

Abstract
Epidemiological evidence has demonstrated a reduced risk of prostate cancer associated with cruciferous vegetable intake. Follow-up studies have attributed this protective activity to the metabolic products of glucosinolates, a class of secondary metabolites produced by crucifers. The metabolic products of glucoraphanin and glucobrassicin, sulforaphane, and indole-3-carbinol respectively, have been the subject of intense investigation by cancer researchers. Sulforaphane and indole-3-carbinol inhibit prostate cancer by both blocking initiation and suppressing prostate cancer progression in vitro and in vivo. Research has largely focused on the anti-initiation and cytoprotective effects of sulforaphane and indole-3-carbinol through induction of phases I and II detoxification pathways. With regards to suppressive activity, research has focused on the ability of sulforaphane and indole-3-carbinol to antagonize cell signaling pathways known to be dysregulated in prostate cancer. Recent investigations have characterized the ability of sulforaphane and indole-3-carbinol derivatives to modulate the activity of enzymes controlling the epigenetic status of prostate cancer cells. In this review, we will summarize the well-established, “classic” non-epigenetic targets of sulforaphane and indole-3-carbinol, and highlight more recent evidence supporting these phytochemicals as epigenetic modulators for prostate cancer chemoprevention.
KEY WORDS: epigenetic, I3C, prostate cancer, sulforaphane
INTRODUCTION
Global cancer diagnoses are predicted to increase for the foreseeable future, with a key contributor being an aging world population. Although age is a strong risk factor for cancer, many other variables also influence the relative risk of disease development. Lifestyle and dietary choices are two factors that have a prominent role in cancer risk; however, given the combination of individual genetic variation and variability in lifestyle and dietary choices, it is extremely difficult to identify discrete factors that have a consequential role in disease risk. Population-wide study of individuals whose characteristics vary can provide correlative data that can then be used to develop testable hypotheses. This strategy has proven useful in identifying dietary components associated with decreased cancer risk. A growing number of epidemiological studies have drawn an association between cruciferous vegetable intake and decreased prostate cancer risk (1,2). Further epidemiological analysis stratifying specifically on glucosinolate intake (a class of natural compounds produced by crucifers) identified a significant inverse trend with prostate cancer risk (3). Controlled experimentation with glucosinolate derivatives, such as sulforaphane and indole-3-carbinol (I3C), has characterized inhibitory and cytotoxic activity in prostate cancer cells and animal model systems and has provided a mechanistic explanation for how crucifers are causative in lowering cancer risk.
Prostate cancer is the second most commonly diagnosed cancer in men worldwide. Clinical prostate cancer incidence by nation however shows considerable variability. In general, Western nations tend to have a high incidence of prostate cancer, while Asian nations are characterized by a low incidence. In the USA, prostate cancer is predicted to account for 28.5% of all male cancer diagnoses in 2012, affecting over 240,000 men (154 per 100,000) (4), whereas the rate in Asian nations can be up to tenfold lower (5). Diet and lifestyle are thought to be primary contributors to the difference in prostate cancer rates between Western and Asian nations. The proposed influence of diet on prostate cancer rate is supported by studies showing convergence with Western prostate cancer rates in Asian immigrant communities in the USA (6,7). With regard to cruciferous vegetable intake, Asian nations tend to consume much higher amounts per person than Western nations (8), suggesting that crucifer intake may be an important diet and lifestyle factor contributing to differences in prostate cancer risk.
The cruciferous vegetable family (Brassicaceae) includes many vegetables that are found in the diet—from broccoli, Brussels sprouts, and cauliflower, that are common in the Western diet, to daikon, watercress, and bok choy that are more common in Asian cuisine. Cruciferous vegetables contain a number of glucosinolates whose presence and relative abundance are specific to each species and even to specific cultivars (9). Glucosinolates are the natural plant chemicals (phytochemicals) that give rise to bioactive species. They are cleaved by the endogenous plant enzyme myrosinase to yield active phytochemicals that possess varying degrees of anti-cancer activity. Two phytochemicals that have drawn a significant amount of attention are sulforaphane and I3C. In this review, we will highlight the ability of these phytochemicals to inhibit prostate cancer, focusing on their post-initiation suppressive activity. Finally, we will discuss more recent data characterizing activity as epigenetic modulators.
METABOLISM AND BIOACTIVITY OF SULFORAPHANE AND INDOLE-3-CARBINOL
Following consumption, glucosinolates are cleaved by plant-derived myrosinase when the plant wall is disrupted by chewing, and, to a lesser extent, by gut microbial myrosinases to release sulforaphane and I3C from their precursors. Sulforaphane and I3C then undergo further post-consumption modification, with sulforaphane undergoing enzymatic metabolism via the mercapturic acid pathway, and I3C undergoing spontaneous self-condensation and polymerization in the gut and possibly in the plasma (Fig. 1). It is the post-consumption products of these glucosinolates that possess anti-cancer activity. Experimentation has demonstrated multi-targeted inhibitory effects that both block cancer formation and suppress prostate cancer growth.
Fig. 1.
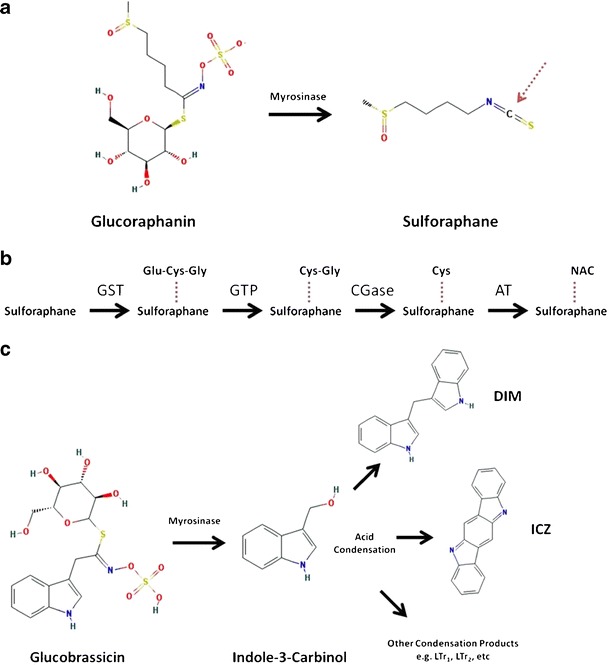
Metabolism of glucoraphanin and glucobrassicin to biologically active metabolites. a Sulforaphane is released from glucoraphanin by the plant enzyme myrosinase. Red dashed arrow marks the reactive carbon atom subject to glutathione conjugation. b Sulforaphane is metabolized via the mercapturic acid pathway into active metabolites. Glutathione-S-transferase (GST) first conjugates a GSH molecule (Glu-Cys-Gly) to the reactive carbon on sulforaphane. Glutamate is then removed by γ-glutamyltranspeptidase (GTP), followed by removal of the glycine residue by cysteinylglycinase (CGase). Cysteine is then acetylated by an acetyltransferase (AT) to sulforaphane-N-acetylcysteine, which is excreted in the urine. c Indole-3-carbinol is released from the glucosinolate glucobrassicin by myrosinase and undergoes spontaneous condensation in the acidic environment of the gut. Diindolylmethane (DIM) is the most abundant post-absorption acid condensation product. Acid condensation products can be modified further post-absorption. LTr 1 linear trimer 1, LTr 2 linear trimer 2. Structures from PubChem at National Center for Biotechnology Information (NCBI)
Sulforaphane
Sulforaphane and its metabolites are the principal bioactive phytochemicals derived from broccoli and broccoli sprouts. Sulforaphane is present as the glucosinolate glucoraphanin in cruciferous vegetables. Glucoraphanin is cleaved by the endogenous plant enzyme myrosinase into sulforaphane and glucose when the enzyme and glucosinolate are brought into contact (Fig. 1a). Once released, sulforaphane is available for uptake in the human gut. Sulforaphane is then metabolized through the mercapturic acid pathway, producing several metabolic products (Fig. 1b).
Human feeding studies have determined the absorption and kinetics of sulforaphane metabolism in vivo. Sulforaphane is rapidly taken up and metabolized by the body, reaching a plasma concentration peak within ∼2 h of consumption (10). Absorption and kinetics in animal models is consistent with the human data; both human and animal feeding studies have shown clearance of sulforaphane and its metabolites from the plasma within 24 h of ingestion, and evidence in animal models suggests that tissue accumulation may be possible following repeated ingestion (10–12).
Although post-consumption sulforaphane levels in human prostate tissue have not yet been evaluated, there are several lines of evidence that suggest sulforaphane does reach the prostate and causes changes in cellular processes. Work in rodent models has demonstrated that sulforaphane and its metabolites reach prostate tissue after oral administration. Clarke et al. showed the presence of sulforaphane metabolites in the prostates of mice 2 and 6 h after ingesting 20 μmol sulforaphane (13). Similarly, Veeranki et al. showed an increase in sulforaphane metabolites in rat prostate tissue 1.5 h after ingesting 150 μmol/kg sulforaphane (12). In both the transgenic adenocarcinoma of mouse prostate (TRAMP) mouse model (transformed prostate tissue) and a prostate specific PTEN deletion mouse model, broccoli sprout or sulforaphane treatment caused prostate-specific changes in gene expression, suggesting sulforaphane or its metabolites reach the prostate (14–16). In the TRAMP model, supplementation with broccoli sprouts stimulated nuclear factor E2-related factor 2 (nrf2) controlled gene expression and decreased Akt signaling in prostate tissue, whereas in the PTEN deletion model sulforaphane treatment reversed gene expression changes caused by PTEN loss in the prostate.
Importantly, oral administration of broccoli sprouts and sulforaphane inhibited prostate tumor progression in the TRAMP and PTEN-null mouse models, demonstrating the therapeutic potential of the natural product sulforaphane in the prostate. These mouse models are currently the closest simulation of human prostate cancer progression and provide strong pre-clinical evidence of sulforaphane bioactivity against prostate cancer progression.
In one human-feeding study, men with high-grade prostatic intraepithelial neoplasia, a pre-cancerous condition characterized by foci of abnormal prostate epithelial cell proliferation, supplemented their diets with broccoli or peas for 12 months and then submitted prostate tissue samples for analysis during routine tissue biopsy (16). The analysis found changes in gene expression related to TGFβ, insulin signaling, and EGF signaling, suggesting broccoli (i.e., sulforaphane) supplementation can affect signaling pathways related to cell growth in prostate tissue. Further work in human subjects quantifying sulforaphane metabolite levels in the prostate after acute versus long-term exposure will help guide both dietary recommendations and the development of sulforaphane as a natural agent for prostate health.
Indole-3-Carbinol
I3C is released from its glucosinolate precursor glucobrassicin when brought into contact with myrosinase. Like glucoraphanin, glucobrassicin is found in cruciferous vegetables, with exceptionally high concentrations in Brussels sprouts and garden cress. In an acidic environment like the human stomach, I3C is rapidly converted into an array of acid condensation products and modified derivatives (17).
In vivo assessment of I3C and its products suggest that diindolylmethane (DIM), an I3C acid-condensation product, is one of the major bioactive compounds responsible for the benefits associated with I3C. I3C undergoes condensation and modification after oral administration in mice, with the parent compound undetectable in plasma within 1 h (18). A separate human-feeding study did not detect I3C in the plasma of study participants administered the pure indole, but instead detected only DIM (19). Because DIM was the only acid condensation product detected in human plasma after oral administration of I3C, these data support the dimer DIM as the key mediator of prostate protection.
It is important to note that in vitro and in vivo studies have shown an anti-cancer effect associated with I3C. These studies utilize pure I3C as the treatment compound in vitro by dosing cultured prostate cancer cell lines, or in vivo by direct injection (20). Relatively few of these investigations assess the post-treatment derivatives of I3C, making it difficult to determine whether treatment effects are in response to I3C or specific condensation products. One study has found significant spontaneous conversion of I3C to DIM in culture media and simulated peritoneal fluid (21), which supports a model where I3C is converted to DIM or other condensation products after in vitro dosing or intraperitoneal administration. Our discussion will therefore focus on the mechanisms through which DIM may block or suppress prostate cancer. Discussion involving purified I3C as treatment will also be presented, but we will be working under the assumption that it is converted to DIM (For further reading supporting I3C conversion, see Bradlow Review (22)).
In vivo work in mouse models supports DIM as an inhibitor of prostate cancer progression. Dietary supplementation of DIM significantly inhibited the progression of prostate cancer in the TRAMP model (23). DIM decreased tumor growth (as measured by genitourinary weight), decreased proliferating cell markers, and increased cell death effectors. The authors also note that DIM supplementation had no significant effect on cell markers in normal mice. Though DIM supplementation did not lead to complete eradication of transformed cells and prostate tumors in the TRAMP model, there is clear evidence that dietary intervention with the natural product DIM is a strategy worth pursuing.
Application of In Vitro and In Vivo Exposure to Dietary Intake
Pharmacokinetic studies suggest peak plasma concentrations of sulforaphane and DIM may be below those achieved in controlled pre-clinical experiments. Plasma sulforaphane level reached over 7 μM in subjects eating “SuperBroccoli” soup (24), and a phase I clinical dose escalation study found DIM to reach levels just over 1 μM in men supplemented with 300 mg of an enhanced-bioavailability formulation of DIM (BR-DIM) (25). Neither study suggests these are maximum achievable plasma concentrations, and no (or very limited) adverse effects were noted, suggesting higher plasma levels are reachable and tolerable. The effects of long-term, low-level dietary exposure are not as well understood, but there is some evidence of tissue-specific sulforaphane accumulation over time in rats (12). In humans, few studies have characterized bioavailability and concentrations of sulforaphane, DIM, or their metabolites in tissues. Further work characterizing tissue-specific acute versus repeated exposure is needed to fully understand the effects of these phytochemicals when attained from the diet.
CHEMOPREVENTION MECHANISMS
Pre-initiation Blocking Activity
Sulforaphane
Sulforaphane has been extensively investigated as a cancer-blocking agent because of its ability to induce phase II enzymes (26,27). The expression of these enzymes is controlled by transcription factor nrf2. Under basal conditions, nrf2 is sequestered in the cytoplasm by redox-sensitive protein Kelch-like ECH-associated protein 1 (KEAP1). However, under redox stress, KEAP1 releases nrf2 which then translocates to the nucleus and binds antioxidant response elements in the promoters of target genes, stimulating their expression. Upregulation of phase II enzymes, such as heme oxygenase I or NADPH quinone oxidoreductase 1, greatly increases a cell’s detoxification capacity. Phase II enzymes conjugate moieties to reactive molecules, thus decreasing their ability to cause cellular damage and enhancing their solubility for excretion. Sulforaphane is a strong glutathione-S-transferase inducer, which conjugates glutathione to electrophiles and neutralizes their reactivity. The first step in sulforaphane metabolism also involves glutathione conjugation, followed by enzymatic reactions in the mercapturic acid pathway (Fig. 1b). For more thorough reviews of sulforaphane and phase II blocking activity, see Fahey and Talalay (27) and Guerrero-Beltran et al (28).
Indole-3-Carbinol
DIM can also be considered a prostate cancer blocking agent through its ability to stimulate cellular detoxification pathways. DIM is a reported aryl-hydrocarbon receptor (Ahr) agonist in multiple cell lines (29). Ahr is a nuclear receptor transcription factor that stimulates the expression of detoxification enzymes in the cytochrome P450 family (phase I) and increases the capacity of cells to deal with xenobiotic stress. DIM treatment also stimulates the nrf2-mediated phase II response, which enhances reactive molecule metabolism and excretion of genotoxic agents (29–31). Through the activation of Ahr and nrf2 signaling pathways, DIM effectively increases the cells detoxification potential and blocks what otherwise could be tumor-initiating events.
An ancillary benefit to enhanced phase I expression also seems to be changes in steroid hormone profile (32). Because hormones are extensively processed and modified through oxidation/reduction reactions, changes in phase I enzyme levels could alter hormone profiles. Sex hormones have a large role in prostate cancer progression and have been found to be altered with I3C or DIM supplementation in men and women (33–36). Hormone-sensitive prostate cancer responds to estradiol (E2) in vitro (37), and I3C can reduce E2 levels in men (34). Though these studies have not yet looked at male hormones that drive hormone-sensitive prostate cancer, changes in estrogen hormone levels raises the possibility that male sex hormone levels are also altered and could therefore influence the growth of transformed cells early in the process of prostate cancer development.
Post-initiation Suppressive Activity
Aside from their blocking activity, sulforaphane and DIM are also able to suppress cancer growth post-initiation (Fig. 2). This effect has been demonstrated in prostate cancer cell lines and in the TRAMP model. The ability to inhibit growth and stimulate apoptosis of transformed cells is suggestive that sulforaphane and DIM have activity outside of the phase I/phase II response. These findings are particularly interesting with respect to prostate cancer because a majority of men will develop hyperplasia and localized neoplasia as a natural part of the aging process. Any treatment that can keep these growths localized and inhibited could substantially decrease the number of advanced prostate cancer cases. Thus, sulforaphane and I3C effects outside the phase I/phase II response have been an area of great interest to prostate cancer researchers. Recent investigations have attributed suppressive activity to antagonism of signaling pathways known to be important for prostate cancer progression, such as the Akt signaling axis, as well as modulation of epigenetic enzymes, both of which contribute to growth arrest and induction of apoptosis.
Fig. 2.
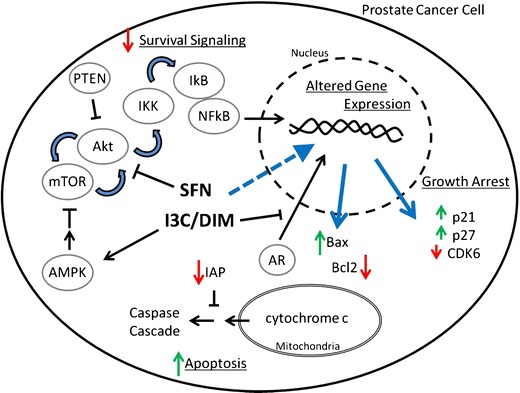
Selected non-epigenetic effects of sulforaphane and I3C/DIM on prostate cancer cells. Sulforaphane (SFN) and I3C/DIM inhibit the Akt signaling axis, a signaling pathway often hyperactive in prostate cancer. Inhibition of this pathway decreases pro-survival signaling by mTOR, Akt, and NFkB. Sulforaphane and I3C/DIM treatment also lead to changes in gene expression (blue arrow) that trigger growth arrest and apoptosis. The expression of proteins controlling the cell cycle (e.g., p21, p27, and CDK6) is altered to effect growth arrest, and apoptosis is finally induced through the mitochondrial pathway. Abbreviations: AR androgen receptor, CDK6 cyclin-dependant kinase 6, IAP inhibitors of apoptosis
Sulforaphane
Attenuation of Akt/NFkB Signaling and Induction of Apoptosis
Enhanced Akt signaling is a common acquisition in transformed prostate tissue (38–40), and inhibiting the Akt signaling axis is potentially a good therapeutic target for suppressing prostate cancer growth and survival (41,42). Traka et al. were able to show that Akt signaling was attenuated in prostate tissue of human subjects in response to long-term dietary consumption of sulforaphane-rich broccoli (15). Studies in animal models and cultured prostate cancer cells using purified sulforaphane have also shown attenuated Akt signaling. In the TRAMP mouse model, Keum et al. showed a decrease in Akt activation in transformed prostate tissue, whereas Traka et al. showed an attenuation of induced gene expression pursuant to the loss of the Akt suppressor PTEN (14,15). In vitro analysis of PC3 prostate cancer cells treated with purified d,l-sulforaphane showed a decrease in Akt phosphorylation, decreased phosphorylation of mTOR target proteins, and a decrease in cellular protein translation (43), supporting a specific activity for sulforaphane in inhibiting the Akt signaling pathway.
Akt signaling is involved in many cellular processes (44–47) and could explain how sulforaphane treatment leads to decreases in the expression of multiple pathways known to support cancer growth. A decrease in NFκB transcriptional activity has been noted in prostate cancer cell lines in response to sulforaphane treatment (46,48,49). Sulforaphane caused the observed decrease by inhibiting NFκB translocation from the cytoplasm to the nucleus. Inhibition of the Akt signaling pathway could lead to sequestration of NFκB in the cytoplasm by decreasing mTOR complex activity and IKK activity (50), a signaling pathway delineated in PTEN null prostate cancer cells.
The net effect of decreased Akt/NFκB signaling could be to tip the cell fate scales toward apoptosis in prostate cancer cells. A decrease in NFκB-dependent inhibitor of apoptosis (IAP) proteins by sulforaphane may provide the stimulus for transformed cells to undergo intrinsic, mitochondrial-mediated apoptosis (51–53). A decrease in IAP by antisense RNA is able to increase basal apoptosis in prostate cancer cells (54). The observation that mitochondria are necessary for at least a portion of sulforaphane-induced cell death supports a model of stimulation of intrinsic apoptosis as an important process in prostate cancer cell killing (55). Sulforaphane treatment in vitro and in the TRAMP model indeed increases the Bax/Bcl-2 protein ratio and triggers a caspase cleavage cascade that results in cell death (56,57).
Inhibition of Akt signaling and stimulation of growth arrest and apoptosis are two key sulforaphane effects in transformed prostate tissue. This is not to say that sulforaphane does not influence many other signaling pathways in prostate cancer cells. In fact, it is likely that other known sulforaphane effects contribute to suppression. For information detailing sulforaphane effects outside of those mentioned here, see Clarke et al. (58) and Juge et al. (59).
Indole-3-Carbinol
Induction of Apoptosis
Initial experiments investigating the potential of I3C to inhibit prostate cancer growth focused on controlled in vitro experimentation using the advanced prostate cancer cell line PC3 (60). I3C treatment led to cell cycle arrest and induction of apoptosis. I3C treatment was able to cause this inhibition by decreasing the expression/activity of pro-cell cycle progression kinase CDK6 and by upregulating the expression of cell cycle inhibitors p21 and p27 independent of p53 (Fig. 2). Intrinsic apoptosis was triggered by a shift in Bax and Bcl2 expression toward a ratio favoring cell death and was evidenced by PARP cleavage and DNA laddering. A decrease in NFκB activation was also noted (60). Further investigation using a range of representative androgen-dependent and androgen-independent prostate cancer cell lines (LNCaP, DU145, and PC3) confirmed a decrease in cell growth and induction of apoptosis in response to I3C and DIM treatment (61,62); however, there are conflicting reports concerning the mechanism responsible for induction of apoptosis (63). Further work characterizing cell death in response to I3C and DIM at physiologically relevant doses will be necessary to understand how these phytochemicals inhibit prostate cancer growth in vivo.
Attenuation of Akt/NFkB Signaling
Subsequent studies in androgen-independent DU145 cells using equimolar I3C and DIM treatment characterized G1 cell cycle arrest and a decrease in Akt and PI3K proteins associated only with DIM treatment (64). DIM also decreases phophorylated (activated) Akt, as well as nuclear NFκB, NFκB DNA binding, and NFκB transcription activity (65–68). DIM may decrease Akt signaling by activating upstream regulator AMPK: a recent report showed DIM activated AMPK both in vitro and in vivo and was associated with mTOR and androgen receptor (AR) inhibition (Fig. 2) (69).
Inhibition of Androgen Receptor Signaling
A comparison of prostate cancer cell I3C/DIM sensitivity between studies and within studies utilizing different prostate cancer cell lines has shown decreased sensitivity of more advanced, AR-negative PC3 cells, and, importantly, prostate cancer cells seem to be much more sensitive to DIM treatment than nontransformed cells (68). DIM may specifically interfere with prostate cancer growth at the initial stages by suppressing the androgen signaling pathway (70), which would explain increased sensitivity of AR-positive cancers. DIM treatment decreased AR-controlled gene expression in prostate cancer cells in vitro by inhibiting translocation of AR to the nucleus (70). Bhuiyan et al. also found decreased androgen signaling in response to DIM treatment but showed that this effect was the result of not only a failure of AR to translocate to the nucleus but also because of a decrease in AR expression (65). This is an important inhibitory activity since hyperactive androgen receptor is one of the most common and early events in prostate cancer development.
Epigenetic Activity
Prostate cancer cells—and cancer cells in general—display epigenetic abnormalities that are thought to enhance the cancer phenotype. Transformed cells show global DNA hypomethylation, site-specific DNA hypermethylation, altered cellular histone deacetylase (HDAC) activity, and altered miRNA expression (71,72). Genes that inhibit cancer cell growth, such as cell cycle inhibitors or pro-apoptotic genes, are frequently silenced epigenetically. DNA methyltransferases (DNMTs; enzymes that methylate DNA cytosine residues) and HDACs often work together in larger protein complexes to strip chromatin of active acetylation marks and lay down DNA methylation for stable gene repression. Targeting the enzymes that regulate the epigenetic signature of prostate cancer cells has proven to be a viable target in cancer prevention and cancer therapeutic research. Currently, there are several clinical trials aimed at determining the tolerance and efficacy of HDAC and DNMT inhibitors in human subjects (73). Importantly, sulforaphane and DIM have been characterized as diet-based modulators of epigenetic enzymes (Fig. 3).
Fig. 3.
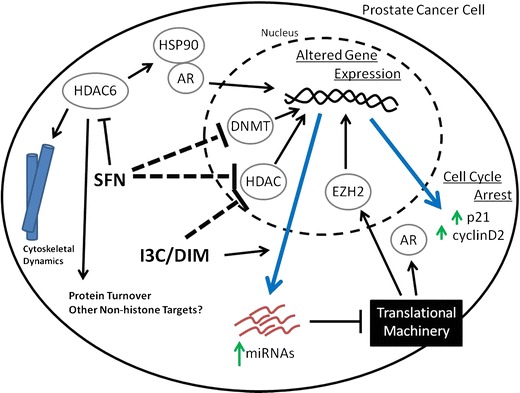
Sulforaphane and I3C/DIM suppress prostate cancer through epigenetic modulation. Sulforaphane (SFN) decreases cellular DNA methyltransferase (DNMT) and histone deacetylase (HDAC) activity, leading to altered chromatin structure and gene expression (blue arrow). Sulforaphane also directly inhibits cytoplasmic HDAC6, which controls the acetylation of many nonhistone proteins, leading to a decrease in AR signaling, altered cytoskeletal dynamics, disrupted protein turnover, and, ultimately, increased cell stress. I3C/DIM also decreases cellular HDAC activity, leading to changes in gene expression (blue arrow). The expression levels of microRNAs (miRNAs) are also disrupted and lead to changes in protein levels of chromatin-associated protein EZH2 and nuclear receptor AR through post-transcriptional regulation
Sulforaphane
Sulforaphane and specifically its metabolites sulforaphane-GSH and sulforaphane-Cys have been characterized as HDAC inhibitors (Fig. 1) (74). HDAC overexpression is frequently observed in prostate cancer (75), and knockdown of HDAC enzymes with small RNAs leads to a decreased in cancer cell growth and alterations in the expression of genes associated with prostate cancer progression (76,77). Treatment of prostate cancer cells with sulforaphane leads to a decrease in cellular HDAC activity and a global increase in histone acetylation (78,79). Increases in histone acetylation also occur within the promoters of silenced tumor suppressor genes and are accompanied by increased gene expression. Tumor suppressor gene p21 is often silenced in prostate cancer cells. Treatment with sulforaphane leads to an increase in promoter acetylation and an increase in p21 expression. This effect was even observed in the p53 null prostate cancer cell line PC3 (80) and suggests epigenetic reactivation.
Sulforaphane also decreases DNMT protein levels in prostate cancer cells by decreasing DNMT1, 3a, and 3b gene expression (81). Hsu et al. showed that the sulforaphane induced decrease in DNMT levels are associated with a global decrease in DNA methylation (81). A more targeted analysis of the cyclin D2 promoter, an epigenetically silenced gene in prostate cancer cells (82), showed a local decrease in DNA methylation associated with increased cyclin D2 transcript levels. Similar findings have recently been reported in breast cancer cells looking at the human telomerase reverse transcriptase (hTERT) gene (83). However, in this study the authors found a decrease in DNMT expression associated with a decrease in DNA methylation and, surprisingly, a decrease in hTERT gene expression. Demethylation, in this instance, appears to allow transcriptional repressors to recognize and bind DNA elements previously unavailable. It is likely that similar phenomena occur in sulforaphane treated prostate cancer cells and that the relationship between DNA methylation and gene expression in transformed tissue is more complicated than a simple inverse association.
Sulforaphane induced changes in chromatin modifications and gene expression plays a large role in mediating its cytotoxic effects but do not account for all its activity. HDAC enzymes also localize outside the nucleus, where they target non-histone proteins and participate in cellular processes beyond chromatin regulation. HDAC6 is a class II HDAC localized primarily in the cytoplasm. It is a critical regulator of the cytoskeletal network and also plays a role in chaperoning ubiquitin-tagged proteins to the perinuclear aggresome for turnover (84). HDAC6 appears to be an important sulforaphane target given that overexpressing HDAC6 in PC3 prostate cancer cells can abrogate a sulforaphane-induced decrease in cell viability (80).
HDAC6 is directly inhibited by sulforaphane (76), leading to increased tubulin acetylation and filament stabilization. In addition to decreasing tubulin dynamics (85), sulforaphane treatment leads to an increase in insoluble tubulin (86). Although these findings were investigated in breast and lung cancer cells, and in a cell-free system, increased tubulin acetylation has been noted in prostate cancer cells treated with sulforaphane (76). Similar changes in tubulin dynamics and solubility are therefore likely to be occurring in prostate cancer cells. One report noted that sulforaphane can directly bind tubulin but does not lead to the collapse of the microtubule network (87); however, a direct binding effect may be unlikely in vivo due to extremely rapid glutathione conjugation. Microtubule stabilization caused by HDAC6 inhibition may be the mechanism behind the anti-metastatic and cell cycle stress properties associated with sulforaphane (57,88).
HDAC6 is also involved in AR and other nuclear receptor signaling pathways through its regulation of heat shock protein 90 (HSP90) acetylation. Deacetylation of HSP90 by HDAC6 releases AR, allowing it to translocate into the nucleus and modulate gene expression (89). Androgen signaling is a strong driver of prostate cancer growth and is initially hormone dependent. As prostate cancer progresses, androgen signaling however becomes hormone independent, and thus clinicians lose a valuable target to suppress prostate cancer growth. HDAC6 is required for hormone-independent nuclear localization in advanced prostate cancer, and HDAC6 inhibition or knock-down decreases AR signaling (89). A separate report showed that sulforaphane-mediated inhibition of HDAC6 leads to AR degradation and decreased androgen signaling (76). HDAC6 inhibition may be a good target in advanced prostate cancer that is no longer sensitive to anti-androgen therapy.
Indole-3-Carbinol
DIM has recently been shown to significantly decrease cellular HDAC activity in prostate cancer cell lines (90). DIM does not directly inhibit HDAC activity but leads to a decrease in HDAC2 protein level. These findings are consistent with an earlier report in colon cancer cells detailing class I HDAC degradation in response to DIM (91).
DIM also alters the expression of other epigenetic modulators, including the enzymes controlling histone methylation and microRNAs. In a small group of prostate cancer patient samples, Kong et al. found a correlation between decreasing Let family microRNA expression and increasing expression of histone methyltransferase EZH2 (92), a marker associated with poor prognosis (93,94). Forced expression of Let 7 family members in prostate cancer cell lines decreased EZH2 expression and inhibited colony growth, demonstrating a causative link between Let 7 and EZH2 expression, and prostate cancer growth. An in vivo assessment of these findings from a prostate cancer study population supplemented with BR-DIM confirmed in vitro findings. Study participants supplemented with BR-DIM for 2 to 4 weeks showed increased Let 7 expression and decreased EZH2 expression. In a related study, this same group showed decreased microRNA miR-34a associated with increased AR expression and signaling (95). BR-DIM supplementation again led to modest re-expression of the silenced miR-34a and decreased AR expression in vivo. These are exciting findings in that they demonstrate in vivo DIM effects that inhibit cancer growth and reverse changes associated with prostate cancer progression and poor clinical outcomes. They also confirm in vitro experimental data and provide a foundation for understanding how DIM intake is associated with prostate cancer inhibition in humans.
FUTURE DIRECTIONS
The biologically active phytochemicals sulforaphane and DIM have well-established suppressive activity in vitro and growing evidence supports activity inhibiting prostate cancer progression in vivo. A number of clinical trials are currently investigating SFN and DIM in prostate cancer cases to determine tolerance and efficacy utilizing an array of sources, including administration of purified sulforaphane, broccoli sprout extract pills, I3C-rich food, and BR-DIM (see www.clinicaltrials.gov). Study endpoints for on-going sulforaphane and BR-DIM investigations include quantitation and characterization of treatment metabolites in prostate tissue, a critical piece of data that will shape study design moving forward.
Despite their very different chemical structures, sulforaphane and DIM share some common targets and treatment endpoints. One explanation for this overlap is that both chemicals target cancer epigenetically: histone modifications, DNA methylation, and microRNA expression are dysregulated in cancer, and may explain why cancer cells are hypersensitive to sulforaphane and DIM treatment relative to normal tissue. Importantly, sulforaphane and DIM do not directly induce DNA damage or disrupt chromatin structure. The multiple, overlapping molecular targets suggest very broad effects governing cell homeostasis and genome stability.
Both phytochemicals alter cellular HDAC activity, and while sulforaphane decreases DNA methyltransferase activity, DIM alters microRNA and histone methyltransferase EZH2 expression in vivo. This last finding is an excellent demonstration of the connectivity and interrelationship between the different systems that regulate the epigenetic characteristics of cancer cells. Furthermore, these findings are likely the tip of the iceberg—a growing body of research consistently finds that the phytochemicals discussed here target an array of cancers arising from disparate tissues (96,97). This again supports sulforaphane and DIM as working through an epigenetic mechanism, targeting cancer cells no matter the underlying mutations that feed unregulated cell growth and survival.
Although the phase I/phase II induction and blocking activity associated with sulforaphane and I3C are not typically thought of as being under epigenetic control, a recent report has demonstrated the importance of epigenetics in the nrf2 response in transformed prostate cancer cells (98). Nrf2 expression is dampened in prostate cancer cells, and treatment with trichostatin A, a pharmaceutical HDAC inhibitor, removes epigenetic marks associated with gene silencing. The effect is particularly strong when used in combination with the DNMT inhibitor 5-aza-2′-deoxycytidine. Although these effects were shown with pharmacological compounds, sulforaphane and I3C may have a similar effect; a decrease in HDAC activity in combination with decreased DNMT activity in response to sulforaphane, accompanied by the innate ability of sulforaphane and/or I3C to induce the phase I/phase II response, could help explain why these natural products are strong inducers of the detoxification response in transformed prostate cancer cells. Combination therapies that exploit the coordinated activity of classic genetic targets and epigenetic regulators will be an important area of research going forward.
Future investigations into the effects of inhibition of HDAC and histone methyltransferase activity should focus on connecting changes in post-translational acetylation/methylation of non-histone proteins to changes in protein activity. The recent publication of the human “acetylome” and the identification of proteins and protein complexes sensitive to HDAC inhibitors identified many transcription factors and chromatin binding complexes as being affected by acetylation (99,100). Altered transcription factor acetylation or chromatin associated protein complex stability could explain the vast changes in gene expression and signaling networks induced by sulforaphane and DIM treatment. Aside from changes in gene expression, miRNA expression in response to sulforaphane and DIM is largely unmapped in prostate cancer. Global analyses of changes in miRNA profile, and subsequent work identifying specific functional RNAs responsive to sulforaphane or DIM, will provide further insight into how changes in the activity of chromatin modifiers and transcriptional profile contribute to prostate cancer inhibition. Moving forward, it will be imperative that we characterize these changes and their downstream effects in order to understand how sulforaphane and DIM may lead to tumor suppression and identify potential new molecular targets for prostate cancer therapy.
CONCLUSIONS
The natural products sulforaphane and I3C inhibit prostate cancer through both blocking of tumor initiation and suppression of transformed cell growth. They affect tumor suppression by inhibiting signaling networks known to have a role in prostate cancer growth and by triggering cell cycle arrest and apoptosis. More recent work has characterized activity as epigenetic modulators in prostate cancer cells in vitro and in vivo. Further investigation into the anti-cancer activity of sulforaphane and I3C will give us a better understanding of how these natural products are associated with decreased prostate cancer risk and uncover new targets for therapeutic intervention.
ACKNOWLEDGMENTS
Our work is funded by NIH grants CA90890, CA65525, CA122906, CA122959, and CA80176 and by National Institute of Environmental Health Sciences (NIEHS) Center grant P30 ES00210.
REFERENCES
- 1.Richman EL, Carroll PR, Chan JM. Vegetable and fruit intake after diagnosis and risk of prostate cancer progression. Int J Cancer. 2011;131(1):201–210. doi: 10.1002/ijc.26348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu B, Mao Q, Cao M, Xie L. Cruciferous vegetables intake and risk of prostate cancer: a meta-analysis. Int J Urol. 2012;19(2):134–141. doi: 10.1111/j.1442-2042.2011.02906.x. [DOI] [PubMed] [Google Scholar]
- 3.Steinbrecher A, Nimptsch K, Husing A, Rohrmann S, Linseisen J. Dietary glucosinolate intake and risk of prostate cancer in the EPIC-Heidelberg cohort study. Int J Cancer. 2009;125(9):2179–2186. doi: 10.1002/ijc.24555. [DOI] [PubMed] [Google Scholar]
- 4.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, et al., editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). 2012.
- 5.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v2.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10 (Internet). International Agency for Research on Cancer. 2010. Available from: http://globocan.iarc.fr. Accessed 12/11/2012
- 6.Shimizu H, Ross RK, Bernstein L, Yatani R, Henderson BE, Mack TM. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br J Cancer. 1991;63(6):963–966. doi: 10.1038/bjc.1991.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Demissie K, Lu SE, Rhoads GG. Cancer incidence among Korean-American immigrants in the United States and native Koreans in South Korea. Cancer Control. 2007;14(1):78–85. doi: 10.1177/107327480701400111. [DOI] [PubMed] [Google Scholar]
- 8.IARC . IARC: cruciferous vegetables, isothiocyanates and indoles. Lyon: IARC; 2004. [Google Scholar]
- 9.McNaughton SA, Marks GC. Development of a food composition database for the estimation of dietary intakes of glucosinolates, the biologically active constituents of cruciferous vegetables. Br J Nutr. 2003;90(3):687–697. doi: 10.1079/BJN2003917. [DOI] [PubMed] [Google Scholar]
- 10.Hanlon N, Coldham N, Gielbert A, Sauer MJ, Ioannides C. Repeated intake of broccoli does not lead to higher plasma levels of sulforaphane in human volunteers. Cancer Lett. 2009;284(1):15–20. doi: 10.1016/j.canlet.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Hauder J, Winkler S, Bub A, Rufer CE, Pignitter M, Somoza V. LC-MS/MS quantification of sulforaphane and indole-3-carbinol metabolites in human plasma and urine after dietary intake of selenium-fortified broccoli. J Agric Food Chem. 2011;59(15):8047–8057. doi: 10.1021/jf201501x. [DOI] [PubMed] [Google Scholar]
- 12.Veeranki OL, Bhattacharya A, Marshall JR, Zhang Y. Organ-specific exposure and response to sulforaphane, a key chemopreventive ingredient in broccoli: implications for cancer prevention. Br J Nutr. 2012;2:1–8. doi: 10.1017/S0007114512000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, et al. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm Res. 2011;28(12):3171–3179. doi: 10.1007/s11095-011-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keum YS, Khor TO, Lin W, Shen G, Kwon KH, Barve A, et al. Pharmacokinetics and pharmacodynamics of broccoli sprouts on the suppression of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: implication of induction of Nrf2, HO-1 and apoptosis and the suppression of Akt-dependent kinase pathway. Pharm Res. 2009;26(10):2324–2331. doi: 10.1007/s11095-009-9948-5. [DOI] [PubMed] [Google Scholar]
- 15.Traka MH, Spinks CA, Doleman JF, Melchini A, Ball RY, Mills RD, et al. The dietary isothiocyanate sulforaphane modulates gene expression and alternative gene splicing in a PTEN null preclinical murine model of prostate cancer. Mol Cancer. 2010;9:189. doi: 10.1186/1476-4598-9-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Traka M, Gasper AV, Melchini A, Bacon JR, Needs PW, Frost V, et al. Broccoli consumption interacts with GSTM1 to perturb oncogenic signalling pathways in the prostate. PLoS One. 2008;3(7):e2568. doi: 10.1371/journal.pone.0002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grose KR, Bjeldanes LF. Oligomerization of indole-3-carbinol in aqueous acid. Chem Res Toxicol. 1992;5(2):188–193. doi: 10.1021/tx00026a007. [DOI] [PubMed] [Google Scholar]
- 18.Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, et al. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res. 2004;10(15):5233–5241. doi: 10.1158/1078-0432.CCR-04-0163. [DOI] [PubMed] [Google Scholar]
- 19.Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3′-diindolylmethane. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2477–2481. doi: 10.1158/1055-9965.EPI-06-0396. [DOI] [PubMed] [Google Scholar]
- 20.Souli E, Machluf M, Morgenstern A, Sabo E, Yannai S. Indole-3-carbinol (I3C) exhibits inhibitory and preventive effects on prostate tumors in mice. Food Chem Toxicol. 2008;46(3):863–870. doi: 10.1016/j.fct.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Bradlow HL, Zeligs MA. Diindolylmethane (DIM) spontaneously forms from indole-3-carbinol (I3C) during cell culture experiments. In Vivo (Athens, Greece) 2010;24(4):387–391. [PubMed] [Google Scholar]
- 22.Bradlow HL. Review. Indole-3-carbinol as a chemoprotective agent in breast and prostate cancer. In Vivo (Athens, Greece) 2008;22(4):441–445. [PubMed] [Google Scholar]
- 23.Cho HJ, Park SY, Kim EJ, Kim JK, Park JH. 3,3′-diindolylmethane inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate model. Mol Carcinog. 2011;50(2):100–112. doi: 10.1002/mc.20698. [DOI] [PubMed] [Google Scholar]
- 24.Gasper AV, Al-Janobi A, Smith JA, Bacon JR, Fortun P, Atherton C, et al. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am J Clin Nutr. 2005;82(6):1283–1291. doi: 10.1093/ajcn/82.6.1283. [DOI] [PubMed] [Google Scholar]
- 25.Heath EI, Heilbrun LK, Li J, Vaishampayan U, Harper F, Pemberton P, et al. A phase I dose-escalation study of oral BR-DIM (BioResponse 3,3′-diindolylmethane) in castrate-resistant, non-metastatic prostate cancer. Am J Trans Res. 2010;2(4):402–411. [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks JD, Paton VG, Vidanes G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol Biomarkers Prev. 2001;10(9):949–954. [PubMed] [Google Scholar]
- 27.Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of phase II detoxication enzymes. Food Chem Toxicol. 1999;37(9-10):973–979. doi: 10.1016/S0278-6915(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 28.Guerrero-Beltran CE, Calderon-Oliver M, Pedraza-Chaverri J, Chirino YI. Protective effect of sulforaphane against oxidative stress: recent advances. Exp Toxicol Pathol. 2012;64(5):503–508. doi: 10.1016/j.etp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Wang TT, Schoene NW, Milner JA, Kim YS. Broccoli-derived phytochemicals indole-3-carbinol and 3,3′-diindolylmethane exerts concentration-dependent pleiotropic effects on prostate cancer cells: comparison with other cancer preventive phytochemicals. Mol Carcinog. 2012;51(3):244–256. doi: 10.1002/mc.20774. [DOI] [PubMed] [Google Scholar]
- 30.Ernst IM, Schuemann C, Wagner AE, Rimbach G. 3,3′-diindolylmethane but not indole-3-carbinol activates Nrf2 and induces Nrf2 target gene expression in cultured murine fibroblasts. Free Radic Res. 2011;45(8):941–949. doi: 10.3109/10715762.2011.571683. [DOI] [PubMed] [Google Scholar]
- 31.Saw CL, Cintron M, Wu TY, Guo Y, Huang Y, Jeong WS, et al. Pharmacodynamics of dietary phytochemical indoles I3C and DIM: induction of Nrf2-mediated phase II drug metabolizing and antioxidant genes and synergism with isothiocyanates. Biopharm Drug Dispos. 2011;32(5):289–300. doi: 10.1002/bdd.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed GA, Peterson KS, Smith HJ, Gray JC, Sullivan DK, Mayo MS, et al. A phase I study of indole-3-carbinol in women: tolerability and effects. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1953–1960. doi: 10.1158/1055-9965.EPI-05-0121. [DOI] [PubMed] [Google Scholar]
- 33.Dalessandri KM, Firestone GL, Fitch MD, Bradlow HL, Bjeldanes LF. Pilot study: effect of 3,3′-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr Cancer. 2004;50(2):161–167. doi: 10.1207/s15327914nc5002_5. [DOI] [PubMed] [Google Scholar]
- 34.Michnovicz JJ, Adlercreutz H, Bradlow HL. Changes in levels of urinary estrogen metabolites after oral indole-3-carbinol treatment in humans. J Natl Cancer Inst. 1997;89(10):718–723. doi: 10.1093/jnci/89.10.718. [DOI] [PubMed] [Google Scholar]
- 35.Michnovicz JJ, Bradlow HL. Altered estrogen metabolism and excretion in humans following consumption of indole-3-carbinol. Nutr Cancer. 1991;16(1):59–66. doi: 10.1080/01635589109514141. [DOI] [PubMed] [Google Scholar]
- 36.Fowke JH, Longcope C, Hebert JR. Brassica vegetable consumption shifts estrogen metabolism in healthy postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2000;9(8):773–779. [PubMed] [Google Scholar]
- 37.Castagnetta LA, Miceli MD, Sorci CM, Pfeffer U, Farruggio R, Oliveri G, et al. Growth of LNCaP human prostate cancer cells is stimulated by estradiol via its own receptor. Endocrinology. 1995;136(5):2309–2319. doi: 10.1210/en.136.5.2309. [DOI] [PubMed] [Google Scholar]
- 38.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268–273. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24(50):7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 40.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15(15):4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 41.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Investig. 2008;118(9):3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009;9(2):237–249. doi: 10.2174/156800909787580999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiczk A, Hofman D, Konopa G, Herman-Antosiewicz A. Sulforaphane, a cruciferous vegetable-derived isothiocyanate, inhibits protein synthesis in human prostate cancer cells. Biochim Biophys Acta. 2012;1823(8):1295–1305. doi: 10.1016/j.bbamcr.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 44.Martelli AM, Tabellini G, Bressanin D, Ognibene A, Goto K, Cocco L, et al. The emerging multiple roles of nuclear Akt. Biochim Biophys Acta. 2012;1823(12):2168–2178. doi: 10.1016/j.bbamcr.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24(28):4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 47.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11(6):701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 48.Xu C, Shen G, Yuan X, Kim JH, Gopalkrishnan A, Keum YS, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27(3):437–445. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 49.Choi S, Lew KL, Xiao H, Herman-Antosiewicz A, Xiao D, Brown CK, et al. d,l-sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis. 2007;28(1):151–162. doi: 10.1093/carcin/bgl144. [DOI] [PubMed] [Google Scholar]
- 50.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22(11):1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi S, Singh SV. Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent. Cancer Res. 2005;65(5):2035–2043. doi: 10.1158/0008-5472.CAN-04-3616. [DOI] [PubMed] [Google Scholar]
- 52.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94(19):10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188(1):211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McEleny K, Coffey R, Morrissey C, Williamson K, Zangemeister-Wittke U, Fitzpatrick JM, et al. An antisense oligonucleotide to cIAP-1 sensitizes prostate cancer cells to fas and TNFalpha mediated apoptosis. Prostate. 2004;59(4):419–425. doi: 10.1002/pros.10371. [DOI] [PubMed] [Google Scholar]
- 55.Xiao D, Powolny AA, Antosiewicz J, Hahm ER, Bommareddy A, Zeng Y, et al. Cellular responses to cancer chemopreventive agent d,l-sulforaphane in human prostate cancer cells are initiated by mitochondrial reactive oxygen species. Pharm Res. 2009;26(7):1729–1738. doi: 10.1007/s11095-009-9883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. 2004;25(1):83–90. doi: 10.1093/carcin/bgg178. [DOI] [PubMed] [Google Scholar]
- 57.Singh SV, Warin R, Xiao D, Powolny AA, Stan SD, Arlotti JA, et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009;69(5):2117–2125. doi: 10.1158/0008-5472.CAN-08-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008;269(2):291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 2007;64(9):1105–1127. doi: 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20(23):2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 61.Nachshon-Kedmi M, Yannai S, Haj A, Fares FA. Indole-3-carbinol and 3,3′-diindolylmethane induce apoptosis in human prostate cancer cells. Food Chem Toxicol. 2003;41(6):745–752. doi: 10.1016/S0278-6915(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 62.Vivar OI, Lin CL, Firestone GL, Bjeldanes LF. 3,3′-diindolylmethane induces a G(1) arrest in human prostate cancer cells irrespective of androgen receptor and p53 status. Biochem Pharmacol. 2009;78(5):469–476. doi: 10.1016/j.bcp.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nachshon-Kedmi M, Yannai S, Fares FA. Induction of apoptosis in human prostate cancer cell line, PC3, by 3,3′-diindolylmethane through the mitochondrial pathway. Br J Cancer. 2004;91(7):1358–1363. doi: 10.1038/sj.bjc.6602145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garikapaty VP, Ashok BT, Tadi K, Mittelman A, Tiwari RK. 3,3′-diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem Biophys Res Commun. 2006;340(2):718–725. doi: 10.1016/j.bbrc.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 65.Bhuiyan MM, Li Y, Banerjee S, Ahmed F, Wang Z, Ali S, et al. Down-regulation of androgen receptor by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells. Cancer Res. 2006;66(20):10064–10072. doi: 10.1158/0008-5472.CAN-06-2011. [DOI] [PubMed] [Google Scholar]
- 66.Kong D, Banerjee S, Huang W, Li Y, Wang Z, Kim HR, et al. Mammalian target of rapamycin repression by 3,3′-diindolylmethane inhibits invasion and angiogenesis in platelet-derived growth factor-d-overexpressing PC3 cells. Cancer Res. 2008;68(6):1927–1934. doi: 10.1158/0008-5472.CAN-07-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong D, Li Y, Wang Z, Banerjee S, Sarkar FH. Inhibition of angiogenesis and invasion by 3,3′-diindolylmethane is mediated by the nuclear factor-kappaB downstream target genes MMP-9 and uPA that regulated bioavailability of vascular endothelial growth factor in prostate cancer. Cancer Res. 2007;67(7):3310–3319. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
- 68.Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by AKT and NF-kappaB pathways in prostate cancer cells. Front Biosci. 2005;10:236–243. doi: 10.2741/1523. [DOI] [PubMed] [Google Scholar]
- 69.Chen D, Banerjee S, Cui QC, Kong D, Sarkar FH, Dou QP. Activation of AMP-activated protein kinase by 3,3′-diindolylmethane (DIM) is associated with human prostate cancer cell death in vitro and in vivo. PLoS One. 2012;7(10):e47186. doi: 10.1371/journal.pone.0047186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le HT, Schaldach CM, Firestone GL, Bjeldanes LF. Plant-derived 3,3′-diindolylmethane is a strong androgen antagonist in human prostate cancer cells. J Biol Chem. 2003;278(23):21136–21145. doi: 10.1074/jbc.M300588200. [DOI] [PubMed] [Google Scholar]
- 71.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3(2):166–179. [PMC free article] [PubMed] [Google Scholar]
- 74.Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27(2):344–349. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98(3):604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gibbs A, Schwartzman J, Deng V, Alumkal J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc Natl Acad Sci U S A. 2009;106(39):16663–16668. doi: 10.1073/pnas.0908908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, et al. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28(14):1714–1724. doi: 10.1038/onc.2009.19. [DOI] [PubMed] [Google Scholar]
- 78.Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27(4):811–819. doi: 10.1093/carcin/bgi265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood, NJ) 2007;232(2):227–234. [PMC free article] [PubMed] [Google Scholar]
- 80.Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res. 2011;55(7):999–1009. doi: 10.1002/mnfr.201000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin Epigenetics. 2011;3:3. doi: 10.1186/1868-7083-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, et al. Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin Cancer Res. 2003;9(13):4730–4734. [PubMed] [Google Scholar]
- 83.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5(7):e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 85.Azarenko O, Okouneva T, Singletary KW, Jordan MA, Wilson L. Suppression of microtubule dynamic instability and turnover in MCF7 breast cancer cells by sulforaphane. Carcinogenesis. 2008;29(12):2360–2368. doi: 10.1093/carcin/bgn241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mi L, Xiao Z, Hood BL, Dakshanamurthy S, Wang X, Govind S, et al. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem. 2008;283(32):22136–22146. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiao Z, Mi L, Chung FL, Veenstra TD. Proteomic analysis of covalent modifications of tubulins by isothiocyanates. J Nutr. 2012;142(7):1377S–1381S. doi: 10.3945/jn.111.152041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shankar S, Ganapathy S, Srivastava RK. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin Cancer Res. 2008;14(21):6855–6866. doi: 10.1158/1078-0432.CCR-08-0903. [DOI] [PubMed] [Google Scholar]
- 89.Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, et al. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol (Baltimore, Md.) 2009;23(12):1963–1972. doi: 10.1210/me.2009-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3′-diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicol Appl Pharmacol. 2012;263(3):345–351. doi: 10.1016/j.taap.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70(2):646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012;7(3):e33729. doi: 10.1371/journal.pone.0033729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 94.van Leenders GJ, Dukers D, Hessels D, van den Kieboom SW, Hulsbergen CA, Witjes JA, et al. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52(2):455–463. doi: 10.1016/j.eururo.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 95.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, et al. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am J Transl Res. 2012;4(1):14–23. [PMC free article] [PubMed] [Google Scholar]
- 96.Fimognari C, Hrelia P. Sulforaphane as a promising molecule for fighting cancer. Mutat Res. 2007;635(2-3):90–104. doi: 10.1016/j.mrrev.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 97.Weng JR, Tsai CH, Kulp SK, Chen CS. Indole-3-carbinol as a chemopreventive and anti-cancer agent. Cancer Lett. 2008;262(2):153–163. doi: 10.1016/j.canlet.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One. 2010;5(1):e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (New York, NY) 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 100.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29(3):255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]