

Abstract
During bone formation, osteoblasts deposit an extracellular matrix (ECM) that is mineralized via a process involving production and secretion of highly specialized matrix vesicles (MVs). Activin A, a transforming growth factor-β (TGF-β) superfamily member, was previously shown to have inhibitory effects in human bone formation models through unclear mechanisms. We investigated these mechanisms elicited by activin A during in vitro osteogenic differentiation of human mesenchymal stem cells (hMSC). Activin A inhibition of ECM mineralization coincided with a strong decline in alkaline phosphatase (ALP1) activity in extracellular compartments, ECM and matrix vesicles. SILAC-based quantitative proteomics disclosed intricate protein composition alterations in the activin A ECM, including changed expression of collagen XII, osteonectin and several cytoskeleton-binding proteins. Moreover, in activin A osteoblasts matrix vesicle production was deficient containing very low expression of annexin proteins. ECM enhanced human mesenchymal stem cell osteogenic development and mineralization. This osteogenic enhancement was significantly decreased when human mesenchymal stem cells were cultured on ECM produced under activin A treatment. These findings demonstrate that activin A targets the ECM maturation phase of osteoblast differentiation resulting ultimately in the inhibition of mineralization. ECM proteins modulated by activin A are not only determinant for bone mineralization but also possess osteoinductive properties that are relevant for bone tissue regeneration.
The quality of bone tissue is determined by the balanced action of the anabolic bone cells, the osteoblasts, and their catabolic counterparts, the osteoclasts. This process of bone remodeling occurs throughout life and can be influenced by a wide variety of molecules, having ultimately an impact on the quality of bone (1, 2). Activins and inhibins are members of the TGF-β superfamily with predominant antagonistic effects in their classically known target tissues, such as in gonadotropin producing cells in the pituitary and their role in reproduction (3, 4). Like other TGF-β member, activins elicit biological responses by binding to type I and II serine/threonine kinase receptors at the cell surface. Upon ligand binding, signaling is further transduced in the cytoplasm by phosphorylated Smad protein complexes that once in the nucleus regulate gene expression. This signaling pathway is highly complex because of crosstalk between different ligands (Activins, BMPs, TGF-β) binding to multiple serine/threonine kinase receptors that activate different Smad proteins signaling to the nucleus. Activin is known to signal using type II receptors ACVR2A or ACVR2B and the type I receptor ACVRIB (shared with BMPs) activating Smad2 and 3 proteins (shared with TGF-β). Inhibins exert their inhibitory effects on activin by competitive binding to the activin receptors in the presence of betaglycan. This signaling regulates a wide array of biological activities from cell proliferation, differentiation to tumor development and endocrine signaling (5, 6) in many cell lineages like hematopoietic (7, 8) and monocyte/macrophage (9, 10). Several consequences of these reproductive hormones, especially those of activin A, are also described in relation to bone metabolism. Activin A is present in bone tissue (11, 12) affecting both osteoclasts and osteoblasts. While having a consistent pro-osteoclastogenic effect (9, 13), the activin A impact on osteoblast differentiation is more controversial (see (14) for review) Several reports support a stimulatory effect of activin A on osteoblast differentiation and mineralization in vitro and in vivo (9, 15, 16). On the other hand, two different studies, using rat and human bone formation models, have demonstrated that activin A treatment has a coherent inhibitory influence on osteogenesis leading to significant reduction of the mineralization capacity (11, 17). These opposing effects of activin A on osteoblastogenesis may simply reflect species differences, however, it may be also driven by heterogeneity of the used cell model or the stage of osteoblast differentiation (14). Nevertheless, a negative role of activin A in bone formation is also supported by other in vivo studies in mice and primates in which blockage of activin signaling resulted in increased bone mass (18, 19). Moreover, transgenic mice overexpressing human inhibin A showed increased bone formation (20).
The extracellular compartment is crucial for bone because it determines most of the bone quality properties (21, 22), including its strength, stability, and integrity. Interestingly, a mature extracellular matrix (ECM) is characterized by the capacity to mineralize even in the absence of further osteoblast activity (11, 23). This biomineralization process is complex and not fully elucidated but it is thought to be started within MVs (24). Osteoblasts in bone and other cells in mineralization competent tissues, such as cartilage (25), tendon (26), teeth (27), and calcifying vasculature (28) produce and release from their plasma membrane these vesicles with diameters ranging between 50 and 200 nm. It is inside these membrane-enclosed particles that first crystals of mineral are formed and grow, before the vesicle membrane is permeated and the mineral crystallization advances into the ECM (29, 30). In this context, proteins that can mobilize calcium and inorganic phosphate (Pi), the backbone of the hydroxyapatite crystals present in bone, are of utmost importance. Pi donor proteins found in MVs include alkaline phosphatase (ALP) and inorganic pyrophosphatases (31) whereas the annexin family of proteins is postulated to be crucial for calcium influx into the vesicles (32–34).
In this study we investigated the inhibitory effect of activin A on human mesenchymal stem cells (hMSC) derived osteoblast differentiation and mineralization. We have previously shown that in human osteoblast cultures activin A influences the expression of many ECM genes altering ECM maturity (11). Thus, we focused our analysis on extracellular environment changes, namely the ECM and matrix vesicles (MVs). The characterization of these compartments was done using the state-of-the-art quantitative proteomics tools including SILAC metabolic labeling and mass spectrometry. Furthermore, the importance of ECM composition for osteoblast differentiation was also determined.
EXPERIMENTAL PROCEDURES
Cell Culture
Human bone marrow-derived mesenchymal stem cells (hMSC; PT-2501, Lonza, Walkersville, MD) from two different donors, at passages 4 and 5, were cultured as described previously (11) in 12-well (3.8 cm2) plates (Greiner bio-one, Frickenhausen, Germany), 75 or 175 cm2 flasks (Greiner bio-one). The hMSC culture medium was freshly supplemented with 100 nm dexamethasone (DEX, Sigma) and 10 mm β-glycerophosphate (Sigma, St. Louis, MO) for the osteogenic (vehicle) condition. Activin A condition contained an extra supplement of 25 ng/ml of activin A (R&D Systems, Minneapolis, MN). For quantitative mass spectrometry analysis, hMSC were cultured similarly in the presence of SILAC medium: arginine- and lysine-free Dulbecco's modified Eagle's medium (DMEM)/F12-Flex supplemented with 10% dialyzed fetal bovine serum (FBS), 4 mm l-glutamine, penicillin-streptomycin, 4.5 g/L glucose, 20 mm HEPES (Sigma), 1.8 mm CaCl2.2H2O (Sigma), pH 7.5. Cells were expanded in both light medium, supplemented with normal l-lysine HCL (12C6,14N2-Lys) and l-arginine (12C6,14N4-Arg), and heavy medium, containing heavy isotopes of the same amino acids (13C6,14N2-Lys and 13C6,15N4-Arg). Complete incorporation of the amino acid isotopes occurred within 3 weeks of cell culture in SILAC medium (supplemental Fig. S1). Vehicle and activin A treated cells were cultured in light and heavy isotope medium reciprocally allowing an optimal differentiation of artifacts and contaminants from biological variation (35). All SILAC reagents were purchased from Invitrogen (Carlsbad, CA) unless stated otherwise.
Devitalization of Cell Cultures
Cell devitalization was performed as described previously (11) using cells cultured in 12-well plates. Briefly, just before the onset of mineralization cell cultures were washed twice in PBS (Invitrogen, Carlsbad, CA), air dried and frozen at −20 °C for at least 24 h. The stage just before the onset of mineralization is the period preceding the detection of significant mineral deposition in culture, occurring between day 10 and 12 or day 15 and 17, depending on the hMSC donor. Next, the devitalized cultures containing an acellular 2D ECM synthesized during vehicle (vehicle ECM) or activin A stimulus (activin A ECM) were incubated in osteogenic medium only (+ medium). In another set of experiments, the devitalized cultures were incubated with freshly seeded undifferentiated hMSC in osteogenic medium (+hMSC) without further addition of activin A. In parallel, as a control for these experiments, hMSC in osteogenic medium were seeded on standard plastic plates (plastic). All experiments were followed further until mineralization. At the end of the cultures we performed mineralization assays.
Alkaline Phosphatase Activity, Protein, and Mineralization Assays
ALP activity and calcium content in cell extracts or isolated MVs were determined as described previously (36). Briefly, ALP activity was assayed by determining the release of paranitrophenol from paranitrophenylphosphate (20 mm diethanolamine buffer supplemented with 1 mm MgCl2 at pH 9.8) in the cell lysates and in MVs for 10 and 70 min at 37 °C respectively. Adsorption was measured at 405 nm. For calcium measurements, cell lysates were incubated overnight in 0.24 m HCl at 4 °C. Calcium content was colorimetrically determined with a calcium assay kit (Sigma) according to the manufacturer's instructions. For mineralization staining, cell cultures were fixed for 60 min with 70% ethanol on ice. After fixation, cells were washed twice with PBS and stained for 10 min with Alizarin Red solution (saturated Alizarin Red in demineralized water was titrated to pH 4.2 using 0.5% ammonium hydroxide). For protein concentration measurements, a BCA kit (Pierce Biotechnology, Rockford, IL, USA) was used following the manufacturer's instructions.
Extracellular Matrix Isolation
The ECM isolation method was adapted from (37). To extract proteins from the ECM, vehicle and activin A treated hMSC were cultured in 175 cm2 flasks. After removing the medium, cells were washed three times in PBS and incubated in collagenase/dispase (1 mg/ml; Roche, Mannheim, Germany) for 90 min at 37 °C. After centrifugation at 500 × g for 10 min, to remove cells, the supernatant containing the ECM protein extract was obtained and stored at −80 °C until analysis.
Matrix Vesicle Isolation
MVs were isolated from the medium of 75 or 175 cm2 culture flasks for subsequent FACS or proteomics analysis respectively, as described previously (38). Briefly, the culture medium was first centrifuged at 20,000 × g for 30 min at 4 °C, to remove cell debris. The supernatant was collected and further centrifuged at 100,000 × g for 60 min at 4 °C. After discarding the supernatant, the pellet containing MVs was dissolved in PBS. All ultracentrifugation steps were performed on an Ultracentrifuge l-70 (Beckmann Coulter).
Quantitative Mass Spectrometry Analysis
Protein extracts from SILAC cultures were mixed in a 1:1 ratio of the light (Vehicle or activin A) and heavy (Vehicle or activin A) condition for both the ECM and MVs isolated. The combined light:heavy samples were resolved by one-dimensional SDS-PAGE (NuPAGE 4–12% Bis-Tris Gel, Invitrogen) in duplicate. Protein bands were visualized with Coomassie staining (Bio-safe Coomassie, Bio-Rad, Hercules, CA). SDS-PAGE gel lanes were cut into 2-mm slices using an automatic gel slicer and subjected to in-gel reduction with dithiothreitol, alkylation with iodoacetamide and digestion with trypsin (Promega, sequencing grade), essentially as described by Wilm et al. (39). Nanoflow LC-MS/MS was performed on an 1100 series capillary LC system (Agilent Technologies) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo) operating in positive mode and equipped with a nanospray source. Peptide mixtures were trapped on a ReproSil C18 reversed phase column (Dr Maisch GmbH; column dimensions 1.5 cm × 100 μm, packed in-house) at a flow rate of 8 μl/min. Peptide separation was performed on ReproSil C18 reversed phase column (Dr Maisch GmbH; column dimensions 15 cm × 50 μm, packed in-house) using a linear gradient from 0 to 80% B (A = 0.1% formic acid; B = 80% (v/v) acetonitrile, 0.1% formic acid) in 120 min and at a constant flow rate of 200 nl/min using a splitter. The column eluent was directly sprayed into the ESI source of the mass spectrometer. Mass spectra were acquired in continuum mode; fragmentation of the peptides was performed in data-dependent mode. Data analysis was performed either by using the Mascot search algorithm or the MaxQuant suite. For Mascot searches, peak lists were automatically created from raw data files using the Mascot Distiller software (version 2.2; MatrixScience). The Mascot search algorithm (version 2.2, MatrixScience) was used for searching against the International Protein Index (IPI) database (IPI human release 06/11/2009, version 3.66). The peptide tolerance was typically set to 10 ppm and the fragment ion tolerance to 0.8 Da. A maximum number of 2 missed cleavages by trypsin were allowed and carbamidomethylated cysteine and oxidized methionine were set as fixed and variable modifications, respectively. The Mascot score cut-off value for a positive protein hit was set to 65. Individual peptide MS/MS spectra with Mascot scores below 35 were checked manually and either interpreted as valid identifications or discarded. For quantitative analysis, the mass spectrometric raw data from MaxQuant software suite (version 1.1.1.25) was used (40). A false discovery rate (FDR) of 0.01 for proteins and peptides and a minimum peptide length of six amino acids were required. The mass accuracy of the precursor ions was improved by the time-dependent recalibration algorithm of MaxQuant. The Andromeda search engine was used to search the MS/MS spectra against the IPI human database concatenated with the reversed versions of all sequences. A maximum of two missed cleavages were allowed. The fragment mass tolerance was set to 0.6 Da. Enzyme specificity was set to trypsin. Further modifications were cysteine carbamidomethylation (fixed) as well as protein N-terminal acetylation, methionine oxidation and lysine ubiquitination (variable). Only proteins identified with at least two peptides and two quantification events were considered for analysis. MaxQuant automatically quantified SILAC peptides and proteins. SILAC protein ratios were calculated as the median of all peptide ratios assigned to the protein. In addition a posterior error probability for each MS/MS spectrum below or equal to 0.1 was required. In case the identified peptides of two proteins were the same or the identified peptides of one protein included all peptides of another protein, these proteins were combined by MaxQuant and reported as one protein group. Before statistical analysis, known contaminants (keratins and abundant proteins from bovine serum) and reverse hits were removed (41).
Flow Cytometry Analysis and Western Blot
The number of cell-secreted MVs and ALP+ MVs was determined by flow cytometry as described elsewhere (42). Briefly, 12.5 μl of freshly isolated MVs were incubated for 20 min in the dark with 12.5 μl ELF-97 staining solution (0.2 m ELF-97 (Invitrogen) in 1.1 m acetic acid, 0.011 m NaNO2, pH 8.0). ELF-97 is a phosphatase substrate, which at pH 8 detects alkaline phosphatase. As negative control, the ELF-97 staining solution was replaced by PBS (unstained MV) and MVs were replaced by PBS or culture medium processed as described for MV isolation. Gating for ALP+ population was set using the unstained MV samples. For each of these mixes 125 μl PBS were added. Vesicles were measured in a Becton Dickinson FACS-Canto (BD Bioscience) for diffraction of light in a right angle, side scatter (SSC) measuring granularity, and for ELF-97 fluorescence signal, AmCyan-A channel (488 nm). For Western blotting experiments MVs isolated from conditioned medium of vehicle and activin A treated cultures were loaded, separated by SDS-PAGE, and transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Biosciences, Buckinghamshire, U.K.). As a negative control vesicles isolated from plain medium were used. After blocking nonspecific signal with 5% BSA in TBS/0.1% Tween-20, the membrane was incubated with antibodies against Annexin A2 (1:1000, rabbit polyclonal to ANXA2, Abcam, Cat. Ab41803). Membranes were probed with secondary antibody, goat antirabbit IgG conjugated with IRDye 800CW (1:5000, LI-COR, Cat. 926–32211). Immunoreactive bands were visualized using the LI-COR Infrared Imaging System according to the manufacturer's instructions (Odyssey Lincoln, NE).
Gene Ontology Analysis
Gene Ontology (GO) analyses were obtained using DAVID Bioinformatics Resources 6.7 (43, 44). Only significantly (p < 0.05) enriched terms in comparison to whole genome background (DAVID default) were selected.
Statistical Analysis
All experiments were repeated at least three times. Values are mean ± S.D. and significance was calculated using a Student's t test.
RESULTS
Activin A Alters the Mineralization Competence of the ECM
Activin A signaling was previously shown to inhibit mineralization of human osteoblast cultures by altering the ECM maturity (11). The results from our current study corroborate this data. Activin A treatment resulted in decreased ALP activity followed by impaired mineral deposition in culture (Fig. 1A) but it did not alter the total number of cells in culture when compared with vehicle treated cultures (data not shown). The maturity of the ECM produced under the activin A stimulus was tested by evaluating whether mineralization could occur in the absence of living cells in acellular 2D ECMs. To this end, vehicle and activin A treated osteoblast cultures were devitalized just before the onset of mineralization (Fig. 1B; scheme), withdrawing living cells but keeping the ECM built up to that moment intact (11). We observed that supplementation of medium to the ECM was enough to induce mineralization of vehicle ECM (Fig. 1B). In contrast, ECM synthesized under activin A treatment failed to become mineralized (Fig. 1B). To evaluate the extracellular effects of activin A during osteoblast differentiation, we isolated ECM as well as MVs from vehicle and activin A osteoblast cultures.
Fig. 1.

Effect of activin A in osteoblast differentiation and mineralization. hMSC were cultured in the presence of osteogenic medium (vehicle) and osteogenic medium containing activin A. A, ALP activity and calcium content in these cultures (n = 3). At the end of culture the calcium content was also visualized by Alizarin Red S staining. B, Vehicle and activin A treated cells were cultured until just before the onset of mineralization, occurring between days 10 and 12. At this stage cell cultures were fixed. Devitalized cells were further incubated in osteogenic medium (n = 4). At the end of the culture (day 17), calcium content was measured and visualized by Alizarin Red S staining. Value means ± S.D. * p < 0.05; ** p < 0.01.
Activin A Significantly Reduces ALP Activity in ECM and in MVs
ECM and MVs from osteoblast cultures at the onset of mineralization were initially accessed for ALP activity. We observed that ALP activity was significantly reduced in ECM and MVs of activin A treated osteoblasts when compared with their vehicle-treated counterparts (Fig. 2). This result supported further the concept that activin A has a significant impact on the osteoblast extracellular milieu. Thus, we set out to analyze and compare the ECM and MVs proteome from vehicle and activin A treated cultures using a quantitative SILAC-based mass spectrometry approach.
Fig. 2.

Effect of activin A in ECM and MV ALP activity. A, ECM and (B) MVs isolated at the onset of mineralization from vehicle and activin A treated cultures were assayed for ALP activity (n = 3). Value means ± S.D. * p < 0.05 ** p < 0.01.
Activin A Induces Changes in ECM Composition and Strongly Reduces the Expression of MVs Markers
The analysis of the ECM resulted in the identification of 293 proteins (supplemental Table S1). We used Gene Ontology (GO) annotation enrichment analysis to categorize the identified proteins for their cellular localization (Fig. 3). The GO term ECM (GO:0044420) was found to be 3.7-fold enriched validating the ECM isolation method. Other over-represented protein groups included proteins associated with membrane structures, like organelle envelope proteins (4.8-fold; GO:0031967) or in close proximity to the membrane, like cell leading edge (4.3-fold; GO:0031252) and cell surface (3.9-fold; GO:0009986) proteins. Interestingly, also vesicle proteins (GO:0031982) were identified to be 4.3-fold over-represented in the ECM samples.
Fig. 3.

Over-represented cellular compartment annotations for the 293 proteins identified in the ECM. Numbers in front of the bars indicate fold-enrichment level. Numbers within bars indicate proteins identified as belonging to each cellular compartment term and within brackets those that were found to be up-/down-regulated in activin A condition in the ECM. Only significantly (p < 0.05) enriched gene ontology terms were considered for analysis.
In quantitative terms mass spectrometry analyses resulted in identification of 104 proteins over 1.5-fold regulated following activin A treatment. More than half of these proteins, 74 proteins, were down-regulated by activin A whereas only 30 proteins were up-regulated. The 10 strongest up and down-regulated proteins are shown in Table I. Collagen, type XII, alpha 1 (COL12A1) was the most up-regulated ECM protein (2.7-fold). Osteonectin (ON), fibronectin 1 (FN1) and fibrillin-1 (FBN) were also up-regulated by more than 1.9-fold (Table I). The strongest down-regulated proteins include enzymes participating in carbohydrate catabolism such as UDP-glucose pyrophosphorylase 2 (UGP2; 0.28-fold), phosphoglucomutase 1 (PGM1; 0.30-fold) and phosphogluconate dehydrogenase (PGD; 0.37-fold; Table I).
Table I. Proteins regulated by activin A signaling in the ECM. ECM was isolated at the onset of mineralization from SILAC labeled cultures and protein regulation was determined by mass spectrometry measurements. Ratios are averages of reciprocal SILAC experiments. A = Activin A; V = Vehicle. # peptides = minimum number of unique peptides identified for the protein. Ratio count = number of SILAC pairs used for quantitation. From the 293 proteins identified in the ECM 104 were found to be over 1.5-fold regulated but only the 20 strongest regulated proteins are shown (see supplementary Table 1 for the complete list of proteins identified and respective regulation).
| # | Protein name | IPI identifier | Gene symbol | Ratio A/V | # Peptides | Ratio count |
|---|---|---|---|---|---|---|
| >1.5-fold UP-regulated by activin A | ||||||
| 1 | Collagen alpha-1(XII) chain | IPI00329573 | COL12A1 | 2.66 | 52 | 89 |
| 2 | Developmentally-regulated endothelial cell locus 1 protein | IPI00306046 | DEL1 | 2.29 | 11 | 16 |
| 3 | CFR-1 | IPI00414717 | CFR1 | 2.24 | 10 | 11 |
| 4 | Basement-membrane protein 40 | IPI00014572 | ON | 2.20 | 2 | 4 |
| 5 | FN1 protein | IPI00845263 | FN1 | 2.11 | 23 | 57 |
| 6 | Breast epithelial antigen BA46 | IPI00966900 | MFGE8 | 2.07 | 2 | 26 |
| 7 | cDNA FLJ58980, highly similar to Sideroflexin-3 | IPI00871988 | hCG_24661 | 2.01 | 2 | 3 |
| 8 | 130 kDa leucine-rich protein | IPI00783271 | LRP130 | 2.00 | 6 | 12 |
| 9 | Fibrillin-1 | IPI00328113 | FBN | 1.97 | 13 | 19 |
| 10 | CD49 antigen-like family member E | IPI00306604 | FNRA | 1.93 | 7 | 16 |
| >1.5-fold DOWN-regulated by activin A | ||||||
| 1 | cDNA, FLJ95012, highly similar to Homo sapiens UDP-glucose pyrophosphorylase 2 | IPI00873223 | UGP2 | 0.28 | 6 | 7 |
| 2 | Glucose phosphomutase 1 | IPI00219526 | PGM1 | 0.30 | 4 | 4 |
| 3 | UDP-glucose 6-dehydrogenase | IPI00031420 | UGDH | 0.30 | 2 | 3 |
| 4 | 70 kDa lamin | IPI00021405 | LMN1 | 0.31 | 6 | 9 |
| 5 | Collapsin response mediator protein 2 | IPI00257508 | CRMP2 | 0.31 | 5 | 6 |
| 6 | Cell migration-inducing gene 10 protein | IPI00169383 | MIG10 | 0.34 | 12 | 22 |
| 7 | Triosephosphate isomerase | IPI00465028 | hCG_25936 | 0.34 | 6 | 9 |
| 8 | Calgizzarin | IPI00013895 | MLN70 | 0.34 | 2 | 3 |
| 9 | BPG-dependent PGAM 1 | IPI00549725 | CDABP0006 | 0.35 | 5 | 5 |
| 10 | 6-phosphogluconate dehydrogenase, decarboxylating | IPI00219525 | PGD | 0.37 | 5 | 4 |
In MVs we have identified 27 proteins (supplemental Table 2). In total, 12 proteins were ≥1.5-fold regulated by activin A (Table II). Of those 12, only two proteins were increased by activin A, hyaluronan binding protein CRTL1 (1.76-fold) and bone proteoglycan II (DCN; 1.51-fold). Among the 10 down-regulated proteins, five were calcium-dependent phospholipid binding proteins known as annexins. Despite markedly down-regulated in MVs (<0.25-fold; Table II), annexins were unchanged in the ECM. This MV specific modulation and the fact that these proteins are known to be present and functional in MVs (32–34) led us to consider that activin A cultures had severely altered or even compromised MV production.
Table II. Proteins regulated by activin A signaling in MVs. MV samples were isolated at the onset of mineralization from SILAC labeled cultures and protein regulation was determined by mass spectrometry measurements. Ratios are averages of reciprocal SILAC experiments. A = Activin A; V = Vehicle. # peptides = minimum number of unique peptides identified for the protein. Ratio count = number of SILAC pairs used for quantitation. From the 27 proteins identified in MVs 12 were found to be over 1.5-fold regulated (see supplementary Table 2 for the complete list of proteins identified and respective regulation).
| # | Protein name | IPI identifier | Gene symbol | Ratio A/V | # Peptides | Ratio count |
|---|---|---|---|---|---|---|
| >1.5-fold UP-regulated by activin A | ||||||
| 1 | Cartilage-linking protein 1 | IPI00023601 | CRTL1 | 1.76 | 4 | 4 |
| 2 | Bone proteoglycan II | IPI00012119 | DCN | 1.51 | 4 | 6 |
| >1.5-fold DOWN-regulated by activin A | ||||||
| 1 | Annexin A1 | IPI00218918 | ANX1 | 0.11 | 10 | 10 |
| 2 | Anchorin CII | IPI00329801 | ANX5 | 0.14 | 12 | 13 |
| 3 | 35-beta calcimedin | IPI00793199 | ANX4 | 0.15 | 2 | 3 |
| 4 | 67 kDa calelectrin | IPI00221226 | ANX6 | 0.23 | 20 | 38 |
| 5 | Annexin A2 | IPI00418169 | ANX2 | 0.24 | 16 | 30 |
| 6 | l-Lactate dehydrogenase | IPI00947127 | LDHA | 0.36 | 3 | 4 |
| 7 | 18 kDa phosphoprotein | IPI00012011 | CFL | 0.39 | 3 | 3 |
| 8 | Alanyl aminopeptidase | IPI00221224 | ANPEP | 0.46 | 9 | 12 |
| 9 | Lung resistance-related protein | IPI00000105 | LRP | 0.50 | 4 | 3 |
| 10 | Cytotactin | IPI00031008 | HXB | 0.60 | 7 | 7 |
Activin A Treated Cells Have Impaired MV Biogenesis at the Onset of Mineralization
To address the hypothesis that MV production is suppressed in osteoblasts following activin treatment, we isolated and counted MV number in vehicle and activin A treated osteoblast cultures. Number of total MVs and ALP positive MVs (ALP+ MVs) was determined using FACS. The production of MVs, total and ALP+, did not differ between vehicle and activin A conditions before the onset of mineralization (Fig. 4A). However, at the onset of mineralization the production of ALP+ MVs in vehicle osteoblasts increased sharply whereas in activin A condition this was significantly suppressed (Fig. 4A). At the onset of mineralization, only 8% of the total MVs were ALP+ in the activin A condition compared with the 72% in the vehicle condition (Fig. 4A and 4B). Immunodetection of annexin 2 in MVs (Fig. 5) confirmed the results obtained by quantitative proteomics and indicates that the differences measured are MV-specific and not derived from serum microvesicles. In summary, our results indicate a bimodal effect of activin A by changing the ECM composition and suppressing the provision of the bio-mineralization initiators, the MVs.
Fig. 4.

Activin A effect in osteoblast MV production. A, Number of MVs and ALP+ MVs secreted by vehicle and activin A treated cultures before and at the onset of mineralization. B, FACS plots used to calculate the number of MVs in vehicle and activin A samples, before and at the onset of mineralization (four leftmost panels). A histogram with the distribution of the ALP+ MV population before mineralization (n = 9) and at the onset of mineralization (n = 3) for vehicle and activin A cultures is also shown (right panel). The gating was set using ELF-97 unstained MV samples (bottom left panel). Other negative controls are shown for PBS and culture medium (bottom middle and bottom right panel respectively). SSC-A is the side scatter-area and AmCyan-A is the channel detecting fluorescence signal from stained vesicles. * p < 0.05; ** p < 0.01.
Fig. 5.
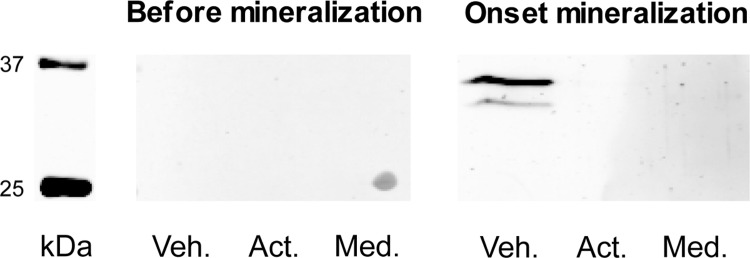
Immunodetection of annexin 2 in MVs. MV preparations from vehicle (Veh.) and activin A (Act.) cultures, before and at the onset of mineralization, were probed for annexin 2 expression. Med. = control sample prepared following the MV preparation protocol from plain medium is also shown.
Activin A ECM is Capable of Signaling to Osteoprogenitor Cells Modulating Their Mineralization
Next, we investigated further the impact of ECM composition in osteogenesis. For these experiments, vehicle and activin A treated cultures were devitalized just before the onset of mineralization similar as shown in Fig. 1B. However, this time besides only osteogenic medium we also seeded undifferentiated hMSCs in osteogenic medium on top of vehicle and activin A ECMs. In parallel as control cultures, hMSC in osteogenic medium were also seeded on plastic in standard culture plates (experimental scheme in Fig. 6A). Seeding of hMSC onto an existing ECM enhanced and anticipated the mineralization (Fig. 6B, vehicle or activin A ECM compared with plastic plates), now occurring already after 6–8 days instead of the 14 days for control cultures on plastic (Fig. 1A). Despite significantly less mineralized than hMSC on vehicle ECM this was also observed when hMSC were seeded on an activin A ECM, which was unable to mineralize in absence of hMSC (Fig. 6B).
Fig. 6.

Effect of the ECM in osteoprogenitor cell mineralization. A, Vehicle and activin A treated cultures were devitalized just before the onset of mineralization. The remaining ECMs were cultured with osteogenic medium (+ medium) and undifferentiated hMSC in osteogenic medium (+ hMSC). In parallel, hMSCs were also seeded directly on standard plastic cultures plates. B, Calcium content was assayed at the end of culture (n = 3). Value means ± S.D. ** p < 0.01; *** p < 0.001.
Altogether, these results demonstrate that the osteoblast ECM contains potent signaling clues to osteoprogenitor cells. In this respect, the altered composition of activin A ECM did not represent a complete block for mineralization by osteoprogenitor cells.
DISCUSSION
We have previously shown that human osteoblast mineralization is strongly inhibited in the presence of activin A and that these cells are particularly sensitive to this hormone at stages preceding the onset of mineralization (11). In this study we provide evidence for changes in protein production during activin A suppression of osteoblast mineralization. We demonstrate that activation of activin A signaling during bone formation has detrimental effects on the ECM production and maturation phase. Furthermore, negative effects of activin A are extensible to the mineralization phase by impairing MV production.
Activin A effect on the ECM proteome was predominantly suppressive, with two-times more down-regulated than up-regulated proteins. Among the latter we identified collagen XII alpha-1 (COL12A1; IPI00329573), osteonectin (ON; IPI00014572), fibronectin (FN1; IPI00845263), and fibrillin-1 (FBN; IPI00328113). All these proteins have been directly implicated in osteoblast differentiation and bone formation (45–48). For example, ON deficiency is responsible for compromised osteoblast differentiation and decreased bone formation (45, 49). Activin A modulation of ON gene expression in osteoblasts (11) and other tissues (50) is consistent with the ON protein regulation we observed despite failing to explain the inhibitory effects of activin A in osteoblast differentiation and mineralization. The complexity of activin A signaling is probably the reason behind these conflicting observations with other proteins overruling ON action during osteoblast differentiation. One of these proteins is fibrillin-1 (FBN). Mutations in the FBN gene are responsible for an increased skeletal size phenotype (51). FBN is a structural component of the ECM thought to regulate osteoblast maturation with its ability to sequester and control latent TGF-β and BMP bioavailability (48, 52). Being both TGF-β and BMP potent osteogenic factors (53, 54), elevated levels of FBN in the activin A ECM is likely to be determinant for the inhibition of osteoblast differentiation and mineralization observed in activin A.
Linking ECM proteins down-regulated by activin A to bone-related functions was more difficult than for those up-regulated. Many of the proteins inhibited by activin were cytoskeleton-binding proteins (e.g. WDR1, IPI00873622; PLS3, IPI00216694; TPM3, IPI00218319) and proteins involved in carbohydrate metabolism (UGDH, IPI00031420; MIG10, IPI00169383; LDHA, IPI00947127). The detection of proteins that bind to the cytoskeleton is possibly related to the ECM control of cytoskeleton mechanisms (55) recently recognized as important during osteoblast differentiation (56, 57). On the other hand, the detection of glucose metabolism proteins in the ECM is more enigmatic but corroborated in the osteoblast ECM proteome listed by Xiao et al. (58). Moreover, several other studies indicate the existence of extracellular glycolytic enzymes contained within different types of secreted vesicles (59, 60) including MVs (58). Despite disclosing an extracellular location for these proteins, their function outside the cell remains unknown. Nevertheless, the fact that they are down-regulated by activin A identifies them as potential modulators of bone formation.
ECM preparations contained various intracellular proteins and we cannot exclude that these represent contamination from cells. Despite not being able to exclude this hypothesis, we believe our extraction method is adequate because we find ECM proteins to be highly enriched (Fig. 3). Other hypothesis for the detection of intracellular proteins in the ECM could be the presence of MVs bound to the ECM (37, 61). Proteins of vesicle origin were enriched in our ECM extracts (Fig. 3). Due to their composition and intracellular origin, MVs represent a likely explanation to the increased numbers intracellular but also of the membrane-related proteins identified.
In general, activin A effect on the ECM proteome was vast with many proteins being targeted. The fact that no single protein was strikingly altered highlights the importance of considering ECM as an integrated compartment. In other words, the function of ECM is determined by the combination of all proteins present rather than individual proteins. Investigating the functions and interactions of these proteins during osteoblast differentiation is of significance to understand and identify therapeutic targets for bone formation. The more so considering that several proteins we identified have not yet been linked to bone.
Osteoblasts under the influence of activin A secreted significantly less MVs containing an altered protein composition. In terms of protein identifications the MV proteome was more limited than that of the ECM. This can be attributed to the relatively low abundance of MVs produced by osteoblasts and the lower complexity of the MV proteome in comparison to that of ECM. Nevertheless, in contrast to the ECM proteome, the MV proteome comprised more pronounced differences between activin A and vehicle cultures. Proteome analysis showed only two proteins as being more than 1.5-fold up-regulated by activin A in MVs, bone proteoglycan II (DCN; IPI00012119) and cartilage-linking protein 1 (CRTL1; IPI00023601) proteoglycans that bind to type I collagen fibrils (62) and hyaluronic acid (63) respectively. Increased expression of proteoglycans within MVs fit with the negative role of activin A in osteoblast mineralization. The negative charge of their glycosaminoglycan chains is able to compete with phosphate for hydroxyapatite crystallization inhibiting mineral growth (64, 65). DCN, for example is confined to the uncalcified osteoid area being removed from the ECM so calcification can take place (66).
Among the most down-regulated proteins in MVs, we found proteins directly associated with their mineralization competences. Annexin family members (I, II, IV, V and V) were the most noticeable, proteins usually high abundant in MVs where they act as calcium channels (32–34). Interestingly, annexins appear to be specifically regulated within MVs because no differences were found in the activin A ECM relative to its vehicle counterpart. MV biogenesis is still controversial (61) but the fact we detected proteins modulated within these vesicles that were not altered in the ECM is coherent with a selective and tightly regulated MV formation process.
Before onset of mineralization, the ECM produced by vehicle osteoblast cultures was mature enough to mineralize independently of further cellular activity. A possible explanation for this is the presence of MVs already attached to the fibrillar collagenous matrix (29), something corroborated by the detection of vesicle proteins in the ECM (Fig. 3). These MVs would allow mineral crystals to grow and propagate further to the ECM without additional osteoblast interference. The fact that activin A osteoblasts had impaired MV biogenesis explains the inability of activin A ECM to mineralize.
Osteoblast differentiation is a sequential order of interdependent events (67) thus the MV impairment is likely to be determined by a primary effect on the previous phase of ECM maturation. Furthermore, we believe that this effect on the ECM is unlikely to be because of a delay in osteoblast differentiation because differentiation marker genes remain unchanged by activin signaling in this bone formation model (11). Devitalization experiments in which acellular 2D ECMs were cultured with osteoprogenitor cells provided more information about maturity of the activin A ECM. Despite less effectively than vehicle, activin A ECM was found capable of enhancing osteoprogenitor cell mineralization, suggesting that activin A ECM still retained, albeit diminished compared with control ECM, capacity to enhance osteoblast differentiation. Because of its inability to support the late stages of mineralization we hypothesize that activin A ECM specifically stimulates the early stages of osteogenesis. Interestingly, collagen and FN1 are two proteins up-regulated in activin A ECM that fit particularly well in this role. Culture plates coated with collagen or FN1 have been shown to enhance osteoblast differentiation (68, 69).
In summary, we demonstrated that activin A is a strong modulator of the extracellular microenvironment of osteoblasts by (1) changing the ECM composition and maturity and (2) impairing production of MVs responsible for the start of bio-mineralization. This supports the concept that osteoblast differentiation is a synergistic process, with disruption of a specific stage (ECM maturation) having consequences for down-stream events (MVs biogenesis) and end point phenotype (mineralization). Furthermore, ECMs produced by osteoblasts were found to contain potent osteoinductive properties of particular interest for regenerative medicine. Proteins identified in the ECM to be regulated by activin A constitute potential targets to modulate mineralization and bone tissue quality.
Supplementary Material
Acknowledgments
We thank Marijke Koedam and Tanja Strini for the technical assistance provided with cell culture experiments.
Footnotes
* This work was supported by an ECTS Postdoctoral Fellowship award granted to M. Eijken in 2008, NWO-ZON (contract grant #91206069) and the Erasmus Medical Center, Rotterdam, The Netherlands.
 This article contains supplemental Fig. S1 and S2 and Tables S1 and S2.
This article contains supplemental Fig. S1 and S2 and Tables S1 and S2.
1 The abbreviations used are:
- ALP
- alkaline phosphatase
- BCA
- bicinchoninic acid
- DAVID
- Database for Annotation, Visualization and Integrated Discovery
- DEX
- dexamethasone
- DMEM/F12-Flex
- Dulbecco's Modified Eagle Medium: nutrient mixture F-12
- ECM
- extracellular matrix
- FBS
- fetal bovine serum
- FDR
- false discovery rate
- GO
- Gene Ontology
- hMSC
- human Mesenchymal Stem Cells
- IPI
- international protein index
- MV
- matrix vesicle
- PBS
- phosphate buffered saline
- Pi
- inorganic phosphate
- SILAC
- stable isotope labeling by amino acids in cell culture
- TGF-β
- transforming growth factor-β.
REFERENCES
- 1. Canalis E., McCarthy T., Centrella M. (1988) Growth factors and the regulation of bone remodeling. J. Clin. Invest. 81, 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manolagas S. C., Jilka R. L. (1995) Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med. 332, 305–311 [DOI] [PubMed] [Google Scholar]
- 3. Ling N., Ying S. Y., Ueno N., Esch F., Denoroy L., Guillemin R. (1985) Isolation and partial characterization of a Mr 32,000 protein with inhibin activity from porcine follicular fluid. Proc. Natl. Acad. Sci. U.S.A. 82, 7217–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vale W., Rivier J., Vaughan J., McClintock R., Corrigan A., Woo W., Karr D., Spiess J. (1986) Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 321, 776–779 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y. G., Wang Q., Lin S. L., Chang C. D., Chuang J., Ying S. Y. (2006) Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp Biol Med (Maywood). 231, 534–544 [DOI] [PubMed] [Google Scholar]
- 6. Risbridger G. P., Schmitt J. F., Robertson D. M. (2001) Activins and inhibins in endocrine and other tumors. Endocr. Rev. 22, 836–858 [DOI] [PubMed] [Google Scholar]
- 7. Broxmeyer H. E., Lu L., Cooper S., Schwall R. H., Mason A. J., Nikolics K. (1988) Selective and indirect modulation of human multipotential and erythroid hematopoietic progenitor cell proliferation by recombinant human activin and inhibin. Proc. Natl. Acad. Sci. U.S.A. 85, 9052–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lebrun J. J., Vale W. W. (1997) Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol. Cell. Biol. 17, 1682–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaddy-Kurten D., Coker J. K., Abe E., Jilka R. L., Manolagas S. C. (2002) Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology. 143, 74–83 [DOI] [PubMed] [Google Scholar]
- 10. Yamada R., Suzuki T., Hashimoto M., Eto Y., Shiokawa K., Muramatsu M. (1992) Induction of differentiation of the human promyelocytic cell line HL-60 by activin/EDF. Biochem. Biophys. Res. Commun. 187, 79–85 [DOI] [PubMed] [Google Scholar]
- 11. Eijken M., Swagemakers S., Koedam M., Steenbergen C., Derkx P., Uitterlinden A. G., van der Spek P. J., Visser J. A., de Jong F. H., Pols H. A., van Leeuwen J. P. (2007) The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 21, 2949–2960 [DOI] [PubMed] [Google Scholar]
- 12. Ogawa Y., Schmidt D. K., Nathan R. M., Armstrong R. M., Miller K. L., Sawamura S. J., Ziman J. M., Erickson K. L., de Leon E. R., Rosen D. M., et al. (1992) Bovine bone activin enhances bone morphogenetic protein-induced ectopic bone formation. J. Biol. Chem. 267, 14233–14237 [PubMed] [Google Scholar]
- 13. Fuller K., Bayley K. E., Chambers T. J. (2000) Activin A is an essential cofactor for osteoclast induction. Biochem. Biophys. Res. Commun. 268, 2–7 [DOI] [PubMed] [Google Scholar]
- 14. Nicks K. M., Perrien D. S., Akel N. S., Suva L. J., Gaddy D. (2009) Regulation of osteoblastogenesis and osteoclastogenesis by the other reproductive hormones, Activin and Inhibin. Molecular and cellular endocrinology. 310, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Centrella M., McCarthy T. L., Canalis E. (1991) Activin-A binding and biochemical effects in osteoblast-enriched cultures from fetal-rat parietal bone. Mol. Cell. Biol. 11, 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakai R., Fujita S., Horie T., Ohyama T., Miwa K., Maki T., Okimoto N., Nakamura T., Eto Y. (2000) Activin increases bone mass and mechanical strength of lumbar vertebrae in aged ovariectomized rats. Bone. 27, 91–96 [DOI] [PubMed] [Google Scholar]
- 17. Ikenoue T., Jingushi S., Urabe K., Okazaki K., Iwamoto Y. (1999) Inhibitory effects of activin-A on osteoblast differentiation during cultures of fetal rat calvarial cells. J. Cell. Biochem. 75, 206–214 [DOI] [PubMed] [Google Scholar]
- 18. Pearsall R. S., Canalis E., Cornwall-Brady M., Underwood K. W., Haigis B., Ucran J., Kumar R., Pobre E., Grinberg A., Werner E. D., Glatt V., Stadmeyer L., Smith D., Seehra J., Bouxsein M. L. (2008) A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl. Acad. Sci. U.S.A. 105, 7082–7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lotinun S., Pearsall R. S., Davies M. V., Marvell T. H., Monnell T. E., Ucran J., Fajardo R. J., Kumar R., Underwood K. W., Seehra J., Bouxsein M. L., Baron R. (2010) A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone. 46, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 20. Perrien D. S., Akel N. S., Edwards P. K., Carver A. A., Bendre M. S., Swain F. L., Skinner R. A., Hogue W. R., Nicks K. M., Pierson T. M., Suva L. J., Gaddy D. (2007) Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 148, 1654–1665 [DOI] [PubMed] [Google Scholar]
- 21. Baum J., Brodsky B. (1999) Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol. 9, 122–128 [DOI] [PubMed] [Google Scholar]
- 22. Fantner G. E., Birkedal H., Kindt J. H., Hassenkam T., Weaver J. C., Cutroni J. A., Bosma B. L., Bawazer L., Finch M. M., Cidade G. A., Morse D. E., Stucky G. D., Hansma P. K. (2004) Influence of the degradation of the organic matrix on the microscopic fracture behavior of trabecular bone. Bone. 35, 1013–1022 [DOI] [PubMed] [Google Scholar]
- 23. Fratzl-Zelman N., Fratzl P., Hörandner H., Grabner B., Varga F., Ellinger A., Klaushofer K. (1998) Matrix mineralization in MC3T3-E1 cell cultures initiated by beta-glycerophosphate pulse. Bone. 23, 511–520 [DOI] [PubMed] [Google Scholar]
- 24. Anderson H. C., Reynolds J. J. (1973) Pyrophosphate stimulation of calcium uptake into cultured embryonic bones. Fine structure of matrix vesicles and their role in calcification. Dev. Biol. 34, 211–227 [DOI] [PubMed] [Google Scholar]
- 25. Anderson H. C. (1969) Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 41, 59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arsenault A. L., Frankland B. W., Ottensmeyer F. P. (1991) Vectorial sequence of mineralization in the turkey leg tendon determined by electron microscopic imaging. Calcif. Tissue Int. 48, 46–55 [DOI] [PubMed] [Google Scholar]
- 27. Bernard G. W. (1972) Ultrastructural observations of initial calcification in dentine and enamel. J. Ultrastruct. Res. 41, 1–17 [DOI] [PubMed] [Google Scholar]
- 28. Tanimura A., McGregor D. H., Anderson H. C. (1983) Matrix vesicles in atherosclerotic calcification. Proc. Soc. Exp. Biol. Med. 172, 173–177 [DOI] [PubMed] [Google Scholar]
- 29. Anderson H. C., Garimella R., Tague S. E. (2005) The role of matrix vesicles in growth plate development and biomineralization. Front. Biosci. 10, 822–837 [DOI] [PubMed] [Google Scholar]
- 30. Morris D. C., Masuhara K., Takaoka K., Ono K., Anderson H. C. (1992) Immunolocalization of alkaline phosphatase in osteoblasts and matrix vesicles of human fetal bone. Bone Miner. 19, 287–298 [DOI] [PubMed] [Google Scholar]
- 31. Ali S. Y., Sajdera S. W., Anderson H. C. (1970) Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. U.S.A. 67, 1513–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen N. X., O'Neill K. D., Chen X., Moe S. M. (2008) Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J. Bone Miner. Res. 23, 1798–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genge B. R., Wu L. N., Wuthier R. E. (1989) Identification of phospholipid-dependent calcium-binding proteins as constituents of matrix vesicles. J. Biol. Chem. 264, 10917–10921 [PubMed] [Google Scholar]
- 34. Kirsch T., Harrison G., Golub E. E., Nah H. D. (2000) The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J. Biol. Chem. 275, 35577–35583 [DOI] [PubMed] [Google Scholar]
- 35. Wang Y. K., Ma Z., Quinn D. F., Fu E. W. (2002) Inverse 15N-metabolic labeling/mass spectrometry for comparative proteomics and rapid identification of protein markers/targets. Rapid Commun. Mass Spectrom. 16, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 36. Eijken M., Koedam M., van Driel M., Buurman C. J., Pols H. A., van Leeuwen J. P. (2006) The essential role of glucocorticoids for proper human osteoblast differentiation and matrix mineralization. Mol. Cell. Endocrinol. 248, 87–93 [DOI] [PubMed] [Google Scholar]
- 37. Xiao Z., Camalier C. E., Nagashima K., Chan K. C., Lucas D. A., de la Cruz M. J., Gignac M., Lockett S., Issaq H. J., Veenstra T. D., Conrads T. P., Beck G. R., Jr. (2007) Analysis of the extracellular matrix vesicle proteome in mineralizing osteoblasts. J. Cell. Physiol. 210, 325–335 [DOI] [PubMed] [Google Scholar]
- 38. Johnson K., Moffa A., Chen Y., Pritzker K., Goding J., Terkeltaub R. (1999) Matrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J. Bone Mineral Res. 14, 883–892 [DOI] [PubMed] [Google Scholar]
- 39. Wilm M., Shevchenko A., Houthaeve T., Breit S., Schweigerer L., Fotsis T., Mann M. (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469 [DOI] [PubMed] [Google Scholar]
- 40. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 41. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., Mann M. (2009) A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 [DOI] [PubMed] [Google Scholar]
- 42. Woeckel V. J., Alves R. D., Swagemakers S. M., Eijken M., Chiba H., van der Eerden B. C., van Leeuwen J. P. (2010) 1Alpha,25-(OH)2D3 acts in the early phase of osteoblast differentiation to enhance mineralization via accelerated production of mature matrix vesicles. J. Cell. Physiol. 225, 593–600 [DOI] [PubMed] [Google Scholar]
- 43. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 44. Hosack D. A., Dennis G., Jr., Sherman B. T., Lane H. C., Lempicki R. A. (2003) Identifying biological themes within lists of genes with EASE. Genome Biol. 4, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Delany A. M., Kalajzic I., Bradshaw A. D., Sage E. H., Canalis E. (2003) Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology 144, 2588–2596 [DOI] [PubMed] [Google Scholar]
- 46. Izu Y., Sun M., Zwolanek D., Veit G., Williams V., Cha B., Jepsen K. J., Koch M., Birk D. E. (2011) Type XII collagen regulates osteoblast polarity and communication during bone formation. J. Cell Biol. 193, 1115–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moursi A. M., Globus R. K., Damsky C. H. (1997) Interactions between integrin receptors and fibronectin are required for calvarial osteoblast differentiation in vitro. J. Cell Sci. 110, 2187–2196 [DOI] [PubMed] [Google Scholar]
- 48. Nistala H., Lee-Arteaga S., Smaldone S., Siciliano G., Carta L., Ono R. N., Sengle G., Arteaga-Solis E., Levasseur R., Ducy P., Sakai L. Y., Karsenty G., Ramirez F. (2010) Fibrillin-1 and -2 differentially modulate endogenous TGF-beta and BMP bioavailability during bone formation. J. Cell Biol. 190, 1107–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delany A. M., Amling M., Priemel M., Howe C., Baron R., Canalis E. (2000) Osteopenia and decreased bone formation in osteonectin-deficient mice. J. Clin. Invest. 105, 915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Damjanovski S., Huynh M. H., Motamed K., Sage E. H., Ringuette M. (1998) Regulation of SPARC expression during early Xenopus development: evolutionary divergence and conservation of DNA regulatory elements between amphibians and mammals. Dev. Genes Evol. 207, 453–461 [DOI] [PubMed] [Google Scholar]
- 51. Green M. C., Sweet H. O., Bunker L. E. (1976) Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am. J. Pathol. 82, 493–512 [PMC free article] [PubMed] [Google Scholar]
- 52. Chaudhry S. S., Cain S. A., Morgan A., Dallas S. L., Shuttleworth C. A., Kielty C. M. (2007) Fibrillin-1 regulates the bioavailability of TGFbeta1. J. Cell Biol. 176, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Erlebacher A., Filvaroff E. H., Ye J. Q., Derynck R. (1998) Osteoblastic responses to TGF-beta during bone remodeling. Mol. Biol. Cell. 9, 1903–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Katagiri T., Yamaguchi A., Komaki M., Abe E., Takahashi N., Ikeda T., Rosen V., Wozney J. M., Fujisawa-Sehara A., Suda T. (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 127, 1755–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang N., Ingber D. E. (1994) Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 66, 2181–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Higuchi C., Nakamura N., Yoshikawa H., Itoh K. (2009) Transient dynamic actin cytoskeletal change stimulates the osteoblastic differentiation. J. Bone Miner. Metab. 27, 158–167 [DOI] [PubMed] [Google Scholar]
- 57. Drabek K., van de Peppel J., Eijken M., van Leeuwen J. P. (2011) GPM6B regulates osteoblast function and induction of mineralization by controlling cytoskeleton and matrix vesicle release. J. Bone Miner. Res. 26, 2045–2051 [DOI] [PubMed] [Google Scholar]
- 58. Xiao Z., Blonder J., Zhou M., Veenstra T. D. (2009) Proteomic analysis of extracellular matrix and vesicles. J Proteomics. 72, 34–45 [DOI] [PubMed] [Google Scholar]
- 59. Graner M. W., Alzate O., Dechkovskaia A. M., Keene J. D., Sampson J. H., Mitchell D. A., Bigner D. D. (2009) Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23, 1541–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ikemoto A., Bole D. G., Ueda T. (2003) Glycolysis and glutamate accumulation into synaptic vesicles. Role of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase. J. Biol. Chem. 278, 5929–5940 [DOI] [PubMed] [Google Scholar]
- 61. Anderson H. C. (1995) Molecular biology of matrix vesicles. Clin. Orthop. Relat. Res. 266–280 [PubMed] [Google Scholar]
- 62. Svensson L., Heinegård D., Oldberg A. (1995) Decorin-binding sites for collagen type I are mainly located in leucine-rich repeats 4–5. J. Biol. Chem. 270, 20712–20716 [DOI] [PubMed] [Google Scholar]
- 63. Franzén A., Björnsson S., Heinegård D. (1981) Cartilage proteoglycan aggregate formation. Role of link protein. Biochem. J. 197, 669–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen C. C., Boskey A. L. (1985) Mechanisms of proteoglycan inhibition of hydroxyapatite growth. Calcif. Tissue Int. 37, 395–400 [DOI] [PubMed] [Google Scholar]
- 65. Chen C. C., Boskey A. L., Rosenberg L. C. (1984) The inhibitory effect of cartilage proteoglycans on hydroxyapatite growth. Calcif. Tissue Int. 36, 285–290 [DOI] [PubMed] [Google Scholar]
- 66. Hoshi K., Kemmotsu S., Takeuchi Y., Amizuka N., Ozawa H. (1999) The primary calcification in bones follows removal of decorin and fusion of collagen fibrils. J. Bone Miner. Res. 14, 273–280 [DOI] [PubMed] [Google Scholar]
- 67. Aubin J. E. (2001) Regulation of osteoblast formation and function. Rev. Endocr. Metab. Disord. 2, 81–94 [DOI] [PubMed] [Google Scholar]
- 68. Thaler R., Karlic H., Spitzer S., Klaushofer K., Varga F. (2010) Extra-cellular matrix suppresses expression of the apoptosis mediator Fas by epigenetic DNA methylation. Apoptosis 15, 728–737 [DOI] [PubMed] [Google Scholar]
- 69. Kim T. I., Jang J. H., Chung C. P., Ku Y. (2003) Fibronectin fragment promotes osteoblast-associated gene expression and biological activity of human osteoblast-like cell. Biotechnol. Lett. 25, 2007–2011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.