

The monoterpene indole alkaloids, which are usually comprised of a tryptamine moiety appended to a single C9- or C10-terpenoid unit, constitute one of the largest known classes of natural products.[1] In 2004, Kam and Choo isolated a new type of monoterpenoid indole alkaloid, angustilodine (1), which contains a unique rearranged skeleton, from the leaves of the Malayan plant Alstonia angustiloba (Figure 1).[2] The structure of angustilodine was determined by detailed spectroscopic analysis to include an indole appended to a cis-fused 2-azadecalin ring system bearing a 7-membered ring ether bridge. An interesting conformational feature of this molecule established by 2D NMR studies is the observation that the piperidine ring exists as a boat. More recently, Morita and coworkers discovered the N-demethyl congener alstilobanine E (2), along with alstilobanine A (3), which lacks the bridging oxepane ring found in 1 and 2, in the same plant.[3] Unlike alkaloids 1 and 2, it was proposed that alstilobanine A has the piperidine ring in a chair conformation as shown in Figure 1. Alstilobanines A and E were found to possess modest relaxant activity against phenylephrine-induced contractions of thoracic rat aortic rings with endothelium. In this communication we describe the first approach to these alkaloids, culminating in a convergent total synthesis of racemic alstilobanine A (3).
Figure 1.
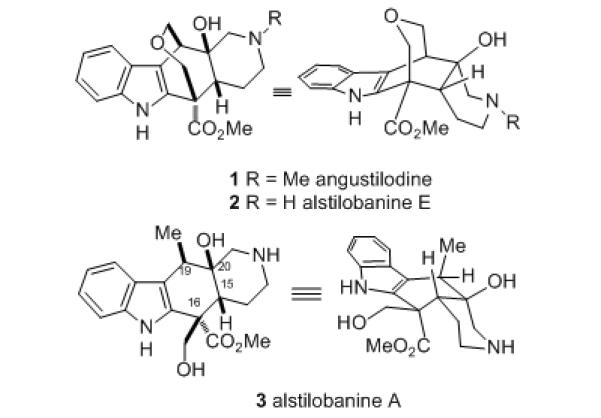
Structures of the alstilobanine alkaloids.
Our synthetic strategy was predicated upon effecting two key carbon-carbon single bond constructions. The first planned transformation involved an intermolecular conjugate addition of an indole ester enolate to a 3-piperidone-derived nitrosoalkene to form the C-15,16 bond of the alkaloid.[4] The second pivotal step was to apply the methodology of Romo, et al. for intramolecular β-lactone formation[5] to generate the requisite cis-2-azadecalin moiety via C-19,20 bond formation, along with the necessary functionality and three contiguous stereocenters at C-15,19,20. The implementation of this strategy is outlined here.[6]
Thus, indole 2-acetic acid methyl ester (4)[7] was first acylated at C-3 with oxalyl chloride, followed by in situ treatment of the resulting α-keto acid chloride with 2-trimethylsilylethanol to afford keto diester 5 (Scheme 1). In order to generate the C-15,16 bond of 3, indole ester 5 was first converted to the dianion 6 using two equivalents of lithium hexamethyldisilazide. Addition of one equivalent of the α-chlorooxime 7 derived from N-tosyl-3-piperidone[8] to the dianion led to the desired coupled product as a 1.2:1 mixture of diastereomers 11a and 11b which could be separated for characterization, each as a single oxime E-geometric isomer, in high total yield. It should be noted that the C-16 mixture obtained in the conjugate addition reaction is of no consequence since it is subsequently corrected (vide infra).
Scheme 1.
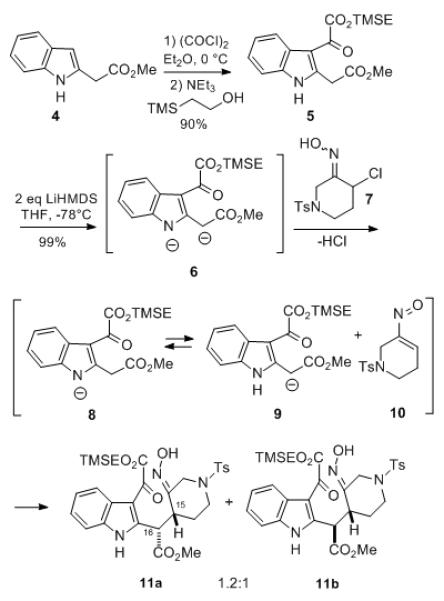
Nitrosoalkene conjugate addition.
We believe that this novel transformation involves the initial dehydrohalogenation of α-chlorooxime 7 by dianion 6 to generate a transient nitrosoalkene 10 along with a monoanion derived from the indole ester. It seems likely that this intermediate is probably an equilibrium mixture of resonance stabilized anions 8 and 9, but the conjugate addition to nitrosoalkene 10 occurs exclusively via the latter form.
To continue the synthesis, the oxime moiety of the mixture of indole ester adducts 11a and 11b was first protected as the TBS ether 12 (Scheme 2). At this stage, the α-ketoester 12 was deoxygenated via a modification of the method of Hlasta, et al.[9] Thus, the ketone was first reduced to the alcohol which was then converted to the corresponding acetate, followed by catalytic hydrogenation using Pd/C in t-butanol/triethylamine to afford a mixture of diastereomeric diesters 13 and 14 which were easily separated by column chromatography.[10]
Scheme 2.
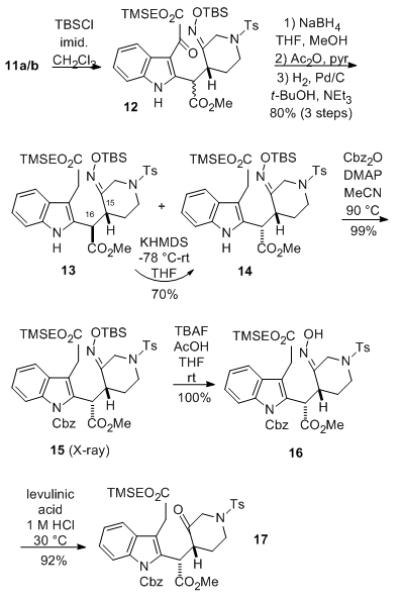
Preparation of Intermediate 17
Since it was later found that these two diastereomeric C-15/16 systems behave differently during the key Romo cyclization (vide infra), isomer 13 was epimerized cleanly to 14 in 70% isolated yield by treatment with potassium hexamethyldisilazide, followed by quenching with aqueous ammonium chloride. The indole nitrogen of 14 was then protected with a Cbz group to afford derivative 15 whose structure was established by X-ray analysis, confirming both the C-15/16 relative stereochemistry and the O-silyloxime E-geometry.[11] At this point, O-silyloxime 15 was converted to oxime 16 with TBAF, followed by acidic cleavage to give the corresponding ketone 17.[12]
Initial attempts to convert the trimethylsilylethyl ester 17 to the corresponding carboxylic acid with fluoride sources failed,[6] but this transformation could be performed with trifluoroacetic acid in methylene chloride to afford the desired keto acid 18 in high yield without affecting the methyl ester (Scheme 3).[13] With compound 18 in hand, we were prepared to attempt the second key step, a Romo formal ketene/ketone-[2+2]-intramolecular cycloaddition to generate the requisite fused β-lactone cis-2-azadecalin system.
Scheme 3.
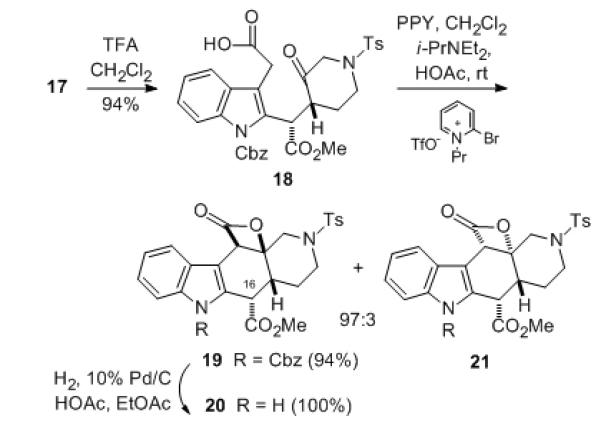
Romo cyclization of keto acid 18 to pentacyclic β-lactone 19
Therefore, treatment of the keto acid 18 with 2-bromo-N-propylpyridinium triflate, 4-pyrrolidinopyridine (PPY) and Hunig’s base in CH2Cl2 containing 1.2 equivalents of acetic acid to avoid epimerization of the C-16 ester led to the desired fused β-lactone system 19 needed for alstilobanine A along with a trace of the trans-2-azadecalin 21 (97:3 NMR ratio) in high yield.[5b] Interestingly, when the Romo cyclization was conducted on the C-16 ester epimer of keto acid 18 prepared from intermediate 13, the major product was the undesired trans-2-azadecalin.
With key intermediate 19 now in hand, we began to investigate introduction of a hydroxymethyl group at C-16. Since we had found in earlier work with various other intermediates that it was not possible to generate a C-16 ester enolate if a Cbz protecting group is in place on the indole nitrogen,[6,14] this group was removed from 19 via hydrogenolysis to afford NH-indole ester 20. This compound could be successfully deprotonated with two equivalents of lithium hexamethyldisilazide to generate the dianion 22, followed by alkylation with monomeric formaldehyde[15] from the least hindered face to produce α-hydroxymethyl ester 23 as a single diastereomer having the configuration needed for alkaloid 3 (Scheme 4).[16] The stereochemistry of compound 23 was established by 2D NMR analysis and was also confirmed by the fact that the β-lactone underwent acyl migration to afford the bridged seven-membered lactone 24 upon treatment with triethylamine in methylene chloride.
Scheme 4.
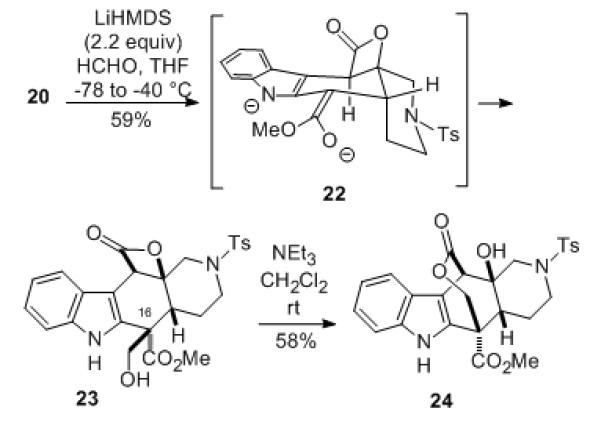
Stereoselective C-16 hydroxymethylation
Since it was observed that the hydroxymethyl group of 23 tends to be lost under a variety of conditions via a retro-aldol process, this functionality was protected as the TBS ether 25 (Scheme 5). The β-lactone moiety of 25 could then be reduced selectively with LiBH4 in THF to yield the diol ester 26, that was converted to iodo alcohol 27 via an Appel reaction.[17] Subsequent catalytic hydrogenation of this compound at atmospheric pressure using 10% Pd/C in a 1:1 mixture of ethyl acetate:t-butanol cleanly led to the desired methyl compound 28.[17] The N-tosyl protecting group of intermediate 28 was cleaved using magnesium metal turnings in methanol under sonication to afford piperidine 29 in good yield.[18] Once, again the structure and stereochemistry of this intermediate were confirmed by 2D NMR analysis (see Supporting Information). Finally, removal of the TBS protecting group with HCl in methanol/chloroform afforded racemic alstilobanine A (3), isolated as its hydrochloride salt, having proton and carbon NMR spectra as reported for the natural alkaloid.[3,19]
Scheme 5.
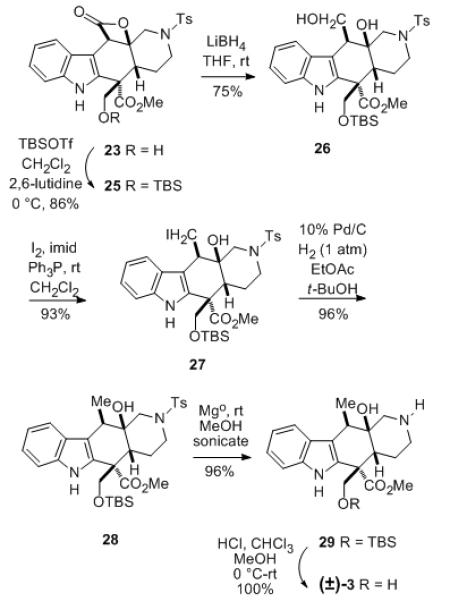
Completion of the synthesis of (±) alstilobanine A (3).
In summary, we have devised a convergent approach to a total synthesis of the novel indole alkaloid alstilobanine A (3). The synthesis of 3 requires about twenty operations starting from indole methyl ester 4. Key steps in the route include an unprecedented conjugate addition of an indole acetate ester enolate to a nitrosoalkene, and an intramolecular Romo cyclization to generate a β-lactone fused to the requisite cis-2-azadecalin needed for the alkaloid. Work is currently underway using the intermediates described here for construction of the bridging oxepane ring directed towards syntheses of the congeneric alkaloids 1 and 2.[20]
Supplementary Material
Footnotes
We are grateful to the National Institutes of Health (GM-087733) and the National Science Foundation (CHE-1105653)for financial support of this research.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1] (a).For reviews see: Cordell GA. Introduction to Alkaloids: A Biogenetic Approach. Wiley-Interscience; New York: 1981. Pelletier SW. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 1. Wiley; New York: 1983. Bisset NG. In: Indoles and Biogenetically Related Alkaloids. Phillipson JD, Zenk MH, editors. Academic Press; London: 1980. Herbert RB. In: The Chemistry of Heterocyclic Compounds. Indoles Part 4, The Monoterpenoid Indole Alkaloids. Saxton JE, editor. Wiley; New York: 1983. Atta-ur-Rahman A. Basha. Biosynthesis of Indole Alkaloids. Clarendon Press; Oxford: 1983. Kutchan TM. Phytochemistry. 1993;32:493. doi: 10.1016/s0031-9422(00)95128-8.
- [2].Kam T-S, Choo Y-M. Helv. Chim. Acta. 2004;87:366. [Google Scholar]
- [3].Koyama K, Hirasawa Y, Zaima K, Hoe TC, Chan K-L, Morita H. Bioorg. Med. Chem. 2008;16:6483. doi: 10.1016/j.bmc.2008.05.033. [DOI] [PubMed] [Google Scholar]
- [4] (a).For reviews of nitrosoalkenes see: Gilchrist TL. Chem. Soc. Rev. 1983;12:53. Lyapkalo IM, Ioffe SL. Russ. Chem. Rev. 1998;67:467.
- [5] (a).Oh SH, Cortez GS, Romo D, Org J. Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (b) Henry-Riyad H, Lee C, Purohit VC, Romo D. Org. Lett. 2006;8:4363. doi: 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]; (c) Ma G, Nguyen H, Romo D. Org. Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Purohit VC, Matla AS, Romo D. J. Am. Chem. Soc. 2008;130:10478. doi: 10.1021/ja803579z. [DOI] [PubMed] [Google Scholar]; (e) Nguyen H, Ma G, Romo D. Chem. Commun. 2010;46:4803. doi: 10.1039/c0cc00607f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Morris KA, Arendt KM, Oh SH, Romo D. Org. Lett. 2010;12:3764. doi: 10.1021/ol101388h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nguyen H, Ma G, Gladysheva T, Fremgen T, Romo D. J. Org. Chem. 2011;76:2. doi: 10.1021/jo101638r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Taken in part from: Majireck MM. Ph.D. Thesis, The Pennsylvania State University. 2011.
- [7].Mahboobi S, Bernauer K. Helv. Chim. Acta. 1988;71:2034. [Google Scholar]
- [8].Chauhan PS, Majireck MM, Weinreb SM. Heterocycles. 2012;84:577. [Google Scholar]
- [9].Hlasta DJ, Luttinger D, Perrone MH, Silbernagel MJ, Ward SJ, Haubrich DR. J. Med. Chem. 1987;30:1555. doi: 10.1021/jm00392a005. [DOI] [PubMed] [Google Scholar]
-
[10].This transformation presumably occurs via an azafulvene intermediate i derived from the indole. Evidence for this supposition is that if the hydrogenation is conducted in ethanol rather than t-butanol, a significant amount of product ii results where the acetate group is replaced by ethoxyl via interception of azafulvene i by the solvent. This compound is resistant to further catalytic reduction.[6]
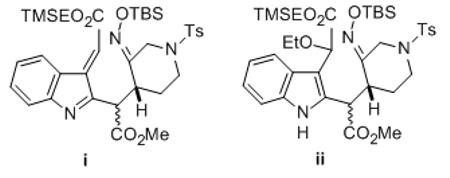
- [11].We thank Dr. Hemant Yennawar (Penn State Small Molecule X-Ray Crystallographic Facility) for the crystal structure determination.
- [12].DePuy CH, Ponder BW. J. Am. Chem. Soc. 1959;81:4629. [Google Scholar]
- [13].Van den Broek LAGM, Breuer ML, Liskamp RMJ, Ottenheijm HCJ. J. Org. Chem. 1987;52:1511. [Google Scholar]
- [14].We believe the bulky N-Cbz group causes the adjacent ester to assume a conformation which is not favorably disposed stereoelectronically for α-proton removal by base.
- [15].Schlosser M, Jenny T, Guggisberg Y. Synlett. 1990;11:704. [Google Scholar]
- [16].Martin Cf. C. L., Overman LE, Rohde JM. J. Am. Chem. Soc. 2010;132:4894. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].See for example: Huscroft IT, Carlson EJ, Chicchi GG, Kurtz MM, London C, Raubo P, Wheeldon A, Kulagowski JJ. Bioorg. Med. Chem. Lett. 2006;16:2008. doi: 10.1016/j.bmcl.2005.12.069.
- [18].Nyasse B, Grehn L, Ragnarsson U. Chem. Commun. 1997:1017. [Google Scholar]
- [19].We are grateful to Professor H. Morita (Hoshi University) for copies of the NMR spectra of the TFA salt of the natural alkaloid.
- [20].Although alkaloids 1 and 2 in principle could be prepared via removal of the lactone oxygen of intermediate 24, to date we have been unable to execute this transformation. Researchis continuing on solving this problem and/or finding another approach to angustilodine and alstilobanine E.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.