 (to J.M.B. and J.M.L.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Horton RE. McLaren PJ. Fowke K. Kimani J. Ball TB. Cohorts for the study of HIV-1-exposed but uninfected individuals: Benefits and limitations. J Infect Dis. 2010;202(Suppl 3):S377–381. doi: 10.1086/655971. [DOI] [PubMed] [Google Scholar]
- 2.Dunkle KL. Stephenson R. Karita E, et al. New heterosexually transmitted HIV infections in married or cohabitating couples in urban Zambia and Rwanda: An analysis of survey and clinical data. Lancet. 2008;371:2183–2191. doi: 10.1016/S0140-6736(08)60953-8. [DOI] [PubMed] [Google Scholar]
- 3.Kaul R. Rowland-Jones SL. Kimani J, et al. Late seroconversion in HIV-resistant Nairobi prostitutes despite pre-existing HIV-specific CD8+ responses. J Clin Invest. 2001;107(3):341–349. doi: 10.1172/JCI10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clerici M. Levin JM. Kessler HA, et al. HIV-specific T-helper activity in seronegative health care workers exposed to contaminated blood. JAMA. 1994;271(1):42–46. [PubMed] [Google Scholar]
- 5.Rowland-Jones SL. Nixon DF. Aldhous MC, et al. HIV-specific cytotoxic T-cell activity in an HIV-exposed but uninfected infant. Lancet. 1993;341(8849):860–861. doi: 10.1016/0140-6736(93)93063-7. [DOI] [PubMed] [Google Scholar]
- 6.Quillent C. Oberlin E. Braun J, et al. HIV-1-resistance phenotype conferred by combination of two separate inherited mutations of CCR5 gene. Lancet. 1998;351(9095):14–18. doi: 10.1016/S0140-6736(97)09185-X. [DOI] [PubMed] [Google Scholar]
- 7.Ranki A. Mattinen S. Yarchoan R, et al. T-cell response towards HIV in infected individuals with and without zidovudine therapy, and in HIV-exposed sexual partners. AIDS. 1989;3(2):63–69. doi: 10.1097/00002030-198902000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Piacentini L. Fenizia C. Naddeo V. Clerici M. Not just sheer luck! Immune correlates of protection against HIV-1 infection. Vaccine. 2008;26(24):3002–3007. doi: 10.1016/j.vaccine.2007.11.062. [DOI] [PubMed] [Google Scholar]
- 9.Miyazawa M. Lopalco L. Mazzotta F. Lo Caputo S. Veas F. Clerici M. The 'immunologic advantage' of HIV-exposed seronegative individuals. AIDS. 2009;23(2):161–175. doi: 10.1097/QAD.0b013e3283196a80. [DOI] [PubMed] [Google Scholar]
- 10.Lo Caputo S. Trabattoni D. Vichi F, et al. Mucosal and systemic HIV-1-specific immunity in HIV-1-exposed but uninfected heterosexual men. AIDS. 2003;17(4):531–539. doi: 10.1097/00002030-200303070-00008. [DOI] [PubMed] [Google Scholar]
- 11.Tran HK. Chartier L. Troung LX, et al. Systemic immune activation in HIV-1-exposed uninfected Vietnamese intravascular drug users. AIDS Res Hum Retroviruses. 2006;22(3):255–261. doi: 10.1089/aid.2006.22.255. [DOI] [PubMed] [Google Scholar]
- 12.Schenal M. Lo Caputo S. Fasano F, et al. Distinct patterns of HIV-specific memory T lymphocytes in HIV-exposed uninfected individuals and in HIV-infected patients. AIDS. 2005;19(7):653–661. doi: 10.1097/01.aids.0000166088.85951.25. [DOI] [PubMed] [Google Scholar]
- 13.Pallikkuth S. Wanchu A. Bhatnagar A. Sachdeva RK. Sharma M. Human immunodeficiency virus (HIV) gag antigen-specific T-helper and granule-dependent CD8 T-cell activities in exposed but uninfected heterosexual partners of HIV type 1-infected individuals in North India. Clin Vaccine Immunol. 2007;14(9):1196–1202. doi: 10.1128/CVI.00488-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Migueles SA. Laborico AC. Shupert WL, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. 2002;3(11):1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 15.Buseyne F. Scott-Algara D. Porrot F, et al. Frequencies of ex vivo-activated human immunodeficiency virus type 1-specific gamma-interferon-producing CD8+ T cells in infected children correlate positively with plasma viral load. J Virol. 2002;76(24):12414–12422. doi: 10.1128/JVI.76.24.12414-12422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheibenbogen C. Romero P. Rivoltini L, et al. Quantitation of antigen-reactive T cells in peripheral blood by IFNgamma-ELISPOT assay and chromium-release assay: A four-centre comparative trial. J Immunol Methods. 2000;244(1–2):81–89. doi: 10.1016/s0022-1759(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 17.Lajoie J. Juno J. Burgener A, et al. A distinct cytokine and chemokine profile at the genital mucosa is associated with HIV-1 protection among HIV-exposed seronegative commercial sex workers. Mucosal Immunol. 2012;5(3):277–287. doi: 10.1038/mi.2012.7. [DOI] [PubMed] [Google Scholar]
- 18.Suresh P. Wanchu A. Bhatnagar A. Sachdeva RK. Sharma M. Spontaneous and antigen-induced chemokine production in exposed but uninfected partners of HIV type 1-infected individuals in North India. AIDS Res Hum Retroviruses. 2007;23(2):261–268. doi: 10.1089/aid.2006.0146. [DOI] [PubMed] [Google Scholar]
- 19.Casazza JP. Brenchley JM. Hill BJ, et al. Autocrine production of beta-chemokines protects CMV-specific CD4 T cells from HIV infection. PLoS Pathog. 2009;5(10):e1000646. doi: 10.1371/journal.ppat.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Card CM. McLaren PJ. Wachihi C. Kimani J. Plummer FA. Fowke KR. Decreased immune activation in resistance to HIV-1 infection is associated with an elevated frequency of CD4(+)CD25(+)FOXP3(+) regulatory T cells. J Infect Dis. 2009;199(9):1318–1322. doi: 10.1086/597801. [DOI] [PubMed] [Google Scholar]
- 21.McLaren PJ. Ball TB. Wachihi C, et al. HIV-exposed seronegative commercial sex workers show a quiescent phenotype in the CD4+ T cell compartment and reduced expression of HIV-dependent host factors. J Infect Dis. 2010;202(Suppl 3):S339–344. doi: 10.1086/655968. [DOI] [PubMed] [Google Scholar]
- 22.Songok EM. Luo M. Liang B, et al. Microarray analysis of HIV resistant female sex workers reveal a gene expression signature pattern reminiscent of a lowered immune activation state. PLoS One. 2012;7(1):e30048. doi: 10.1371/journal.pone.0030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell DJ. Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2010;11(2):119–130. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lund JM. Hsing L. Pham TT. Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320(5880):1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angin M. King M. Altfeld M. Walker BD. Wucherpfennig KW. Addo MM. Identification of HIV-1-specific regulatory T-cells using HLA class II tetramers. AIDS. 2012;26(16):2112–2115. doi: 10.1097/QAD.0b013e328358cc75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legrand FA. Nixon DF. Loo CP, et al. Strong HIV-1-specific T cell responses in HIV-1-exposed uninfected infants and neonates revealed after regulatory T cell removal. PLoS One. 2006;1:e102. doi: 10.1371/journal.pone.0000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F. Malhotra U. Gilbert PB, et al. Peptide selection for human immunodeficiency virus type 1 CTL-based vaccine evaluation. Vaccine. 2006;24(47–48):6893–6904. doi: 10.1016/j.vaccine.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Defawe OD. Fong Y. Vasilyeva E, et al. Optimization and qualification of a multiplex bead array to assess cytokine and chemokine production by vaccine-specific cells. J Immunol Methods. 2012;382:117–128. doi: 10.1016/j.jim.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venken K. Thewissen M. Hellings N, et al. A CFSE based assay for measuring CD4+ CD25+ regulatory T cell mediated suppression of auto-antigen specific and polyclonal T cell responses. J Immunol Methods. 2007;322(1–2):1–11. doi: 10.1016/j.jim.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 30.Du P. Kibbe WA. Lin SM. lumi: A pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 31.Smyth GK. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; New York: 2005. [Google Scholar]
- 32.Reiner A. Yekutieli D. Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19(3):368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 33.Tusher VG. Tibshirani R. Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomescu C. Abdulhaqq S. Montaner LJ. Evidence for the innate immune response as a correlate of protection in human immunodeficiency virus (HIV)-1 highly exposed seronegative subjects (HESN) Clin Exp Immunol. 2011;164(2):158–169. doi: 10.1111/j.1365-2249.2011.04379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capobianchi MR. Abbate I. Antonelli G. Turriziani O. Dolei A. Dianzani F. Inhibition of HIV type 1 BaL replication by MIP-1alpha, MIP-1beta, and RANTES in macrophages. AIDS Res Hum Retroviruses. 1998;14(3):233–240. doi: 10.1089/aid.1998.14.233. [DOI] [PubMed] [Google Scholar]
- 36.Wasik TJ. Bratosiewicz J. Wierzbicki A, et al. Protective role of beta-chemokines associated with HIV-specific Th responses against perinatal HIV transmission. J Immunol. 1999;162(7):4355–4364. [PubMed] [Google Scholar]
- 37.Alimonti JB. Kimani J. Matu L, et al. Characterization of CD8 T-cell responses in HIV-1-exposed seronegative commercial sex workers from Nairobi, Kenya. Immunol Cell Biol. 2006;84(5):482–485. doi: 10.1111/j.1440-1711.2006.01455.x. [DOI] [PubMed] [Google Scholar]
- 38.Alimonti JB. Koesters SA. Kimani J, et al. CD4+ T cell responses in HIV-exposed seronegative women are qualitatively distinct from those in HIV-infected women. J Infect Dis. 2005;191(1):20–24. doi: 10.1086/425998. [DOI] [PubMed] [Google Scholar]
- 39.Jennes W. Vuylsteke B. Borget MY, et al. HIV-specific T helper responses and frequency of exposure among HIV-exposed seronegative female sex workers in Abidjan, Cote d'Ivoire. J Infect Dis. 2004;189(4):602–610. doi: 10.1086/381454. [DOI] [PubMed] [Google Scholar]
- 40.Sriwanthana B. Hodge T. Mastro TD, et al. HIV-specific cytotoxic T lymphocytes, HLA-A11, and chemokine-related factors may act synergistically to determine HIV resistance in CCR5 delta32-negative female sex workers in Chiang Rai, northern Thailand. AIDS Res Hum Retroviruses. 2001;17(8):719–734. doi: 10.1089/088922201750236997. [DOI] [PubMed] [Google Scholar]
- 41.Perez CL. Hasselrot K. Bratt G. Broliden K. Karlsson AC. Induction of systemic HIV-1-specific cellular immune responses by oral exposure in the uninfected partner of discordant couples. AIDS. 2010;24(7):969–974. doi: 10.1097/qad.0b013e328337aff8. [DOI] [PubMed] [Google Scholar]
- 42.Beretta A. Furci L. Burastero S, et al. HIV-1-specific immunity in persistently seronegative individuals at high risk for HIV infection. Immunol Lett. 1996;51(1–2):39–43. doi: 10.1016/0165-2478(96)02553-9. [DOI] [PubMed] [Google Scholar]
- 43.Murashev BV. Nazarenko OV. Akulova EB, et al. The high frequency of HIV type 1-specific cellular immune responses in seronegative individuals with parenteral and/or heterosexual HIV type 1 exposure. AIDS Res Hum Retroviruses. 2012;28(12):1598–1605. doi: 10.1089/aid.2011.0335. [DOI] [PubMed] [Google Scholar]
- 44.Wawer MJ. Gray RH. Sewankambo NK, et al. Rates of HIV-1 transmission per coital act, by stage of HIV-1 infection, in Rakai, Uganda. J Infect Dis. 2005;191(9):1403–1409. doi: 10.1086/429411. [DOI] [PubMed] [Google Scholar]
- 45.Restrepo C. Rallon NI. del Romero J, et al. Low-level exposure to HIV induces virus-specific T cell responses and immune activation in exposed HIV-seronegative individuals. J Immunol. 2010;185(2):982–989. doi: 10.4049/jimmunol.1000221. [DOI] [PubMed] [Google Scholar]
- 46.Goh WC. Markee J. Akridge RE, et al. Protection against human immunodeficiency virus type 1 infection in persons with repeated exposure: Evidence for T cell immunity in the absence of inherited CCR5 coreceptor defects. J Infect Dis. 1999;179(3):548–557. doi: 10.1086/314632. [DOI] [PubMed] [Google Scholar]
- 47.Addo MM. Altfeld M. Brainard DM, et al. Lack of detectable HIV-1-specific CD8(+) T cell responses in Zambian HIV-1-exposed seronegative partners of HIV-1-positive individuals. J Infect Dis. 2011;203(2):258–262. doi: 10.1093/infdis/jiq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma MD. Hou DY. Baban B, et al. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33(6):942–954. doi: 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang XO. Nurieva R. Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29(1):44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Addo MM. Altfeld M. Brainard DM, et al. Lack of detectable HIV-1-specific CD8(+) T cell responses in Zambian HIV-1-exposed seronegative partners of HIV-1-positive individuals. J Infect Dis. 2011;203(2):258–262. doi: 10.1093/infdis/jiq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw JM. Hunt PW. Critchfield JW, et al. Increased frequency of regulatory T cells accompanies increased immune activation in rectal mucosae of HIV-positive noncontrollers. J Virol. 2011;85(21):11422–11434. doi: 10.1128/JVI.05608-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss L. Piketty C. Assoumou L, et al. Relationship between regulatory T cells and immune activation in human immunodeficiency virus-infected patients interrupting antiretroviral therapy. PLoS One. 2010;5(7):e11659. doi: 10.1371/journal.pone.0011659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackelprang RD. Baeten JM. Donnell D, et al. Quantifying ongoing HIV-1 exposure in HIV-1-serodiscordant couples to identify individuals with potential host resistance to HIV-1. J Infect Dis. 2012;206(8):1299–1308. doi: 10.1093/infdis/jis480. [DOI] [PMC free article] [PubMed] [Google Scholar]