

Abstract
Human ether-á-go-go (eag)-related gene (hERG) potassium channels play a critical role in cardiac repolarization and are characterized by unusually slow closing (deactivation) kinetics. The N-terminal “eag” domain and a C-terminal C-linker/cyclic nucleotide–binding homology domain (CNBHD) are required for regulation of slow deactivation. The region between the S4 and S5 transmembrane domains (S4–S5 linker) is also implicated in this process, but the mechanism for regulation of slow deactivation is unclear. Here, using an eag domain–deleted channel (hERG Δeag) fused to Citrine fluorescent protein, we found that most channels bearing individual alanine mutations in the S4–S5 linker were directly regulated by recombinant eag domains fused to a cyan fluorescent protein (N-eag-CFP) and had robust Förster resonance energy transfer (FRET). Additionally, a channel bearing a group of eight alanine residues in the S4–S5 linker was not measurably regulated by N-eag-CFP domains, but robust FRET was measured. These findings demonstrate that the eag domain associated with all of the S4–S5 linker mutant channels. In contrast, channels that also lacked the CNBHD (hERG Δeag ΔCNBHD-Citrine) were not measurably regulated by N-eag-CFP nor was FRET detected, suggesting that the C-linker/CNBHD was required for eag domains to directly associate with the channel. In a FRET hybridization assay, N-eag-CFP had robust FRET with a C-linker/CNBHD-Citrine, suggesting a direct and specific interaction between the eag domain and the C-linker/CNBHD. Lastly, coexpression of a hERG subunit lacking the CNBHD and the distal C-terminal region (hERG ΔpCT-Citrine) with hERG Δeag-CFP subunits had FRET and partial restoration of slow deactivation. Collectively, these findings reveal that the C-linker/CNBHD, but not the S4–S5 linker, was necessary for the eag domain to associate with the channel, that the eag domain and the C-linker/CNBHD were sufficient for a direct interaction, and that an intersubunit interaction between the eag domain and the C-linker/CNBHD regulated slow deactivation in hERG channels at the plasma membrane.
INTRODUCTION
The human ether-á-go-go (eag)-related gene (hERG) voltage-gated potassium channel is the primary pore-forming subunit of the rapidly activating delayed-rectifier potassium current (IKr) in the heart (Warmke and Ganetzky, 1994; Sanguinetti et al., 1995; Trudeau et al., 1995). The physiological role of cardiac IKr is to repolarize myocytes during the terminal phase of cardiac action potentials (Sanguinetti and Jurkiewicz, 1990, 1991). Loss of function in hERG, either caused by inheritable mutations or pharmacological block, causes long QT syndrome, which can develop into ventricular arrhythmias and sudden cardiac death (Curran et al., 1995; Sanguinetti et al., 1995).
hERG is a member of the KCNH family of voltage-activated potassium (K+) channels, which are closely related to CNG and hyperpolarization-activated, cyclic nucleotide–modulated (HCN) channels (Guy et al., 1991; Warmke and Ganetzky, 1994). Like other voltage-gated potassium channels, KCNH channels are tetrameric, and each subunit has six transmembrane domains, a pore-loop domain, and intracellular amino and carboxy termini (Warmke and Ganetzky, 1994). However, unlike other voltage-activated K+ channels, KCNH channels have an eag domain in the N-terminal region and a C-terminal domain that contains a C-linker and cyclic nucleotide–binding homology domain (CNBHD), which shares structural homology with the C-linker and CNBHDs from HCN2 channels (Warmke et al., 1991; Warmke and Ganetzky, 1994; Morais Cabral et al., 1998). The N-terminal region has a conserved eag domain (residues 1–135), which contains a Per-Arnt-Sim (PAS) domain (26–135) that is preceded by a shorter PAS-CAP region (1–25) (Morais Cabral et al., 1998; Li et al., 2010; Muskett et al., 2011; Ng et al., 2011).
hERG channels have unique gating properties, in particular, fast inactivation relative to activation and unusually slow closing (deactivation) kinetics, compared with the other voltage-gated potassium channels (Trudeau et al., 1995). Previous work revealed that the N-terminal eag domain is a key regulator of the characteristic slow deactivation gating in hERG channels. Channels with deletions of the eag domain exhibit deactivation kinetics that are approximately fivefold faster than that of WT hERG channels (Spector et al., 1996; Morais Cabral et al., 1998; Wang et al., 1998, 2000; Gustina and Trudeau, 2009; Fernández-Trillo et al., 2011). Additionally, channels with mutations (including long QT syndrome–associated mutations) in the eag domain have significantly accelerated deactivation kinetics (Morais Cabral et al., 1998; Chen et al., 1999; Gustina and Trudeau, 2009; Gianulis and Trudeau, 2011). Expression of the eag domain as either a purified peptide or an isolated gene fragment restored slow deactivation kinetics to hERG channels bearing deletions of the eag domain or point mutations in the eag domain, demonstrating a direct regulatory role of the eag domain in channel deactivation (Morais Cabral et al., 1998; Gustina and Trudeau, 2009, 2011, 2013; Fernández-Trillo et al., 2011; Gianulis and Trudeau, 2011). Förster resonance energy transfer (FRET) revealed that isolated eag domains were in close proximity to N-truncated hERG channels at the plasma membrane, suggesting a direct interaction of the eag domain with the rest of the channel (Gustina and Trudeau, 2009).
The mechanism by which the eag domain regulates hERG channel deactivation remains poorly understood. Although functional and optical results strongly suggest that the eag domain makes a direct interaction with another region of the channel to regulate gating (Morais Cabral et al., 1998; Gustina and Trudeau, 2009), the identity of potential interacting partners is somewhat unclear. One potential candidate for an interaction is the S4–S5 linker. Point mutations in the S4–S5 linker produced channels with accelerated deactivation kinetics, similar to those seen in N-truncated hERG channels (Sanguinetti and Xu, 1999; Alonso-Ron et al., 2008; Ng et al., 2012). Mutagenesis studies of the S4–S5 linker suggested that it may act as a “signal integrator” and mediate the effects of eag domain binding to the rest of the hERG channel (Ng et al., 2012), whereas other work proposed a more direct eag domain interaction with the S4–S5 linker (Wang et al., 1998; Li et al., 2010; de la Peña et al., 2011; Fernández-Trillo et al., 2011; Ng et al., 2012). Intriguingly, because a channel with a mutation at one site (Y542) in the S4–S5 linker completely disrupted FRET with the eag domain, it was proposed that a part of the S4–S5 linker domain interacted with the eag domain (Fernández-Trillo et al., 2011).
Another putative site for an interaction is the C-terminal C-linker/CNBHD, a domain whose function is unclear in KCNH channels. Instead of binding to cyclic nucleotides, the CNBHD in KCNH channels is bound by an intrinsic ligand formed from the β9 helix in the CNBHD itself (Brelidze et al., 2012; Marques-Carvalho et al., 2012). The CNBHD in hERG has been proposed to be involved in channel deactivation (Al-Owais et al., 2009; Gustina and Trudeau, 2011; Muskett et al., 2011). Point mutations in the CNBHD partially sped up channel deactivation, and channels with a deletion of the CNBHD had fast deactivation kinetics that were similar to those seen in N-truncated hERG channels, suggesting a shared mechanism (Al-Owais et al., 2009; Gustina and Trudeau, 2011; Muskett et al., 2011). Previously, an interaction between the eag domain and the C-linker/CNBHD was inferred from electrophysiology studies of channels with point mutations and deletions (Al-Owais et al., 2009; Gustina and Trudeau, 2011; Muskett et al., 2011) and detected with in vitro biochemical pulldown interactions between eag domains and C-linker/CNBHDs (Gustina and Trudeau, 2011).
Here, our goal was to distinguish the contributions of the S4–S5 linker and the C-linker/CNBHD in channel deactivation gating and as putative determinants of a global interaction with the eag domain. To carry out these experiments, we generated channels tagged with fluorescent proteins and bearing deletions of the eag domain plus either additional mutations in the S4–S5 linker or deletion of the CNBHD, and used a combination of electrophysiology to measure function and FRET spectroscopy to measure structural interactions in functional channels at the cell surface. We found that mutation of individual S4–S5 linker residues to alanine caused significant alterations in channel deactivation kinetics; however, FRET measurements showed that the eag domain remained associated with these channels. Furthermore, we showed that complete replacement of the S4–S5 linker residues with alanines severely altered hERG channel gating but did not diminish FRET, suggesting that the eag domain was still associated with this channel. Instead, we found that the CNBHD was necessary for eag domains to regulate gating and for eag domains to associate with the channel. Using a FRET-based hybridization assay (Erickson et al., 2003), we found that isolated eag domains and isolated C-linker/CNBHDs were sufficient for a direct interaction in a mammalian cellular environment. The findings from this study reveal that the CNBHD was required for the eag domain interaction with the channel, and that the C-linker/CNBHD was sufficient for a direct interaction with the eag domain, suggesting that this interaction was necessary for eag domain–dependent regulation of gating in functional channels at the plasma membrane.
MATERIALS AND METHODS
Molecular biology and cell culture
The hERG S4–S5 mutant constructs, including hERG Δeag[S4–S5]Alacomplete, were synthesized as gene fragments (Bio Basic, Inc.) and cloned into hERG-mCitrine.pcDNA3.1. The hERG deletion constructs (hERG Δeag, hERG ΔCNBHD, and hERG Δeag ΔCNBHD), as well as the N-eag-CFP gene fragment (encoding amino acids 1–135 fused to cyan fluorescent protein [CFP]), were created using overlap extension PCR with custom-made primers, as described previously (Gustina and Trudeau, 2009, 2011). The C-linker/CNBHD-CFP (666–872-CFP) gene fragment and the hERG ΔpCT-Citrine construct were synthesized in the pcDNA3.1 mammalian expression vector (BioInnovatise, Inc.). The C-linker/CNBHD-Citrine (666–872-Citrine) and C-linker/CNBHD-YFP (666–872-YFP) gene fragments, as well as the rCB1-YFP construct, were provided by W.N. Zagotta (University of Washington, Seattle, WA), and the YFP-calmodulin (CaM)1234 was provided by J. Adelman (Vollum Institute, Portland, OR) and S. Gordon (University of Washington). All constructs were subcloned into the pcDNA3.1 mammalian expression vector (Invitrogen). Each hERG channel construct, including WT hERG, contained the S620T point mutation, which removes channel inactivation without affecting eag domain regulation of gating and increases ionic current (Herzberg et al., 1998; Wang et al., 1998; Gustina and Trudeau, 2011), and was genetically fused in-frame to either monomeric Citrine (mCitrine) or monomeric enhanced CFP (mCFP) at its carboxy terminus. For clarity, mCFP and mCitrine are referred to as “CFP” and “Citrine,” respectively, in this paper.
Human embryonic kidney 293 (HEK293) cell culture and transfection were performed as described previously (Gianulis and Trudeau, 2011). Cells were transfected with the appropriate cDNA using the TransIT-LT1 transfection reagent (Mirus) according to the manufacturer’s protocol.
Electrophysiology and analysis
HEK293 cells were plated on 35-mm cell culture dishes and transiently transfected with 1 µg hERG channel cDNA plus 1 µg N-eag-CFP (or CFP) cDNA. After ∼24–48 h, cells were analyzed by whole-cell patch clamp using an EPC10 patch-clamp amplifier and PatchMaster Data Acquisition Software, version 2.0 (HEKA). Recordings were performed at room temperature (22 ± 2°C). All data were recorded with a sampling rate of 1 kHz unfiltered. Patch pipettes had resistances of 2–4 MΩ when filled with the internal pipette solution. The internal pipette solution contained (mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, and 10 HEPES, pH 7.2 with KOH. The external bath solution contained (mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, 5 tetraethylammonium, and 10 HEPES, pH 7.4 with NaOH. For recording hERG Δeag[S4–S5]Alacomplete, an external bath solution with an elevated concentration of K+ was used (mM: 131 NaCl, 10 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, 5 tetraethylammonium, and 10 HEPES, pH 7.4 with NaOH) and shown to increase the amplitude of hERG current (Sanguinetti et al., 1995; Zhou et al., 1998). Series resistance was compensated such that the voltage error was <5 mV. No leak subtraction was used.
Ionic currents were measured using standard voltage command protocols (see Figs. 2, 4, 5, and 8) with a holding potential of −80 mV. All recorded data were analyzed using the IgorPro Software (version 5.03; WaveMetrics). Current relaxation with repolarization from a depolarized potential (deactivation) was fit with a double-exponential function (y = A1e−t/τ1 + A2e−t/τ2), where t is time and τ is the time constant of deactivation. The I-V relationship was measured by plotting the current at the end of the test pulse normalized to the peak outward current for that cell versus voltage. The steady-state voltage dependence of activation (G-V) was measured by plotting the tail current amplitude versus the previous test pulse voltage and fit with a Boltzmann function: y = 1/(1 + exp[(V1/2 − V)/k]), where V1/2 is the half-maximal activation potential and k is the slope factor. All data are presented as mean ± SEM, and n represents the number of cells. Statistical analyses were performed using a one-way ANOVA with a Tukey’s post-hoc test for pairwise comparisons. P < 0.05 was considered statistically significant.
Figure 2.

N-eag-CFP regulates deactivation gating in most of the hERG [S4–S5]Alaind mutant channels. Representative current recordings from HEK293 cells expressing each hERG Δeag [S4–S5]Alaind (A) alone or (B) coexpressed with N-eag-CFP. The voltage command protocol used to record ionic currents is shown on the bottom; the inset represents the voltage command protocol used to record hERG Δeag D540A alone and with N-eag-CFP coexpression. (C and D) G-V relationships for each hERG Δeag [S4–S5]Alaind mutant channel alone and with N-eag-CFP coexpression. Plotted points were fit with a Boltzmann function to yield the V1/2 and k values (averaged data are given in Table S1). (C) The G-V relationships for each hERG Δeag [S4–S5]Alaind mutant channel, except for hERG Δeag D540A. (D) The G-V relationship for hERG Δeag D540A. Blue squares represent the G-V relationship for WT hERG in both C and D. n ≥ 3 for each. (E and F) Box plots of the time constants of deactivation derived from a double-exponential fit to the tail current produced during the −50-mV pulse from 60 mV to yield the τfast values (E) and the τslow values (F). The middle line is the mean, the top and bottom lines are the 75th and 25th percentile, respectively, and the straight lines are the 90th and 10th percentiles. (G) Box plot of the time constants of deactivation at −100 mV for hERG Δeag D540A alone and with N-eag-CFP coexpression. Tail currents produced during the −100-mV pulse from 60 mV were fit with a double-exponential function to yield the τfast and τslow values. Blue squares represent the τfast and τslow for WT hERG in E–G. n ≥ 4 for each. All data are plotted as mean ± SEM. *, P < 0.05; **, P < 0.01 (ANOVA).
Figure 4.
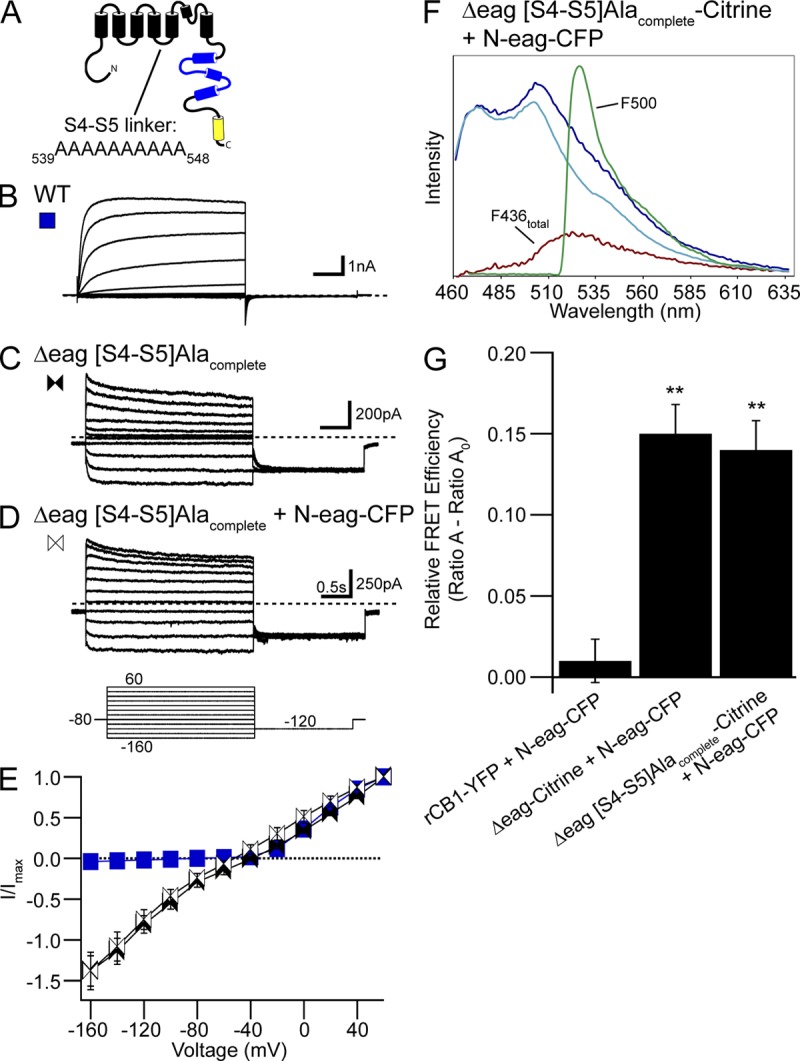
Replacement of the hERG S4–S5 linker with alanines disrupted eag domain regulation of gating but not interaction with the channel. (A) hERG channel schematic illustrating the hERG Δeag[S4–S5]Alacomplete mutant channel in which all the residues in the S4–S5 linker were replaced with alanines. Representative current recordings from cells expressing (B) WT hERG, (C) hERG Δeag[S4–S5]Alacomplete, or (D) hERG Δeag[S4–S5]Alacomplete + N-eag-CFP. The inset represents the voltage command protocol used to record the currents. (E) I-V relationships for WT hERG and hERG Δeag[S4–S5]Alacomplete with or without N-eag-CFP expression. Data are plotted as mean ± SEM. n ≥ 4 for each. (F) Representative emission spectra from cells expressing hERG Δeag[S4–S5]Alacomplete-Citrine + N-eag-CFP. The emission spectra are color-coded as follows: dark blue trace, total emission with 436-nm excitation; cyan trace, CFP emission with 436-nm excitation; red trace, subtracted spectrum (difference between the cyan and the dark blue traces), which contains Citrine emission with 436-nm excitation; green trace, Citrine emission with 500-nm excitation. (G) Histogram of Ratio A–Ratio A0 values. Data are presented as mean ± SEM and are given in Table S3. **, P < 0.01 versus rCB1-YFP + N-eag-CFP (ANOVA). n ≥ 10 for each.
Figure 5.

Regulation of slow deactivation by the eag domain requires the presence of the CNBHD in the hERG C-terminal region. Representative current recordings from HEK293 cells expressing WT hERG, hERG Δeag, hERG ΔCNBHD, or hERG Δeag ΔCNBHD in the absence (A) or presence (B) of N-eag-CFP domains. The inset represents the voltage command protocol used. (C and D) Box plots of the time constants of deactivation at −50 mV. Tail currents produced during the −50-mV pulse from 60 mV were fit with a double-exponential function to yield the τfast (C) and τslow (D) time constants of deactivation. The middle line represents the mean, the top and bottom lines represent the 75th and 25th percentiles, respectively, and the straight lines represent the 90th and 10th percentiles. **, P < 0.01 versus WT hERG (ANOVA). n ≥ 3 for each.
Figure 8.

hERG Δeag and hERG ΔpCT subunits form heterotetrameric channels. (A) Representative current recordings from cells expressing WT hERG, hERG Δeag, hERG ΔpCT, or hERG Δeag + hERG ΔpCT. The voltage command protocol used to elicit the ionic currents is shown. (B and C) Box plots of time constants of deactivation at −50 mV. Tail currents produced during the −50-mV pulse from 60 mV were fit with a double-exponential function to yield the τfast (B) and τslow (C) time constants of deactivation. **, P < 0.01 versus hERG Δeag alone and hERGΔpCT alone (ANOVA). n ≥ 5 for each. Representative emission spectra from cells expressing (D) hERG-CFP + hERG-Citrine or (E) hERG Δeag-CFP + hERG ΔpCT-Citrine. In both D and E, the emission spectra are color-coded as follows: dark blue trace, total emission with 436-nm excitation; cyan trace, CFP emission with 436-nm excitation; red trace, subtracted spectrum (difference between the cyan and the dark blue traces), which contains Citrine emission with 436-nm excitation; green trace, Citrine emission with 500-nm excitation. (F) Histogram of the Ratio A–Ratio A0 values. Data are plotted as mean ± SEM and are given in Table S3. **, P < 0.01 versus rCB1-YFP + N-eag-CFP (ANOVA). n ≥ 9 for each.
FRET
HEK293 cells were plated on 35-mm poly-d-lysine–coated glass-bottom dishes (MatTek Corporation) and transiently transfected with Citrine-tagged hERG cDNA constructs plus N-eag-CFP to achieve a ratio of donor to acceptor fluorophores (Fc/Fy; see below) of ∼1. rCB1-YFP plus N-eag-CFP were cotransfected and used as a negative control, as in previous work (Gustina and Trudeau, 2009). The donor used in these experiments was mCFP, and the acceptor was Citrine (Heikal et al., 2000; Griesbeck et al., 2001). Approximately 24–48 h after transfection, FRET measurements were performed using an inverted epifluorescence microscope (TE2000-U; Eclipse; Nikon) and a 60× oil-immersion objective (NA 1.45; Nikon). The excitation light was generated using a 120-W lamp (X-Cite 120; Lumen Dynamics), and the duration of light was controlled by a mechanical shutter driver (VMM-D3; Uniblitz). Fluorescence emission and spectroscopic measurements were performed using a spectrograph (SpectraPro 2150i; Acton Research Corporation) and a camera (CCD97; Roper Scientific). Fluorescence imaging and analysis were performed with Metamorph software (version 6.3r7; Universal Imaging). For both cell and spectroscopic imaging, two filter cubes (Chroma Technology Corp.) were used (excitation, dichroic, emission): a YFP cube (HQ500/20, Q515lp, HQ520lp) and a CFP cube (D436/20, 455dclp, D460lp). Two spectroscopic images were obtained from each cell: one using the CFP cube and the other using the YFP cube. From these, the total emission spectrum and the Citrine emission spectrum were measured, respectively. Emission spectra from cells transfected with donor only, or with acceptor only, were also measured. Emission spectra were obtained from the edge of each cell by positioning the spectrograph input slit over a region corresponding to the plasma membrane. Therefore, the same slit position applies to the spectra taken with both CFP cube and the Citrine cube, thus preserving the spectral and positional information. Spectra were corrected for background light, which was determined from a blank area of the image.
Calculation of relative FRET efficiency
To measure FRET, we used a spectral separation approach, termed “spectra FRET” (Selvin, 1995; Zheng et al., 2002; Takanishi et al., 2006; Cheng et al., 2007; Gustina and Trudeau, 2009). Spectra FRET offers several advantages. First, it corrects for overlaps between the donor and acceptor emission spectra, referred to as “bleed-through.” Second, it corrects for cross-talk, which is the direct excitation of the acceptor by the donor-specific excitation wavelength. Finally, it eliminates errors from variability in quantum yield of the acceptor, as well as variations in expression levels of the donor and acceptor molecules. To correct for bleed-through, a CFP spectrum was measured from cells expressing donor only. This CFP spectrum was subtracted from the total emission spectrum recorded with excitation of the CFP cube from cells expressing both donor and acceptor; this yielded a subtracted Citrine spectrum (F436total) free of donor contamination. F436total contained two components: one caused by direct excitation of Citrine at 436 nm (F436direct) and one caused by FRET (F436FRET). The F436total spectrum was normalized to the Citrine emission spectrum with excitation at 500 nm (F500), termed “Ratio A” (Eq. 1):
| (1) |
To solve for F436FRET, the ratio of F500direct to F436, termed “Ratio A0” (Eq. 2), was calculated from cells expressing only acceptor. This ratio represents the degree of excitation of the acceptor fluorophore at 436 nm relative to the peak acceptor excitation at 500 nm, thus accounting for cross-talk.
| (2) |
The calculated difference between Ratio A and Ratio A0 (Eq. 3) yields F436FRET/F500 and is a value directly proportional to FRET efficiency (the relative FRET efficiency):
| (3) |
Correction of Fc
In these experiments, some of the observed CFP fluorescence intensities were reduced because of the transfer of energy to Citrine as a result of FRET. This FRET-associated CFP signal reduction was corrected using a method as described previously (Erickson et al., 2001; Zheng and Zagotta, 2004; Gustina and Trudeau, 2009). First, the FRET ratio (FR) was calculated as:
| (4) |
From the FRET ratio, the effective FRET efficiency (Eeff) was calculated as:
| (5) |
where εCitrine436 and εCFP436 are the molar extinction coefficients for Citrine and CFP, respectively. The true CFP emission (Fc) could then be calculated as:
| (6) |
Using the FC values, the ratio of CFP to Citrine fluorescence intensities (Fc/Fy) was calculated and reported in Table S3.
Online supplemental material
We measured properties of hERG S620T channels with individual mutations in the S4–S5 linker domain (Fig. S1). We showed a representative family of currents from (A) hERG S620T, (B) hERG S620T Δeag, and (C) hERG S620T Δeag + N-eag-CFP channels, each bearing a D540A mutant in the S4–S5 linker domain (Fig. S2), with a tail current at −50 mV. Data with −50-mV tails was used to generate G-V curves in Fig. 2 D and Fig. S1 C. We characterized deactivation in hERG S620T Δeag channels bearing selected S4–S5 mutants in detail by recording a family of tail currents (Fig. S3). The online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201310995/DC1.
RESULTS
Individual S4–S5 alanine mutations produce alterations in channel gating
The sequence of the S4–S5 linker of hERG consists of amino acids 539–548 (see Fig. 1). To examine the role of the S4–S5 linker in channel gating, we first performed alanine-scanning mutagenesis in which each individual residue was replaced with an alanine (hERG [S4–S5]Alaind). The two existing alanines in the S4–S5 linker were left unchanged. Each hERG [S4–S5]Alaind mutant channel contained a serine to threonine mutation at amino acid 620 (S620T), which removes channel inactivation and increases ionic current, to more directly measure channel deactivation (Herzberg et al., 1998; Wang et al., 1998; Gustina and Trudeau, 2011). We expressed each hERG [S4–S5]Alaind mutant channel in HEK293 cells and measured ionic currents using whole-cell patch-clamp analysis (Fig. S1). From a holding potential of −80 mV, channels were activated by a series of test potentials ranging from −80 to 60 mV in 10-mV increments, followed by a repolarizing pulse to −50 or −100 mV (for hERG D540A; Fig. S1 A, bottom, inset) to produce a tail current. Additionally, an alternate protocol was used to generate the voltage dependence of activation curve for hERG D540A in which the initial test pulse from the holding potential ranged from −160 to 60 mV in 20-mV increments, followed by a shorter pulse to −50 mV to produce a tail current (Fig. S2 A). We found that all of the hERG [S4–S5]Alaind mutant channels produced robust currents (with the exception of hERG G546A from which we were not able to record any measurable current). We observed significant hyperpolarizing shifts in the steady-state voltage dependence of activation (G-V) curves (Fig. S1, B and C, and Table S1) for hERG D540A, R541A, S543A, and Y545A (P < 0.01 for hERG D540A and S543A; P < 0.05 for hERG R541A and Y545A). hERG D540A had the largest shift (−40 mV) and a much shallower G-V curve compared with WT hERG channels, with a slope factor (k) that was significantly larger than that for WT hERG (P < 0.01). We detected no significant change in the G-V relationships for the remaining hERG [S4–S5]Alaind mutant channels (L539A, Y542A, and E544A; P > 0.05).
Figure 1.
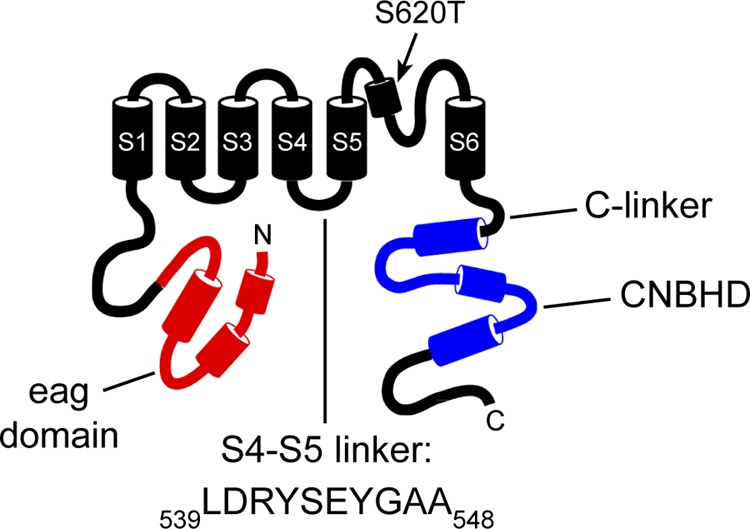
Schematic of the hERG potassium channel highlighting the S4–S5 linker. The eag domain is shown in red, and the CNBHD is shown in blue. The point mutation, S620T, is indicated. The intracellular loop between the S4 and S5 transmembrane domains, referred to as the “S4–S5 linker,” consists of ∼10 amino acids beginning with L539 and ending with A548.
To determine the effect of the individual alanine mutations on deactivation kinetics, we fit the peak tail current with a double-exponential function to yield a fast (τfast) and a slow (τslow) time constant of deactivation (Fig. S1, D–F). We found that each of the hERG [S4–S5]Alaind mutant channels exhibited significant alterations in the kinetics of deactivation compared with WT hERG (P < 0.01). Each hERG [S4–S5]Alaind mutant channel had faster kinetics of deactivation than those of WT hERG, as measured by a marked reduction in both τfast and τslow. In contrast, hERG S543A had significantly slower kinetics of deactivation as measured by an increase in both τfast and τslow. In summary, replacement of each residue in the S4–S5 linker with alanine altered the kinetics of slow deactivation and shifted the G-V relationships in some of the mutant channels, albeit with varying levels of severity. These data support the notion that the S4–S5 linker is a key determinant in hERG channel gating.
N-eag-CFP domain regulates deactivation gating in most of the hERG Δeag[S4–S5]Alaind mutant channels
To determine whether the S4–S5 linker played a role in the mechanism of eag domain–dependent regulation of channel gating, we investigated each of the S4–S5 linker alanine mutations in hERG channels lacking the eag domain (hERG Δeag[S4–S5]Alaind) alone (Fig. 2 A) and after coexpression with the N-eag-CFP domain (Fig. 2 B). As positive controls, we measured currents from hERG Δeag channels, which had significantly faster deactivation kinetics than WT hERG channels, and from hERG Δeag channels coexpressed with a genetically encoded eag domain fused to CFP (N-eag-CFP), which restored slow deactivation to values that were similar to those in WT hERG channels (Fig. 2, A, B, E, and F). We found that all of the hERG Δeag[S4–S5]Alaind channels, except for hERG Δeag G546A, produced robust currents (Fig. 2 A), and each of the hERG Δeag[S4–S5]Alaind mutant channels exhibited deactivation kinetics that were more rapid than those in the corresponding hERG [S4–S5]Alaind channels (see Fig. S1) and similar to that in hERG Δeag channels (Fig. 2, A and E–G). Coexpression with N-eag-CFP significantly slowed deactivation kinetics in most of the channels, including hERG Δeag L539A, hERG Δeag D540A, hERG Δeag R541A, hERG Δeag S543A, and hERG Δeag Y545A, but it did not significantly change the deactivation kinetics for two of the hERG Δeag[S4–S5]Alaind mutant channels, hERG Δeag Y542A and hERG Δeag E544A (Fig. 2, B and E–G). We observed significant hyperpolarizing shifts in the G-V relationships for hERG Δeag D540A and hERG Δeag S543A compared with hERG Δeag channels (P < 0.01; Fig. 2, C and D, and Table S1), which were not measurably different after coexpression with N-eag-CFP. Additionally, similar to full-length hERG D540A, hERG Δeag D540A had a shallower G-V curve than both WT hERG and hERG Δeag channels, with significantly larger k values (P < 0.01). There was no significant change in the G-V relationships for any of the other hERG Δeag[S4–S5]Alaind mutant channels (L539A, R541A, Y542A, E544A, and Y545A) both in the absence or presence of N-eag-CFP (P > 0.01). These data show that most of the hERG Δeag[S4–S5]Alaind mutant channels were regulated by N-eag-CFP domains, suggesting that the eag domain made an interaction with these channels. In contrast, the eag domain did not measurably slow deactivation seen in the hERG Δeag Y542 or hERG Δeag E544A channels, suggesting that these residues might play a role in eag domain–dependent deactivation.
To further examine the effect of individual S4–S5 linker alanine mutations on channel deactivation, and test whether N-eag-CFP could induce slow deactivation in the hERG Δeag[S4–S5]Alaind mutant channels at different voltages, we measured kinetics of deactivation over a range of potentials using a two-pulse voltage command protocol (Fig. S3 A). First, channels were activated by a step to 20 mV, followed by a series of repolarizing pulses from −120 to −40 mV in 20-mV increments to elicit a family of tail currents. Each tail current was fit with a double-exponential function to yield the τfast and τslow time constants of deactivation (Fig. S3 and Table S2). As a positive control, hERG Δeag channels exhibited accelerated deactivation kinetics in both the τfast and the τslow values, and coexpression with N-eag-CFP significantly slowed the deactivation kinetics in hERG Δeag channels at all potentials tested to values that were closer to those for WT hERG (Fig. S3, A, B, and F, and Table S2). We found that each hERG Δeag[S4–S5]Alaind mutant channel exhibited faster deactivation kinetics than WT hERG channels at all voltages tested (Fig. S3, A, C–E, and G–I and Table S2). Consistent with the results shown in Fig. 2, the hERG Δeag[S4–S5]Alaind mutant channels were regulated by N-eag-CFP domains at all voltages tested here, with the exception of hERG Δeag Y542A and hERG Δeag E544A (Fig. S3, C and G, and Table S2).
Individual S4–S5 linker mutations do not disrupt N-eag-CFP association with the channel
Our findings that N-eag-CFP modulated gating for most of the hERG Δeag[S4–S5]Alaind mutant channels suggested that the eag domain likely associates with most of the mutant channels; however, the situation was less clear for hERG Δeag Y542A and hERG Δeag E544A channels, which were not measurably regulated by N-eag-CFP domains. To test for global eag domain association with the rest of the hERG channel directly, we used FRET spectroscopy, which is the transfer of light energy from a donor to an acceptor when the two are in close (1–10-nm) proximity (Stryer, 1978; Clegg, 1992; Selvin, 1995). HEK293 cells were transfected with one of the hERG Δeag[S4–S5]Alaind mutant channels tagged with a Citrine (which was the acceptor) plus N-eag-CFP (which was the donor). We also expressed hERG-Citrine with hERG-CFP as a positive control. As a negative control, cells were cotransfected with the cannabinoid-1 receptor tagged with a YFP (rCB1-YFP) and N-eag-CFP, as was done previously (Gustina and Trudeau, 2009). We used a spectral separation approach, “spectra FRET,” to measure FRET (described in Materials and methods). We took fluorescent images of the cells using an epifluorescence microscope with either Citrine (Fig. 3 A) or CFP (Fig. 3 B) excitation. The spectrographic input slit was then positioned along a portion of the cell that corresponded to the plasma membrane (Fig. 3, A and B, white rectangle). Two spectroscopic images were taken from the region within the slit, one with Citrine excitation (Fig. 3 C) and another with CFP excitation (Fig. 3 D). In each of the spectroscopic images, the y axis represents the cell position visible through the input slit, and the x axis represents wavelength. A horizontal line was then drawn across each spectroscopic image along a region where the fluorescent signal comes specifically from the plasma membrane (Fig. 3, C and D, red line), which yielded the emission spectrum (Fig. 3 E). Excitation of CFP yielded the total emission spectrum (Fig. 3, E–H, dark blue trace), which contained emission from both CFP and Citrine. From this, we subtracted CFP emission from cells expressing donor only (Fig. 3, E–H, cyan trace), which yielded the Citrine component (red trace, F436total). The F436total spectrum contains Citrine emission as a result of both direct excitation of Citrine (F436direct) and FRET (F436FRET). The F436total spectrum was normalized to the maximal Citrine emission spectrum with excitation at 500 nm (F500) to yield the Ratio A value. From this, we subtracted the Ratio A0 value, which was calculated from cells expressing acceptor only, to isolate F500FRET. This difference provides a measurement of the relative FRET efficiency, where values greater than zero indicate FRET (Fig. 3 I and Table S3).
Figure 3.
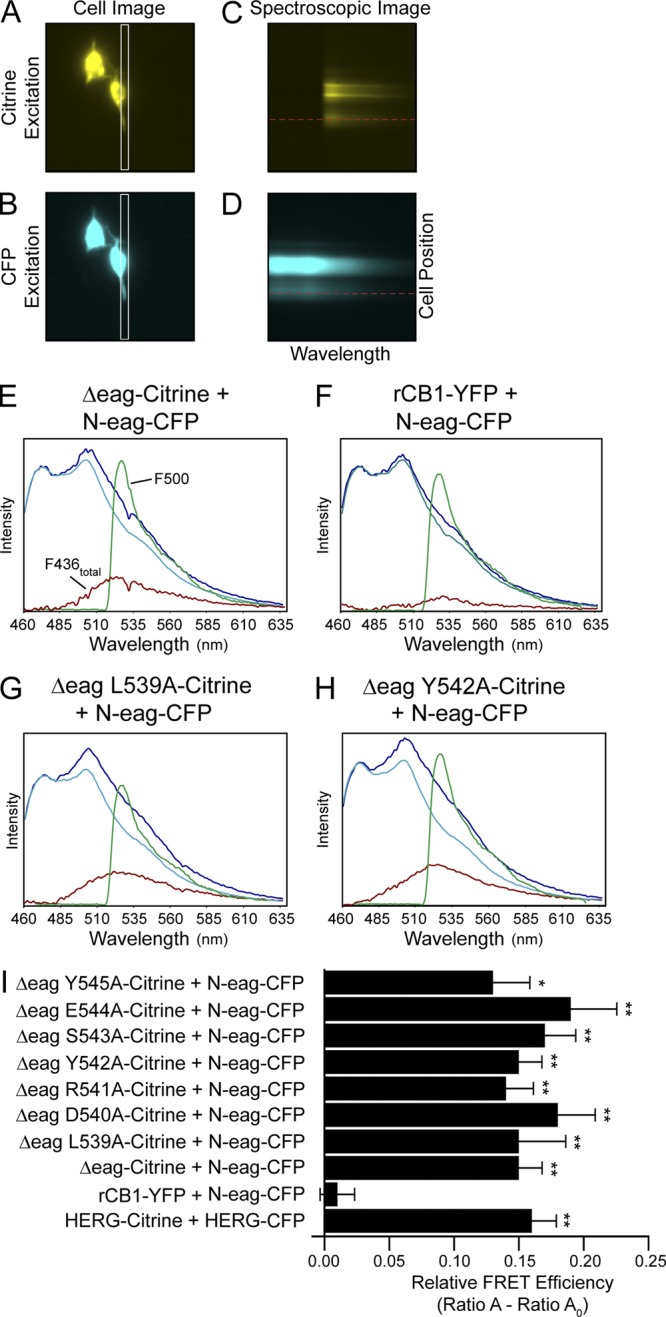
FRET spectroscopy shows that individual mutations in the hERG S4–S5 linker do not disrupt eag domain association with the channel. A single HEK293 cell expressing hERG Δeag-Citrine + N-eag-CFP was imaged with either (A) Citrine excitation at 500 nm or (B) CFP excitation at 436 nm. The spectrographic input slit (A and B, white rectangle) was positioned over a region of the cell that corresponded to the plasma membrane, and spectroscopic images were taken from the area within the slit with excitation of either Citrine (C) or CFP (D). In each spectroscopic image, the x axis represents the wavelength, and the y axis represents the position of the cell within the slit. A horizontal line drawn across each spectroscopic image (C and D, red line) indicates the region from which the emission spectra were measured, which are plotted in E. Representative emission spectra from HEK293 cells expressing (E) hERG Δeag-Citrine + N-eag-CFP, (F) rCB1-YFP + N-eag-CFP, (G) hERG Δeag L539A-Citrine + N-eag-CFP, or (H) hERG Δeag Y542A-Citrine + N-eag-CFP. The total emission spectrum from excitation at 436 nm is shown in dark blue. The extracted spectrum (red trace, F436total) is the CFP emission (cyan trace) subtracted from the total emission spectrum and contains the Citrine emission with excitation at 436 nm. The green trace is the Citrine emission with excitation at 500 nm (F500). Ratio A was determined as the ratio of the red trace (F436total) to the green trace (F500). As a control, cells expressing acceptor only (Citrine or YFP constructs) were excited at 436 nm (F436) and at 500 nm (F500), and Ratio A0 was calculated as the ratio of F436 emission to F500 emission. (I) Histogram of Ratio A–Ratio A0 values, a value that is directly proportional to the relative FRET efficiency (averaged data are also given in Table S3). Data are presented as mean ± SEM. *, P < 0.05 versus rCB1-YFP + N-eag-CFP; **, P < 0.01 versus rCB1-YFP + N-eag-CFP (ANOVA). n ≥ 6 for each.
We detected a robust FRET signal from positive controls, which were cells expressing hERG Δeag-Citrine + N-eag-CFP (P < 0.01 vs. rCB1-YFP + N-eag-CFP) or hERG-Citrine + hERG-CFP. In contrast, we did not detect a FRET signal from negative controls, which were cells expressing rCB1-YFP + N-eag-CFP (Fig. 3, E, F, and I, and Table S3). We calculated the donor to acceptor ratio (Fc/Fy) and found that the donor to acceptor fluorescence was similar in the negative control cells, indicating that the lack of FRET was not caused by a lack of donor fluorescence (Table S3). We next measured FRET between each of the hERG Δeag[S4–S5]Alaind mutant channels and N-eag-CFP and found that for each mutant channel, we observed a positive and significant FRET signal (P < 0.01 vs. rCB1-YFP + N-eag-CFP; P < 0.05 for hERG Δeag Y545A-Citrine) that was not measurably different from either of the two positive controls, hERG Δeag-Citrine + N-eag-CFP or hERG-Citrine + hERG-CFP (Fig. 3, G–I, and Table S3). These data indicate that individual alanine mutations introduced in the S4–S5 linker did not disrupt the global association of the eag domain with the rest of the channel.
Replacement of the S4–S5 linker with a series of alanine residues disrupts eag regulation of gating but not association with the channel
The finding that point mutations in the S4–S5 linker did not eliminate FRET between the eag domain and the channel could mean that individual mutations may not be sufficient to disrupt a global interaction of the eag domain with the rest of the channel. To address this, we created a hERG Δeag channel in which all the residues in the S4–S5 linker were mutated to alanine (hERG Δeag[S4–S5]Alacomplete; Fig. 4 A). We first measured functional expression of hERG Δeag[S4–S5]Alacomplete channels using a modified pulse protocol (Fig. 4, inset) in which cells were stepped from a holding potential of −80 mV to a series of potentials ranging from −160 to 60 mV in 20-mV increments, followed by a pulse to −120 mV to elicit a tail current. Additionally, recordings of hERG Δeag[S4–S5]Alacomplete were performed using an elevated (10-mM K+) bath solution to increase the amplitude of hERG current, as shown previously for WT hERG (Sanguinetti et al., 1995; Zhou et al., 1998). We found hERG Δeag[S4–S5]Alacomplete channels exhibited currents with unique properties (Fig. 4 C). Depolarization to potentials more positive than −40 mV elicited an outward current, whose I-V relationship was similar to that for WT hERG channels (Fig. 4, B, C, and E). However, at more hyperpolarized potentials (more negative than −40 mV), hERG Δeag[S4–S5]Alacomplete channels exhibited a large inward current that reached a steady-state amplitude by the end of the 3-s hyperpolarizing pulse, whereas WT hERG channels produced no current at potentials more negative than −40 mV (P < 0.01 vs. WT hERG; Fig. 4, B, C, and E). For WT hERG channels, a step to −120 mV from depolarized potentials (more positive than −40 mV) elicited an inward tail current that relaxed to zero current. In contrast, hERG Δeag[S4–S5]Alacomplete channels had an inward tail current that did not relax to zero, suggesting that channels remained open during the −120-mV pulse. We next asked whether N-eag-CFP could rescue the aberrant gating properties of hERG Δeag[S4–S5]Alacomplete channels. Coexpression with N-eag-CFP resulted in no measurable changes in the current properties of hERG Δeag[S4–S5]Alacomplete channels (Fig. 4, D and E). These data indicate that (a) replacement of the S4–S5 linker with alanine residues produced channels that were open at most test potentials; and (b) N-eag-CFP exhibited no measurable functional effect on hERG Δeag[S4–S5]Alacomplete channels, suggesting that the eag domain modulation of gating was completely impaired at the voltages we could test.
Based on the findings that N-eag-CFP had no measurable effect on hERG Δeag[S4–S5]Alacomplete channel properties, we next asked whether these channels interacted with the eag domain. To test this, cells were transfected with hERG Δeag[S4–S5]Alacomplete channels tagged with a Citrine (acceptor) plus N-eag-CFP tagged with a CFP (donor), and we measured the emission spectrum (shown in Fig. 4 F) and calculated FRET using spectral analysis (Fig. 4 G and Table S3). We observed a positive and significant FRET signal between hERG Δeag[S4–S5]Alacomplete-Citrine channels and N-eag-CFP (P < 0.01 vs. rCB1-YFP + N-eag-CFP) that was not significantly different from hERG Δeag-Citrine + N-eag-CFP. In contrast, we did not detect FRET from cells expressing rCB1-YFP + N-eag-CFP. The calculated ratio of donor to acceptor (Fc/Fy) indicated that the levels of donor to acceptor fluorescence were similar (Table S3). Together with the functional results, these data indicate that replacing the S4–S5 linker with alanines disrupts regulation of channel gating but does not disrupt a global interaction of the eag domain with the channel.
Eag domain regulation of channel gating requires the CNBHD in the C-terminal region
To examine the role of the CNBHD in eag domain–dependent deactivation gating, we generated hERG channels (all bearing the S620T mutant and fused to Citrine) with targeted deletions of either the eag domain (amino acids 2–135; hERG Δeag), the CNBHD (amino acids 749–872; hERG ΔCNBHD), or both domains (amino acids 2–135 and 749–872; hERG Δeag ΔCNBHD) and expressed them in HEK293 cells. We measured ionic currents from each of these hERG channels using whole-cell patch-clamp recordings (Fig. 5 A). From a holding potential of −80 mV, channels were activated by a series of depolarizing pulses ranging from −80 to 60 mV, followed by a repolarizing pulse to −50 mV to elicit an outward tail current. The tail current after the pulse to 60 mV was fit with a double-exponential function and the fast (τfast) and slow (τslow) time constants of deactivation were plotted in Fig. 5 (C and D). We found that deletion of either the eag domain (hERG Δeag) or the CNBHD (hERG ΔCNBHD) resulted in channels with markedly accelerated deactivation kinetics compared with WT hERG channels (Fig. 5, A, C, and D). Dual deletion of the eag domain and the CNBHD (hERG Δeag ΔCNBHD) produced channels with similarly fast deactivation kinetics, which were not significantly different from either of the individual deletions (Fig. 5, A, C, and D). Coexpression of a gene fragment encoding the eag domain tagged with a CFP (N-eag-CFP) with each hERG channel slowed the kinetics of deactivation only in hERG Δeag channels, in which the CNBHD was intact, but not in either hERG ΔCNBHD or hERG Δeag ΔCNBHD channels (Fig. 5, B–D). N-eag-CFP coexpressed with WT hERG channels had no significant effect on the kinetics of deactivation. These results support previous results reported in Xenopus laevis oocytes in channels lacking fluorescent proteins (Gustina and Trudeau, 2011), and demonstrate that the presence of the CNBHD in the C-terminal region is necessary for the eag domain to regulate channel gating.
FRET reveals that CNBHD is necessary for the eag domain to associate with the channel
To directly examine whether the CNBHD was required for a global interaction of the eag domain with the rest of the hERG channel, we used FRET spectroscopy (see Materials and methods). We transiently transfected HEK293 cells with the N-eag-CFP fragments (which were the donors), and hERG channels were tagged with Citrine (which were the acceptors). As a negative control, we used N-eag-CFP and the cannabinoid-1 receptor tagged with YFP (rCB1-YFP), as in Fig. 3 (Gustina and Trudeau, 2009). The emission spectra from cells expressing N-eag-CFP + hERG Δeag-Citrine or N-eag-CFP + hERG Δeag ΔCNBHD-Citrine are shown in Fig. 6 (A and B, respectively). Calculating the relative FRET efficiency for each revealed a positive and significant (P < 0.01 vs. N-eag-CFP + rCB1-YFP) FRET signal in cells expressing N-eag-CFP + hERG Δeag-Citrine, but not in cells expressing N-eag-CFP + hERG Δeag ΔCNBHD-Citrine (Fig. 6 C and Table S3). In cells expressing N-eag-CFP + rCB1-YFP, as a negative control, we did not detect a FRET signal. It is important to note that the donor to acceptor ratios (Fc/Fy) were similar, indicating that the lack of FRET observed was not caused by a low donor to acceptor ratio (Table S3). These results demonstrate that the eag domain is in close proximity to hERG channels at the cell surface in channels that contain CNBHDs in the C-terminal region. This suggests that the CNBHD is necessary for the eag domain to associate with the channel, and that the CNBHD is a site of interaction for the eag domain in functional channels.
Figure 6.
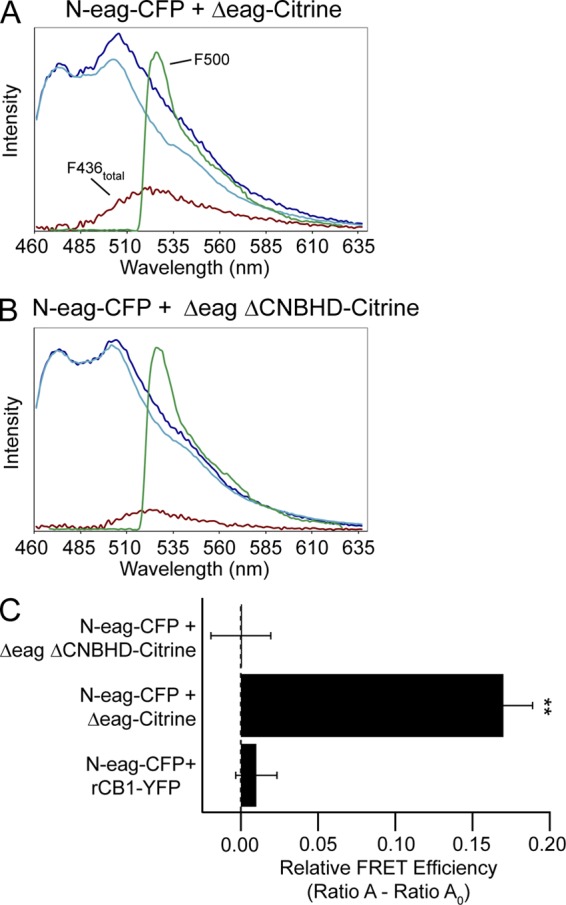
FRET spectroscopy reveals that association of the eag domain with the channel requires the CNBHD in the hERG C-terminal region. Representative emission spectra from cells expressing (A) N-eag-CFP + hERG Δeag-Citrine and (B) N-eag-CFP + hERG Δeag ΔCNBHD-Citrine. The dark blue trace represents the total emission spectrum with excitation at 436 nm. The cyan trace is the CFP emission with excitation at 436 nm taken from cells expressing donor only (N-eag-CFP). The red trace represents the Citrine emission with excitation at 436 nm (F436total) and was calculated by subtracting the cyan trace from the dark blue trace. The green trace represents the Citrine emission with excitation at 500 nm (F500). (C) Histogram of Ratio A–Ratio A0 values, which are proportional to the relative FRET efficiency. Data are plotted as mean ± SEM and are also given in Table S3. **, P < 0.01 versus N-eag-CFP + rCB1-YFP (ANOVA). n = 11 for each.
The eag domain directly interacts with the C-linker/CNBHD
To test for direct interactions between the eag domain and the CNBHD, we used a FRET two-hybrid assay (Erickson et al., 2003). In this method, cells were transfected with a “bait” construct tagged with a CFP and a “prey” construct tagged with a Citrine and tested for FRET. This method offers several advantages to determine specific domain–domain interactions: (a) direct interactions between two protein domains is measured in the in situ mammalian cellular environment, providing a “biological cuvette” for an interaction to occur and, thus, a low false-positive rate; (b) several combinations of potential interacting partners can be tested by creating different prey constructs; and (c) attaching fluorescent probes to smaller protein domains yields a low false-negative rate because typical limiting factors of FRET sensitivity, including fluorophore distance and orientation, are overcome when attached to small protein domains.
To carry out these experiments, we cotransfected cells with N-eag-CFP (bait) and C-linker/CNBHD tagged with either Citrine or YFP (prey), as outlined in Fig. 7 A. The C-linker was included with the CNBHD based on previous analyses from HCN2, zELK, and mEAG, which showed that the presence of the C-linker helped the CNBHD to fold (Zagotta et al., 2003; Brelidze et al., 2009, 2012; Marques-Carvalho et al., 2012). We measured the emission spectrum of cells expressing bait and prey and calculated the relative FRET efficiency. As a negative control, we coexpressed N-eag-CFP with the Ca2+-insensitive mutant CaM tagged with YFP (YFP-CaM1234) (Xia et al., 1998; Erickson et al., 2001), and its emission spectrum is shown in Fig. 7 B. The emission spectrum of N-eag-CFP + C-linker/CNBHD-Citrine is shown in Fig. 7 C. We observed a positive and significant FRET signal between N-eag-CFP and C-linker/CNBHD-Citrine domains (P < 0.01 vs. N-eag-CFP + YFP-CaM1234), whereas there was no FRET observed between N-eag-CFP and YFP-CaM1234 (Fig. 7 D and Table S3). To determine whether having a different acceptor fluorophore tagged to the C-linker/CNBHD had an effect on the FRET signal, we tested the N-eag-CFP with C-linker/CNBHD tagged with YFP (C-linker/CNBHD-YFP). We found that there was a similar level of FRET (P < 0.01 vs. N-eag-CFP + YFP-CaM1234) that was not significantly different from N-eag-CFP coexpressed with C-linker/CNBHD-Citrine (Fig. 7 D and Table S3). Switching the fluorophores between the two domains so that the eag domain gene fragment was tagged with Citrine (N-eag-Citrine) and C-linker/CNBHD was tagged with CFP (C-linker/CNBHD-CFP) similarly revealed a significant FRET signal (P < 0.01 vs. N-eag-CFP + YFP-CaM1234; Fig. 7 D and Table S3), demonstrating that any potential variations in fluorophore orientation do not influence the FRET signal. Collectively, these results indicate that the eag domain forms a direct and specific interaction with the C-linker/CNBHD in a live cell environment, and that the isolated eag domain and the isolated C-linker/CNBHD were sufficient to make an interaction.
Figure 7.
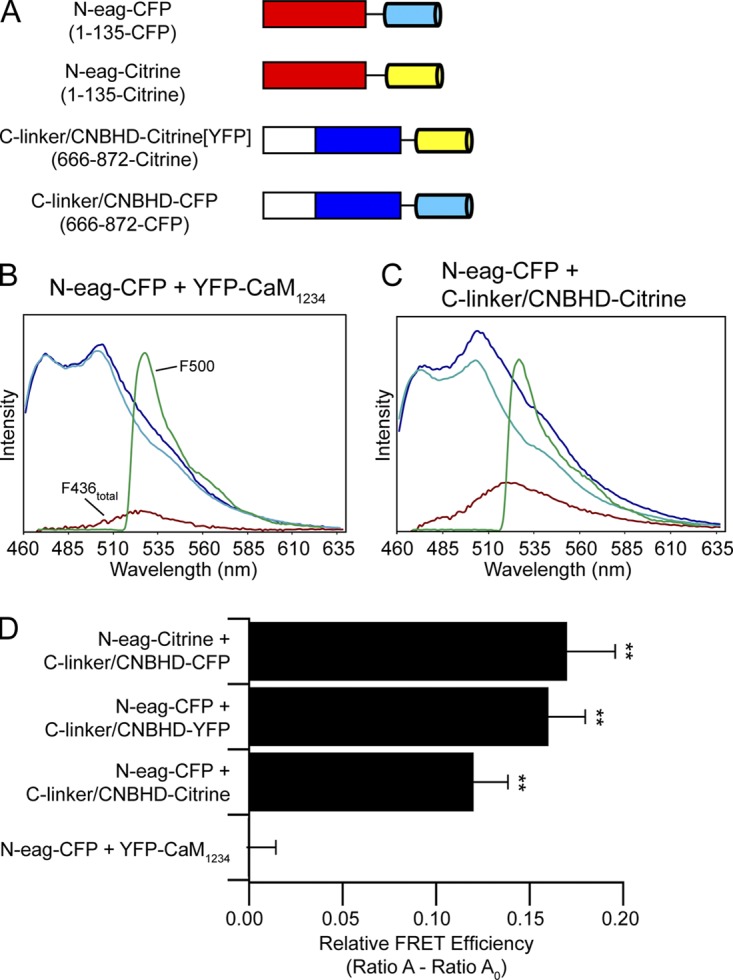
FRET two-hybrid analysis reveals that the eag domain directly interacts with the CNBHD. (A) Schematic illustrating hERG gene fragments used in the FRET two-hybrid assay. CFP is shown in cyan, and Citrine (or YFP) is shown in yellow. Representative emission spectra from cells expressing the bait-prey pairs (B) N-eag-CFP + YFP-CaM1234 or (C) N-eag-CFP + C-linker/CNBHD-Citrine. The dark blue trace represents the total emission spectrum with 436-nm excitation. The cyan trace represents the CFP emission with 436-nm excitation taken from cells expressing “bait” only (N-eag-CFP). The red trace is the subtracted spectrum (difference between the dark blue and cyan traces) and represents the Citrine emission with 436-nm excitation. The green trace represents Citrine emission with 500-nm excitation. (D) Histogram of Ratio A–Ratio A0 values of each bait-prey pair. Data are plotted as mean ± SEM and are given in Table S3. **, P < 0.01 versus N-eag-CFP + YFP-CaM1234 (ANOVA). n ≥ 6 for each.
We tested the possibility of intersubunit interactions between eag domains and C-linker/CNBHDs. HEK293 cells were transiently transfected with WT hERG, hERG Δeag, hERG ΔpCT (Δ816–1159), or hERG Δeag coexpressed with hERG ΔpCT (Fig. 8 A). Whereas deactivation was rapid for hERG Δeag channels or hERG ΔpCT channels, coexpression of hERG Δeag with hERG ΔpCT resulted in channels with deactivation kinetics that were markedly slowed (Fig. 8, A–C). Optical measurements showed robust FRET between hERG Δeag-CFP subunits and hERG ΔpCT-Citrine subunits, which was similar to that detected for hERG-CFP and hERG-Citrine positive controls (Fig. 8, D–F). Collectively, these data strongly suggest that the hERG Δeag subunits and hERG ΔpCT subunits assemble into heteromeric hERG Δeag/hERG ΔpCT channels, and that the eag domain and the CNBHD from different subunits make intersubunit interactions that regulate slow deactivation gating in hERG channels.
DISCUSSION
In this study, we investigated the role of the S4–S5 linker in hERG channel slow deactivation gating. We report here that most channels lacking the eag domain and bearing alanine mutations in the S4–S5 linker region (including L539A, D540A, E541A, S543A, and Y545A) were regulated by N-eag-CFP domains. We found that a few S4–S5 mutant channels (Y542A, E544A, or those with a continuous series of alanine substitutions) were not measurably regulated by recombinant N-eag-CFP domains. Using FRET spectroscopy, we showed that N-eag-CFP domains were in close physical proximity to all the channels with individual alanine mutations or channels with a continuous series of alanine residues in place of the S4–S5 linker region. These results indicate that most channels with S4–S5 linker mutants were regulated by and associated with eag domains, and that the apparent loss of eag domain–regulated gating in Y542A, E544A, and hERG Δeag[S4–S5]Alacomplete channels was not simply caused by a global loss of the association of the eag domain with the channel. Instead, we showed that N-eag-CFP domains did not measurably regulate gating in channels lacking the CNBHD (Fig. 5), and that FRET was not detected between N-eag-CFP and channels lacking the CNBHD (Fig. 6). However, we observed channel regulation and FRET when the N-eag-CFP domain was coexpressed with channels containing an intact CNBHD (Figs. 5 and 6). We used a FRET two-hybrid technique to measure protein interactions (Erickson et al., 2003) and found that N-eag-CFP domains and C-linker/CNBHDs produced FRET, indicating that eag domains and CNBHDs directly interacted in a mammalian expression system. Collectively, these results provide strong evidence that the N-terminal eag domain directly interacts with the C-terminal C-linker/CNBHD in the intact hERG channel, and that the CNBHD is necessary for eag domains to associate with the channel and regulate deactivation gating.
We propose a scheme to explain the mechanism for slow eag domain–dependent deactivation gating in hERG channels that has similarities to basic schemes for gating in HCN and BK channels (Horrigan and Aldrich, 2002; Craven and Zagotta, 2004) (Fig. 9 A). In this scheme, we propose that the voltage-sensor domains (VSDs), which comprise the S1–S4 transmembrane domains, are coupled (a, arrow) to the pore domains (S5-P-S6 domains), and that the VSD and pore are coupled (b, arrow) to the CNBHD (Fig. 9 A). The VSD can exist in resting (R) or active (A) positions, and the pore can be open (O) or closed (C). The CNBHD can be bound to the eag domain (as in WT channels) or unbound (as in Δeag or ΔCNBHD channels). VSDs in the active position and CNBHDs in the “eag-bound” position favor the O state of the pore. hERG channels lacking the eag domain (or lacking the CNBHD) are proposed to gate using only the VSD and pore modules. We propose that in WT hERG channels, information that the eag domain and the CNBHD interact is reported (Fig. 9 A, arrow b) to the VSD and pore domain modules, which work to favor the open state during repolarization, thus making deactivation slow. The role of the S4–S5 residues falls into different categories. D540A and S543A mutations have a large effect on gating in hERG Δeag channels (both have a large left-shift in the G-V curve). These two channels are robustly regulated by eag domains and interact with eag domains. Similarly, Y545A has a modest effect on the G-V of hERG Δeag channels, is robustly regulated by eag domains, and interacts with eag domains. Therefore, we propose that the role of D540, S543, and Y545 is primarily to influence the transitions in the VSD and pore modules or the coupling between the modules (Fig. 9 A, arrow a) but with a minimal influence on the CNBHD module. In contrast, Y542A and E544A have little measurable effect in the background of hERG Δeag channels and are not regulated by eag domains, but the eag domain remains bound to these channels. Thus, we propose that Y542A and E544A greatly reduced the coupling (i.e., greatly reduce arrow b in Fig. 9 A) between the CNBHD and the VSD and pore modules. The L539A and R541A mutations have little effect in the background of hERG Δeag channels, undergo partial regulation by eag domains, and remain bound by eag domains. Thus, we propose that these two mutants partially reduce the coupling (Fig. 9 A, arrow b) between the pore and the CNBHD, and that this accounts for the speeding of slow deactivation. Because of the slight effect on the G-V by R541A, we propose that this site might also weakly couple directly to the VSD and pore domains.
Figure 9.

Model of eag domain–dependent regulation of gating. (A) Schematic illustrating the proposed mechanism of hERG channel slow deactivation gating. The VSD module (VSD or S1–S4 transmembrane domains; yellow rectangle) is coupled (arrow a) to the pore module (S5-P-S6 domains; green rectangle). Vertical arrows depict movement between the active (A) and resting (R) state of the VSD and the closed (C) or open state (O) of the pore. Together, the VSD and pore modules are coupled (arrow b) to the interdomain interaction between the C-terminal C-linker/CNBHD (blue) and the N-terminal eag domain (red). Deletion of either the eag domain or the CNBHD disrupts the eag domain–C-linker/CNBHD interaction and its regulation of gating at the channel pore. Residues in the S4–S5 linker, such as D540, S543, and Y545, primarily alter gating by influencing the VSD and pore modules, whereas residues such as Y542 and E544 are primarily involved in the coupling pathway (arrow b) between the eag domain–C-linker/CNBHD interaction and the channel pore. (B) Two hERG subunits indicating that eag domain–dependent regulation of gating occurs through a direct intersubunit interaction between the N-terminal eag domain (red) and the C-terminal CNBHD (blue).
Here, we show evidence that eag domains in one subunit can form a direct interaction with CNBHDs in another subunit, providing evidence for the formation of heterotetrameric hERG channels with intersubunit interactions (see Fig. 9 B). We demonstrate that hERG Δeag subunits coexpressed with hERG ΔpCT subunits, each of which forms channels with fast deactivation kinetics when expressed alone, exhibited slower deactivation kinetics when coexpressed (Fig. 8). Further, we demonstrate that hERG Δeag and hERG ΔpCT subunits directly interact as measured by FRET spectroscopy, providing evidence for the formation of heterotetrameric channels. These results suggest that the C-terminal CNBHD in hERG subunits lacking an eag domain (hERG Δeag) and the N-terminal eag domain in hERG subunits lacking the majority of the CNBHD as well as the distal C terminus (hERG ΔpCT) are capable of coassembling and forming an intersubunit interaction to functionally regulate channel gating.
In this study, we concluded that the S4–S5 linker region was not sufficient for the association of the eag domain with the rest of the channel. Based on our results here, we cannot strictly rule out that S4–S5 linker residues may make some interactions with the eag domain. However, the results shown here indicate that hERG channels lacking a CNBHD but containing an intact S4–S5 linker were neither regulated by N-eag-CFP domains nor interacted with N-eag-CFP domains, demonstrating that the S4–S5 linker was not sufficient to make a stable interaction with the eag domain.
Our findings are in contrast with that of a previous study (Fernández-Trillo et al., 2011) in which the eag domain was proposed to interact with the amino terminal end of the S4–S5 linker because FRET was not observed between hERG Δeag Y542C-CFP channels and N-eag-YFP domains, which was interpreted to mean that mutation at the Y542 site completely disrupted an interaction with the eag domain. In contrast, in a similar experiment performed here, we found that a channel with a mutation at the same site, hERG Δeag Y542A-Citrine, had robust FRET with the N-eag-CFP domain. There are several variations between the experimental details of our work and that of the previous study. First, in our experiments, hERG channels were labeled with Citrine and the N-eag-CFP domains were labeled with CFP; in contrast, the previous study used CFP to label hERG channels and YFP to label the N-eag-CFP domain. Second, our work used an alanine mutant at the Y542 site, whereas the previous work used a cysteine mutant. Finally, the method used to measure FRET was different; whereas our work used spectra FRET, the previous study used donor de-quenching under TIRF conditions. Although it is possible that the difference between our observations and those of the previous study may be caused by these somewhat minor variations in methodology, we do not think this is likely because of our findings with other S4–S5 linker mutants. In particular, here we showed robust FRET between N-eag-CFP domains and hERG-Citrine channels with individual alanine mutations adjacent to Y542, as well as hERG channels in which the entire S4–S5 linker was replaced with alanines (see Figs. 3 and 4). Therefore, we propose that the eag domain makes a robust interaction with the rest of the channel that does not require the S4–S5 linker.
Our results from hERG S4–S5 linker experiments fit well with existing models of the role of the S4–S5 linker in gating of Kv channels. Structural data from Kv1.2 channels shows that the S4–S5 linker is a helix that links the S1–S4 (VSD) to the pore (S5-P-loop-S6) domains by connecting the S4 to the S5 domain (Long et al., 2005). The Kv1.2 structures also show that the S4–S5 linker sits above the S6 gate, and structural models suggest that, to close Kv channels, the VSD pushes down on the S4–S5 linker, which in turn pushes on the S6 domain to close the pore (Yarov-Yarovoy et al., 2006). The S4–S5 linker and the S6 gate likely interact in Kv channels (including hERG) as shown by complementation studies and cysteine cross-linking (Lu et al., 2002; Tristani-Firouzi et al., 2002; Ferrer et al., 2006). In particular, D540C in the S4–S5 linker was chemically cross-linked to L666C in the S6 domain of hERG channels (Ferrer et al., 2006). The structural location of the S4–S5 linker fits well with our results. In particular, the close proximity to the S4 and S6 domains may explain the effects of S543 and D540 on voltage-dependent gating, whereas the proximity of the S4–S5 linker to the lower S6 domain might explain why mutations at Y542 and E544 inhibit how information from the CNBHD is communicated to the VSD and pore. Our interpretation agrees with and extends existing models of S4–S5 linker function in the Kv family of channels. Finally, eag domains can interact with hERG Δeag channels in closed states (Gustina and Trudeau, 2009), and because eag domains remained associated with hERG channels (e.g., hERG Δeag[S4–S5]Alacomplete) that were open at very negative potentials, we propose that eag domains can also associate with channels in an open state.
Supplementary Material
Acknowledgments
We thank Dr. W.N. Zagotta for rCB1-YFP, Dr. S. Gordon and Dr. J. Adelman for calmodulin-YFP, and Dr. A. Gustina and C. Gallagher for helpful discussions.
This work was supported by a National Institutes of Health R01 grant (HL-083121 to M.C. Trudeau), the Interdisciplinary Training Program in Muscle Biology (T32-AR007592 to E.C. Gianulis), and a gift from the Helen Pumphrey Denit Trust.
Kenton J. Swartz served as editor.
Footnotes
Abbreviations used in this paper:
- CaM
- calmodulin
- CFP
- cyan fluorescent protein
- CNBHD
- cyclic nucleotide–binding homology domain
- eag
- ether-á-go-go
- FRET
- Förster resonance energy transfer
- HCN
- hyperpolarization-activated, cyclic nucleotide–modulated
- HEK293
- human embryonic kidney 293
- hERG
- human eag-related gene
- VSD
- voltage-sensor domain
References
- Alonso-Ron C., de la Peña P., Miranda P., Domínguez P., Barros F. 2008. Thermodynamic and kinetic properties of amino-terminal and S4-S5 loop HERG channel mutants under steady-state conditions. Biophys. J. 94:3893–3911 10.1529/biophysj.107.116731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Owais M., Bracey K., Wray D. 2009. Role of intracellular domains in the function of the herg potassium channel. Eur. Biophys. J. 38:569–576 10.1007/s00249-009-0408-2 [DOI] [PubMed] [Google Scholar]
- Brelidze T.I., Carlson A.E., Zagotta W.N. 2009. Absence of direct cyclic nucleotide modulation of mEAG1 and hERG1 channels revealed with fluorescence and electrophysiological methods. J. Biol. Chem. 284:27989–27997 10.1074/jbc.M109.016337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelidze T.I., Carlson A.E., Sankaran B., Zagotta W.N. 2012. Structure of the carboxy-terminal region of a KCNH channel. Nature. 481:530–533 10.1038/nature10735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zou A., Splawski I., Keating M.T., Sanguinetti M.C. 1999. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J. Biol. Chem. 274:10113–10118 10.1074/jbc.274.15.10113 [DOI] [PubMed] [Google Scholar]
- Cheng W., Yang F., Takanishi C.L., Zheng J. 2007. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 129:191–207 10.1085/jgp.200709731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg R.M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388 10.1016/0076-6879(92)11020-J [DOI] [PubMed] [Google Scholar]
- Craven K.B., Zagotta W.N. 2004. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J. Gen. Physiol. 124:663–677 10.1085/jgp.200409178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran M.E., Splawski I., Timothy K.W., Vincent G.M., Green E.D., Keating M.T. 1995. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 80:795–803 10.1016/0092-8674(95)90358-5 [DOI] [PubMed] [Google Scholar]
- de la Peña P., Alonso-Ron C., Machín A., Fernández-Trillo J., Carretero L., Domínguez P., Barros F. 2011. Demonstration of physical proximity between the N terminus and the S4-S5 linker of the human ether-a-go-go-related gene (hERG) potassium channel. J. Biol. Chem. 286:19065–19075 10.1074/jbc.M111.238899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson M.G., Alseikhan B.A., Peterson B.Z., Yue D.T. 2001. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 31:973–985 10.1016/S0896-6273(01)00438-X [DOI] [PubMed] [Google Scholar]
- Erickson M.G., Liang H., Mori M.X., Yue D.T. 2003. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 39:97–107 10.1016/S0896-6273(03)00395-7 [DOI] [PubMed] [Google Scholar]
- Fernández-Trillo J., Barros F., Machín A., Carretero L., Domínguez P., de la Peña P. 2011. Molecular determinants of interactions between the N-terminal domain and the transmembrane core that modulate hERG K+ channel gating. PLoS ONE. 6:e24674 10.1371/journal.pone.0024674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T., Rupp J., Piper D.R., Tristani-Firouzi M. 2006. The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-a’-go-go-related gene (hERG) K+ channel. J. Biol. Chem. 281:12858–12864 10.1074/jbc.M513518200 [DOI] [PubMed] [Google Scholar]
- Gianulis E.C., Trudeau M.C. 2011. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-á-go-go-related gene potassium channels. J. Biol. Chem. 286:22160–22169 10.1074/jbc.M110.205948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O., Baird G.S., Campbell R.E., Zacharias D.A., Tsien R.Y. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276:29188–29194 10.1074/jbc.M102815200 [DOI] [PubMed] [Google Scholar]
- Gustina A.S., Trudeau M.C. 2009. A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels. Proc. Natl. Acad. Sci. USA. 106:13082–13087 10.1073/pnas.0900180106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina A.S., Trudeau M.C. 2011. hERG potassium channel gating is mediated by N- and C-terminal region interactions. J. Gen. Physiol. 137:315–325 10.1085/jgp.201010582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina A.S., Trudeau M.C. 2013. The eag domain regulates hERG channel inactivation gating via a direct interaction. J. Gen. Physiol. 141:229–241 10.1085/jgp.201210870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy H.R., Durell S.R., Warmke J., Drysdale R., Ganetzky B. 1991. Similarities in amino acid sequences of Drosophila eag and cyclic nucleotide-gated channels. Science. 254:730 10.1126/science.1658932 [DOI] [PubMed] [Google Scholar]
- Heikal A.A., Hess S.T., Baird G.S., Tsien R.Y., Webb W.W. 2000. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: coral red (dsRed) and yellow (Citrine). Proc. Natl. Acad. Sci. USA. 97:11996–12001 10.1073/pnas.97.22.11996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg I.M., Trudeau M.C., Robertson G.A. 1998. Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J. Physiol. 511:3–14 10.1111/j.1469-7793.1998.003bi.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrigan F.T., Aldrich R.W. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305 10.1085/jgp.20028605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Gayen S., Chen A.S., Huang Q., Raida M., Kang C. 2010. NMR solution structure of the N-terminal domain of hERG and its interaction with the S4-S5 linker. Biochem. Biophys. Res. Commun. 403:126–132 10.1016/j.bbrc.2010.10.132 [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., Mackinnon R. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908 10.1126/science.1116270 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A.M., Ramu Y. 2002. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120:663–676 10.1085/jgp.20028696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Carvalho M.J., Sahoo N., Muskett F.W., Vieira-Pires R.S., Gabant G., Cadene M., Schönherr R., Morais-Cabral J.H. 2012. Structural, biochemical, and functional characterization of the cyclic nucleotide binding homology domain from the mouse EAG1 potassium channel. J. Mol. Biol. 423:34–46 10.1016/j.jmb.2012.06.025 [DOI] [PubMed] [Google Scholar]
- Morais Cabral J.H., Lee A., Cohen S.L., Chait B.T., Li M., Mackinnon R. 1998. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 95:649–655 10.1016/S0092-8674(00)81635-9 [DOI] [PubMed] [Google Scholar]
- Muskett F.W., Thouta S., Thomson S.J., Bowen A., Stansfeld P.J., Mitcheson J.S. 2011. Mechanistic insight into human ether-à-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 286:6184–6191 10.1074/jbc.M110.199364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C.A., Hunter M.J., Perry M.D., Mobli M., Ke Y., Kuchel P.W., King G.F., Stock D., Vandenberg J.I. 2011. The N-terminal tail of hERG contains an amphipathic α-helix that regulates channel deactivation. PLoS ONE. 6:e16191 10.1371/journal.pone.0016191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C.A., Perry M.D., Tan P.S., Hill A.P., Kuchel P.W., Vandenberg J.I. 2012. The S4-S5 linker acts as a signal integrator for HERG K+ channel activation and deactivation gating. PLoS ONE. 7:e31640 10.1371/journal.pone.0031640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M.C., Jurkiewicz N.K. 1990. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96:195–215 10.1085/jgp.96.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M.C., Jurkiewicz N.K. 1991. Delayed rectifier outward K+ current is composed of two currents in guinea pig atrial cells. Am. J. Physiol. 260:H393–H399 [DOI] [PubMed] [Google Scholar]
- Sanguinetti M.C., Xu Q.P. 1999. Mutations of the S4-S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J. Physiol. 514:667–675 10.1111/j.1469-7793.1999.667ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M.C., Jiang C., Curran M.E., Keating M.T. 1995. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 81:299–307 10.1016/0092-8674(95)90340-2 [DOI] [PubMed] [Google Scholar]
- Selvin P.R. 1995. Fluorescence resonance energy transfer. Methods Enzymol. 246:300–334 10.1016/0076-6879(95)46015-2 [DOI] [PubMed] [Google Scholar]
- Spector P.S., Curran M.E., Zou A., Keating M.T., Sanguinetti M.C. 1996. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 107:611–619 10.1085/jgp.107.5.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L. 1978. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 47:819–846 10.1146/annurev.bi.47.070178.004131 [DOI] [PubMed] [Google Scholar]
- Takanishi C.L., Bykova E.A., Cheng W., Zheng J. 2006. GFP-based FRET analysis in live cells. Brain Res. 1091:132–139 10.1016/j.brainres.2006.01.119 [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M., Chen J., Sanguinetti M.C. 2002. Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J. Biol. Chem. 277:18994–19000 10.1074/jbc.M200410200 [DOI] [PubMed] [Google Scholar]
- Trudeau M.C., Warmke J.W., Ganetzky B., Robertson G.A. 1995. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 269:92–95 10.1126/science.7604285 [DOI] [PubMed] [Google Scholar]
- Wang J., Trudeau M.C., Zappia A.M., Robertson G.A. 1998. Regulation of deactivation by an amino terminal domain in human ether-à-go-go–related gene potassium channels. J. Gen. Physiol. 112:637–647 10.1085/jgp.112.5.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Myers C.D., Robertson G.A. 2000. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K+ channels. J. Gen. Physiol. 115:749–758 10.1085/jgp.115.6.749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke J.W., Ganetzky B. 1994. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA. 91:3438–3442 10.1073/pnas.91.8.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke J., Drysdale R., Ganetzky B. 1991. A distinct potassium channel polypeptide encoded by the Drosophila eag locus. Science. 252:1560–1562 10.1126/science.1840699 [DOI] [PubMed] [Google Scholar]
- Xia X.-M., Fakler B., Rivard A., Wayman G., Johnson-Pais T., Keen J.E., Ishii T., Hirschberg B., Bond C.T., Lutsenko S., et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 395:503–507 10.1038/26758 [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., Baker D., Catterall W.A. 2006. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. USA. 103:7292–7297 10.1073/pnas.0602350103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta W.N., Olivier N.B., Black K.D., Young E.C., Olson R., Gouaux E. 2003. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 425:200–205 10.1038/nature01922 [DOI] [PubMed] [Google Scholar]
- Zheng J., Zagotta W.N. 2004. Stoichiometry and assembly of olfactory cyclic nucleotide-gated channels. Neuron. 42:411–421 10.1016/S0896-6273(04)00253-3 [DOI] [PubMed] [Google Scholar]
- Zheng J., Trudeau M.C., Zagotta W.N. 2002. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 36:891–896 10.1016/S0896-6273(02)01099-1 [DOI] [PubMed] [Google Scholar]
- Zhou Z., Gong Q., Ye B., Fan Z., Makielski J.C., Robertson G.A., January C.T. 1998. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 74:230–241 10.1016/S0006-3495(98)77782-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.