

Abstract
Protein homooligomers afford several important benefits for the cell; they mediate and regulate gene expression, activity of many enzymes, ion channels, receptors, and cell–cell adhesion processes. The evolutionary and physical mechanisms of oligomer formation are very diverse and are not well understood. Certain homooligomeric states may be conserved within protein subfamilies and between different subfamilies, therefore providing the specificity to particular substrates while minimizing interactions with unwanted partners. In addition, transitions between different oligomeric states may regulate protein activity and support the switch between different pathways. In this chapter, we summarize the biological importance of homooligomeric assemblies, physicochemical properties of their interfaces, experimental methods for their identification, their evolution, and role in human diseases.
1. FUNCTIONAL ROLES OF HOMOOLIGOMERS IN A CELL
Only a small fraction of proteins function in isolation and many soluble and membrane-bound proteins form oligomeric complexes defined as those having identical or very similar homologous chains (called “homooligomers” hereafter).1–3 It is difficult to overestimate the functional importance of protein homooligomers which provide the diversity and specificity of many pathways, regulate their cross talk, and may mediate and regulate gene expression, activity of enzymes, ion channels, receptors, and cell–cell adhesion processes.4 Moreover, large assemblies consisting of many identical subunits have advantageous regulatory properties as they can undergo sensitive phase transitions.3,5 The molecular structure of homooligomers can also provide sites for allosteric regulation, generate new binding sites at interfaces to increase specificity, and diversity.6 In addition, oligomerization allows proteins to form large assemblies without increasing genome size and may enhance stability, while the reduced surface area of the components in a complex can offer protection against denaturation.2,5,7
There are certain characteristic protein regions (discussed in detail in the following sections) that might be important for providing a given oligomeric state. These features can function as specificity determinants and be responsible for high binding specificity to certain interacting partners while minimizing interactions with other unwanted partners. Some protein regions modulating oligomerization may be preserved in evolution only within a specific protein subfamily which can be characterized by a well-defined oligomeric state. Such an evolutionary mechanism would be essential for the separation of functional pathways of close paralogs, preventing the possible usage of similar surface regions interacting with the same or very similar partners or facilitating through specific features the interactions with the novel partners. As shown in Table 1.1, proteins in different oligomeric states may have various binding affinities and functional activities. Figure 1.1 and the two following examples illustrate the development of new protein specificities through homooligomerization in different organisms or in paralogs from the same organism.
Table 1.1.
Experimental examples of proteins regulated through transitions between different oligomeric states
| Protein name | Regulation of transition | Function of one oligomeric state | Function of the other oligomeric state | Reference |
|---|---|---|---|---|
| Pyruvate kinase M2 | Tetramer–dimer ratio is regulated by fructose 1,6-P2, serine, and phosphorylation | Inactive dimer leads to accumulation of phosphometabolites in tumor cells | Active tetramer associates with glycolytic enzyme complex to perform its functions in normal cell | 8 |
| Rad53, DNA damage checkpoint kinase in yeast | Assembly/disassembly is regulated through phosphorylation | Rad53 forms oligomers upon DNA damage. Rad53/Chk2 homooligomerization is a mechanism to activate signal in DNA damage responses | 9 | |
| Human thymidylate synthase | Conformational changes accompanying dimer/monomer transition lead to exposure of Cys residues of monomer and binding to TS mRNA and autoregulation of translation by its protein product | Dimeric form is an active thymidylate synthase | 10 | |
| Estrogen receptor | Phosphorylation of Tyr547 by Src tyrosine kinases controls receptor dimerization | Monomer probably does not bind DNA | Dimerization induces DNA binding | 11 |
| Hexokinase PII | Reversible phosphorylation is carbon source dependent. Glucose promotes dephosphorylation | When monomer is phosphorylated, cells cannot provide glucose repression of invertase | Dimer is dephosphorylated | 12 |
| Mammalian STE20-like kinase (MST) | Okadaic acid induces phosphorylation in the activation loop and monomerization | Phosphorylated active monomeric enzyme is translocated into nucleus | Dimerization contributes to the cytoplasmic retention of MST | 13 |
| Bacterial enhancer-binding protein nitrogen regulatory protein C (NtrC) | Phosphorylation induces oligomerization and changes interdomain interactions | Oligomerization activates the ATPase activity | 14 | |
| C-Terminal Src kinase (Csk) | SH3 domains form homodimers and bury recognition surface for SH3 interactors including tyrosine phosphatase | 15 | ||
| D-Hydantoinase | Removal of C-terminal Arg induces monomer formation | Monomer shows 40% activity reduction | Dimer and tetramers are active | 16 |
| 14-3-3 Proteins | Phosphorylation of Ser58 disrupts dimers | Monomer–dimer transition modulates target protein activity | Monomer–dimer transition modulates target protein activity | 17 |
Adapted from Ref. 3.
Figure 1.1.

Illustration for the development of novel protein specificities and regulation of protein activity through homooligomerization. Adapted from Ref. 3.
The first example involves proteins from the human p53 C-terminal domain family which function as homotetramers. Two of these family members (p63 and p73) can also form mixed heterotetramers; however, p53 protein cannot associate with either p63 or p73.18 Interestingly, a recent study showed that p63 and p73 are different from p53 in that the former have an additional alpha-helix which stabilizes the tetramer.19 The absence of this helix in p53 explains its inability to oligomerize with p63/p73, resulting in the separation of the p53 pathway from the paralogous p63/p73 pathways. Another example is the LIM-domain-binding protein Ldb1, a nuclear adaptor protein which interacts with diverse proteins containing LIM domains and plays essential roles in development and cellular differentiation.20,21 Humans have two close paralogs Ldb1 and Ldb2. Since the loss of Ldb1 causes severe developmental defects in embryos that are not compensated for by Ldb2, Ldb1 and Lbd2 are likely to participate in different pathways. It is known that Ldb1 forms a trimer while Ldb2 exists in a monomer–tetramer–octamer equilibrium, suggesting that the oligomeric differences might enable Ldb1 and Ldb2 to interact with different partners in different pathways.22
2. EXPERIMENTAL CHARACTERIZATION AND COMPUTATIONAL PREDICTION
Experimental characterization of homooligomeric structures, their dynamic equilibrium between different oligomeric states, and computational inference of the biologically relevant oligomeric form from the crystalline state have always been very challenging. Various experimental and computational methods have been devised over the years to meet these challenges. They aim to accurately identify biological interactions and distinguish them from nonbiological crystal packing or nonspecific aggregation. The main experimental techniques include X-ray and neutron scattering, mass spectrometry, gel filtration, dynamic light scattering, analytical ultracentrifugation, and fluorescence resonance energy transfer (FRET)23 (see also references for Table 1.1). For instance, analytical centrifugation and gel filtration chromatography provide data on molecular mass distribution, the subunit stoichiometry of the complexes, and equilibrium constants. FRET characterizes the kinetics and dynamics of complex formation, monitoring the extent of energy transfer between donor and acceptor, whereas X-ray and neutron scattering offer the atomic details of interaction interfaces. Nowadays, proteins are being crystallized using high-throughput techniques and very often without the extensive biochemical or biophysical characterization of their oligomeric states. This raises the importance of computational methods to reconstruct biological assemblies from crystalline states.24–26 These algorithms apply crystallographic symmetry operations and then attempt to differentiate the biological from the crystal packing interfaces by computational criteria. For example, the PISA algorithm applies graph theory to find the set of stable assemblies, which fill all the crystal space in a regular manner, with nodes and edges in the graph corresponding to protein monomers and interfaces between them.25 To distinguish “biologically relevant” from crystal packing interactions, one can also use ad-hoc scoring schemes which are determined by interface area, amino acid composition, number of contacts, topological complementarity, hydrogen bonding, and other characteristics.27–31
3. PHYSICOCHEMICAL PROPERTIES OF HOMOOLIGOMERIC INTERFACES
The amino acid composition of homooligomeric interfaces differs from those of crystal packing interfaces, heterooligomeric interfaces, and solvent-exposed surfaces and largely depends on the type of homooligomeric complexes.27–30,32,33 Moreover, besides interface composition, allosteric mutations may also affect intersubunit geometry and contribute to the evolution of oligomeric states.34 Obligate homooligomers (complexes where monomers are unstable and are prone to unfold upon isolation) are characterized by the large fraction of hydrophobic and, to a lesser extent, aromatic residues on their interfaces, while nonobligate (transient) complexes include more polar and charged residues.28,32,35 Interfaces of obligate homooligomers are usually larger28 but contain fewer hydrogen bonds per residue compared to heterooligomers.33 The analysis of kinetic and equilibrium data on dimeric proteins shows that per-residue interface and surface areas of “three-state dimers” (monomers are stable on their own) are significantly smaller than that of “two-state dimers” (monomers are not stable when separated from the complex).36
As can be seen in Fig. 1.2, homooligomers in the Protein Databank (PDB) mostly form dimers (37%), then trimers (5%) and tetramers (7%) with predominantly cyclic or dihedral symmetries. Moreover, eukaryotes have a considerably smaller fraction of homooligomers (40%) compared to eubacteria and archaea (60%). This might be explained by previous observations that eukaryotic proteins evolved through the more extensive domain fusion and shuffling events and contain more heterooligomers and multi-domain proteins.37 The self-attraction and symmetry in homooligomers might occur due to stability, foldability, and evolutionary optimization.38,39 The physical effect of a statistically enhanced self-attraction was recently modeled to show that interactions between identical random surfaces are stronger than attractive interactions between different random surfaces of the same size.38 Furthermore, it was demonstrated that the efficiency of co-aggregation between different monomers and protein domains decreases with decreasing sequence identity.40 Binding arrangements involving isologous homooligomeric interfaces with a two-fold symmetry axis seemed to be more frequently conserved in evolution compared to nonisologous interfaces.41 In addition, symmetrical isologous dimers were shown to contain more residues in disordered regions than heterodimers or non-symmetrical dimer interfaces,42 which might modulate the high specificity of interactions between the dimer complexes and their interacting partners and play an important role in allosteric regulation.43
Figure 1.2.
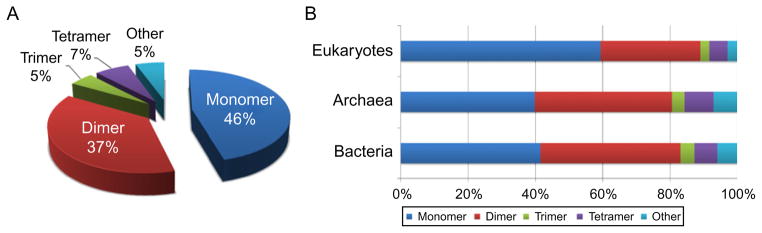
Distribution of different homooligomeric states in a nonredundant set of Protein Data Bank (PDB) structures (A) and in Eukaryotes, Archaea, and Eubacteria (B). The nonredundant set of structures was obtained using the criteria of BLAST p value <10−7 on all PDB chains and oligomeric state annotations were taken from PDB.
4. EVOLUTIONARY MECHANISMS TO FORM HOMOOLIGOMERS
A number of evolutionary mechanisms of homooligomerization have been proposed. According to some of them, evolutionary pathways may follow kinetic pathways of two-state or three-state folding.44,45 At the same time, assembly pathways of oligomers may mimic evolutionary pathways, and it was suggested and verified experimentally that homomeric proteins with dihedral symmetry may evolve and assemble through their cyclic intermediates.46,47 One of the major mechanisms of oligomerization is gene duplication with subsequent diversification where one copy of the gene retains its original function whereas the other gene copy is under relaxed evolutionary constraints, allowing it to develop new functional specificities. This mechanism may lead to the formation of oligomeric paralogs and may create protein complexes with novel specificities.41,48–51 While in S. cerevisiae up to 20% of complexes evolved by stepwise partial duplications,48 this mechanism was found to be less prevalent in Escherichia coli.49
Similarity in protein sequences, folds, and functions between two orthologous proteins does not necessarily imply that they will have the same interacting partners.52 Although homooligomeric states and binding interfaces are usually conserved within the clades on phylogenetic trees, they can only be reliably transferred from very close homologs (sharing more than 30% sequence identity for oligomeric states and sharing more than 50–70% identity for binding modes inference).41,46 In some cases, the oligomeric state can be evolutionarily conserved while the binding arrangement can be markedly different. This points to the possibility that interactions between monomers are not necessarily inherited from the ancestral homooligomer but rather can develop anew in evolution. For example, proteins from the glycosyltransferase family may function as monomers, dimers, or tetramers in different organisms. Although the dimeric and tetrameric proteins appear to be as ancient as the monomers, the binding modes can differ even between very similar proteins, for instance, engaging completely opposite sites on the molecule.50 Despite this diversity, there are certain characteristics of evolutionarily conserved homooligomeric binding modes. For example, binding modes with larger interfaces are more frequently conserved, while smaller interfaces are acquired more recently in evolution.41,46
In the next few sections, we will consider several evolutionary mechanisms that play key roles in homooligomerization: domain swapping, formation of Leucine zippers, amino acid substitutions, and insertions/deletions.
4.1. Domain swapping
Domain swapping includes opening up of the monomeric conformation and exchanging identical regions between two monomers. Although the term “domain swapping” was introduced by Eisenberg and coworkers in 1994,53 the first example of this type of oligomer, bovine pancreatic ribonuclease (RNase A, Fig. 1.3A), was found by Crestfield et al. in 1962.54 Later on, it was shown that RNase A exchanges N-termini upon dimerization. Originally, domain swapping was applied only in the context of homooligomers; however, the usage of the term has been extended lately and it now encompasses different cases including those where swapping may occur between regions of closely homologous but nonidentical monomers. Overall, about 300 examples of domain swapping are reported in protein structures from the Protein Data Bank.55,56 The length of the swapped region may vary considerably, from a single secondary structure element to regions of over 100 residues. Although swapping of terminal regions seems to be more common, the regions can be located anywhere in the structure, and on occasion, two or more distinct regions may be swapped.55,57 For example, RNase A has two swapped regions at the N- and C- termini and can form a dimer, trimer, or higher-order oligomers (Fig. 1.3A).58
Figure 1.3.

Different mechanisms of homooligomerization. (A) Domain swapping of RNase A: monomer (1RTB) and domain-swapped dimer (1A2W) of RNase A. The swapped regions and domain linker regions are shown as red and yellow, respectively. (B) Leu-zipper of GCN4 (1YSA). Leu at position “d” is shown as stick model and colored in red. (C) Amino acid substitutions for designed homooctamer of L-rhamnulose-1-phosphatase aldolase (2UYU). The original protein exists as a tetramer which is shifted to the octamer upon a single substitution (A88F, shown in red). (D) Insertions of N-acetyl-L-glutamate kinase hexamer (2BUF). Inserted N-terminal helix shown in red enables the hexamer formation. Adapted from Ref. 3.
Interdomain linker regions have been studied in order to identify regions responsible for domain swapping.59–61 The importance of proline residues in linker regions was recognized by several experimental studies,59,60 and recently, the amino acid proline has been shown to have the highest propensity in hinge regions, similar to loop structures.62 However, the roles and number of these proline residues varied between the proteins. Cell cycle regulatory protein, p13suc1, for example, has two prolines in the hinge loop which control its monomer–dimer equilibrium.59 Interestingly, the replacement of the first proline by alanine results in the shift of monomer–dimer equilibrium toward the monomer, whereas the replacement of the second proline shifts the equilibrium toward the dimer. In another example, in the hinge region of cyanovirin-N, the substitution of proline by glycine stabilizes both the monomeric and dimeric states, while the replacement of serine by proline results in an exclusively dimeric protein.60 The length of the domain linker itself may also affect the monomer–dimer equilibrium. For instance, interleukin-5 (IL-5) belongs to the short-chain helical cytokine subfamily and forms a domain-swapped homodimer although other members in the family form monomers. The loop between helices C and D is shorter in IL-5 compared to other family members and the engineered lengthening of the loop enabled IL-5 to form an active monomer.63
There are computational and theoretical studies which aim to examine the mechanism of domain swapping. Using a model which incorporates only the monomeric conformation in an energy function, it has been shown that the intermediate states represent open-ended, domain-swapped dimers, and it also appears that overall monomer topology is a main determinant of the structure of domain-swapped dimers.64 According to another mechanism for domain swapping, the swapping starts from the C-terminus and progresses by exchanging an increasing portion of the chains until a stable conformational state is reached. In contrast to the previous study, this process does not include complete monomer unfolding.65
4.2. Structural oligomerization motifs
There are several known structural motifs used by proteins for oligomerization, the alpha-helical coiled-coil being the most common one among them. The coiled-coil motif appears in proteins with different functions, for example, alpha-keratin and vimentin (intermediate filaments of the cytoskeleton), myosin and kinesin (motor proteins), SNARE proteins (mediation of vesicle fusion), and the b-Zip family (transcription factor).66 For instance, the b-Zip family uses the Leu-zipper motif for DNA binding (Fig. 1.3B). The coiled-coil motif is observed as a series of continuous heptad repeats (abcdefg)n in protein sequences.67 When the motif forms an alpha-helical structure, hydrophobic residues at the “a” and “d” positions interact with each other to form a helical bundle. The “Leu-zipper” motif is a type of coiled-coil structure where leucine is frequently observed at the “d” position (Fig. 1.3B).68 The coiled-coil motifs may form different oligomeric states depending on the particular amino acids in the heptad repeat. For example, the yeast transcription factor GCN4 forms a dimeric coiled coil; however, the replacements of Ile by Leu at the “a” and Leu by Ile at the “d” positions enables GCN4 to form a tetramer.69 Other combinations of Leu, Ile, and Val at the “a” and “d” positions may also cause trimerization,69,70 whereas replacements in positions “e” or “g” can lead to higher-order oligomers such as heptamers.71 Moutevelis and Woolfson organized coiled-coil structures in a “periodic table”72 where coiled-coil structures were arranged in columns with increasing numbers of alpha-helices. According to the authors’ results, 74% of coiled-coil structures were dimers and 12% formed trimers.
4.3. Amino acid substitutions
Amino acid substitutions introduced on the protein surface/interface may cause association or dissociation of homooligomers. A simple rule for amino acid substitutions that mediate the oligomeric states was proposed recently: the replacement of solvent-exposed residues by more hydrophobic and/or larger protruding residues tends to shift the equilibrium toward the formation of oligomers.73 Another study found that as few as two substitutions could shift the average composition of a surface patch to that of an equivalent interface.74 The artificial design of new oligomers by amino acid substitutions was performed on four protein assemblies75 (Fig. 1.3C). Some of these proteins needed only a single amino acid replacement to associate to a higher-order complex, and it was discovered that introducing large nonpolar side chains, such as phenylalanines or tryptophans, facilitated the complex formation.
Residues or sites with a large contribution to binding energy are sometimes called “binding hotspots.” Originally, hotspots were recognized by alanine scanning of the interface region between human growth hormone and the extracellular domain of its receptor as those few residues which caused the largest reduction in the binding affinity.76 Bogan and Thorn compiled a database of alanine mutants and described some important properties of hotspot residues.77 It was shown that hotspots were located near the center of the interface completely occluded from bulk solvent, with an abundance of arginine, tryptophan, and tyrosine.77,78 Additionally, it was suggested that some tightly packed binding hot spots could form a cluster on an interface.79
Amino acid substitutions on homooligomer interfaces may change the functional activity of a protein and have been implicated in several diseases. For example, mutations leading to the disease fructose intolerance are shown to destabilize the tetramer of the enzyme fructose-1,6-biphosphate aldolase A and thereby decrease its activity,80 while certain substitutions in glutamate receptors may stabilize their dimer interface and reduce desensitization.81
4.4. Insertions and deletions
Insertions and deletions at oligomer interfaces provide another important mechanism to modulate different functional specificities and protein oligomeric states in evolution. The protein N-acetyl-L-glutamate kinase (NAGK) from Pseudomonas aeruginosa is such an example; it catalyzes the second step in arginine biosynthesis. P. aeruginosa NAGK has an extra alpha-helix on its N-terminus and forms a homohexamer (trimers of dimers), although the same kinase from E. coli does not have this helix and exists as a homodimer (Fig. 1.3D). The homohexameric NAGK has a built-in negative feedback mechanism; binding of arginine causes a conformational change which inhibits the enzyme function by stabilizing an enlarged active center conformation. The arginine binding site is formed by the N-terminal helix and C-terminal lobe, flanking the junction between dimers, thus, hexamerization mediated by the N-terminal helix would be essential for this inhibition.82
The inspection of inserted and deleted regions among homologous proteins existing in different oligomeric states has revealed that about a quarter of them are located on interaction interfaces83 and many of them are responsible for enabling or disabling the formation of oligomers.50,84,85 Indeed, removal of enabling regions from protein structures may result in the complete or partial loss of oligomer stability.83,85 According to these investigations, the insertions and deletions are located preferentially in loop regions and to a lesser extent on alpha-helices and beta-strands. Furthermore, the insertions and deletions modulating oligomerization usually have a lower aggregation propensity and contain a larger fraction of polar and charged residues compared to conventional interfaces and protein surfaces.83 The residue composition of enabling regions depends on the size of the interface area; large insertions (>500 Å2 on interface area) tend to be hydrophobic, similar to conventional interfaces, but small insertions have polar or small residues such as Asn, Ser, Gly, or Pro. This tendency can be explained from the viewpoint of structural restraints for these insertions; the small insertions mainly form turns in the protein secondary structure, thus residues with high turn propensities are required to maintain their local structures.85 Interestingly, different oligomerization mechanisms can be employed within different proteins from the same family. For example, in the dihydrofolate reductase family, the enabling loop of bacteriophage T4 leads to the formation of a homodimer,84 while dimerization of the same enzyme from Thermotoga maritima is achieved by amino acid substitutions.73
5. REGULATION OF PROTEIN ACTIVITY THROUGH OLIGOMERIZATION
Cellular processes are extremely complex, requiring many factors to provide desired outcomes and prevent inefficiency. This control might be applied at the level of gene expression or protein–protein interactions, by changing protein activity, posttranslational modifications, or other means. Moreover, proteins can exist in dynamic equilibrium between different oligomeric states which can be controlled by physiological conditions (pH, certain ionic strength, temperature), ligands, and posttranslational modifications.86 Protein activity may be also regulated through the dynamic transitions between oligomeric states with differential activity. It has also been shown that certain mutations may induce changes in oligomeric state and activity but do not compromise stability.16 In addition, reversible transitions between discrete conformations and oligomeric states might account for protein cooperative binding properties and allosteric mechanisms in signal transduction.6
Here we present different scenarios which can explain how shifting the equilibrium between different oligomeric states might serve as a regulatory mechanism. These scenarios include: -homooligomerization may be important for protein self-activation when the active or binding site occupies the oligomeric binding interface; -conformational changes accompanying oligomerization could lead to exposure/suppression of the active or protein binding sites; -the formation of an oligomer may inhibit binding of a monomer to its substrate through competitive binding even without conformational changes; -the posttranslational modifications or binding of small molecules at or near the oligomeric interface can shift the equilibrium between different oligomeric states. In addition to examples from the previous study,86 we manually compiled a list of experimentally verified examples of the above-mentioned mechanisms in Table 1.1. As can be seen from this table, phosphorylation is a very widespread mechanism used by the cell to control protein activity through oligomerization. Indeed, recent work has shown that phosphorylation sites tend to be located on binding interfaces in heterooligomeric and weak transient homooligomeric complexes, and phosphorylation may potentially mediate or in many cases disrupt the complex formation.87
We will describe a specific example, which shows how the transition between the dimeric and tetrameric forms of pyruvate kinase can be implicated in tumor formation. Pyruvate kinase is a key glycolytic enzyme and its activity is consistently altered during tumorigenesis. It has been noted that, during tumor formation, the M2 isoform of pyruvate kinase is over-expressed.8 The active tetrameric form of this enzyme from normal cells has high affinity to its substrate phosphoenolpyruvate (PEP), associates with the glycolytic enzyme complex, and produces high levels of ATP. The dimeric form has low affinity to PEP, does not associate with the glycolytic enzyme complex, and is accompanied by low levels of ATP. Having the inactive dimeric form is advantageous for tumor cells as phosphometabolites above pyruvate kinases in the glycolytic cycle accumulate in tumor cells and are then available as precursors for synthesis required by tumorigenesis. The tetramer–dimer ratio is regulated by different factors, for example, by fructose 1,6-P2 and serine concentrations. Moreover, Rous sarcoma virus may also phosphorylate M2-PK and lead to its dimerization and disassociation from the glycolytic enzyme complex.88
6. OLIGOMERIZATION, PROTEIN AGGREGATION, AND RELATED DISEASES
Protein aggregation and amyloid fibril formation are associated with a number of devastating human diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Amyloid fibril is an insoluble protein fibril, which usually represents a straight and unbranched polymer of several thousand angstroms in length and is deposited mainly in extracellular regions.89 The major difference from other biological polymers is that amyloid fibers have very high stability, are highly insoluble in detergent, and have a considerable amount of β-sheet structure.90 The mechanism of amyloidogenesis has been extensively studied, and several models have been proposed. The prevalent model to explain the self-assembly process is nucleation-dependent polymerization.91 According to this model, a nucleus is required to initiate the fibrillation process. After the formation of a nucleus, the elongation of fiber becomes thermodynamically favorable and a large percentage of the starting protein material is assembled into fibrils. Another mechanism is double-concerted fibrillation.92 In this model, the amyloid fibril formation is achieved via two consecutive processes. The first process involves concerted associations of monomers, forming oligomeric granular species. In the second process, the oligomeric species act as a growing unit and form fibrils. In addition, a number of theoretical and computational approaches have been developed to elucidate the detailed process of amyloid fibril formation.93 An example of this is a molecular dynamics simulation showing the aggregation process of eight SH3 domains, where the initial step of aggregation is the dimerization, followed by the formation of one aggregate that consists of all eight proteins.94
An important question is why disease-related amyloids are toxic. In the case of Alzheimer’s disease, which is one of the most common neurodegenerative diseases, many studies suggest that a high concentration of amyloid-β (Aβ) proteins causes neuronal alterations.95 Interestingly, not only assembled amyloid fibrils but also prefibrillar aggregates including dimers, trimers, and relatively short oligomers show cytotoxicity.96 In particular, the Aβ dimers extracted directly from brains of Alzheimer’s disease patients inhibited long-term potentiation, enhanced long-term depression, and reduced dendritic spine density.97 In contrast, the same study demonstrated that amyloid plaque cores did not impair long-term potentiation unless Aβ dimers were released from the amyloid. This suggests that the amyloid core is not toxic by itself but the dynamic release of oligomers from the amyloid is toxic.
It remains unclear why natively soluble proteins aggregate under some specific conditions. Certainly, one of the factors is mutation, which could trigger diseases or increase the risk of diseases. For example, myofibrillar myopathies are a group of neuromuscular disorders, characterized by the cytoplasmic aggregation of multiple proteins, resulting in slowly progressive weakening of limb muscles. Desmin, one of the aggregated proteins in this disorder, is a muscle-specific filament protein, which plays an important role as a structural component of the muscle cytoarchitecture. A genetic study identified three different missense mutations in the desmin protein, namely R350P, E413K, and R454W.98 These mutations have a profound effect on the stability of the dimer and tetramer, which impairs the proper filament assembly. A nonsense mutation was found in the gene encoding another aggregated protein, FLNC. The mutation causes the truncation of a part of the dimerization domain, leading to the inability to form a proper dimer.99 Overall, more than 20 missense mutations that cause Alzheimer’s disease have been identified in or near the coding region of Aβ protein,96 which enhance the aggregation of the peptides.
On the other hand, the ability to form amyloid fibril is not limited to a small group of disease-related proteins.100,101 There is a growing list of functional amyloids, where proteins natively form filamentous aggregates and fulfill specific biological functions.90 A recent study demonstrated that more than 30 human peptide hormones can be spontaneously assembled and form amyloids, which function as a storage of hormones.102 This study also showed that the amyloid fibrils formed by the peptide hormones were able to release active monomeric hormones despite the high stability of the amyloids. Although the mechanism of the isolation of the monomers from the amyloid fibrils is still unknown, the secretion of peptide hormones is a highly regulated process. Other examples of functional amyloids include the Pmel17 protein forming intramelanosome fibrils through normal biological proteolytic processing associated with skin pigmentation103 and the curli fiber of E. coli, which are involved in adhesion to surfaces and biofilm formation.104 These functional amyloids are regulated and contribute to cellular biology without causing significant cytotoxicity. In addition, oligomerization is used in nature to counterbalance the destabilizing effects of mutations or higher temperatures and protect against nonspecific aggregation.105,106
7. CONCLUSIONS
Analysis of high-throughput protein–protein interaction networks shows that there are significantly more self-interacting proteins than expected by chance.107 Despite the importance and abundance of homooligomers in a cell, they are usually neglected in studies of protein–protein interaction networks. An explanation comes from the ambiguity of the experimental characterization of homooligomers and the difficulty of their computational prediction. In this review, we attempted to summarize the biological importance of homooligomeric assemblies, their evolution, and physicochemical properties. We also outlined the regulatory properties of homooligomers which might exist in dynamic equilibrium between different oligomeric states and can be controlled by physiological conditions, ligands, and post-translational modifications. The disruption of their regulatory functions may lead to many human diseases, namely certain mutations may induce changes in oligomeric state and protein activity and in some cases trigger non-specific protein aggregation and amyloid fibril formation.
Acknowledgments
This work was supported by the Intramural Research Program of the National Library of Medicine at the United States.
References
- 1.Cornish-Bowden AJ, Koshland DE., Jr The quaternary structure of proteins composed of identical subunits. J Biol Chem. 1971;246:3092–102. [PubMed] [Google Scholar]
- 2.Jones S, Thornton JM. Protein-protein interactions: a review of protein dimer structures. Prog Biophys Mol Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto K, Nishi H, Bryant S, Panchenko AR. Caught in self-interaction: evolutionary and functional mechanisms of protein homooligomerization. Phys Biol. 2011;8:035007. doi: 10.1088/1478-3975/8/3/035007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali MH, Imperiali B. Protein oligomerization: how and why. Bioorg Med Chem. 2005;13:5013–20. doi: 10.1016/j.bmc.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 5.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–53. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 6.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–8. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 7.Miller S, Lesk AM, Janin J, Chothia C. The accessible surface area and stability of oligomeric proteins. Nature. 1987;328:834–6. doi: 10.1038/328834a0. [DOI] [PubMed] [Google Scholar]
- 8.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Jia-Lin Ma N, Stern DF. Regulation of the Rad53 protein kinase in signal amplification by oligomer assembly and disassembly. Cell Cycle. 2008;7:808–17. doi: 10.4161/cc.7.6.5595. [DOI] [PubMed] [Google Scholar]
- 10.Lin X, Liu J, Maley F, Chu E. Role of cysteine amino acid residues on the RNA binding activity of human thymidylate synthase. Nucleic Acids Res. 2003;31:4882–7. doi: 10.1093/nar/gkg678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnold SF, Vorojeikina DP, Notides AC. Phosphorylation of tyrosine 537 on the human estrogen receptor is required for binding to an estrogen response element. J Biol Chem. 1995;270:30205–12. doi: 10.1074/jbc.270.50.30205. [DOI] [PubMed] [Google Scholar]
- 12.Randez-Gil F, Sanz P, Entian KD, Prieto JA. Carbon source-dependent phosphorylation of hexokinase PII and its role in the glucose-signaling response in yeast. Mol Cell Biol. 1998;18:2940–8. doi: 10.1128/mcb.18.5.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KK, Yonehara S. Phosphorylation and dimerization regulate nucleocytoplasmic shuttling of mammalian STE20-like kinase (MST) J Biol Chem. 2002;277:12351–8. doi: 10.1074/jbc.M108138200. [DOI] [PubMed] [Google Scholar]
- 14.Hwang I, Thorgeirsson T, Lee J, Kustu S, Shin YK. Physical evidence for a phosphorylation-dependent conformational change in the enhancer-binding protein NtrC. Proc Natl Acad Sci USA. 1999;96:4880–5. doi: 10.1073/pnas.96.9.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levinson NM, Visperas PR, Kuriyan J. The tyrosine kinase Csk dimerizes through its SH3 domain. PLoS One. 2009;4:e7683. doi: 10.1371/journal.pone.0007683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devenish SR, Gerrard JA. The role of quaternary structure in (beta/alpha)(8)-barrel proteins: evolutionary happenstance or a higher level of structure-function relationships? Org Biomol Chem. 2009;7:833–9. doi: 10.1039/b818251p. [DOI] [PubMed] [Google Scholar]
- 17.Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J Biol Chem. 2003;278:36323–7. doi: 10.1074/jbc.M304689200. [DOI] [PubMed] [Google Scholar]
- 18.Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem. 1999;274:18709–14. doi: 10.1074/jbc.274.26.18709. [DOI] [PubMed] [Google Scholar]
- 19.Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci USA. 2009;106:17705–10. doi: 10.1073/pnas.0905867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews JM, Visvader JE. LIM-domain-binding protein 1: a multifunctional cofactor that interacts with diverse proteins. EMBO Rep. 2003;4:1132–7. doi: 10.1038/sj.embor.7400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews JM, Bhati M, Craig VJ, Deane JE, Jeffries C, Lee C, et al. Competition between LIM-binding domains. Biochem Soc Trans. 2008;36:1393–7. doi: 10.1042/BST0361393. [DOI] [PubMed] [Google Scholar]
- 22.Cross AJ, Jeffries CM, Trewhella J, Matthews JM. LIM domain binding proteins 1 and 2 have different oligomeric states. J Mol Biol. 2010;399:133–44. doi: 10.1016/j.jmb.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Dafforn TR. So how do you know you have a macromolecular complex? Acta Crystallogr D Biol Crystallogr. 2007;63:17–25. doi: 10.1107/S0907444906047044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–61. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Domingues FS, Sommer I, Lengauer T. NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics. 2006;7:27. doi: 10.1186/1471-2105-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponstingl H, Henrick K, Thornton JM. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Bahadur RP, Chakrabarti P, Rodier F, Janin J. Dissecting subunit interfaces in homodimeric proteins. Proteins. 2003;53:708–19. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- 29.Janin J, Rodier F. Protein-protein interaction at crystal contacts. Proteins. 1995;23:580–7. doi: 10.1002/prot.340230413. [DOI] [PubMed] [Google Scholar]
- 30.Dasgupta S, Iyer GH, Bryant SH, Lawrence CE, Bell JA. Extent and nature of contacts between protein molecules in crystal lattices and between subunits of protein oligomers. Proteins. 1997;28:494–514. doi: 10.1002/(sici)1097-0134(199708)28:4<494::aid-prot4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 31.Shoemaker BA, Panchenko AR, Bryant SH. Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci. 2006;15:352–61. doi: 10.1110/ps.051760806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ofran Y, Rost B. Analysing six types of protein-protein interfaces. J Mol Biol. 2003;325:377–87. doi: 10.1016/s0022-2836(02)01223-8. [DOI] [PubMed] [Google Scholar]
- 33.Zhanhua C, Gan JG, Lei L, Sakharkar MK, Kangueane P. Protein subunit interfaces: heterodimers versus homodimers. Bioinformation. 2005;1:28–39. doi: 10.6026/97320630001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perica T, Chothia C, Teichmann SA. Evolution of oligomeric state through geometric coupling of protein interfaces. Proc Natl Acad Sci USA. 2012;109:8127–32. doi: 10.1073/pnas.1120028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dey S, Pal A, Chakrabarti P, Janin J. The subunit interfaces of weakly associated homodimeric proteins. J Mol Biol. 2010;398:146–60. doi: 10.1016/j.jmb.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 36.Gunasekaran K, Tsai CJ, Nussinov R. Analysis of ordered and disordered protein complexes reveals structural features discriminating between stable and unstable monomers. J Mol Biol. 2004;341:1327–41. doi: 10.1016/j.jmb.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Lynch M. The evolution of multimeric protein assemblages. Mol Biol Evol. 2012;29:1353–66. doi: 10.1093/molbev/msr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukatsky DB, Shakhnovich BE, Mintseris J, Shakhnovich EI. Structural similarity enhances interaction propensity of proteins. J Mol Biol. 2007;365:1596–606. doi: 10.1016/j.jmb.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andre I, Strauss CE, Kaplan DB, Bradley P, Baker D. Emergence of symmetry in homooligomeric biological assemblies. Proc Natl Acad Sci USA. 2008;105:16148–52. doi: 10.1073/pnas.0807576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright CF, Teichmann SA, Clarke J, Dobson CM. The importance of sequence diversity in the aggregation and evolution of proteins. Nature. 2005;438:878–81. doi: 10.1038/nature04195. [DOI] [PubMed] [Google Scholar]
- 41.Dayhoff JE, Shoemaker BA, Bryant SH, Panchenko AR. Evolution of protein binding modes in homooligomers. J Mol Biol. 2010;395:860–70. doi: 10.1016/j.jmb.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fong JH, Shoemaker BA, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV, Panchenko AR. Intrinsic disorder in protein interactions: insights from a comprehensive structural analysis. PLoS Comput Biol. 2009;5:e1000316. doi: 10.1371/journal.pcbi.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon SM, Sousa FJ, Mohana-Borges R, Walker GC. Regulation of Escherichia coli SOS mutagenesis by dimeric intrinsically disordered umuD gene products. Proc Natl Acad Sci USA. 2008;105:1152–7. doi: 10.1073/pnas.0706067105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu D, Tsai CJ, Nussinov R. Mechanism and evolution of protein dimerization. Protein Sci. 1998;7:533–44. doi: 10.1002/pro.5560070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiana G, Broglia RA. Design and folding of dimeric proteins. Proteins. 2002;49:82–94. doi: 10.1002/prot.10196. [DOI] [PubMed] [Google Scholar]
- 46.Levy ED, Boeri Erba E, Robinson CV, Teichmann SA. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–5. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villar G, Wilber AW, Williamson AJ, Thiara P, Doye JP, Louis AA, et al. Self-assembly and evolution of homomeric protein complexes. Phys Rev Lett. 2009;102:118106. doi: 10.1103/PhysRevLett.102.118106. [DOI] [PubMed] [Google Scholar]
- 48.Pereira-Leal JB, Teichmann SA. Novel specificities emerge by stepwise duplication of functional modules. Genome Res. 2005;15:552–9. doi: 10.1101/gr.3102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reid AJ, Ranea JA, Orengo CA. Comparative evolutionary analysis of protein complexes in E. coli and yeast. BMC Genomics. 2010;11:79. doi: 10.1186/1471-2164-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hashimoto K, Madej T, Bryant SH, Panchenko AR. Functional states of homooligomers: insights from the evolution of glycosyltransferases. J Mol Biol. 2010;399:196–206. doi: 10.1016/j.jmb.2010.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereira-Leal JB, Levy ED, Kamp C, Teichmann SA. Evolution of protein complexes by duplication of homomeric interactions. Genome Biol. 2007;8:R51. doi: 10.1186/gb-2007-8-4-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–18. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci USA. 1994;91:3127–31. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crestfield AM, Stein WH, Moore S. On the aggregation of bovine pancreatic ribonuclease. Arch Biochem Biophys Suppl. 1962;1:217–22. [PubMed] [Google Scholar]
- 55.Gronenborn AM. Protein acrobatics in pairs: dimerization via domain swapping. Curr Opin Struct Biol. 2009;19:39–49. doi: 10.1016/j.sbi.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shameer K, Shingate PN, Manjunath SC, Karthika M, Pugalenthi G, Sowdhamini R. 3DSwap: curated knowledgebase of proteins involved in 3D domain swapping. Database (Oxford) 2011;2011:bar042. doi: 10.1093/database/bar042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–99. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Gotte G, Libonati M, Eisenberg D. A domain-swapped RNase A dimer with implications for amyloid formation. Nat Struct Biol. 2001;8:211–4. doi: 10.1038/84941. [DOI] [PubMed] [Google Scholar]
- 59.Rousseau F, Schymkowitz JW, Wilkinson HR, Itzhaki LS. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc Natl Acad Sci USA. 2001;98:5596–601. doi: 10.1073/pnas.101542098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barrientos LG, Louis JM, Botos I, Mori T, Han Z, O’Keefe BR, et al. The domain-swapped dimer of cyanovirin-N is in a metastable folded state: reconciliation of X-ray and NMR structures. Structure. 2002;10:673–86. doi: 10.1016/s0969-2126(02)00758-x. [DOI] [PubMed] [Google Scholar]
- 61.Orlikowska M, Jankowska E, Kolodziejczyk R, Jaskolski M, Szymanska A. Hinge-loop mutation can be used to control 3D domain swapping and amyloidogenesis of human cystatin C. J Struct Biol. 2011;173:406–13. doi: 10.1016/j.jsb.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Shingate P, Sowdhamini R. Analysis of domain-swapped oligomers reveals local sequence preferences and structural imprints at the linker regions and swapped interfaces. PLoS One. 2012;7:e39305. doi: 10.1371/journal.pone.0039305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dickason RR, Huston DP. Creation of a biologically active interleukin-5 monomer. Nature. 1996;379:652–5. doi: 10.1038/379652a0. [DOI] [PubMed] [Google Scholar]
- 64.Yang S, Cho SS, Levy Y, Cheung MS, Levine H, Wolynes PG, et al. Domain swapping is a consequence of minimal frustration. Proc Natl Acad Sci USA. 2004;101:13786–91. doi: 10.1073/pnas.0403724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malevanets A, Sirota FL, Wodak SJ. Mechanism and energy landscape of domain swapping in the B1 domain of protein G. J Mol Biol. 2008;382:223–35. doi: 10.1016/j.jmb.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 66.Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–8. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 67.Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. Chembiochem. 2004;5:170–6. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 68.O’Shea EK, Rutkowski R, Kim PS. Evidence that the leucine zipper is a coiled coil. Science. 1989;243:538–42. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- 69.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–7. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 70.Harbury PB, Kim PS, Alber T. Crystal structure of an isoleucine-zipper trimer. Nature. 1994;371:80–3. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 71.Liu J, Zheng Q, Deng Y, Cheng CS, Kallenbach NR, Lu M. A seven-helix coiled coil. Proc Natl Acad Sci USA. 2006;103:15457–62. doi: 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moutevelis E, Woolfson DN. A periodic table of coiled-coil protein structures. J Mol Biol. 2009;385:726–32. doi: 10.1016/j.jmb.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 73.Nishi H, Ota M. Amino acid substitutions at protein-protein interfaces that modulate the oligomeric state. Proteins. 2010;78:1563–74. doi: 10.1002/prot.22673. [DOI] [PubMed] [Google Scholar]
- 74.Levy ED. A simple definition of structural regions in proteins and its use in analyzing interface evolution. J Mol Biol. 2010;403:660–70. doi: 10.1016/j.jmb.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 75.Grueninger D, Treiber N, Ziegler MO, Koetter JW, Schulze MS, Schulz GE. Designed protein-protein association. Science. 2008;319:206–9. doi: 10.1126/science.1150421. [DOI] [PubMed] [Google Scholar]
- 76.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 77.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 78.Chakrabarti P, Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–43. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 79.Keskin O, Ma B, Nussinov R. Hot regions in protein-protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–94. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 80.Malay AD, Allen KN, Tolan DR. Structure of the thermolabile mutant aldolase B, A149P: molecular basis of hereditary fructose intolerance. J Mol Biol. 2005;347:135–44. doi: 10.1016/j.jmb.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 81.Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–53. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- 82.Ramon-Maiques S, Fernandez-Murga ML, Gil-Ortiz F, Vagin A, Fita I, Rubio V. Structural bases of feed-back control of arginine biosynthesis, revealed by the structures of two hexameric N-acetylglutamate kinases, from Thermotoga maritima and Pseudomonas aeruginosa. J Mol Biol. 2006;356:695–713. doi: 10.1016/j.jmb.2005.11.079. [DOI] [PubMed] [Google Scholar]
- 83.Hashimoto K, Panchenko AR. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc Natl Acad Sci USA. 2010;107:20352–7. doi: 10.1073/pnas.1012999107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akiva E, Itzhaki Z, Margalit H. Built-in loops allow versatility in domain-domain interactions: lessons from self-interacting domains. Proc Natl Acad Sci USA. 2008;105:13292–7. doi: 10.1073/pnas.0801207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nishi H, Koike R, Ota M. Cover and spacer insertions: small nonhydrophobic accessories that assist protein oligomerization. Proteins. 2011;79:2372–9. doi: 10.1002/prot.23084. [DOI] [PubMed] [Google Scholar]
- 86.Nooren IM, Thornton JM. Structural characterisation and functional significance of transient protein-protein interactions. J Mol Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 87.Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in protein-protein binding: effect on stability and function. Structure. 2011;19:1807–15. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Presek P, Reinacher M, Eigenbrodt E. Pyruvate kinase type M2 is phosphorylated at tyrosine residues in cells transformed by Rous sarcoma virus. FEBS Lett. 1988;242:194–8. doi: 10.1016/0014-5793(88)81014-7. [DOI] [PubMed] [Google Scholar]
- 89.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17:101–4. doi: 10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- 90.Hammer ND, Wang X, McGuffie BA, Chapman MR. Amyloids: friend or foe? J Alzheimers Dis. 2008;13:407–19. doi: 10.3233/jad-2008-13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xue WF, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci USA. 2008;105:8926–31. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhak G, Choe YJ, Paik SR. Mechanism of amyloidogenesis: nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep. 2009;42:541–51. doi: 10.5483/bmbrep.2009.42.9.541. [DOI] [PubMed] [Google Scholar]
- 93.Straub JE, Thirumalai D. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Annu Rev Phys Chem. 2011;62:437–63. doi: 10.1146/annurev-physchem-032210-103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ding F, Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Molecular dynamics simulation of the SH3 domain aggregation suggests a generic amyloidogenesis mechanism. J Mol Biol. 2002;324:851–7. doi: 10.1016/s0022-2836(02)01112-9. [DOI] [PubMed] [Google Scholar]
- 95.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011;3:7. doi: 10.1101/cshperspect.a004457. Pii: a004457. http://dx.doi.org/10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levin J, Bulst S, Thirion C, Schmidt F, Botzel K, Krause S, et al. Divergent molecular effects of desmin mutations on protein assembly in myofibrillar myopathy. J Neuropathol Exp Neurol. 2010;69:415–24. doi: 10.1097/NEN.0b013e3181d71305. [DOI] [PubMed] [Google Scholar]
- 99.Vorgerd M, van der Ven PF, Bruchertseifer V, Lowe T, Kley RA, Schroder R, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304. doi: 10.1086/431959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 101.Shewmaker F, McGlinchey RP, Wickner RB. Structural insights into functional and pathological amyloid. J Biol Chem. 2011;286:16533–40. doi: 10.1074/jbc.R111.227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–32. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–33. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–47. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma BG, Goncearenco A, Berezovsky IN. Thermophilic adaptation of protein complexes inferred from proteomic homology modeling. Structure. 2010;18:819–28. doi: 10.1016/j.str.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 106.Bershtein S, Mu W, Shakhnovich EI. Soluble oligomerization provides a beneficial fitness effect on destabilizing mutations. Proc Natl Acad Sci USA. 2012;109:4857–62. doi: 10.1073/pnas.1118157109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ispolatov I, Yuryev A, Mazo I, Maslov S. Binding properties and evolution of homodimers in protein-protein interaction networks. Nucleic Acids Res. 2005;33:3629–35. doi: 10.1093/nar/gki678. [DOI] [PMC free article] [PubMed] [Google Scholar]