

Abstract
Induction of heme oxygenase-1, a stress-inducible enzyme with anti-inflammatory activity, reduces the immunogenicity of therapeutic factor VIII in experimental hemophilia A. In humans, heme oxygenase-1 expression is modulated by polymorphisms in the promoter of the heme oxygenase-1-encoding gene (HMOX1). We investigated the relationship between polymorphisms in the HMOX1 promoter and factor VIII inhibitor development in severe hemophilia A. We performed a case-control study on 99 inhibitor-positive patients and 263 patients who did not develop inhibitors within the first 150 cumulative days of exposure to therapeutic factor VIII. Direct sequencing and DNA fragment analysis were used to study (GT)n polymorphism and single nucleotide polymorphisms located at −1135 and −413 in the promoter of HMOX1. We assessed associations between the individual allele frequencies or genotypes, and inhibitor development. Our results demonstrate that inhibitor-positive patients had a higher frequency of alleles with large (GT)n repeats (L: n≥30), which are associated with lesser heme oxygenase-1 expression (odds ratio 2.31; 95% confidence interval 1.46–3.66; P<0.001]. Six genotypes (L/L, L/M, L/S, M/M, M/S and S/S) of (GT)n repeats were identified (S: n<21; M: 21≤n<30). The genotype group including L alleles (L/L, L/M and L/S) was statistically more frequent among inhibitor-positive than inhibitor-negative patients, as compared to the other genotypes (33.3% versus 17.1%) (odds ratio 2.21, 95% confidence interval 1.30–3.76; P<0.01). To our knowledge, this is the first association identified between HMOX1 promoter polymorphism and development of anti-drug antibodies. Our study paves the way towards modulation of the endogenous anti-inflammatory machinery of hemophilia patients to reduce the risk of inhibitor development
Introduction
The development of inhibitory anti-factor VIII (FVIII) antibodies is the major complication of replacement therapy in patients with hemophilia A, a rare X-linked recessive hemorrhagic disorder.1 The reasons for such immunogenicity of FVIII concentrates, as compared to other therapeutic proteins, remain unclear. Different risk factors have been associated with the appearance of FVIII inhibitors, including the type of F8 mutations responsible for hemophilia A, the HLA haplotypes and polymorphisms in TNFα, IL10 and CTLA-4 genes.2–5 Inflammatory events in act or occurring at the time of therapeutic FVIII administration have also been proposed as potential risk factors. Thus, repeated joint bleeds create a chronic inflammation favoring the local recruitment and activation of antigen-presenting cells and immune effectors.3,6 Likewise, surgery, which, in conjuncture with intensive FVIII treatment, has been proposed as a risk factor for inhibitor development,7 induces acute inflammation. Besides, the very administration of therapeutic FVIII has been controversially proposed to bring about inflammatory signals by virtue of the capacity of FVIII to induce a burst of thrombin generation, that in turn triggers proteinase-activated receptors.8,9
Heme oxygenase (HO) is an essential enzyme for the catabolism of heme and has been shown to have potent anti-oxidant, cytoprotective, immunosuppressive and anti-inflammatory properties via the production of bile pigments, carbon monoxide (CO) and the induction of ferritin.10,11 Two isoforms of HO have been identified:12,13 HO-2 is produced constitutively, whereas HO-1 is inducible. Thus, HO-1 is normally undetectable in resting cells, but may be induced in vivo as a result of inflammation or oxidative stress, by various stimuli, such as pro-oxidative compounds, pro-inflammatory cytokines, toxins or toll-like receptor ligands.10,12,14 In animal models, the pharmacological induction of HO-1 ameliorates chronic and severe inflammation,15,16 has beneficial effects in various autoimmune conditions17,18 and improves graft survival.16,19 HO-1 was demonstrated to participate in the resolution of physiological inflammation and in wound healing.14,20 Accordingly, congenital defects in HO-1 expression are associated with systemic inflammation in both mice and humans.20 Recently, we demonstrated that the induction of HO-1 before the administration of FVIII to FVIII-deficient mice protects against the anti-FVIII immune response.21 The protective effect of HO-1 induction was reverted by tin-mesoporphyrin, an inhibitor of HO-1, and was reproduced by the administration of the end-degradation products of heme by HO-1, i.e., CO and bilirubin.
The human HO-1-encoding gene (HMOX1) has been mapped to chromosome 22q12.22 The transcriptional control of this gene is regulated by multiple elements, which are localized in the 5′ flanking region of the promoter. To date, three polymorphisms in the 5′ flanking region have been described: two single nucleotide polymorphisms −413 T>A (rs2071746) and −1135 G>A (rs2071749) and a (GT)n repeat dinucleotide length polymorphism.23 The number of (GT) repeats modulates gene transcription;24 long (GT)n repeats are associated with low levels of HO-1 expression in response to a given stimulus, while short (GT)n repeats are associated with high expression levels.25 For instance, umbilical endothelial cells (HUVEC) from healthy donors with fewer than 23 GT repeats produce more HO-1 in vitro than HUVEC from healthy donors with ≥32 GT repeats following stimulation with H2O2.26 Polymorphisms in the promoter of the HMOX1 gene that result in greater inducibility of the enzyme have been associated with positive outcomes in a number of human pathologies characterized by cellular/tissue damage and inflammation.22 We hypothesized that polymorphisms in the HMOX1 promoter may confer different genetic predispositions to the induction of the immune response against exogenous FVIII among patients with hemophilia A by differentially influencing the capacity to modulate the inflammatory status of the patients. To test our hypothesis, we analyzed polymorphisms in the HMOX1 gene promoter of a large international cohort of patients with severe hemophilia A, and correlated the polymorphisms present with the development of FVIII inhibitors.
Methods
Study population
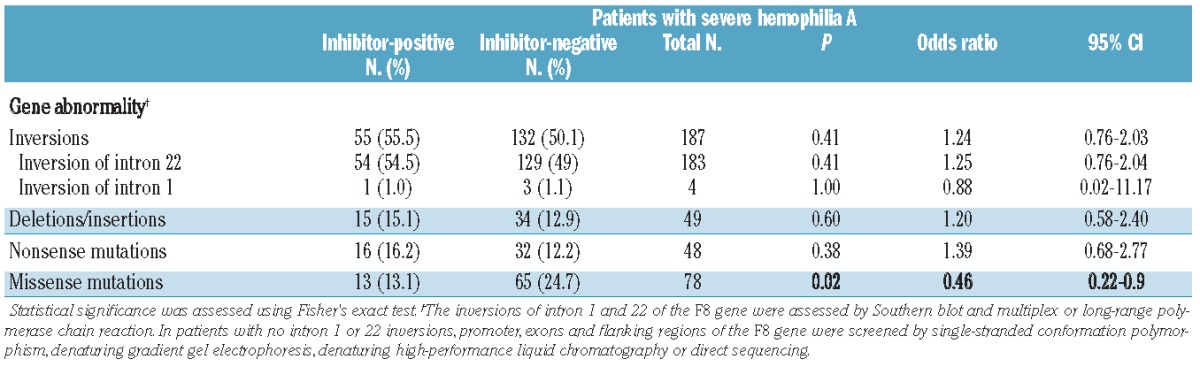
Our study included 362 patients with severe hemophilia A from different hemophilia centers in France (Caen, Kremlin-Bicêtre, Paris, Rennes) and Germany (Bonn). Ninety-nine patients had been diagnosed with a FVIII inhibitor. The 263 inhibitor-negative patients matched with inhibitor-positive patients for mutation type except for missense mutations (Table 1). The selection criterion was severe hemophilia A (FVIII:C<1%). Inhibitor historical peak titers were documented for 75 of the 99 inhibitor-positive patients: 26 patients had a historical peak titer <5 Bethesda units (BU)/mL (mean 2.6; range 1.0–4.8) and 49 patients had a historical peak titer ≥5 BU/mL (mean 1259; range 5 – 50000). Patients who had never developed an inhibitor after 150 cumulative exposure days (CED) or more were defined as inhibitor-negative patients. Approval for these studies was obtained from the Caen University institutional review board. Written informed consent was provided by each patient according to the Declaration of Helsinki.
Table 1.
Characteristics of the studied population.
Factor VIII activity and factor VIII inhibitory titers
FVIII activity was assessed using standard techniques. The original Bethesda method and the Nijmegen modification of the Bethesda assay were used to test for the presence or absence of FVIII-specific inhibitors in patients with hemophilia A.27
Genotyping of the variable (GT)n polymorphism in the promoter of the HO-1 gene
Genomic DNA was obtained from blood, anticoagulated with ethylene-diamine-tetra-acetic acid (EDTA), using commercial DNA isolation kits and a standard salting out procedure.28 The 5′-flanking region of the HO-1 gene containing the (GT)n dinucleotide repeat was amplified as described elsewhere.23 A polymerase chain reaction (PCR) was performed using sense primer 5′-AGCAAAATCACACCCAGAGC-3′, carrying a 6-FAM fluorescent label and downstream primer 5′-CCCTTGGGAAACAAAGTCTG-3′,23 and using Amplitaq Gold buffer (Applied Biosystems, Courtaboeuf, France) with 2.5 mM MgCl2, 200 μM dNTP (Roche Diagnostics, Mannheim, Germany), 10 pmol of each oligonucleotide primer, 100–200 ng of genomic DNA and 1 U AmpliTaq Gold Taq polymerase (Applied Biosystems). After an initial denaturation for 10 min at 94 °C, 30 cycles (94°C for 20 s, 60°C for 10 s and 72°C for 20 s) were carried out and followed by a final extension at 72 °C for 5 min, in the presence of the GeneScan 500 Rox (Applera France, Villebon Sur Yvette, France) size standard and analyzed on an automated 3700 DNA fragments analyzer (Applied Biosystems). Each repeat number was calculated with GeneMapper (Applied Biosystems). To confirm the size of (GT)n repeats, selected samples were subjected to sequence analysis.
Statistical analysis
Associations between groups and specific classes of allele, as well as genotypes were analyzed by the two-tailed Fisher’s exact test. Odds ratios (OR) and the associated 95% confidence interval (CI) were calculated to assess the relative risk conferred by a particular allele or genotype. We also performed logistic regression analysis including the hemophilia-causing mutations, using the SAS software for Windows, version 9.3. In the logistic regression analysis, patients with inversions in intron 1 or intron 22 were pooled in the same group to reach a sufficient number of cases.29 Statistical significance was accepted at P<0.05.
Results
In the present study, microsatellite variability in the promoter of HO-1 was analyzed for its association with the development of FVIII inhibitors in 362 patients with severe hemophilia A. This case-controlled study consisted of 99 severe hemophilia A patients with FVIII inhibitors and 263 patients without inhibitors. We first investigated the association of the different severe hemophilia-causing mutations with inhibitor-development in our cohort (Table 1). As previously reported,29,30 missense mutations were associated with a significantly lower risk of inhibitor formation (OR 0.46, 95% CI 0.22–0.9, P=0.02).
Single nucleotide polymorphisms
We characterized the G(−1135)A and T(−413)A polymorphisms in the promoter of HO-1 in 248 patients. The distribution of each genotype complied with the Hardy-Weinberg equilibrium in both cases and controls. Allele and genotype frequencies were similar to those previously described.31 No significant association between the genotypes or the individual allele frequencies, and the development of inhibitors was observed, in agreement with a recently published genome-wide association study by Astermark et al.32 (Online Supplementary Table S1; P>0.05).
Allele frequencies at the polymorphic locus
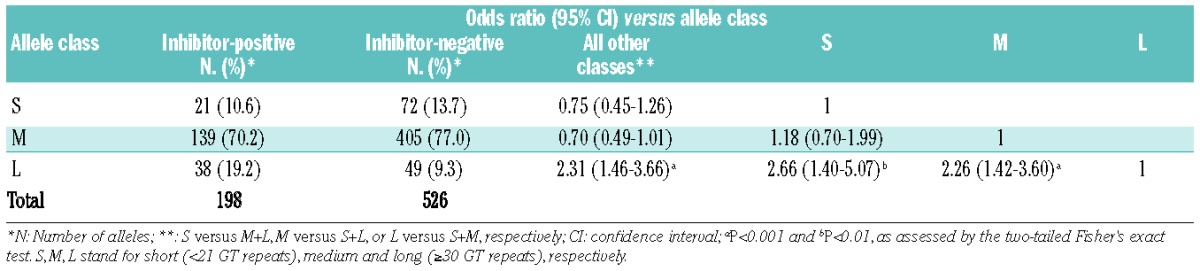
The number of (GT)n repeats in the HMOX1 gene ranged between 14 and 38 among the 362 subjects studied (Figure 1). As described earlier,33 the distribution of the numbers of (GT)n showed a bimodal pattern with two main peaks at 21 GT repeats (15.2% for inhibitor-positive patients and 14.3% for inhibitor-negative patients) and 29 GT repeats (24.7% for inhibitor-positive patients and 24.5% for inhibitor-negative patients). Based on the frequency distribution in our cohort of patients, and as described in previous reports,33–35 we divided the alleles into three subclasses: class S (<21 repeats); class M (21–29 repeats) and class L (≥30 repeats) alleles. The χ2 test for independence between allelic classes confirmed the Hardy-Weinberg equilibrium in both the inhibitor-negative and inhibitor-positive groups. Of the 526 alleles among patients without FVIII inhibitor, 72 (13.7%), 405 (77.0%) and 49 (9.3%) alleles were classes S, M and L, respectively (Table 2), whereas the distribution of the 198 alleles among patients with FVIII inhibitors was 21 (10.6%), 139 (70.2%) and 38 (19.2%) alleles for classes S, M and L, respectively. The comparative results showed a positive association of class L alleles with inhibitor formation (19.2% versus 9.3%). The OR for class L versus all other classes (M+S) was 2.31 (95% CI 1.46–3.66; P<0.001) (Table 2).
Figure 1.
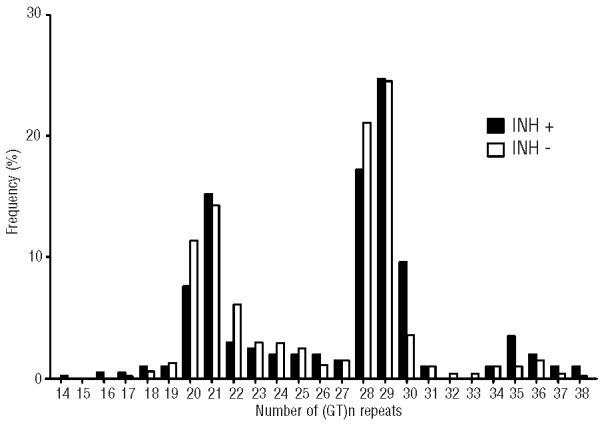
Frequency distribution of the number of GT repeats in patients with severe hemophilia A with or without FVIII inhibitors. Frequency distribution of the number of GT repeats in patients with severe hemophilia A (HA) with (black bars, INH+) or without (whites bars, INH−) FVIII inhibitors. The numbers of GT repeats ranged from 14 to 38 and showed a bimodal distribution, with one peak located at 21 repeats and the other located at 29 repeats.
Table 2.
HO-1 (GT)n microsatellite distribution in patients with hemophilia A.
Genotypic frequencies in patients with or without factor VIII inhibitors
Six genotypes (L/L, L/M, L/S, M/M, M/S and S/S) of (GT)n repeats were identified (Table 3). The L/L genotype was significantly associated with an increased prevalence of inhibitor development (OR 4.9, 95% CI 1.21–23.37; P=0.01), although the results may be biased by the small number of cases in each group (e.g., 7 inhibitor-positive and 4 inhibitor-negative patients in the L/L genotype). In order to reach an acceptable number of cases, genotypes were divided into two subgroups according to their allele subclasses: group I with a class L allele (L/L, L/M and L/S) and group II without a class L allele (M/M, S/S and S/M). The proportion of genotypic frequencies in group I was significantly higher in patients with FVIII inhibitors (31.3% versus 17.1%, OR 2.21, 95% CI 1.30–3.76; P<0.01) (Table 4). Importantly, the association remained significant when we controlled for hemophilia-causing mutations in a multivariable logistic regression (adjusted OR 2.13, 95% CI 1.24–3.64; P=0.006).
Table 3.
Distribution of genotypes.
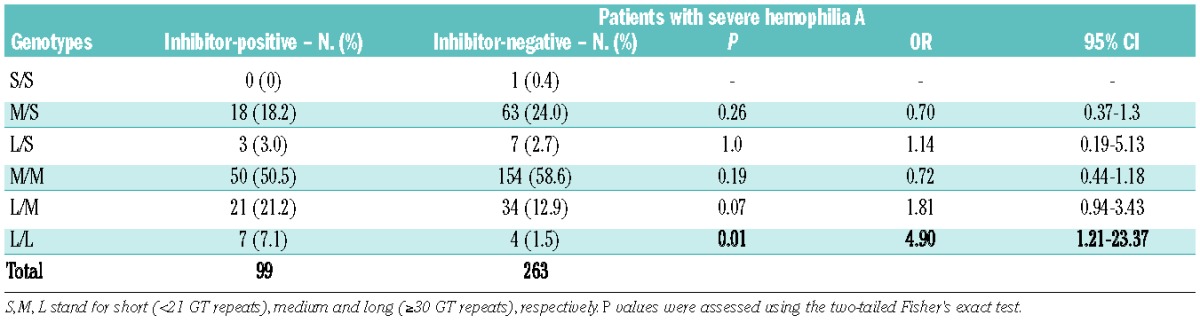
Table 4.
Genotype frequencies at the polymorphic locus.

Discussion
Inhibitory anti-FVIII IgG develop in up to 30% of patients with hemophilia A following replacement therapy with exogenous FVIII. The immune response to exogenous FVIII is not exclusively explainable by the absence of the endogenous FVIII antigen, and by the ensuing defect in negative selection of FVIII-specific naïve T lymphocytes in the thymus and/or generation of regulatory T cells. Indeed, a substantial percentage of patients with mild/moderate hemophilia A, who present with detectable levels of circulating endogenous dysfunctional FVIII antigen, also develop FVIII inhibitors. Initiation of the immune response presumably requires the presence of stimulatory signals, i.e., “danger signals”, which promote the maturation of the antigen-presenting cells, at the time of presentation of FVIII-derived peptides to naïve T lymphocytes. The nature of the signal(s) responsible for the initiation of the anti-FVIII immune response in hemophilia A patients is not known.
A recent genome-wide association study evaluated 13,331 single nucleotide polymorphisms from 1,081 genes using the Illumina iSelect platform for the association with inhibitor development in patients with hemophilia A. The study group included 833 subjects from three independent cohorts. The authors identified 53 single nucleotide polymorphisms as significant predictors of inhibitor status, thus highlighting the complexity of the anti-FVIII immune response.32 However, the genome-wide association study did not find associations of single nucleotide polymorphisms located in the promoter of the HMOX1 gene with the inhibitor status of the patients, which is in agreement with the present study. Of note, the Illunina iSelect platform is not designed to investigate microsatellite polymorphisms, thus explaining why the genome-wide association study failed to identify dinucleotide length polymorphisms associated with the FVIII inhibitor status of the patients.
We and others have recently hypothesized that inflammation, either chronic due to recurrent bleeding in joints or acute during surgery or following FVIII administration, may play an adjuvant role in the anti-FVIII immune response.3,6,7,36 Intra-articular bleeding is the most common clinical manifestation of hemophilia A, and can adversely affect joints and lead to arthropathy.37 The pathogenesis of arthropathy in hemophilia resembles inflammatory and degenerative joint disease, and is characterized by cartilage degeneration and deposition of iron, interleukin-1, interleukin-6 and tumor necrosis factor-α in the synovium.38 Our finding of an association between polymorphisms in the promoter of the HMOX1 gene and the occurrence of FVIII inhibitors supports the hypothesis of a role of inflammation in inhibitor development, and is reminiscent of results obtained in patients with rheumatoid arthritis. Thus, HO-1 (GT)n microsatellite was identified as a genetic marker involved in rheumatoid arthritis.31
The role of HO-1 in the anti-FVIII immune response was recently investigated in FVIII-deficient mice.21 The induction of HO-1 by heme, or the administration of the catabolic heme products (CO, biluribin), drastically reduced the intensity of the anti-FVIII immune response.21 Interestingly, the recurrent bleeds that characterize hemophilia A in humans are not observed in FVIII-deficient mice, and hemophilic mice do not spontaneously develop signs of arthropathy. Furthermore, unlike the mouse model of hemophilia A, in which HO-1 was induced by systemic administration of heme, bleeds in patients take place in localized sites of the body (e.g., joints) and are accompanied by confined inflammation-mediated tissue damage.
HO-1 expression is mainly regulated at the transcriptional level and the capacity to induce HO-1 is modulated, at least in part, by the number of GT repeats in the promoter region of the HMOX1 gene. Although the exact cut-off in the number of GT repeats determining optimal HO-1 induction remains undetermined, in vitro experiments suggest a progressive decrease in levels of expression with increasing numbers of GT repeats in the promoter region. Thus, the transfection of rat aortic smooth muscle cells with the 5′-flanking regions of the human HO-1 gene containing 22 GT repeats induced greater levels of expression of the reporter gene than transfection with the 5′-flanking regions of the gene containing 26 or 30 GT repeats.34 It is possible that, in hemophilia A patients with shorter (GT)n repeats in the promoter region of the HMOX1 gene, mild inflammatory signals at the sites of injury are sufficient to induce HO-1, thus reducing the maturation of antigen-presenting cells and decreasing the probability of initiating an anti-FVIII immune response.39,40 Conversely, similar recurrent joint bleeding in patients with long (GT)n repeats would fail to induce sufficient levels of HO-1, and would leave the patients at a greater risk of developing FVIII inhibitors. The present set of data suggests that the capacity of patients with hemophilia A to cope with inflammation, whether induced by recurrent hemorrhages or by other yet uncharacterized stimuli, may be one of the critical parameters that control the development of the immune response to FVIII. In support of this, direct evidence that long (GT)n repeats are associated with a lesser capacity to express HO-1 has been provided in the case of non-hemophilic individuals.26,41,42
We have demonstrated that the administration of CO-releasing molecules (CORM) to FVIII-deficient mice prior to the injection of therapeutic FVIII reduces the anti-FVIII immune response,21 thus indicating that CO administration to a living organism (under non-toxic conditions) protects against inhibitor development. In vivo, CO may be delivered either as a gas or by the use of CORM. Both gaseous CO and CORM have shown protective effects in animal models of vascular disease, organ transplant and inflammatory syndromes.43 Our results pave the way towards the use of CO or CO-delivering molecules as novel therapeutic tools to prevent inhibitor development.
Acknowledgments
We thank Robert Calvora (Baxter, Maurepas, France) for his trust and support. We also thank Ludivine Launay for statistical analyses. The research leading to these results was conducted as part of the ABIRISK consortium (Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize the risk). For further information please refer to www.abirisk.eu
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Université Pierre et Marie Curie - Paris 6, Agence Nationale de la Recherche (ANR-07-MRAR-028-01), and the Innovative Medicines Initiative Joint Undertaking under grant agreement n. 115303, with resources from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution. JDD and SD were recipients of fellowships from the Fondation de la Recherche Médicale (FRM, Paris). IP was the recipient of a fellowship from the Ministère de la Recherche (France).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ehrenforth S, Kreuz W, Scharrer I, Linde R, Funk M, Güngör T, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992; 339(8793):594–8 [DOI] [PubMed] [Google Scholar]
- 2.Gouw SC, van den Berg HM. The multifactorial etiology of inhibitor development in hemophilia: genetics and environment. Semin Thromb Hemost. 2009;35(8):723–34 [DOI] [PubMed] [Google Scholar]
- 3.Astermark J, Oldenburg J, Carlson J, Pavlova A, Kavakli K, Berntorp E, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. Blood. 2006;108(12): 3739–45 [DOI] [PubMed] [Google Scholar]
- 4.Pavlova A, Delev D, Lacroix-Desmazes S, Schwaab R, Mende M, Fimmers R, et al. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-alpha and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. 2009;7(12):2006–15 [DOI] [PubMed] [Google Scholar]
- 5.Astermark J, Oldenburg J, Pavlova A, Berntorp E, Lefvert AK. Polymorphisms in the IL10 but not in the IL1beta and IL4 genes are associated with inhibitor development in patients with hemophilia A. Blood. 2006;107(8):3167–72 [DOI] [PubMed] [Google Scholar]
- 6.Lacroix-Desmazes S, Navarrete AM, Andre S, Bayry J, Kaveri SV, Dasgupta S. Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood. 2008;112(2):240–9 [DOI] [PubMed] [Google Scholar]
- 7.Eckhardt CL, van der Bom JG, van der Naald M, Peters M, Kamphuisen PW, Fijnvandraat K. Surgery and inhibitor development in hemophilia A: a systematic review. J Thromb Haemost. 2011;9(10):1948–58 [DOI] [PubMed] [Google Scholar]
- 8.Skupsky J, Zhang AH, Su Y, Scott DW. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. 2009;114(21):4741–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meeks SL, Cox CL, Healey JF, Parker ET, Doshi BS, Gangadharan B, et al. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. 2012;120 (12):2512–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–54 [DOI] [PubMed] [Google Scholar]
- 11.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8(3):240–6 [DOI] [PubMed] [Google Scholar]
- 12.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86(2):583–650 [DOI] [PubMed] [Google Scholar]
- 13.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacological reviews. 2008;60(1):79–127 [DOI] [PubMed] [Google Scholar]
- 14.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, et al. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. 2003;55 (3):551–71 [DOI] [PubMed] [Google Scholar]
- 15.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2(1):87–90 [DOI] [PubMed] [Google Scholar]
- 16.Soares MP, Marguti I, Cunha A, Larsen R. Immunoregulatory effects of HO-1: how does it work? Curr Opin Pharmacol. 2009;9 (4):482–9 [DOI] [PubMed] [Google Scholar]
- 17.Takeda Y, Takeno M, Iwasaki M, Kobayashi H, Kirino Y, Ueda A, et al. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin Exp Immunol. 2004;138(2):237–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117 (2):438–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. 1998;4(9): 1073–7 [DOI] [PubMed] [Google Scholar]
- 20.Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165(3):1045–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimitrov JD, Dasgupta S, Navarrete AM, Delignat S, Repesse Y, Meslier Y, et al. Induction of heme oxygenase-1 in factor VIII-deficient mice reduces the immune response to therapeutic factor VIII. Blood. 2010;115(13):2682–5 [DOI] [PubMed] [Google Scholar]
- 22.Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. 2004;37(8):1097–104 [DOI] [PubMed] [Google Scholar]
- 23.Kimpara T, Takeda A, Watanabe K, Itoyama Y, Ikawa S, Watanabe M, et al. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum Genet. 1997;100 (1):145–7 [DOI] [PubMed] [Google Scholar]
- 24.Okinaga S, Takahashi K, Takeda K, Yoshizawa M, Fujita H, Sasaki H, et al. Regulation of human heme oxygenase-1 gene expression under thermal stress. Blood. 1996;87(12):5074–84 [PubMed] [Google Scholar]
- 25.Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet. 2000;66(1):187–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taha H, Skrzypek K, Guevara I, Nigisch A, Mustafa S, Grochot-Przeczek A, et al. Role of heme oxygenase-1 in human endothelial cells: lesson from the promoter allelic variants. Arterioscler Thromb Vasc Biol. 2010;30(8):1634–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73:247–51 [PubMed] [Google Scholar]
- 28.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gouw SC, van den Berg HM, Oldenburg J, Astermark J, de Groot PG, Margaglione M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119(12):2922–34 [DOI] [PubMed] [Google Scholar]
- 30.Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia. 2002;8 (Suppl 2):23–9 [DOI] [PubMed] [Google Scholar]
- 31.Rueda B, Oliver J, Robledo G, Lopez-Nevot MA, Balsa A, Pascual-Salcedo D, et al. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum. 2007;56(12):3953–8 [DOI] [PubMed] [Google Scholar]
- 32.Astermark J, Donfield SM, Gomperts ED, Schwarz J, Menius ED, Pavlova A, et al. The polygenic nature of inhibitors in hemophilia A: results from the Hemophilia Inhibitor Genetics Study (HIGS) combined cohort. Blood. 2013;121(8):1446–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arredondo M, Jorquera D, Carrasco E, Albala C, Hertrampf E. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with iron status in persons with type 2 diabetes mellitus. Am J Clin Nutr. 2007;86(5):1347–53 [DOI] [PubMed] [Google Scholar]
- 34.Chen YH, Lin SJ, Lin MW, Tsai HL, Kuo SS, Chen JW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet. 2002;111(1):1–8 [DOI] [PubMed] [Google Scholar]
- 35.Hausmann M, Paul G, Kellermeier S, Frey I, Scholmerich J, Falk W, et al. (GT)N dinucleotide repeat polymorphism of haem oxygenase-1 promotor region is not associated with inflammatory bowel disease risk or disease course. Clin Exp Immunol. 2008;153 (1):81–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skupsky J, Su Y, Lei TC, Scott DW. Tolerance induction by gene transfer to lymphocytes. Curr Gene Ther. 2007;7(5):369–80 [DOI] [PubMed] [Google Scholar]
- 37.Hoots WK. Pathogenesis of hemophilic arthropathy. Semin Hematol. 2006;43(1 Suppl 1):S18–22 [DOI] [PubMed] [Google Scholar]
- 38.Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010;8(9): 1895–902 [DOI] [PubMed] [Google Scholar]
- 39.Chauveau C, Remy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106(5):1694–702 [DOI] [PubMed] [Google Scholar]
- 40.Remy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ, et al. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol. 2009;182(4):1877–84 [DOI] [PubMed] [Google Scholar]
- 41.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, et al. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood. 2003;102(5):1619–21 [DOI] [PubMed] [Google Scholar]
- 42.Brydun A, Watari Y, Yamamoto Y, Okuhara K, Teragawa H, Kono F, et al. Reduced expression of heme oxygenase-1 in patients with coronary atherosclerosis. Hypertens Res. 2007;30(4):341–8 [DOI] [PubMed] [Google Scholar]
- 43.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9(9):728–43 [DOI] [PubMed] [Google Scholar]