

Abstract
Transglutaminase-1 (TG1)-deficient autosomal-recessive congenital ichthyosis (ARCI) is a rare and severe genetic skin disease caused by mutations in TGM1. It is characterized by collodion babies at birth, dramatically increased transepidermal water loss (TEWL), and lifelong pronounced scaling. The disease has a tremendous burden, including the problem of stigmatization. Currently, no therapy targeting the molecular cause is available, and the therapeutic situation is deplorable. In this study, we developed the basis for a causative therapy aiming at the delivery of the enzyme to the inner site of the keratinocytes’ plasma membrane. We prepared sterically stabilized liposomes with encapsulated recombinant human TG1 (rhTG1) and equipped with a highly cationic lipopeptide vector to mediate cellular uptake. The liposomes overcame the problems of insufficient cutaneous delivery and membrane penetration and provided excellent availability and activity of rhTG1 in primary keratinocytes. To demonstrate the general feasibility of this therapeutic approach in a humanized context, we used a skin-humanized mouse model. Treatment with rhTG1 liposomes resulted in considerable improvement of the ichthyosis phenotype and in normalization of the regenerated ARCI skin: in situ monitoring showed a restoration of TG1 activity, and cholesterol clefts vanished ultrastructurally. Measurement of TEWL revealed a restoration of epidermal barrier function. We regard this aspect as a major advance over available nonspecific approaches making use of, for example, retinoid creams. We conclude that this topical approach is a promising strategy for restoring epidermal integrity and barrier function and provides a causal cure for individuals with TG1 deficiency.
Introduction
Transglutaminase 1 (TG1) is an intracellular key enzyme for the formation of the cornified envelope (CE), which acts as a mechanical barrier to protect against transepidermal water loss (TEWL) and infectious agents. TG1 is found in the upper differentiated layers of the epidermis and in hair follicles and shows a complex pattern of activities given that it exists in multiple cytosolic and membrane-anchored forms in terminally differentiating keratinocytes.1–3 It catalyzes the covalent crosslinking of numerous structural CE proteins, including involucrin, loricrin, filaggrin, and small proline-rich proteins, through the formation of Nε-(γ-glutamyl) lysine isopeptide bonds. In contrast to other transglutaminases (TGs), TG1 is anchored to the inner plasma membrane of keratinocytes. This membrane localization is thought to be essential for the assembly of CE components.4 Another important function is the stabilization of the lipid lamella structures of the corneocyte lipid envelope via attachment of long-chain ω-hydroxyceramides to involucrin.5
Mutations in TGM1 result in the severe and ultra-rare genetic skin disease autosomal-recessive congenital ichthyosis (ARCI [MIM 242300]).6,7 Individuals with ARCI due to TG1 deficiency are born as collodion babies with pronounced ectropion and are encased in a tight shiny collodion membrane, which sheds during the first weeks of life. After shedding, dark-gray or brownish plate-like scales develop and usually affect the entire integument. Loss of TG1 function results in an impaired epidermal barrier with dramatically increased TEWL, causing the inherent clinical problem of dehydration, which can be life threatening, especially during the first weeks of life. Other TGs (e.g., TG2, TG3, and TG5) are also active in different epidermal layers but fail to compensate for TG1 activity.8 For example, TG3 catalyzes the intramolecular linkage or dimerization of loricrin monomers in the cytoplasm but cannot accomplish the multimerization of loricrin at the inner surface of the plasma membrane catalyzed by TG1.9
In addition to clinical examination and sequencing of TGM1, in situ monitoring of TG1 activity on cryosections is an important tool for rapid diagnosis of TG1 deficiency.10 Further confirming the diagnosis, ultrastructural analysis of the stratum corneum showed cholesterol clefts, which are important markers for TG1-deficient ARCI.11
Currently, no specific therapeutic approach targeting the molecular cause of TG1 deficiency exists. All available treatment modalities are insufficient: they are directed at improving symptoms and can only provide symptomatic relief. Therefore, many affected individuals struggle with this chronic and disfiguring disease and often spend 2–3 hours a day with treatment of their skin. The burden of TG1-deficient ARCI is tremendous.12
Because of the disappointing therapeutic situation, we wanted to develop a targeted topical enzyme-replacement therapy for TG1-deficient ARCI. To overcome the problems of insufficient cutaneous delivery, particularly insufficient membrane penetration of drugs, there is a need for a carrier system tagged with an uptake-facilitating ligand that delivers the enzyme at the site of action into keratinocytes.
Here, we describe the development of sterically stabilized liposomal preparations with encapsulated recombinant human TG1 (rhTG1). A highly cationic, uptake-mediating lipopeptide13 was incorporated into the liposomal surface. We show that the rhTG1 liposomes were internalized into cultured primary keratinocytes derived from individuals with TG1 deficiency and demonstrate the restoration of intracellular TG1 activity. Furthermore, this topical approach was tested in a previously developed skin-humanized mouse model for TG1-deficient ARCI14 with the use of different rhTG1 concentrations. Treatment of these mice resulted in a clearly dosage-dependent and considerable improvement of the ichthyosis phenotype and in normalization of the regenerated ARCI skin: in situ monitoring revealed a restoration of TG1 activity, and cholesterol clefts vanished ultrastructurally. Immunohistochemical staining of TG1 substrates showed a normalization of the distribution patterns. Measurement of TEWL revealed that normal epidermal barrier function was maintained.
Subjects and Methods
Study Approval
This study was conducted according to Declaration of Helsinki principles and was approved by the institutional review board of the University Hospital of Münster. All individuals enrolled gave their informed consent.
All animal studies were approved by the institutional review board of the Centro de Investigaciones Energéticas, Medioambientales, y Tecnológicas (CIEMAT), and all experimental procedures were conducted according to European and Spanish laws and regulations.
Subjects
Affected individuals were recruited in our specialized outpatient clinic and had previously been characterized by clinical, immunohistologic, ultrastructural, biochemical, and molecular genetic means.14 Biopsies were taken under local anesthesia.
Animals
Nude (nu/nu, NMRI background) mice were purchased from Elevage-Janvier and were housed individually in pathogen-free conditions at the CIEMAT Laboratory Animals Facility (Spanish registration number 28079-21 A).
Insect Cell Culture, Recombinant Expression, and Purification of Human TG1
Spodoptera frugiperda cells (Sf9 cells) and BTI-TN-5B1-4 insect cells were cultured according to the manufacturer’s instructions.
A full-length cDNA construct of human TG1, fl hTGk His, was designed for recombinant protein production in Baculovirus-infected insect cells, and this was followed by Ni-NTA chromatography for purification. Successful insertion of the C-terminal 6xHis-tag via PCR and subcloning was verified by sequencing (Seqlab). The vector pENTR3C fl hTGk his was incubated with BaculoDirect linear DNA according to the manufacturer’s instructions. Evolving Baculovirus clones were analyzed by PCR.
For transfection, Sf9 cells were plated in a density of 8 × 105 cells per well in a 6-well plate. The transfection procedure was performed according to the manufacturer’s instructions. Ninety-six hours after transfection, medium was collected and stored at 4°C in the dark. This baculoviral P1 stock was amplified by the infection of 1.4 × 107 Sf9 cells with the P1 stock solution (viral titer: 0.5 × 106 pfu/ml), and cells were incubated for 96 hr in Grace’s medium supplemented with 100 μM ganciclovir for selection of recombinant viruses. P2 viral stock was isolated and stored at 4°C. For definition of the viral titers, plaque assays were performed according to the manufacturer’s instructions.
Protein production was performed in 400 ml suspension cultures of BTI-TN-5B1-4 insect cells growing in ExpressFive SFM in Fernbach-flasks for 96 hr. Cells were infected at a density of 2 × 106 cells/ml with an amplified viral stock at a multiplicity of infection of ∼10. rhTG1 was purified from the media by ammonium sulfate precipitation (65% saturation). Pellets were dissolved in a 1/20 volume of 50 mM NaH2PO4, 300 mM NaCl, pH 8.0 and dialyzed against 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0. Afterward, the protein solution was incubated with Ni-NTA material (QIAGEN) overnight at 6°C. Protein was eluted with 50 mM NaH2PO4, 300 mM NaCl, 120 mM imidazole, pH 8.0 under native conditions.
SDS-PAGE and Immunoblotting
Protein samples derived from purification were assayed for TG1 content by immunoblotting. Samples were subjected to 10% SDS-PAGE under reducing conditions and transferred onto a polyvinylidene difluoride membrane by electroblotting. Membranes were saturated with 5% BSA in TBS, incubated with the first antibody, and later incubated with horseradish-peroxidase-conjugated anti-IgG.
Assay of Enzymatic Activity
Enzymatic activity of rhTG1 was analyzed via fluorescence spectrometry (LS55, Perkin Elmer; FLWINLAB software) at an emission wavelength of 332 nm and an excitation wavelength of 500 nm with a slit of 5.0 nm for 15 min as described previously.15
Liposome Preparation
Small unilamellar vesicles (SUVs) and large unilamellar vesicles (LUVs) were prepared as described elsewhere.13 In brief, dry egg phosphatidylcholine (egg-PC), poly(ethylene glycol)-2000-dipalmitoyl-phosphatidylethanolamine (PEG-PE) (Avanti Polar Lipids), and cholesterol (Sigma-Aldrich) were suspended by vortexing in Tris buffer containing rhTG1 (0.1 mg/ml). The suspensions were sonicated (Labsonic L instrument, B. Braun) or extruded (MiniExtruder, Avestin) for obtaining SUVs or LUVs, respectively. Vesicle diameter was checked on a Coulter N4 Plus particle sizer (BeckmanCoulter). The lipid concentration was determined by phosphorous analysis.16 Appropriate volumes of the liposomal suspension and an aqueous solution of the lipopeptide were mixed for achieving the desired peptide-liposome complexes. For uptake studies, a tiny concentration of rhodamin-labeled dipalmitoyl-phosphatidylethanolamine (Rhod-PE) was used as a lipid marker, and the peptide was carboxyfluorescein labeled as described before.17
Primary Cultures of Normal and TG1-Deficient Human Keratinocytes and Fibroblasts
Primary keratinocytes and fibroblasts were obtained by enzymatic digestion from 3–5 mm punch biopsies according to a standard protocol described previously.14 In brief, biopsies were incubated overnight at 4°C in buffer containing 0.5 mg/ml protease X (Sigma-Aldrich). Epidermal sheets were peeled off the dermis and incubated in 0.25% trypsin and 0.02% EDTA (PAA) for achieving single-cell suspension and were resuspended in serum-free culture medium supplemented with 10 ng/ml EGF, 50 mg/ml bovine pituitary extract (BPE) (all from Life Technologies), and 2 mM glutamine (PAA).
For obtaining fibroblasts, the dermis was incubated in buffer containing 0.5 mg/ml collagenase IA (Sigma-Aldrich), and cells were resuspended in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and 2 mM glutamine (PAA).
For inducing differentiation, keratinocytes were incubated with 1.2 mM CaCl2 for 48 hr.
Confocal Laser Scanning Microscopy
For uptake studies of rhTG1 liposomes with the use of confocal laser-scanning microscopy (CLSM), cells were seeded on poly-L-lysine-coated coverslips (30 mm) positioned in 35 mm plastic culture dishes and cultured in serum-free medium supplemented with 10 ng/ml EGF, 50 mg/ml BPE (all from Life Technologies), and 2 mM glutamine (PAA) for 2 days. Differentiated cells were incubated with 1.2 mM CaCl2. Cells were washed with Dulbecco’s PBS (pH 7.4) supplemented with 1 g/l D-glucose (Biochrom) and exposed to rhTG1 liposomal formulations for 1 hr at 37°C. CLSM pictures were taken with an LSM 510 inverted confocal laser-scanning microscope equipped with a Plan-Neofluar 100×/1.3 oil objective (Carl Zeiss) as described previously.13,18 Carboxyfluorescein and rhodamine were excited with a 200 mW argon laser at 488 nm and a 15 mW HeNe laser at 543 nm, respectively, and pictures were taken with a BP505-530 bandpass filter and a LP650 cutoff filter, respectively. Cell viability was checked by trypan-blue exclusion. The dye was excited at 633 nm with the HeNe laser, and emission was recorded with a LP650 cutoff filter. Image acquisition was done sequentially for minimizing cross-talk between the fluorophores.
Bioengineered Skin Preparation and Grafting to Immunodeficient Mice
Bioengineered skin preparation and grafting to immunodeficient mice were performed as described previously.14,19 In brief, a fibrinogen solution containing 5 × 105 dermal fibroblasts, 500 IU of bovine aprotinin (Trasylol, Bayer), and 0.025 mM CaCl2 with 11 IU of bovine thrombin (Sigma-Aldrich) was prepared. Finally, the mixture was poured in 6-well plates and allowed to clot at 37°C. Keratinocytes derived from individuals with TG1-deficient ARCI (1 × 105 to 5 × 105 cells per well) were seeded on the dermal equivalent so that the epidermal layer could form. When confluent, bioengineered skins were grafted onto immunodeficient mice. Grafting was performed under sterile conditions with 6-week-old female nude mice. Generally, the regenerated human skin became visible within 4–6 weeks after grafting.
Ultrastructural Analysis and Histology of Treated Engrafted Human Skin
All graft specimens were fixed for at least 2 hr at room temperature in a 3% glutaraldehyde solution in 0.1 M cacodylate buffer (pH 7.4), cut into pieces of approximately 1 mm3, washed in buffer, postfixed for 1 hr at 4°C in 1% osmium tetroxide, rinsed in water, dehydrated through graded ethanol solutions, transferred into propylene oxide, and embedded in epoxy resin (glycidether 100). Semithin sections were stained with methylene blue. Ultrathin sections were treated with uranyl acetate and lead citrate. For negative staining of LUVs, a drop of LUV preparation was placed onto formvar-coated copper grids for 2 min, and then almost all solution was removed with filter paper. Grids were placed onto a drop of 1% aqueous uranyl acetate solution for 2 min. Excessive fluid was removed, and the samples were air-dried at room temperature. All specimens were examined with a Zeiss EM900 electron microscope equipped with a high-speed slow-scan CCD camera and ImageSP software (Tröndle).
In Situ Monitoring of Transglutaminase Activity and Immunohistochemistry
In situ monitoring of transglutaminase activity on cryosections was performed as described elsewhere.10 For immunohistochemistry, unfixed or acetone-fixed cryosections were incubated with the following primary antibodies: loricrin (Covance), involucrin (Sigma-Aldrich), filaggrin (Novocastra), and plasminogen activator inhibitor 2 (PAI-2, Santa Cruz Biotechnology). Slides were viewed with an Axioskop 2 microscope, and digital images were taken with an AxioCam HR video camera and AxioVision 3.0 software (Carl Zeiss).
Analysis of Epidermal Permeability Barrier Function
To determine the barrier function, TEWL was measured on the back (graft) of the mice with the use of a noninvasive electronic device, Tewameter TM 300 (Courage & Khazaka). All mice were acclimatized for 15 min under standard conditions (20°C and 45% humidity). Measurements were performed every second day before the next treatment by the same investigator (K.A.).
Results
Recombinant Protein Production and Activity of Full-Length Human TG1
The full-length TG1 his cDNA construct was designed for expression in baculovirus-infected insect cells; subsequently, Ni-NTA-chromatography was performed for purification. Successful insertion of the C-terminal hexahistidine tag (his-tag) was verified by DNA sequencing. After amplification of recombinant baculoviruses, infection of BTI-TN-5B1-4 insect cells resulted in a protein with a molecular mass of about 92 kDa, as determined by SDS-PAGE. This was identified as full-length human TG1 by immunoblotting with TG1 and his-tag-specific antibodies, which gave similar results. rhTG1 could be purified to homogeneity at yields of up to 7 mg/l of cell-culture medium (Figures S1A and S1B, available online). The fluorimetric activity assay using cadaverin as a substrate for rhTG1, which was incorporated into casein, showed a specific activity of around 1,000 U/mg (data not shown).
Development of a Vector-Coupled Liposomal Carrier System for rhTG1
To overcome the problems of insufficient cutaneous delivery and intracellular availability of the enzyme, we encapsulated rhTG1 in liposomes of different diameters: 50 nm for SUVs, 100 and 200 nm for LUVs, and >1,500 nm for multilamellar large vesicles (MLVs), which were sterically stabilized with PEG-PE. The liposomal carriers were modified with a highly cationic peptide sequence derived from apoE.13,20 The lipopeptide rapidly bound to liposomes and served as a vector to facilitate the carrier transport across the membrane of keratinocytes. A schematic presentation of the developed liposomes and their ultrastructure is given in Figures 1A and 1B. Four different liposomal formulations, prepared with BSA as a model protein (5 mg/ml) (Table S1), were used for characterizing the diameter and uniformity, inner volume, amount of encapsulated protein, and stability of the carriers at different temperatures. Properties such as high stability, uniformity, and high theoretical inner volume made LUVs of 200 nm in diameter and MLVs favorable candidates for further cell-culture experiments.
Figure 1.
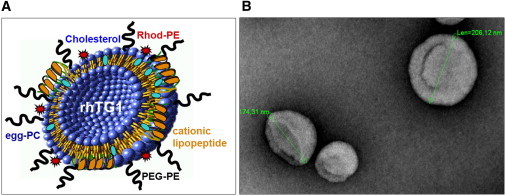
Schematic Presentation and Ultrastructural Appearance of rhTG1 LUVs
(A) The cationic lipopeptide is an apoE-derived dipalmitoylated tandem dimer with or without a fluorescence marker.
(B) Electron microscopy of representative LUVs (∼200 nm; negative staining).
Cellular Uptake of rhTG1 LUV Formulations
Cellular uptake of rhTG1 LUV and rhTG1 MLV formulations into differentiated and undifferentiated keratinocytes of normal individuals and individuals with TG1-deficient ARCI was measured via CLSM (Figure 2A, Figures S2A and S2B, and data not shown). The two fluorescence labels, carboxyfluorescein (green fluorescence) for the lipopeptide and Rhod-PE (red fluorescence) for the lipid matrix, allowed confirmation of the internalization of the complete liposome (Figure 2A, merge). Internalization of the rhTG1 LUVs occurred in differentiated and undifferentiated ARCI cells (TGM1−/−) and in undifferentiated normal cells (TGM1+/+). Uptake into differentiated normal cells in comparison to ARCI cells was markedly reduced. Large rhTG1 MLVs and liposomes without the lipopeptide vector were not internalized into the cells (data not shown). Subsequent immunoblot analysis confirmed the uptake of the enzyme after incubation with rhTG1 LUV preparations (Figure 2B). Using a his-tag antibody that only detects the recombinant his-tagged full-length form of TG1, we found rhTG1 only in cells incubated with rhTG1 LUV formulations. In accordance with the CLSM results, rhTG1 could not be detected in normal differentiated cells.
Figure 2.
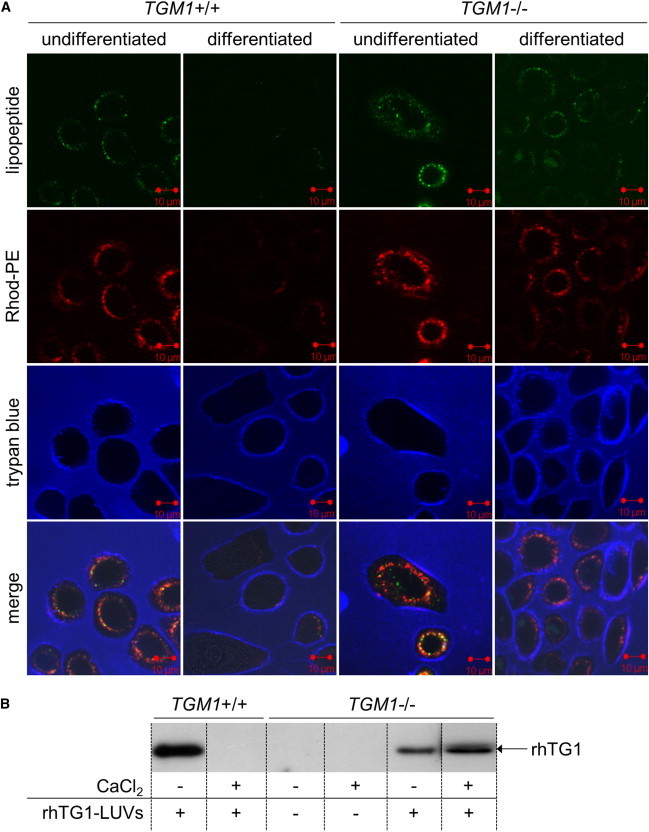
Measurement of Cellular Uptake of rhTG1 LUVs
TG1-deficient cells (TGM1−/−) were isolated from skin biopsies of an individual with TGM1 mutations (c.377G>A [(p.Arg126His] and c.876+2T>C [(p.Glu253Valfs∗2]) associated with generalized TG1-deficient ARCI. Differentiated cells were incubated with 1.2 mM CaCl2 for 48 hr to mimic cells of the upper differentiated cell layers.
(A) Internalization of rhTG1 LUVs into primary keratinocytes was measured via CLSM. Colors are as follows: red, Rhod-PE; green, cationic lipopeptide with fluorescence marker; and blue, trypan-blue staining for visualization of intact cell membranes.
(B) Internalization of rhTG1 LUVs was confirmed by subsequent immunoblot analysis using a his-tag antibody, which only detects the full-length recombinant TG1, for distinguishing rhTG1 from the wild-type TG1 in differentiated normal cells.
Intracellular Activity of rhTG1 in Cultured Primary Keratinocytes
Intracellular activity of rhTG1 after internalization of rhTG1 LUVs into cultured primary keratinocytes was monitored with the in situ activity assay on cryosections (Figure 3). After keratinocytes derived from individuals with TG1-deficient ARCI were incubated with rhTG1 LUV preparations, cells were trypsinized and immediately frozen in liquid nitrogen. The intracellular activity of rhTG1 in differentiated and undifferentiated ARCI keratinocytes was comparable to that detected in normal differentiated keratinocytes.
Figure 3.
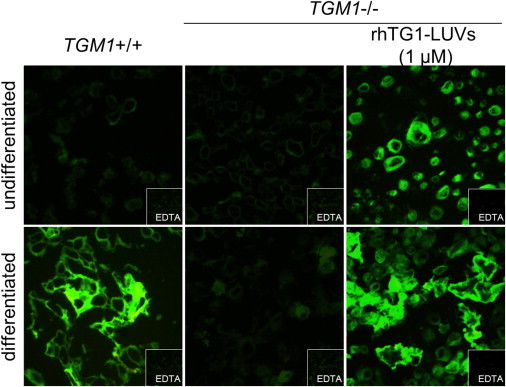
Intracellular Activity of rhTG1 after Internalization into Primary Keratinocytes
Intracellular activity of rhTG1 in cultured keratinocytes was measured after trypsinization. Only internalized rhTG1 was detected. The TG1 activity in cells derived from individuals with TG1-deficient ARCI was restored after incubation with rhTG1 LUV preparations. EDTA was used as a negative control.
In Vivo Testing of rhTG1 LUVs: Evidence of a Dosage-Dependent Effect
For in vivo testing of rhTG1 LUV formulations, we used the previously developed humanized ARCI mouse model.14 This model overcomes the drawback of the classical Tgm1-knockout mouse model, i.e., lethality within 3–4 hr after birth and lack of faithful recapitulation of the human skin phenotype.21 Furthermore, it has the great advantage of testing the rhTG1 LUVs in a humanized context. Eight weeks after grafting, TG1-deficient human grafts were treated with different dosages of liposomal preparations (40 ng rhTG1/cm2 and 2 ng rhTG1/cm2). Empty liposomes without encapsulated rhTG1 and a retinoid cream (0.05%) as a known and potent nonspecific therapeutic for ARCI served as control regimens.
After 14 days of treatment every second day, we observed a considerable change in phenotype (Figure 4). Application of rhTG1 LUVs resulted in a phenotypic change after the second treatment (day 5) at both dosages. Application of the high rhTG1 dosage (40 ng rhTG1/cm2) resulted in a nearly complete normalization of the skin (Figure 4D). Histology in some areas of the sections showed elongated ridges and hyperproliferation, which can also be detected in diseases such as psoriasis and Netherton syndrome (MIM 256500), in which TG1 is overexpressed (Figure 4I). In contrast, with 2 ng rhTG1/cm2, the treated grafts showed residual scaly patches (Figure 4E) but without psoriasiform features (Figure 4J), indicating a dosage-dependent effect. Also, treatment with the retinoid cream after 7 days resulted in a clear normalization (Figures 4B and 4G), whereas treatment with empty liposomes did not have any effect on phenotype or histology (Figures 4C and 4H).
Figure 4.
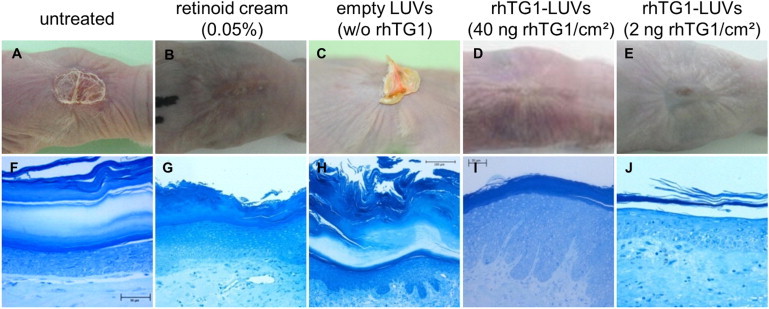
Phenotype and Histology of Regenerated TG1-Deficient Skin Grafts in a Skin-Humanized Mouse Model before and after Application of rhTG1 LUV Preparations
Phenotypic changes in mice treated with 40 ng rhTG1/cm2 (n = 3) were observed after the second application (day 5) (D). Grafts treated with 2 ng rhTG1/cm2 (n = 3) after 14 days showed a minor effect with residual hyperkeratotic patches, suggesting dose dependency (E). Compared to untreated grafts (A and F), regenerated skin treated with empty LUVs showed no change in phenotype or histology (C and H). Treatment with a retinoid cream resulted in a phenotypic change after the first application but also with residual hyperkeratosis (B and G) as described above for treatment with the lower rhTG1 dosage (E). Histology shows a clear reduction of the very thick and packed cornified layer and psoriasiform signs at some sites of the sections only after treatment with 40 ng rhTG1/cm2. The bottom row depicts methylene-blue staining of semithin sections.
Diagnostic Markers: In Situ TG1 Activity and Ultrastructure
After treatment of the skin-humanized mice, biopsies were taken and investigated with regard to diagnostic markers of TG1-deficient ARCI (Figure 5). The in situ activity assay on cryosections (Figures 5A–5E) after treatment with rhTG1 LUV preparations showed a restoration of TG1 activity in both samples (Figures 5D and 5E). In contrast, retinoid-treated samples, as predicted, did not show a restoration of TG1 activity (Figure 5C). As important ultrastructural markers, the cholesterol clefts were no longer detectable in the regenerated skin treated with 40 ng rhTG1/cm2 (Figure 5I). Compared to untreated mice (Figure 5G) or retinoid-treated mice (Figure 5H), mice treated with the lower dosage of 2 ng rhTG1/cm2 showed a marked decrease of cholesterol clefts (Figure 5J). As expected, grafts treated with empty LUVs did not show any change either macroscopically (Figures 5C and 5H) or in TG1 activity or ultrastructure (data not shown).
Figure 5.
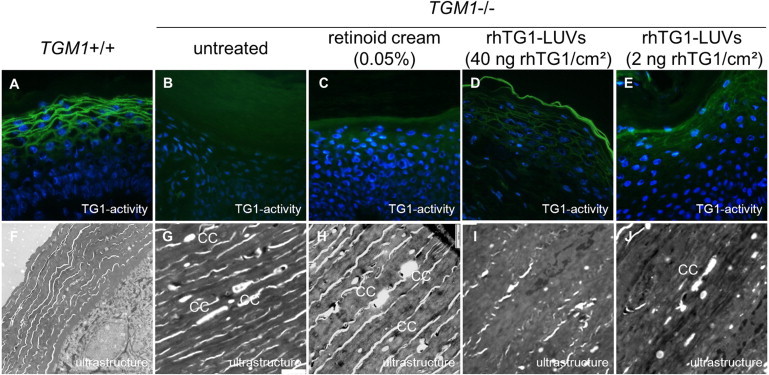
Diagnostic Markers of Normal Human Skin in Comparison to Regenerated Human TG1-Deficient Skin before and after Application of rhTG1 LUV Preparations
Normal human skin showed a pericellular distribution of TG1 activity in the granular layer of the epidermis (A). Cholesterol clefts in the stratum corneum were not present (F). Untreated human skin grafts and grafts treated with a retinoid cream showed cholesterol clefts within the stratum corneum (G and H), whereas TG1 activity was absent (B and C). After treatment of regenerated skin grafts with rhTG1 LUV formulations, the epidermis normalized: TG1 activity was detected with both dosages (D and E), and the formation of cholesterol clefts vanished (I) or was markedly reduced (J). “CC” stands for cholesterol cleft.
Investigation of Differentiation Markers and TG1 Substrates
Investigation of several differentiation markers and TG1 substrates, such as involucrin, filaggrin, PAI-2, and loricrin, with specific antibodies confirmed the results of the in situ monitoring of TG1 activity and ultrastructural investigations. These markers showed a more diffuse and slightly shifted distribution in regenerated TG1-deficient grafts and in human TG1-deficient skin compared to normal skin (Figures S3A, S3F, S3K, and S3P). Compared to staining in untreated mice (Figures S3B, S3G, S3L, and S3Q) or retinoid-treated mice (Figures S3C, S3H, S3M, and S3R), staining of filaggrin (Figures S3D and S3E), involucrin (Figures S3I and S3J), PAI-2 (Figures S3N and S3O), and loricrin (Figures S3S and S3T) in mice treated with rhTG1 LUV preparations revealed a normalization of the distribution patterns.
Measurement of TEWL
To further investigate the effect of the rhTG1 LUV preparations on epidermal barrier function, we performed measurements of TEWL during the 14 days of application (Figure 6). Every second day prior to the next treatment with rhTG1 LUV preparations or the retinoid cream, we measured the change in TEWL. Untreated mice did not show an increase in TEWL (Figure 6, before treatment). After treatment of regenerated TG1-deficient skin grafts with the retinoid cream, TEWL was dramatically increased to a very critical level (40.62 ± 2.11 g/m2h on day 5) but seemed to normalize to some degree during the treatment (16.18 ± 0.46 g/m2h on day 12). Treatment of the skin grafts with both rhTG1 dosages (40 or 2 ng rhTG1/cm2) did not result in any change in TEWL (3.07 ± 1.36 to 5.7 ± 2.28 g/m2h). The values remained in the “very good” range.
Figure 6.
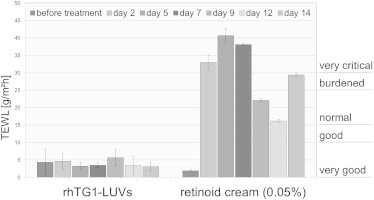
Measurement of TEWL
Measurement of TEWL was done directly before the next treatment of mice with rhTG1 LUV preparations or the retinoid cream (n = 4). As part of a homeostatic response to the barrier disturbance and for the prevention of TEWL, untreated animals developed compact hyperkeratosis and therefore did not show an increase in TEWL before treatment. Treatment with the retinoid cream resulted in dramatically increased TEWL, which seemed to normalize during the treatment process. Compared to untreated regenerated TG1-deficient grafts (before treatment) or normal human skin (7.42 ± 0.13 g/m2h), normal mouse skin (4.91 ± 2.58 g/m2h), and normal regenerated grafts (3.52 ± 0.85 g/m2h) (data not shown), skin treated with the developed rhTG1 LUV preparations maintained the physiological situation and did not show an increase in TEWL. This indicates a restoration of the epidermal barrier function after treatment with rhTG1 LUV preparations. Values are given as means ± SEMs.
Discussion
Individuals who suffer from TG1-deficient ARCI are currently in a deplorable situation. Safe and efficient treatment modalities are lacking. Because of this situation and the fact that TGM1 mutations account for up to 48% (about 55% in the United States) of all ARCI cases,22,23 we have developed the experimental basis for an enzyme-replacement therapy for individuals suffering from TG1-deficient ARCI. We show that a topical approach using a formulation of liposomal-encapsulated rhTG1 is suitable for restoring TG1 activity in the upper stratified layers of the epidermis for reconstituting epidermal integrity and barrier function.
Of particular importance are our in vivo experiments using the skin-humanized mouse model with two different dosages of rhTG1 encapsulated in vector-coupled sterically stabilized liposomes. We have shown that the liposomes used in our experiments overcame the enormous problem of cutaneous delivery and actually penetrated the membrane of keratinocytes, which offers tremendous therapeutic possibilities, as well as for other genodermatoses. The ichthyosis phenotype on skin-humanized mice vanished in a dosage-dependent manner, which even applied to ultrastructural markers of TG1 deficiency, namely the presence of cholesterol clefts in the stratum corneum. Moreover, in situ monitoring of TG1 activity on cryosections demonstrated a restoration of TG1 activity in regenerated skin grafts treated with 40 and 2 ng rhTG1/cm2. Furthermore, distribution patterns of a number of epidermal differentiation markers, such as loricrin, filaggrin, involucrin, and PAI-2, showed normalization. The fact that the higher rhTG1 dosage used was associated with psoriasiform epidermal hyperplasia in some histologic sections is a further convincing argument that this hyperplasia directly relates to restored TG1 activity. Similar histologic features are found in skin diseases characterized by epidermal hyperproliferation and TG1 hyperactivity, such as psoriasis vulgaris (MIM 177900) and Netherton syndrome.24,25 We assume that the used 40 ng rhTG1/cm2 dosage is too high, and we expect even better results from optimizing the liposomal formulation with respect to composition and concentration of rhTG1.
To further investigate the effect of the rhTG1 LUV preparations on epidermal barrier function, we performed measurements of TEWL. The development of ichthyosiform skin in ARCI is thought to be a homeostatic response to compensate for defective permeability barrier function in TG1 deficiency.7 Untreated mice displayed a compact hyperkeratosis and therefore did not show an increase in TEWL.26 Despite the clear reduction of the very thick and packed cornified layer, treatment with both rhTG1 dosages did not result in any change in TEWL, indicating a maintenance of epidermal barrier function and integrity as a result of the restoration of TG1 activity. In contrast, after treatment of regenerated ARCI skin with the retinoid cream, TEWL dramatically increased. This observation is in line with our expectations given that retinoid treatment decreases the thickness of the stratum corneum but does not correct TG1 deficiency and therefore does not restore epidermal barrier function.
The concept of enzyme-replacement therapy has already gained clinical significance in different lysosomal-storage diseases, such as Fabry disease (MIM 301500), mucopolysaccharidosis type II (MIM 309900), and Gaucher disease (MIM 230800).27–29 Uptake of enzymes involved in lysosomal-storage disease is driven by endocytosis. The fate of endocytotic vesicles by the formation of endosomes and lysosomes is well established.30
So that rhTG1 is accessible at the inner site of the plasma membrane of differentiating keratinocytes, there is a need for efficient transmembrane transport. Liposomes have received widespread attention as drug delivery systems. They are capable of incorporating and delivering hydrophilic and hydrophobic drugs (among which are peptides and proteins), are biodegradable, and show good biocompatibility, low toxicity, and minimal activation of the immune system.31–33 By altering the biodistribution and protecting the entrapped materials from inactivation by host defense mechanisms (e.g., extracellular proteases), liposomes can be used for target-specific drug delivery. Natural phospholipids, such as phosphatidylcholine, have been commonly used to form liposomes. Cholesterol reduces the permeability to water-soluble molecules and enhances the stability in biological fluids.34 PEGylation further enhances carrier stability and circulation time and improves pharmacokinetics and pharmacodynamics of encapsulated compounds by protecting from enzymatic degradation, reduced renal clearance, and limited immunogenic and antigenic reactions.35 Our optimized peptide-modified liposomal preparation proved to be highly efficient in the delivery of rhTG1 into keratinocytes and in an in vivo mouse model.
The similarity in lipid composition of the liposomes and membranes of keratinocytes might enable the liposomes to penetrate into the epidermal barrier more efficiently than other application forms.36 Subsequent changes in the hydration spheres of the membrane layer might decrease the retarding action of the skin cell layer with the consequence of improved diffusion of the liposomes. This might result in an increase in drug absorption into the epidermis and a decrease in drug clearance.37 The diameter of liposomes is an important factor in therapeutic applications. We found that MLVs with a size distribution in the range of >1,500 nm were not internalized into keratinocytes. The preferred structures for drug delivery are LUVs with a diameter between 100 and 500 nm.37,38 Our carrier-optimizing experiments confirm these early studies (Table S1).
To mediate cellular uptake, the vesicles were modified with a highly cationic lipopeptide. The apoE-derived sequence bears characteristics of so-called cell-penetrating peptides, which are able to shuttle attached cargos across cellular barriers and comprise binding sites for the LDL receptor and for cell-surface heparan sulfate proteoglycans, two ubiquitous mammalian membrane constituents able to mediate cellular uptake.13,20 In this study, the lipopeptide was proven to be highly efficient in mediating the transport of rhTG1 LUVs across the membrane of keratinocytes. Cells exposed to liposomal preparations without the lipopeptide vector did not show intracellular fluorescence, indicating the importance of the developed vector-coupled system. Internalization into differentiated normal cells (TGM1+/+) was markedly lower than that into ARCI cells (TGM1−/−). The activity of wild-type TG1, which catalyzes the formation of an intact CE, probably prevents the uptake of liposomes.
The cell-culture experiments validated the feasibility of a topical approach, but they were not sufficient to demonstrate that the clinical problems of ARCI can be corrected. We therefore evaluated the reconstitution of epidermal barrier formation in regenerated ARCI skin in vivo and used a previously developed skin-humanized mouse model.14 We had the great advantage of being able to test our liposomal formulations in a humanized context. Noteworthy, after 14 days of treatment with both dosages of rhTG1 LUVs, we could not detect clinical signs of local toxicity. Furthermore, our studies demonstrate an efficient penetration of the vector-coupled rhTG1 LUVs through the thick and packed stratum corneum.
As a control in our experiments, we used a retinoid cream as a known and potent therapeutic agent for ARCI. Considerable improvement of lesions was detected clinically in the treated skin. After treatment with the retinoid cream, we observed a considerable change in phenotype, but cholesterol clefts still remained visible. The clear-cut reduction of the cornified layer after application of the retinoid cream shows that the skin-humanized mouse model also responded very well to a known treatment method. It is known that this treatment, although effective, induces side-effects such as mild pruritus, slight burning, and mild to moderate skin irritation in humans, which limits its use.39,40
Ex vivo gene therapy was reported for TG1 deficiency 15 years ago in an experimental model. Normal gene expression of TGM1 was restored, and a phenotype correction was observed in engrafted lesional skin.41,42 This approach has been abandoned and has never reached the clinic, probably because of biological safety concerns associated with this therapeutic modality.
Acknowledgments
This work was supported by the Bundesministerium für Bildung und Forschung as part of the Network for Ichthyosis and Related Keratinization Disorders (NIRK) (grants 01GM0901 and 01GM0902), the Foundation for Ichthyosis and Related Skin Types, and the Selbsthilfe Ichthyose e.V. F.L. was supported in part by the Instituto de Salud Carlos III (grant PI11/1225) and by grant S2010/BMD-2359 from C.M. M.D.R. was supported by the Ministerio de Ciencia y Innovación (grant SAF2010-16976). The work of M.D. was partially supported by Deutsche Forschungsgemeinschaft (grant DA 324/9-1). We are very grateful to all individuals who participated in the study. Primary normal human keratinocytes for uptake studies were kindly provided by the Institute of Pharmacy of Freie Universität Berlin. The excellent technical assistance of Androniki Kolovou is gratefully acknowledged, and Katrin Jordan is thanked very much for the biophysical characterization of the liposomal formulations. Very special thanks go to Brigitte Willis for her excellent work as the coordinator of the NIRK. The European commission has awarded an orphan drug designation for the therapeutic modality described in this article (http://www.ema.europa.eu/ema).
Contributor Information
Karin Aufenvenne, Email: karin_aufenvenne@yahoo.de.
Heiko Traupe, Email: traupeh@ukmuenster.de.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Goldsmith L.A. Human epidermal transglutaminase. J. Invest. Dermatol. 1983;80:39s–41s. [PubMed] [Google Scholar]
- 2.Rice R.H., Rong X.H., Chakravarty R. Proteolytic release of keratinocyte transglutaminase. Biochem. J. 1990;265:351–357. doi: 10.1042/bj2650351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinert P.M., Chung S.I., Kim S.Y. Inactive zymogen and highly active proteolytically processed membrane-bound forms of the transglutaminase 1 enzyme in human epidermal keratinocytes. Biochem. Biophys. Res. Commun. 1996;221:101–106. doi: 10.1006/bbrc.1996.0552. [DOI] [PubMed] [Google Scholar]
- 4.Candi E., Schmidt R., Melino G. The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005;6:328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 5.Nemes Z., Marekov L.N., Fésüs L., Steinert P.M. A novel function for transglutaminase 1: attachment of long-chain omega-hydroxyceramides to involucrin by ester bond formation. Proc. Natl. Acad. Sci. USA. 1999;96:8402–8407. doi: 10.1073/pnas.96.15.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell L.J., DiGiovanna J.J., Rogers G.R., Steinert P.M., Hashem N., Compton J.G., Bale S.J. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat. Genet. 1995;9:279–283. doi: 10.1038/ng0395-279. [DOI] [PubMed] [Google Scholar]
- 7.Oji V., Tadini G., Akiyama M., Blanchet Bardon C., Bodemer C., Bourrat E., Coudiere P., DiGiovanna J.J., Elias P., Fischer J. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Hitomi K. Transglutaminases in skin epidermis. Eur. J. Dermatol. 2005;15:313–319. [PubMed] [Google Scholar]
- 9.John S., Thiebach L., Frie C., Mokkapati S., Bechtel M., Nischt R., Rosser-Davies S., Paulsson M., Smyth N. Epidermal transglutaminase (TGase 3) is required for proper hair development, but not the formation of the epidermal barrier. PLoS ONE. 2012;7:e34252. doi: 10.1371/journal.pone.0034252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raghunath M., Hennies H.C., Velten F., Wiebe V., Steinert P.M., Reis A., Traupe H. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch. Dermatol. Res. 1998;290:621–627. doi: 10.1007/s004030050362. [DOI] [PubMed] [Google Scholar]
- 11.Laiho E., Niemi K.M., Ignatius J., Kere J., Palotie A., Saarialho-Kere U. Clinical and morphological correlations for transglutaminase 1 gene mutations in autosomal recessive congenital ichthyosis. Eur. J. Hum. Genet. 1999;7:625–632. doi: 10.1038/sj.ejhg.5200353. [DOI] [PubMed] [Google Scholar]
- 12.Kamalpour L., Gammon B., Chen K.H., Veledar E., Pavlis M., Rice Z.P., Chen S.C. Resource utilization and quality of life associated with congenital ichthyoses. Pediatr. Dermatol. 2011;28:512–518. doi: 10.1111/j.1525-1470.2011.01432.x. [DOI] [PubMed] [Google Scholar]
- 13.Sauer I., Nikolenko H., Keller S., Abu Ajaj K., Bienert M., Dathe M. Dipalmitoylation of a cellular uptake-mediating apolipoprotein E-derived peptide as a promising modification for stable anchorage in liposomal drug carriers. Biochim. Biophys. Acta. 2006;1758:552–561. doi: 10.1016/j.bbamem.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Aufenvenne K., Rice R.H., Hausser I., Oji V., Hennies H.C., Rio M.D., Traupe H., Larcher F. Long-term faithful recapitulation of transglutaminase 1-deficient lamellar ichthyosis in a skin-humanized mouse model, and insights from proteomic studies. J. Invest. Dermatol. 2012;132:1918–1921. doi: 10.1038/jid.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aufenvenne K., Oji V., Walker T., Becker-Pauly C., Hennies H.C., Stöcker W., Traupe H. Transglutaminase-1 and bathing suit ichthyosis: molecular analysis of gene/environment interactions. J. Invest. Dermatol. 2009;129:2068–2071. doi: 10.1038/jid.2009.18. [DOI] [PubMed] [Google Scholar]
- 16.Böttcher C.J.F., Van Gent C.M., Pries C. A rapid and sensitive submicro phosphorus determination. Anal. Chim. Acta. 1961;24:203–204. [Google Scholar]
- 17.Leupold E., Nikolenko H., Dathe M. Apolipoprotein E peptide-modified colloidal carriers: the design determines the mechanism of uptake in vascular endothelial cells. Biochim. Biophys. Acta. 2009;1788:442–449. doi: 10.1016/j.bbamem.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 18.Leupold E., Nikolenko H., Beyermann M., Dathe M. Insight into the role of HSPG in the cellular uptake of apolipoprotein E-derived peptide micelles and liposomes. Biochim. Biophys. Acta. 2008;1778:2781–2789. doi: 10.1016/j.bbamem.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Del Rio M., Larcher F., Serrano F., Meana A., Muñoz M., Garcia M., Muñoz E., Martin C., Bernad A., Jorcano J.L. A preclinical model for the analysis of genetically modified human skin in vivo. Hum. Gene Ther. 2002;13:959–968. doi: 10.1089/10430340252939069. [DOI] [PubMed] [Google Scholar]
- 20.Keller S., Sauer I., Strauss H., Gast K., Dathe M., Bienert M. Membrane-mimetic nanocarriers formed by a dipalmitoylated cell-penetrating peptide. Angew. Chem. Int. Ed. Engl. 2005;44:5252–5255. doi: 10.1002/anie.200500519. [DOI] [PubMed] [Google Scholar]
- 21.Matsuki M., Yamashita F., Ishida-Yamamoto A., Yamada K., Kinoshita C., Fushiki S., Ueda E., Morishima Y., Tabata K., Yasuno H. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase) Proc. Natl. Acad. Sci. USA. 1998;95:1044–1049. doi: 10.1073/pnas.95.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodríguez-Pazos L., Ginarte M., Fachal L., Toribio J., Carracedo A., Vega A. Analysis of TGM1, ALOX12B, ALOXE3, NIPAL4 and CYP4F22 in autosomal recessive congenital ichthyosis from Galicia (NW Spain): evidence of founder effects. Br. J. Dermatol. 2011;165:906–911. doi: 10.1111/j.1365-2133.2011.10454.x. [DOI] [PubMed] [Google Scholar]
- 23.Farasat S., Wei M.H., Herman M., Liewehr D.J., Steinberg S.M., Bale S.J., Fleckman P., Toro J.R. Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J. Med. Genet. 2009;46:103–111. doi: 10.1136/jmg.2008.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghunath M., Tontsidou L., Oji V., Aufenvenne K., Schürmeyer-Horst F., Jayakumar A., Ständer H., Smolle J., Clayman G.L., Traupe H. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J. Invest. Dermatol. 2004;123:474–483. doi: 10.1111/j.0022-202X.2004.23220.x. [DOI] [PubMed] [Google Scholar]
- 25.Oji V., Oji M.E., Adamini N., Walker T., Aufenvenne K., Raghunath M., Traupe H. Plasminogen activator inhibitor-2 is expressed in different types of congenital ichthyosis: in vivo evidence for its cross-linking into the cornified cell envelope by transglutaminase-1. Br. J. Dermatol. 2006;154:860–867. doi: 10.1111/j.1365-2133.2005.07109.x. [DOI] [PubMed] [Google Scholar]
- 26.Kuramoto N., Takizawa T., Takizawa T., Matsuki M., Morioka H., Robinson J.M., Yamanishi K. Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J. Clin. Invest. 2002;109:243–250. doi: 10.1172/JCI13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer R.M., Tylki-Szymańska A., Hilz M.J. Enzyme replacement therapy for Fabry disease: a systematic review of available evidence. Drugs. 2009;69:2179–2205. doi: 10.2165/11318300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Okuyama T., Tanaka A., Suzuki Y., Ida H., Tanaka T., Cox G.F., Eto Y., Orii T. Japan Elaprase Treatment (JET) study: idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II) Mol. Genet. Metab. 2010;99:18–25. doi: 10.1016/j.ymgme.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka N., Saito H., Ito T., Momose K., Ishida F., Hora K., Kiyosawa K., Ida H. Initiation of enzyme replacement therapy for an adult patient with asymptomatic type 1 Gaucher’s disease. Intern. Med. 2001;40:716–721. doi: 10.2169/internalmedicine.40.716. [DOI] [PubMed] [Google Scholar]
- 30.De Duve C. Principles of tissue fractionation. J. Theor. Biol. 1964;6:33–59. doi: 10.1016/0022-5193(64)90065-7. [DOI] [PubMed] [Google Scholar]
- 31.Mufamadi M.S., Pillay V., Choonara Y.E., Du Toit L.C., Modi G., Naidoo D., Ndesendo V.M. A review on composite liposomal technologies for specialized drug delivery. J. Drug Deliv. 2011;2011:939851. doi: 10.1155/2011/939851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierre M.B., Dos Santos Miranda Costa I. Liposomal systems as drug delivery vehicles for dermal and transdermal applications. Arch. Dermatol. Res. 2011;303:607–621. doi: 10.1007/s00403-011-1166-4. [DOI] [PubMed] [Google Scholar]
- 33.Fireman S., Toledano O., Neimann K., Loboda N., Dayan N. A look at emerging delivery systems for topical drug products. Dermatol. Ther. 2011;24:477–488. doi: 10.1111/j.1529-8019.2012.01464.x. [DOI] [PubMed] [Google Scholar]
- 34.Damen J., Regts J., Scherphof G. Transfer and exchange of phospholipid between small unilamellar liposomes and rat plasma high density lipoproteins. Dependence on cholesterol content and phospholipid composition. Biochim. Biophys. Acta. 1981;665:538–545. doi: 10.1016/0005-2760(81)90268-x. [DOI] [PubMed] [Google Scholar]
- 35.Li S.D., Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim. Biophys. Acta. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.du Plessis J., Egbaria K., Ramachandran C., Weiner N. Topical delivery of liposomally encapsulated gamma-interferon. Antiviral Res. 1992;18:259–265. doi: 10.1016/0166-3542(92)90059-e. [DOI] [PubMed] [Google Scholar]
- 37.de Leeuw J., de Vijlder H.C., Bjerring P., Neumann H.A. Liposomes in dermatology today. J. Eur. Acad. Dermatol. Venereol. 2009;23:505–516. doi: 10.1111/j.1468-3083.2009.03100.x. [DOI] [PubMed] [Google Scholar]
- 38.Nagayasu A., Uchiyama K., Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv. Drug Deliv. Rev. 1999;40:75–87. doi: 10.1016/s0169-409x(99)00041-1. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann B., Stege H., Ruzicka T., Lehmann P. Effect of topical tazarotene in the treatment of congenital ichthyoses. Br. J. Dermatol. 1999;141:642–646. doi: 10.1046/j.1365-2133.1999.03101.x. [DOI] [PubMed] [Google Scholar]
- 40.Marulli G.C., Campione E., Chimenti M.S., Terrinoni A., Melino G., Bianchi L. Type I lamellar ichthyosis improved by tazarotene 0.1% gel. Clin. Exp. Dermatol. 2003;28:391–393. doi: 10.1046/j.1365-2230.2003.01318.x. [DOI] [PubMed] [Google Scholar]
- 41.Choate K.A., Khavari P.A. Direct cutaneous gene delivery in a human genetic skin disease. Hum. Gene Ther. 1997;8:1659–1665. doi: 10.1089/hum.1997.8.14-1659. [DOI] [PubMed] [Google Scholar]
- 42.Choate K.A., Medalie D.A., Morgan J.R., Khavari P.A. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat. Med. 1996;2:1263–1267. doi: 10.1038/nm1196-1263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.