

Abstract
A facile six-step synthesis (15.2% yield) of ent-17β-estradiol from readily accessible precursors is described. The preparation of analogues with 2-alkyl substitutents, double bond unsaturation in the C-ring, a cis C, D-ring fusion and modified substituents at C17 is also reported.
Keywords: ent-17β-estradiol, enantiomer, neuroprotectants, antioxidants, estradiol analogues
1. Introduction
Phenolic compounds are free-radical scavengers and have antioxidant activity. Estrone and 17β-estradiol, as well as other steroids containing a phenolic A-ring, are antioxidants with neuroprotective properties that are potentially useful for the treatment of Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases [1-6]. Since the antioxidant properties of 17β-estradiol are not dependent on the absolute configuration of this steroid, ent-17β-estradiol also has neuroprotective properties [7]. Additionally, ent-17β-estradiol lacks the feminizing actions of 17β-estradiol [7-9] so that neuroprotection is possible without the complications of feminization or other undesirable actions attributable to the hormonal actions of 17β-estradiol (e.g., stimulation of estrogen-dependent breast cancer). Many different routes have been published for the total syntheses of natural or racemic estrogens [10-23]. However, only two enantioselective syntheses of ent-estrogens have been published. Hutchinson and Money used (+)-3-bromocamphor as a starting material for the synthesis of ent-estrone [24]. We previously reported [7], without discussion of the chemistry methods, that ent-17β-estradiol can be obtained by the aromatization of ent-19-nortestosterone (Scheme 1).
Scheme 1.
Herein we describe an alternative method for the synthesis of ent-17β-estradiol (3, Scheme 2) that does not proceed by aromatization of a 19-norsteroid intermediate. This synthetic route is based on literature analogies used for the synthesis of 7-substituted analogues of 17β-estradiol [25,26]. The synthesis of several structurally related analogues (Scheme 3, 12a,b; Scheme 4, 14a,b; Scheme 5, 17a,b; Scheme 6, 20; Scheme 7, 22) that were part of a previously published structure–activity study [27] is also reported.
Scheme 2.

Scheme 3.
Scheme 4.
Scheme 5.
Scheme 6.
Scheme 7.
2. Experimental methods
2.1 General Methods
Melting points were determined on a Kofler micro hot stage and were uncorrected. NMR spectra were recorded in CDCl3, or (CD3)2CO at 300MHz (1H) or 75MHz (13C). Chemical shifts (δ) were reported downfield from internal Me4Si (δ: 0.00). Mass spectra were obtained using mass spectrometry facilities located at either the Univ. of Wisconsin or Washington University. IR spectra were recorded either as films on a NaCl plate or in KBr. Elemental analyses were carried out by M-H-W laboratories. Phoenix AZ. Chromatography was performed using flash chromatography grade silica gel (32–63μm) purchased from Scientific Adsorbents, Atlanta, GA. Organic extracts were dried over anhydrous Na2SO4.
2.1.1. 2-(3-Methoxyphenyl) ethanol (5a) [28]
To a suspension of LiAlH4 (4.93 g, 123 mmol) in anhydrous Et2O (100 ml), m-methoxyphenylacetic acid (17 g, 102 mmol) dissolved in anhydrous Et2O (100 ml) was added dropwise over 1.5 h at room temperature. Stirring was continued overnight and the reaction flask was cooled with an ice bath, H2O (15 ml) was cautiously added dropwise over 30 min, and then 2 N H2SO4 (200 ml) was added to bring the aqueous layer to neutral pH. The aqueous layer was extracted with Et2O and the combined organic extracts were washed with brine and dried. Solvent removal gave the crude product (16.2 g), which was purified by vacuum distillation to yield alcohol 5a (bp 133 °C/6 mm Hg, 14.6 g, 94%).
2.1.2 Toluene-4-sulfonic acid 2-(3-methoxyphenyl)ethyl ester (5b) [29]
To a solution of m-methoxyphenylethanol (5a, 3 g, 19.7 mmol) in anhydrous pyridine (16 ml), was added p-toluenesulfonyl chloride (4.15 g) at 0 °C. After 30 min, the reaction flask was placed in a cold room (5 °C) and stirred overnight. The reaction mixture was poured onto ice, neutralized with 6 N HCl and extracted with EtOAc. The combined extracts were again washed with 6 N HCl, washed with brine and dried. Solvent removal gave a thick oil (5.6 g) which was purified by chromatography (20% EtOAc in hexanes) to yield compound 5b (4.73 g, 78%): 1H NMR(CDCl3) δ 2.41 (s, 3H, Ar-CH3), 2.91 (t, J = 7.2 Hz, 2H), 3.74 (s, 3H, OCH3), 4.20 (t, J = 7.2 Hz, 2H), 6.62 (s, 1H, Ar-H), 6.69 (d, J = 7.5 Hz, 1H, Ar-H), 6.73–6.77 (dd, J = 8.4 Hz, 1.5 Hz, 1H, Ar-H), 7.16 (t, J = 8.1 Hz, 1H, Ar-H), 7.25–7.28 ( d, J = 8.4 Hz, 2H, Ar-H), 7.66–7.69 (d, J = 8.1 Hz, 2H, Ar-H).
2.1.3. (1R,7aR)-1-(1,1-dimethylethoxy)-1,2,3,6,7,7a-hexhydro-4-[2-(3-methoxyphenyl)ethyl]-7a-methyl-5H-inden-5-one (6)
Under N2, a 60% suspension of NaH (1.21 g, 30.3 mmol) in mineral oil was washed with anhydrous hexanes (2 × 10 ml) to remove the mineral oil. After removal of the hexanes, anhydrous DME (48 ml) and then indenone 4 (4.5 g, 20.3 mmol) were added. The reaction mixture was heated and stirred at 65 °C for 20 h during which time it turned dark brown. Tosylate (5b, 7.09 g, 23.2 mmol) dissolved in DME (40 ml) was then added over 15 min and the reaction mixture was further heated at 65 °C for 20 h. After cooling the reaction flask with an ice bath, saturated aqueous NaH2PO4 (50 ml) was added and the resultant red orange solution was extracted with CH2Cl2. The combined organic extracts were washed with brine and dried. Solvent removal gave deep orange crude product 6 (9.01 g), which was purified by chromatography (6% EtOAc in hexanes). Product 6 (4.33 g, 60%) was obtained as a colorless oil: [α]20D −41.8 (c = 0.46, CHCl3); UV λmax (EtOH) 251nm (ε =15100); IR (neat, cm−1) 1661, 1584, 1258, 1195, 1098; 1H NMR (CDCl3) δ 1.06 (s, 3H, CH3), 1.20 (s, 9H, C(CH3)3 ), 3.48–3.43 (q, J = 10.2 Hz, 7.5 Hz, 1H, HC-OtBu), 3.83 (s, 3H, OCH3), 6.72–6.75 (m, 1H, Ar-H), 6.75–6.80 (m, 2H, Ar-H), 7.21 (t, J = 7.8 Hz, 1H, Ar-H); 13C NMR (CDCl3) δ 198.69, 169.65, 159.42, 143.74, 131.51, 129.02, 121.34, 114.73, 110.84, 79.66, 72.77, 54.98, 44.52, 34.45, 33.93, 33.46, 29.55, 28.45, 27.51, 25.13, 15.54; MS m/z 356 (M+), 300, 222, 179, 166, 148, 135, 122, 107, 91, 57.
2.1.4. (1R,3aR,4S,7aR)-1-(1,1-Dimethylethoxy)octahydro-4-[2-(3-methoxyphenyl)ethyl]-7a-methyl-5H-inden-5-one (7a)
To a solution of compound 6 (3.76 g, 10.6 mmol) in EtOH (360 ml) was added 10% Pd/C (0.96 g) and the reaction mixture was hydrogenated (H2, 3.4 atm) for 1 h. After filtration to remove the catalyst, the solvent was removed and the crude product was purified by chromatography (3.5% EtOAc in hexanes) to yield product 7a (1.96 g, 52%) as a colorless oil: [α20D −24.7 (c = 0.22, EtOH); UV (EtOH) λ max 280 nm (ε = 1800), 273 nm (ε = q930); IR (neat, cm−1) 1705, 1602, 1585, 1259, 1194, 1153, 1118, 1046; 1H NMR (CDCl3) δ 1.01(s, 3H, CH3), 1.13(s, 9H, C(CH3)3), 3.38 (t, J = 8.4 Hz, HC-OtBu), 3.79 (s, 3H, OCH3), 6.72–6.78 (m, 3H, Ar-H), 7.17–7.22 (m 1H, Ar-H); 13C NMR (CDCl3) δ 215.38, 159.68, 143.41, 129.35, 120.91, 114.32, 111.13, 79.60, 72.45, 55.03, 52.86, 47.56, 41.47, 36.06, 34.98, 31.28, 29.20, 28.53, 28.42, 21.67, 12.53; MS m/z 358 (M+), 302, 224, 181, 167, 147, 134, 122, 107, 93, 57.
2.1.5. (1R,3aR,4R,7aR)-1-(1,1-Dimethylethoxy)octahydro-4-[2-(3-methoxyphenyl)ethyl]-7a-methyl-5H-inden-5-one (7b)
Compound 7b (100 mg) was obtained as an oil from the chromatographic separation that yielded compound 7a. Compound 7b had: IR (neat, cm−1) 1706, 1602, 1585, 1259, 1194, 1152, 1119, 1077; 1H NMR (CDCl3) δ 1.02 (s, 3H, CH3), 1.14 (s, 9H, C(CH3)3 ), 3.45 (t, J = 8.7 Hz, HC-OtBu), 3.80 (s, 3H, OCH3), 6.72–6.81 (m, 3H, Ar-H), 7.19 (t, J = 7.8 Hz, 1H, Ar-H); 13C NMR (CDCl3) δ 213.07, 159.74, 144.59, 129.31, 120.89, 114.08, 111.16, 79.40, 72.51, 55.09, 49.99, 49.65, 42.75, 38.01, 35.91, 33.46, 31.70, 28.57, 28.39, 24.48, 11.03; MS m/z 358 (M+), 301, 245, 224, 181, 167, 134, 121, 93, 57.
2.1.6. (8α,13α,14β,17α-17-(1,1-Dimethylethoxy)-3-methoxyestra-1,3,5(10),9(11)-tetraene (8a)
Compound 7a (1.21 g, 3.38 mmol) was dissolved in MeOH (30 ml) and cooled to 0 °C with and ice/salt bath. 10 N HCl (3.2 ml) was quickly added and stirring was continued at 0 °C for 4 h and then at room temperature for an additional 4 h. Product 8a formed as a white precipitate during this time. The reaction was then moved to a cold room (5 °C) and stirring was continued overnight. Filtration gave crude product 8a (0.91g, mp 124–126 °C, this material contains minor amounts of the isomeric tetraene 8b), which was removed by recrystallization from MeOH/CH2Cl2. Product 8a (0.83 g, 72%) had: mp 128–129 °C; [α]20D −109.9 (c = 0.36, CHCl3); UV λ max (EtOH) 263 nm (ε = 14900); IR (KBr, cm−1) 1626, 1604, 1568, 1255, 1197, 1116; 1H NMR (CDCl3) δ 0.78 (s, 3H, C18-CH3), 1.17 (s, 9H, C(CH3)3 ), 3.54 (t, J = 8.7 Hz, C17-H), 3.79 (s, 3H, C3-OCH3), 6.12 (d, J = 5.4 Hz, C11-H), 6.59 (d, J = 2.7 Hz, C4-H), 6.71 (dd, J = 8.7 Hz, 2.7 Hz, 1H, C2-H), 7.53 (d, J = 8.7 Hz, 1H, C1-H); 13 C NMR (CDCl3) δ 158.34, 137.57, 135.06, 127.70, 125.15, 118.02, 113.29, 112.63, 80.79, 72.22, 55.15, 47.30, 41.08, 39.49, 38.89, 31.17, 30.09, 28.68, 28.15, 24.29, 11.55. Anal. Calcd for C23H32O2: C, 81.13; H, 9.47; Found: C, 81.26; H, 9.47.
Compound 7b (140 mg) was similarly converted to product 8a (100 mg, 75%).
2.1.7. (13α,14β,17α)-17-(1,1-Dimethylethoxy)-3-methoxyestra-1,3,5(10),8-tetraene (8b)
The solid (150 mg) recovered from the mother liquors from recrystallizations of several preparations of compound 8a contained both ent-steroids 8a and 8b (~2:3 ratio). Chromatography (successive elution with 1%, 1.5%, 2% and 2.5% Et2O in hexanes) gave product 8b (90 mg eluted in the 2% Et2O fractions after compound 8a eluted): mp 82–83 °C; [α]25D = −118.1 (c = 0.43, CHCl3); 1H NMR (CDCl3) δ 0.93 (s, 3H, C18-CH3), 1.17 (s, 9H, C(CH3)3 ), 2.71 (t, J = 8 Hz, 2H, C6-CH2), 3.56 (t, 1H, C17-H), 3.79 (s, 3H, OCH3), 6.68 (s, 1H, C4-H), 6.71 (d, J = 8.7 Hz, 1H, C2-H), 7.12 (d, J = 8.7 Hz, 1H, C1-H); 13 C NMR (CDCl3) δ 157.83, 137.16, 135.11, 129.77, 123.97, 122.81, 113.41, 110.84, 78.69, 72.61, 55.19, 48.96, 42.46, 33.20, 30.44, 28.86, 28.76, 28.59, 28.03, 22.38, 20.18.
2.1.8. (8α,9β,13α,14β,17α)-17-(1,1-Dimethylethoxy)-3-methoxyestra-1,3,5(10)-triene (9a)
Compound 8a (1.08 g, 3.18 mmol) in EtOAc (74 ml) was hydrogenated in a Parr hydrogenator (H2, 3.4 atm) using a 10% Pd/C (200 mg) catalyst for 6 h. Solvent removal gave a crude product (1.27 g) that was a mixture of products 9a and 9b. Chromatography (1% ether in hexanes) gave product 9a (0.88 g, 81%): mp 90–91 °C; [α20D −61.9 (c = 0.49, CHCl3); UV λmax (EtOH) 273nm (ε = 2310), 277 nm (ε = 2330), 287 nm (ε = 2050); IR (KBr, cm−1) 1611, 1575, 1198; 1H NMR(CDCl3) δ 0.75 (s, 3H, C18-CH3), 1.15 (s, 9H, C(CH3)3, 2.82–2.86 (m, 2H, C6-CH2), 3.45( t, J = 7.8 Hz, C17-H), 3.78 (s, 3H, OCH3), 6.63 (d, J = 2.7 Hz, C4-H), 6.72 (dd, J = 8.7 Hz, 2.7 Hz, C2-H), 7.22 (d, J = 8.4 Hz, C1-H); 13C NMR (CDCl3) δ 157.49, 138.17, 132.99, 126.44, 113.80, 111.47, 80.84, 72.17, 55.15, 49.96, 44.05, 42.69, 38.68, 37.16, 31.14, 29.82, 28.68, 27.19, 26.30, 23.40, 11.49.
2.1.9. (8α,9α,13α,14β,17α)-17-(1,1-Dimethylethoxy)-3-methoxyestra-1,3,5(10)-triene (9b)
Chromatography of the mixture of products 9a and 9b also yielded pure product 9b (200 mg, 18%): [α20D +20.2 (c = 0.51, CHCl3); UV λ max (EtOH) 288 nm (ε = 1580), 279 nm (ε = 1910); IR (KBr, cm−1) 1608, 1575, 1267, 1079, 1059; 1H NMR (CDCl3) δ 0.83 (s, 3H, C18-CH3), 1.06 (s, 9H, C(CH3)3, 3.23 (t, J = 7.8 Hz, C17-H), 3.77 (s, 3H, OCH3), 6.62 (d, J = 2.4 Hz, C4-H), 6.72 (dd, J = 8.7 Hz, 2.7 Hz, C2-H), 7.27 (d, J = 8.4 Hz, C1-H); 13C NMR (CDCl3) δ 157.15, 139.04, 130.75, 127.51, 113.71, 111.80, 80.87, 72.10, 55.00, 42.63, 41.26, 37.27, 33.92, 32.78, 30.52, 28.61, 25.92, 25.28, 24.46, 23.42, 11.09.
2.1.10. (8α,9β,13α,14β,17α)-3-Methoxyestra-1,3,5(10)-trien-17-ol (10)
6 N HCl (3 ml) was added to a solution of compound 9a (0.31 g, 0.91 mmol) dissolved in THF (3 ml) and EtOH (3 ml). The mixture, which became turbid, was refluxed 35 min during which time a clear solution was obtained. After cooling with an ice bath, 6 N NaOH (3 ml) was added. The THF was removed on a rotary evaporator and the remaining aqueous phase was extracted with EtOAc. The combined EtOAc extracts were washed with brine and dried to yield crude product. This material was converted to ent-17β-estradiol (3) without purification. Product 10 had: 1H NMR (CDCl3) δ 0.77 (s, 3H, C18-CH3), 2.85 (m, 2H, C6-CH2), 3.77 (s, 3H, OCH3), 6.63 (d, J = 2.7 Hz, 1H, C4-H), 6.69–6.74 (dd, J = 8.7 Hz, 2.7 Hz, C2-H), 7.21 (d, J = 8.4Hz, C1-H).
2.1.11. (8α,9β,13α,14β,17α)-Estra-1,3,5(10)-trien-3,17-diol (3, ent-17β-estradiol)
Unpurified compound 10 dissolved in anhydrous toluene (8 ml) was added to a 1 M solution of DIBALH (8 ml, 8 mmol) in hexanes under N2. Then the reaction solution was refluxed for 24 h during which time it became pale yellow. After cooling to room temperature, the reaction solution was poured onto ice (50 g). After the oily product 3 solidified, the water was acidified with 3 N HCl and the mixture was extracted with EtOAc. The combined organic extracts were washed with brine and dried. Solvent removal gave crude product 3. Chromatography (30% EtOAc in hexanes) and recrystallization from acetone/hexanes gave purified product 3 (200 mg, 84% from compound 9a): mp 176–177 °C; lit [7] mp 176–177 °C; lit [30] mp 177–178 °C; after second recrystallization, [α25D −82 (c = 0.28, dioxane); UV (EtOH) λ max 281 nm (ε = q250); IR (KBr, cm−1) 3434, 1610, 1586, 1500, 1250, 1056, 1012; δH NMR (CDCl3/acetone-d6) δ 0.79 (s, 3H, C18-CH3), 2.78–2.756 (m, 2H, C6-CH2), 2.96 (s, 1H, OH), 3.60 (d, J = 5.1 Hz, C9-H), 3.69 (m, 1H, C17-H), 6.53(d, J = 2.1 Hz, C4-H), 6.60 (dd, J = 8.4 Hz, 2.7 Hz C2-H), 7.09 (d, J = 8.7 Hz, C1-H), 7.90 (d, J = 5.1 Hz, 1H, OH); 13CNMR(CDCl3/acetone-d6) δ 154.03, 136.67, 130.33, 125.24, 114.15, 111.75, 80.07, 48.97, 42.95, 42.13, 38.00, 35.74, 29.04, 26.19, 25.28, 21.88, 9.77. Anal. Calcd. for C18H24O2: C, 79.37; H, 8.88; Found: C, 79.20; H, 8.95.
2.1.12. (8α,9β,13α,14β,17α)-Estra-1,3,5(10)-trien-3,17-diol 17-acetate (11)
To a solution of compound 10 (70 mg, 0.25 mmol) in glacial HOAc (1.5 ml) was added 48% HBr (0.7 ml). A white solid formed. The reaction mixture was then heated under N2 for 1 h to yield a yellow solution. After cooling to room temperature, ice was added and a pink precipitate formed. The mixture was extracted with Et2O and the combined organic extracts were washed with water, aqueous NaHCO3, brine and dried. Solvent removal gave a crude product, which was purified by chromatography (20% EtOAc in hexanes) to give crystalline product 11 (60 mg, 78%). Further elution (30% EtOAc in hexanes gave compound 3 (10 mg). Product 11 had: 1H NMR (CDCl3/acetone-d6) δ 0.84 (s, 3H, C18-CH3), 2.03 (s, 3H, OCOCH3), 2.77–2.79 (m, 2H, C6-CH2), 4.66 (t, J = 8.1 Hz, C17-H), 6.55 (s, 1H, C4-H), 6.60 (d, J = 8.4 Hz, C2-H), 7.09 (d, J = 8.4 Hz, C1-H), 7.87 (br s, C3-OH); 13C NMR (CDCl3/acetone-d6) δ 170.14, 154.12, 137.02, 130.61, 125.57, 114.61, 112.20, 81.96, 49.16, 43.18, 42.24, 38.08, 36.31, 29.86, 27.00, 26.61, 25.59, 22.52, 20.16, 11.29.
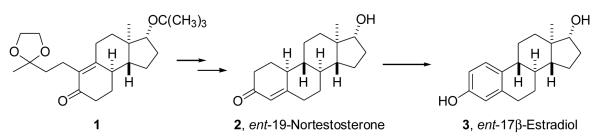
2.1.13. (8α,9β,13α,14β,17α)-2-(1,1-Dimethylethyl)estra-1,3,5(10)-trien-3,17-diol (12a)
A suspension of compound 3 (30 mg, 0.11 mmol) and 2-methyl-2-propanol (0.06 ml, 0.63 mmol) in anhydrous pentane (1 ml) was stirred at room temperature for 15 min and at 0 °C to −5 °C for 20 min. BF3–EtOEt (0.07 ml, 0.56 mmol) was added, stirring was continued at 0 °C to −5 °C for 20 min and then at room temperature. The reaction mixture first became a homogeneous solution and after 15 min a yellow solid formed on the flask wall. After stirring an additional 15 min at room temperature, ice was added. The solid product was filtered, washed with water and dried over P2O5 overnight in a vacuum desiccator. Chromatography (18% EtOAc in hexanes) and recrystallization from acetone/hexanes gave product 12a (20 mg, 55%): mp 177–179 °C; [α]25D −91.3 (c = 0.23, CHCl3); IR (film, cm−1) 3368, 1612, 1511, 1214, 1058, 1011; 1H NMR (CDCl3) δ 0.78 (s, 3H, C18-CH3), 1.40 (s, 9H, C(CH3)3) , 2.74–2.75 (m, 2H, C6-CH2), 3.74 (t, J = 8.4 Hz, C17-H), 6.41 (s, 1H, C4-H), 7.19 (s, 1H, C1-H); 13C NMR (CDCl3) δ 152.05, 135.33, 133.37, 131.81, 124.04, 116.55, 81.98, 50.03, 44.23, 43.26, 38.98, 36.78, 34.47, 30.57, 29.74, 28.91, 27.22, 26.38, 23.12, 11.07; MS m/z 328 (M+), 313, 271, 253, 213, 185, 159, 147, 129, 115, 107, 91, 81, 69; Anal Calcd. for: C22H32O2: C, 80.44; H, 9.82. Found: C, 80.41, H, 9.62.
2.1.14. (8α,9β,13α,14β,17α)-2-(1-adamantyl)estra-1,3,5(10)-trien-3,17-diol (12b)
A suspension of compound 3 (40 mg, 0.15 mmol) and 1-adamantanol (20 mg, 0.13 mmol) in anhydrous pentane (1 ml) was stirred at room temperature for 20 min and then at −5 °C for 15 min. BF3–EtOEt (0.05 ml, 0.40 mmol) was added, stirring was continued at 0 °C to −5 °C for 20 min and gave a pale yellow solution. Stirring was continued at room temperature and after 15 min a precipitate formed on the flask wall. After 45 min, ice was added. The solid product was filtered, washed with water and dried over P2O5 in a vacuum desiccator. Crude product 12b (50 mg) was purified by chromatography (20% EtOAc in hexanes). Product 12b (40 mg, 67%) was recrystallized from CH2Cl2/hexanes and had: mp174–176 °C; [α]24D −198 (c = 0.1, CHCl3); IR (film, cm−1) 3368, 1613, 1511, 1249, 1215, 1121, 1052, 1011; 1H NMR ( CDCl3) δ 0.78 (s, 3H, C18-CH3); 1.77 (br s, 6H, adamantyl-H), 2.07 (br s, 3H, adamantyl-H), 2.11 (br s, 6H, adamantyl-H), 2.75–2.76 (m, 2H, C6-CH2); 3.78 (t, J = 8 Hz, 1H, C17-H); 6.39 (s, 1H, C4-H) 7.15 (s, 1H, C1-H ); 13C NMR (CDCl3) δ 152.16, 135.16, 133.73, 132.13, 124.02, 116.81, 81.98, 50.08, 44.31, 43.29, 40.82, 39.01, 37.10, 36.81, 36.64, 30.65, 29.10, 28.87, 27.22, 26.44, 23.14, 11.07; MS m/z 406 (M+), 306, 293, 271, 253, 183, 159, 135, 107, 91, 79, 67. Anal Calcd. for C28H38O2: C, 82.71; H, 9.42. Found: C, 82.88; H, 9.31.
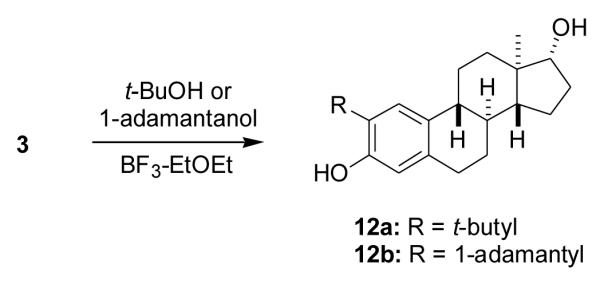
2.1.15. (8α,13α,14β,17α)-3-Methoxyestra-1,3,5(10),9(11)-tetraen-17-ol (13a)
To a stirred solution of compound 8a (100 mg, 0.29 mmol) in anhydrous CH2Cl2 (4 ml) at −10 °C, was quickly added a 1 M solution of TiCl4 in CH2Cl2 (0.38 ml). After 15min, water (4 ml) was added and the heterogeneous solution was extracted with CH2Cl2. The combined extracts were washed with brine and dried. Solvent removal gave crude product 13a (90 mg) as a solid, which was used without further purification. Product 13a had: 1H NMR (CDCl3) δ 0.80 (s, 3H, C18-CH3), 2.84 (m, 2H, C6-CH2), 3.79 (s, 3H, OCH3), 3.82 (m, 1H, C17-H), 6.13 (t, 1H, J = 2.1 Hz, C11-H), 6.60 (d, 1H, J = 2.7 Hz, C4-H), 6.72 (dd, 1H, J = 2.7 Hz, 8.7 Hz, C2-H), 7.54 (d, 1H, J = 8.7 Hz, C1-H); 13C NMR (CDCl3) δ 158.47, 137.58, 135.12, 127.57, 125.23, 117.54, 113.35, 112.69, 82.02, 55.18, 47.35, 41.50, 38.91, 38.80, 30.71, 30.02, 28.15, 23.87, 10.85.
2.1.16. (13α,14β,17α)-3-Methoxyestra-1,3,5(10),8-tetraen-17-ol (13b)
To a solution of compound 8b (90 mg, 0.27 mmol) in anhydrous CH2Cl2 (3 ml) at −5 °C was added a 1 M solution of TiCl4 in CH2Cl2 (0.30 ml, 0.30 mmol). The reaction solution became orange. After 5 min, water was added and the heterogeneous solution was extracted with CH2Cl2. The combined extracts were washed with brine and dried. Solvent removal gave crude product 13b (70 mg) as a solid, which was used without further purification. Product 13b had: 1H NMR (CDCl3) δ 1.00 (s, 3H, C18-CH3), 2.73 (t, J = 8.1 Hz, 2H, C6-CH2), 3.84 (t, J = 6 Hz, 1H, C17-H), 6.69 (s, 1H, C4-H), 6.73 (dd, J = 2.7 Hz, 8.1 Hz, C2-H), 7.13 (d, J = 8.1Hz, C1-H).
2.1.17. (8α,13α,14β,17α)-Estra-1,3,5(10),9(11)-tetraene-3,17-diol (14a)
A solution of compound 13a (120 mg, 0.42 mmol) in anhydrous toluene (4 ml) under N2 was added to DIBALH (1.5 M in toluene, 3 ml, 4.5 mmol). The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 3 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. Solvent removal gave a crude product which was purified by chromatography (35% EtOAc in hexanes) and crystallized from CH2Cl2/hexanes to give product 14a (70 mg, 61%) as white crystals: mp 191–192 °C; lit [31] mp 186-191 °C ; [α25D −138.4 (c = 0.31, dioxane), UV (EtOH) λmax 265 nm (ε = 11900); IR (KBr, cm−1) 3421, 1630, 1614, 1578, 1287, 1246, 1055; 1H NMR (CDCl3/acetone-d6) δ 0.81 (s, 3H, C18-CH3), 2.77–2.82 (m, 2H, C6-CH2), 3.35 (d, 1H, J = 5.1 Hz, C8-H) 3.80 (m, 1H, C17-H), 6.08 (d, 1H, J = 5.1 Hz, C11-H), 6.56 (d, 1H, J = 2.7 Hz, C4-H), 6.65 (dd, 1H, J = 8.7 Hz, 2.7 Hz, C2-H), 7.45 (d, J = 8.7Hz, C1-H); 13C NMR (CDCl3/acetone-d6) δ 155.28, 130.92, 134.67, 125.97, 124.55, 116.25, 114.44, 113.16, 80.82, 46.79, 40.87, 38.39, 38.30, 29.83, 28.29, 27.59, 23.20, 10.18; MS m/z: 270 (M+), 211, 181, 169, 157, 149, 129, 111, 97, 83.69.
2.1.18. (13α,14β,17α)-Estra-1,3,5(10),8-tetraene-3,17-diol (14b)
A solution of compound 13b (70 mg, 0.25 mmol) in anhydrous toluene (3 ml) under N2 was added to DIBALH (1.5 M in toluene, 1.5 ml, 2.25 mmol). The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 2.5 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. Solvent removal gave a pale yellow oil (80 mg), which was purified by chromatography (30% EtOAc in hexanes) to give product 14b (40 mg, 60%) as an oil, which later solidified to a red orange solid that could not be further purified. Product 14b had: mp 86–96 °C; lit [31] mp 130–132 °C; [α]25D −99.5 (c = 0.37, CHCl3), UV (EtOH) λ max 273 nm (ε = 16200); IR (KBr, cm−1) 3447, 1678, 1649, 1615, 1260, 1194, 1098; 1H NMR (CDCl3) δ 1.00 (s, 3H, C18-CH3), 2.68 (t, J = 7.6 Hz, 2H, C6-CH2), 3.86 (t, J = 5.4 Hz, 1H, C17-H), 5.83 (s, OH), 6.61 (s, 1H, C4-H), 6.64 (dd, J = 2.7 Hz, 8.1 Hz, 1H, C2-H), 7.06 (d, J = 8.1 Hz, 1H, C1-H); 13C NMR (CDCl3) δ 153.87, 137.27, 134.28, 129.30, 123.82, 122.97, 114.52, 112.59, 80.86, 48.09, 43.56, 32.17, 29.63, 29.16, 28.72, 28.24, 22.24, 18.49; MS m/z 270 (M+), 237, 227, 211, 197, 181, 172, 157, 145, 137, 129, 111, 97, 81, 69.
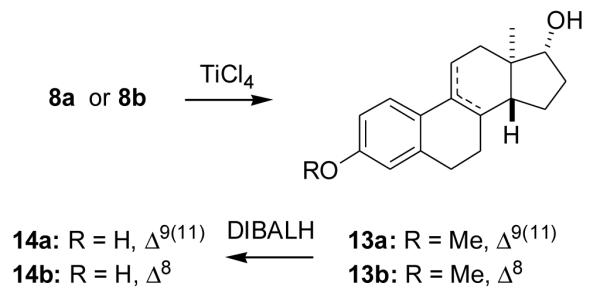
2.1.19. (8α,9β,13α,14β,17β)-3-Methoxyestra-1,3,5(10)-trien-17-ol p-nitrobenzoate (15a)
The mixture of compound 10 (80 mg, 0.28 mmol), p-nitrobenzoic acid (0.12 g, 0.72 mmol), triphenylphosphine (0.15 g, 0.57 mmol) and diethylazodicarboxylate (0.13 g, 0.75 mmol) in anhydrous toluene (2 ml) was heated at 80 °C for 3.5 h. Solvent removal followed by chromatography (10% EtOAc in hexanes) gave product 15a (70 mg, 58%) as a solid: 1H NMR (CDCl3) δ 0.79 (s, 3H, C18-CH3), 2.80 (m, 2H, C6-CH2), 3.69 (s, 3H, OCH3) 5.07 (d, 1H, J = 6 Hz, C17-H), 6.56 (d, 1H, J = 2.4 Hz, C4-H), 6.62 (dd, J = 8.7 Hz, 2.7 Hz, C2-H), 7.11 (d, 1H, J = 8.7 Hz, C1-H), 8.12–8.23 (m, 4H, Ar-H); 13C NMR (CDCl3) δ 164.34, 157.60, 150.55, 137.93, 136.25, 132.35, 130.68, 126.39, 123.59, 113.84, 111.54, 83.82, 55.09, 49.49, 45.39, 43.54, 38.96, 32.08, 30.09, 29.74, 27.94, 25.98, 24.29, 16.60.
2.1.20. (8α,13α,14β,17β)-3-Methoxyestra-1,3,5(10),9(11)-tetraen-17-ol p-nitrobenzoate (15b)
A mixture of compound 13a (0.24 g, 0.85 mmol), p-nitrobenzoic acid (305 mg, 1.84 mmol), triphenylphosphine (0.49 g, 1.87 mmol) and diethylazodicarboxylate (0.45 g, 2.58 mmol) in anhydrous toluene (7 ml) was heated at 80 °C for 6 h. Solvent removal and chromatography (10% EtOAc in hexanes) gave product 15b (150 mg, 41%) as a solid: 1H NMR (CDCl3) δ 0.90 (s, 3H, C18-CH3), 2.87–2.89 (m, 2H, C6-CH2), 3.80 (s, 3H, OCH3), 5.21 (d, J = 6 Hz, C17-H), 6.15 (m, 1H, C11-H), 6.62 (d, 1H, J = 2.7 Hz, C4-H), 6.72 (dd, 1H, J = 8.7 Hz, 2.7 Hz, C2-H), 7.55 (d, 1H, J = 8.7 Hz, C1-H), 8.18–8.31(m, 4H, Ar-H).
2.1.21. (8α,9β,13α,14β,17β)-3-Methoxyestra-1,3,5(10)-trien-17-ol (16a)
Compound 15a (70 mg, 0.16 mmol) dissolved in THF (2 ml) was stirred with 2.8% methanolic KOH (3 ml) at room temperature for 2 h. Solvent removal and chromatography (15% EtOAc in hexanes) gave product 16a (40 mg, 87%): 1H NMR (CDCl3) δ 0.70 (s, 3H, C18-CH3), 2.84–2.87 (m, 2H, C6-CH2), 3.78 (s, 3H, OCH3), 3.82 (m, 1H, C17-H), 6.64 (d, 1H, C4-H), 6.72 (dd, J = 8.7 Hz, 2.7 Hz, C2-H), 7.23 (d, J = 8.7 Hz, C1-H); 13C NMR (CDCl3) δ 157.57, 138.17, 132.87, 126.45, 113.88, 111.53, 80.08, 55.16, 47.71, 45.51, 43.55, 39.03, 32.36, 31.41, 29.84, 27.99, 26.14, 24.18, 16.96.
2.1.22. (8α,13α,14β,17β)-3-Methoxyestra-1,3,5(10),9(11)-tetraen-17-ol(16b)
Compound 15b (150 mg, 0.35 mmol) dissolved in THF (4 ml) was stirred with 3% methanolic KOH (6 ml) at room temperature for 1h. After acidification with 3 N HCl, and solvent removal, the residue was chromatographed (20% EtOAc in hexanes) to give product 16b (60 mg, 61%) as a solid: 1H NMR (CDCl3) δ 0.71 (s, 3H, C18-CH3), 2.82–2.84 (m, 2H, C6-CH2), 3.78 (s, 3H, OCH3), 3.84–3.86 (d, 1H, J = 5.1 Hz, C17-H), 6.17 (t, 1H, J = 2.7 Hz, C11-H), 6.60 (d, 1H, J = 2.4 Hz, C4-H), 6.71 (dd, 1H, J = 8.7 Hz, 2.4 Hz, C2-H), 7.54 (d, 1H, J = 8.7 Hz, C1-H). 13C NMR (CDCl3) δ 158.31, 137.55, 134.79, 127.66, 125.15, 117.90, 113.27, 112.63, 79.49, 55.13, 45.43, 43.89, 38.98, 33.08, 32.66, 30.09, 29.09, 24.92, 17.37.
2.1.23. (8α,9β,13α,14β,17β)-estra-1,3,5(10)-triene-3,17-diol (17a)
A solution of compound 16a (40 mg, 0.14 mmol) in anhydrous toluene (3 ml) under N2 was added to DIBALH (1.5 M in toluene, 1.5 ml, 2.25 mmol). The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 3 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. Solvent removal, chromatography (20% EtOAc in hexanes) and recrystallization from acetone/hexanes gave product 17a (30 mg, 79%): mp 224–225 °C; [α22D −54.9 (c = 0.40, dioxane); IR (KBr, cm−1) 3421, 1611, 1587, 1252, 1234, 1035; 1 D H NMR (CDCl3/acetone-d6) δ 0.70 (s, 3H, C18-CH3), 2.77–2.78 (m, 2H, C6-CH2), 3.75 (q, J = 6 Hz, 1H, C17-H), 6.54 (d, 1H, J = 2.4 Hz, C4-H), 6.59 (dd, 1H, J = 8.4 Hz, 2.4 Hz, C2-H), 7.10 (d, 1H, J = 8.4 Hz, C1-H); 13C NMR (CDCl3/acetone-d6) δ 154.02, 136.72, 130.50, 125.29, 114.18, 111.78, 78.14, 46.51, 44.39, 42.72, 38.30, 31.18, 30.53, 29.01, 27.12, 25.27, 23.08, 15.77. Anal Calcd. for C18H24O2: C, 79.37; H, 8.88. Found: C, 79.18; H, 8.62.
2.1.24. (8α,13α,14β,17β)-Estra-1,3,5(10),9(11)-tetraene-3,17-diol (17b)
A solution of compound 16b (60 mg, 0.21 mmol) in anhydrous toluene (3 ml) under N2 was added to DIBALH (1.5 M in toluene, 1.5 ml, 2.25 mmol). The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 3 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. Solvent removal, chromatography (20% EtOAc in hexanes) and recrystallization from acetone/hexanes gave product 17b (40 mg, 70%): mp 239–241 °C; [α25D −131.3 (c = 0.27, dioxane); UV (EtOH) λmax 263 nm (ε = 15100); IR (KBr, cm−1) 3482, 1634, 1581, 1284, 1236, 1158, 1027; 1H NMR (CD3OD) δ 0.72 (s, 3H, C18-CH3), 3.76 (d, 1H, J = 6 Hz, C17-H), 6.12 (t, 1H, J = 2.4 Hz, C11-H), 6.47 (d, 1H, J = 2.4 Hz, C4-H), 6.55 (dd, 1H, J = 8.7 Hz, 2.4 Hz, C2-H), 7.43 (d, 1H, J = 8.7 Hz, C1-H); 13C NMR (CD3OD) δ 157.41, 138.94, 136.71, 128.10, 126.46, 118.39, 116.07, 114.91, 80.53, 47.01, 45.30, 40.88, 34.54, 33.16, 31.24, 30.82, 26.24, 18.13. Anal Calcd. for C18H22O2 : C, 79.96; H, 8.20. Found: C, 79.77; H, 8.37.
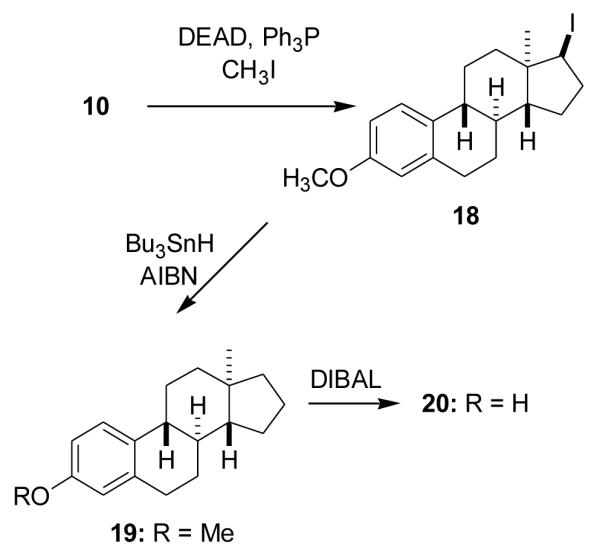
2.1.25. (8α,9β,13α,14β,17β)-17-Iodo-3-methoxyestra-1,3,5(10)-triene (18)
To the stirred solution of compound 10 (120 mg, 0.42 mmol) and triphenylphosphine (140 mg, 0.53 mmol) in anhydrous toluene (3 ml) was added diethylazodicarboxylate (140 mg, 0.80 mmol) and then CH3I (130 mg, 0.92 mmol) dissolved in toluene (1 ml). A precipitate formed. The reaction was stirred at room temperature for 30 min and then refluxed for 15 min. Solvent removal gave a dark brown oil and chromatography (5% EtOAc in hexanes) gave product 18 (70 mg, 41%) as an oil: 1H NMR (CDCl3) δ 0.86 (s, 3H, C18-CH3), 2.84–2.89 (m, 2H, C6-CH2), 3.77 (s, 3H, OCH3), 4.42 (d, 1H, J = 6.9 Hz, - CHI), 6.63 (d, 1H, J = 1.8 Hz, C4-H), 6.71 (dd, 1H, J = 8.4 Hz, 2.4 Hz, C2-H), 7.21 (d, 1H, J = 8.4 Hz, C1-H).
2.1.26. (8α,9β,13α,14β)-3-Methoxyestra-1,3,5(10)-triene (19)
Under N2, AIBN (14 mg, 85 μmol) and (Bu)3SnH (0.3 ml, 1.12 mmol) were added to a solution of compound 18 ( 90 mg, 0.22 mmol) in anhydrous benzene (3 ml). The reaction was refluxed for 1.5 h. Solvent removal and chromatography (5% EtOAc in hexanes) gave product 19 (70 mg) as an oil: 1H NMR (CDCl3) δ 0.74 (s, 3H, C18-CH3), 2.83–2.85 (m, 2H, C6-CH2), 3.72 (s, 3H, OCH3), 6.63 (d, 1H, J = 2.7 Hz, C4-H), 6.71 (dd, 1H, J = 8.7 Hz, 2.7 Hz, C2-H), 7.22 (d, 1H, J = 8.7 Hz, C1-H); 13C NMR (CDCl3) δ 157.50, 138.15, 133.19, 126.40, 113.82, 111.44, 55.11, 53.49, 43.97, 41.00, 40.44, 39.10, 38.75, 29.87, 28.02, 26.67, 25.09, 20.46, 17.43.
2.1. 27. (8α,9β,13α,14β)-Estra-1,3,5(10)-trien-3-ol (20)
A solution of compound 19 (70 mg, 0.26 mmol) in anhydrous toluene (3 ml) was added to DIBALH (1.5 M in toluene, 1.5 ml, 2.25 mmol) under N2. The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 3 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. After solvent removal, chromatography (20% EtOAc in hexanes) gave product 20 (40 mg, 60%): mp 130–131 °C (recrystallized from EtOAc/hexanes); lit [32] mp 134–135 °C; [α25D −100.5 (c = 0.19, CHC13); lit [32] [α20D −92 (c = 1, EtOH); 1H NMR (CDCl3) δ 0.74 (s, 3H, C18-CH3), 2.80–2.81 (m, 2H, C6-CH2), 4.63 (s, OH), 6.56 (s, 1H, C4-H), 6.63 (d, J = 8.4 Hz, C2-H), 7.17 (d, 1H, J = 8.4 Hz, C1-H); 13C NMR (CDCI3) δ 153.27, 138.51, 133.33, 126.64, 115.26, 112.62, 53.51, 43.96, 41.00, 40.46, 39.08, 38.76, 29.68, 27.97, 26.69, 25.11, 20.47, 17.45.
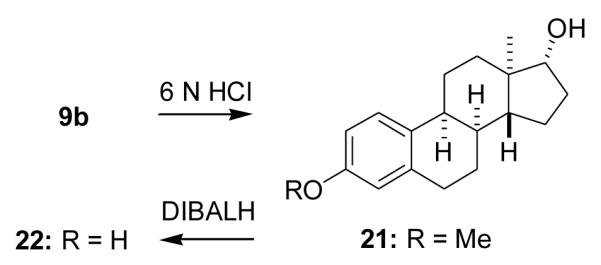
2.1.28. (8α,9α,13α,14β,17α)-3-Methoxyestra-1,3,5(10)-trien-17-ol (21)
To a solution of compound 9b (0.41 g, 1.2mmol) in THF (4 ml) and EtOH (4 ml) was added 6 N HCl (4 ml). The reaction was heated with an oil bath to 100 °C for 1 h, then cooled with an ice bath and neutralized with 6 N NaOH (3.5 ml). The THF was removed and the remaining solution was extracted with EtOAc. The combined extracts were dried and the solvent removed to give crude product 21 as a pale brown solid (0.36 g), which was immediately converted to compound 22 without purification or characterization.
2.1.29. (8α,9α,13α,14β,17α)-Estra-1,3,5(10)-triene-3,17-diol (22)
A solution of compound 21 (90 mg, 0.32 mmol) in anhydrous toluene (4 ml) under N2 was added to DIBALH (1.5 M in toluene, 3 ml, 4.5 mmol). The reaction was refluxed overnight, cooled to room temperature and ice was added. The reaction mixture was then acidified with 3 N HCl and extracted with EtOAc. The combined extracts were washed with brine and dried. Solvent removal and chromatography (30% EtOAc in hexanes) gave product 22 (70 mg, 82%): mp 223–224 °C (crystallized from acetone/hexanes); [α25D +55.3 (c = 0.34, dioxane); IR (KBr, cm−1) 3393, 1610, 1505, 1239, 1230, 1044; 1H NMR (CDCl3/acetone-d6) δ 0.86 (s, 3H, C18-CH3), 3.21 (d, 1H, J = 5.1 Hz, C9-H), 3.5 (m, 1H, C17-H), 6.56 (d, 1H, J = 2.4 Hz, C4-H), 6.63 (dd, J = 2.4 Hz, 8.4 Hz, C2-H), 7.16 (d, J = 8.4 Hz, C1-H). 13C NMR (CDCl3/acetone-d6) δ 153.84, 138.07, 128.49, 126.64, 114.67, 112.54, 80.69, 42.44, 40.59, 36.47, 33.46, 31.65, 29.70, 24.95, 24.64, 23.84, 22.34, 9.85. Anal Calcd. for C18H24O2: C, 79.37; H, 8.88. Found: C, 79.10; H, 8.68.
3. Results and Discussion
3.1. Synthesis of ent-17β-estradiol
Indenone 4 [33], the C,D-ring synthon, was treated with NaH in ethylene glycol dimethyl ether and reacted with tosylate 5b [29] to afford compound 6 (60%). Hydrogenation of compound 6 gave a 52% yield of the indenones 7a (major isomer) and 7b (minor isomer). Treatment of compound 7a with 10 N HCl at 0 °C results in epimerization of the (methoxyphenyl)ethyl group to give 7b and subsequent cyclization leads to Δ9(11) ent-steroid 8a (72%) and a small amount of the isomeric Δ8 ent-steroid 8b as products. Under these reaction conditions, compound 7b also yields products 8a and 8b. The trans ring fusion of the C,D-rings of ent-steroid 8a was established initially by 1H NMR and 13C NMR spectroscopy. The chemical shifts of the 18-Me group protons (δ = 0.78) and the carbon resonance of this group (δ = 11.55) are both characteristic of the trans C,D-ring fusion of ent-steroid 8a [34]. Hydrogenation of compound 8a produced ent-steroids 9a (81%, major product) and 9b (minor product). Removal of the tert-butyl protecting group from the oxygen atom at C17 using 6 N HCl in THF/EtOH converts compound 9a to ent-steroid 10, which was used without purification. Removal of the methyl protecting group from the oxygen atom at C3 using DIBALH [35] converts compound 10 to ent-17β-estradiol 3 (84% yield overall for the 9a to 3 conversion). Alternatively, methyl group removal from the oxygen atom at C3 using 48% HBr [36] in glacial acetic acid gave a mixture of ent-steroids 3 (minor product) and 11 (major product). The overall yield for the conversion of indenone 4 to ent-17β-estadiol is 15.2%.
3.2. Synthesis of ent-17β-estradiol analogues
We have previously shown that 2-(1-adamantyl)-estrogens bind very poorly to estrogen receptors and are more potent neuroprotective agents than estrogens lacking the adamantyl subsitutent [37]. In part, at least, increased neuroprotective potency may be attributed to increased antioxidant potency. The bulky electron donating substituent increases the stability of the free-radical phenoxy radical. This effectively makes it easier for homolytic cleavage of the phenolic OH bond to occur and increases the rate of formation of the hydrogen radical needed to quench lipid hydoperoxy radicals [38]. To extend these studies of the effect of bulky 2-subsitutents on neuroprotective potency into the ent-estrogen series, either t-butyl alcohol or 1-adamantanol in the presence of BF3–EtOEt were reacted with steroid 3 to obtain steroids 12a (55%) and 12b (67%), respectively (Scheme 3).
Since extended conjugation can also increase the stability of a phenoxy radical, we used the Δ9(11) and Δ8 intermediates 8a and 8b, respectively, to prepare ent-17β-estadiol analogues having an additional double bond conjugated to the phenol ring. The t-butyl group was removed from ent-steroids 8a and 8b with TiCl4 to yield compounds 13a and 13b, and these products were then converted without purification into products 14a (61%) and 14b (60%) by cleavage of the C3 methoxy group with DIBALH (Scheme 4).
Also of interest to us are ent-estrogens in which the D-ring C17 substituent is varied. Previous studies with estrogens having the natural absolute configuration show that the substituent at C17 has a minor effect on neuroprotective activity [1,39] and we sought to verify this for the corresponding ent-estrogens. Using a Mitsunobu reaction [40] the C17 α-OH groups of compounds 10 and 13a were inverted to the epimeric p-nitrobenzoates of compounds 15a (58%) and 15b (41%), the esters were hydrolyzed to obtain compounds 16a (87%) and 16b (61%), and the C3 methoxy groups were cleaved with DIBALH to obtain compounds 17a (79%) and 17b (70%) (Scheme 5).
Since estra-1,3,5(10)-trien-3-ol is an effective neuroprotective agent [2], we also removed the C17 substituent to obtain its enantiomer, ent-steroid 20. Compound 10 was first converted into the C17 β-iodo compound 18 (41%) using DEAD, Ph3P, MeI, the iodo group was removed with Bu3SnH and AIBN [41,42] and the C3 methoxy group was cleaved with DIBALH to obtain ent-steroid 20 [32] (60%) (Scheme 6).
Finally, we used compound 9b to obtain ent-steroid 22 (82%), which has a cis B,C-ring fusion, via compound 21 by the previously described two step acid hydrolysis, DIBALH reaction sequence (Scheme 7).
3.3. Biological evaluation
The estrogen receptor binding, antioxidant and neuroprotective properties of compounds 3, 12a,b, 14a,b, 17b, 20, and 22 have been reported in detail elsewhere [27,43]. All of the evaluated ent-steroids bound more weakly to estrogen receptors (α and β forms) than 17β-estradiol. Compounds 12a,b were particularly weak estrogen receptor ligands because of the bulky substituents at C2. All of the ent-steroids were antioxidants and neuroprotective agents. Compounds 12a,b, because of the steric and electronic properties of the C2 substituents, were the most potent antioxidants. Thus, neuroprotection correlated with antioxidant activity rather than with estrogen receptor binding.
5. Conclusion
Starting with indenone 4, ent-17β-estradiol is obtained in six steps with an overall yield of 15.2%. No hazardous Li/liquid NH3 reduction or expensive reagents are used in the reaction sequence and ent-estrogens containing the Δ9(11)-bond are also accessible via this synthetic route.
Acknowledgement
This research was supported by a research grant from Apollo Biopharmaceutics, Inc., prior to its acquisition by MIGENIX Corp. and by NIH grant AG10485.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- [1].Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure–activity relationship. Mol Pharmacol. 1997;51:535–542. [PubMed] [Google Scholar]
- [2].Green PS, Gordon K, Simpkins JW. Phenolic A ring requirement for the neuroprotective effects of steroids. J Steroid Biochem Mol Biol. 1997;63:229–235. doi: 10.1016/s0960-0760(97)00124-6. [DOI] [PubMed] [Google Scholar]
- [3].Miller CP, Jirkovsky I, Hayhurst DA, Adelman SJ. In vitro antioxidant effects of estrogens with a hindered 3-OH function on the copper-induced oxidation of low density lipoprotein. Steroids. 1996;61:305–308. doi: 10.1016/0039-128x(95)00234-h. [DOI] [PubMed] [Google Scholar]
- [4].Romer W, Oettel M, Droesher P, Schwarz S. Novel “scavestrogens” and their radical scavenging effects, iron-chelating, and total antioxidative activities: Δ8,9-dehydro derivatives of 17α-estradiol and 17β-estradiol. Steroids. 1997;62:304–310. doi: 10.1016/s0039-128x(96)00224-3. [DOI] [PubMed] [Google Scholar]
- [5].Romer W, Oettel M, Menzenbach B, Droescher P, Schwarz S. Novel estrogens and their radical scavenging effects, iron-chelating, and total antioxidative activities: 17α-substituted analogs of Δ9(11)-dehydro-17β-estradiol. Steroids. 1997;62:688–694. doi: 10.1016/s0039-128x(97)00068-8. [DOI] [PubMed] [Google Scholar]
- [6].Tang M, Abplanalp W, Ayres S, Subbiah MT. Superior and distinct antioxidant effects of selected estrogen metabolites on lipid peroxidation. Metabolism. 1996;45:411–414. doi: 10.1016/s0026-0495(96)90212-7. [DOI] [PubMed] [Google Scholar]
- [7].Green PS, Yang S-H, Nilsson KR, Kumar AS, Covey DF, Simpkins JW. The nonfeminizing enantiomer of 17β-estradiol exerts protective effects in neuronal cultures and a rat model of cerebral ischemia. Endocrinology. 2001;142:400–406. doi: 10.1210/endo.142.1.7888. [DOI] [PubMed] [Google Scholar]
- [8].Terenius L. Differential inhibition in vitro of 17β-estradiol binding in the mouse uterus and vagina by optical antipodes of estrogen. Mol Pharmacol. 1968;4:301–310. [PubMed] [Google Scholar]
- [9].Edgren RA, Jones RC. An anti-estradiol effect of Ent-estradiol-17β. Steroids. 1969;14:335–341. doi: 10.1016/0039-128x(69)90021-x. [DOI] [PubMed] [Google Scholar]
- [10].Ananchenko SN, Torgov IV. New synthesis of estrone, d,l-8-iso-oestrone and d.l-19-nortestosterone. Tetrahedron Lett. 1963;4:1553–1558. [Google Scholar]
- [11].Oppolzer W, Roberts DA. 177. The enantioselective synthesis of (+)-estradiol from 1,3-dihydrobenzo[c]thiophene-2,2-dioxide by successive thermal SO2-extrusion and cycloaddition reactions. Helv Chim Acta. 1980;63:1703–1706. [Google Scholar]
- [12].Eder U, Gibian H, Haffer G, Neef G, Sauer G, Wiechert R. Total synthesis of optically active steroids, XIV. Synthesis of estradiol. Chem Ber. 1976;109:2948–2953. [Google Scholar]
- [13].Eder U. Total synthesis of natural and non-natural steroid hormones. J Steroid Biochem. 1979;11:55–60. doi: 10.1016/0022-4731(79)90276-0. [DOI] [PubMed] [Google Scholar]
- [14].Collins MA, Jones DN. A total synthesis of estradiol and its 6,6-dimethyl analogue. Tetrahedron Lett. 1995;36:4467–4470. [Google Scholar]
- [15].Kametani T, Matsumoto H, Nemoto H, Fukumoto K. Asymmetric total synthesis of estradiol by an intramolecular cycloaddition of benzocyclobutene derivative. J Am Chem Soc. 1978;100:6218–6220. [Google Scholar]
- [16].Posner GH, Switzer CJ. Total synthesis of natural estrone and estradiol methyl ethers in extremely high enantiomeric purity via an asymmetric Michael addition to an unsaturated sulfoxide. J Am Chem Soc. 1986;108:1239–1244. [Google Scholar]
- [17].Tietze LF, Nobel T, Spescha M. Synthesis of enantiopure estrone via a double Heck reaction. J Am Chem Soc. 1998;120:8971–8977. [Google Scholar]
- [18].Rigby JH, Warshakoon NC, Payen AJ. Studies on chromium(0)-promoted higher-order cycloaddition-based benzannulation. Total synthesis of (+)-estradiol. J Am Chem Soc. 1999;121:8237–8245. [Google Scholar]
- [19].Kametani T, Aizawa M, Nemoto H. A stereoselective total synthesis of 17-O-acetyl-14α-hydroxy-3-O-methyl-11-oxo-estradiol-17β. J. Chem. Soc. Perkin Trans 1. 1980:2793–2796. [Google Scholar]
- [20].Daniewski AR, Kiegiel J. A facile total synthesis of estrogens. J Org Chem. 1988;53:5535–5538. [Google Scholar]
- [21].Pattenden G, Reddy LK, Walter A. A new total synthesis of (±)-oestrone. Tetrahedron Lett. 2004;45:4027–4030. [Google Scholar]
- [22].Micheli RA, Hajos ZG, Cohen N, Parrish DR, Portland LA, Sciamanna W, Scott MA, Wehrli PA. Total synthesis of optically active 19-norsteroids. (+)-Estr-4-ene-3,17-dione and (+)-13β-ethylgon-4-ene-3,17,-dione. J Org Chem. 1975;40:675–681. doi: 10.1021/jo00894a003. [DOI] [PubMed] [Google Scholar]
- [23].Douglas GH, Graves JMH, Hartley D, Hughes GA, McLoughlin BJ, Siddall J, Smith H. Totally synthetic steroid hormones. Part 1. Oestrone and related Oestrapolyenes. J Chem Soc. 1963:5072–5094. [Google Scholar]
- [24].Hutchinson JH, Money T. An enantioselective synthesis of estrone. Tetrahedron Lett. 1985;26:1819–1822. [Google Scholar]
- [25].Cai ZY, Ni Y, Sun JK, Yu XD, Wang Y. Total synthesis of optically active 17β-tbutoxy-3-methoxy-7α- or 7β-18-dimethyl-1,3,5(10)-estratrienes. J. Chem. Soc. Chem. Commun. 1985:1277–1278. [Google Scholar]
- [26].Cai ZY, Ni Y, Sun JK, Yu XD, Wang Y. The synthesis of optically active 17β-tert-butoxy-3-methoxy-7α (or 7β),18-dimethyl-1,3,5(10) estratrienes. Huaxue Xuebao. 1986;44:78–83. [Google Scholar]
- [27].Perez E, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of estratriene analogs: structure–activity relationships and molecular optimization. Drug Development Research. 2005;66:78–92. [Google Scholar]
- [28].Hunter JH, Hogg JA. Synthetic sterols. III. Isomers of 1-ethyl-2-methyl-7-methoxy-1,2,3,4,9,10,11,12-octahydrophenanthrene-2-carboxylic acid. J Am Chem Soc. 1949;71:1922–1925. [Google Scholar]
- [29].Collins DJ, Fallon GD, Skene CE. The structure and function of estrogens. 11. Synthesis of (±)- 7(8→11α) abeo-estradiol and its 9,11-didehydro derivative. Aust J Chem. 1992;45:71–97. [Google Scholar]
- [30].Buzby GC, Jr., Hartley D, Hughes GA, Smith H. Totally synthetic steroid hormones. XIII. The chemical resolution of some racemic estrane, 13β-ethylgonane, and 13β-n-propylgonane derivatives and the preparation of some estrane and 13β-ethylgonane derivatives of unnatural configuration. J Med Chem. 1967;10:199–204. doi: 10.1021/jm00314a015. [DOI] [PubMed] [Google Scholar]
- [31].Preparation of ent-steroids as selectively effective estrogens. German Patent DE 19917930 A1, 18 pp. Chem Abstr. 2000;133:296594.
- [32].Allais A, Paturet M. 3-(β-Dialkylaminoethoxy)estra-1,3,5(10)-trienes. Addition to French Patent 1338308, 2 pp. Chem Abstr. 1969;70:115418e.
- [33].Rychnovsky SD, Mickus DE. Synthesis of ent-cholesterol, the unnatural enantiomer. J Org Chem. 1992;57:2732–2736. [Google Scholar]
- [34].Groth U, Kohler T, Taapken T. Zinc(II)-chloride induced thioalkylation of aluminum enolates: enantioselective synthesis of estradiol-3-methyl-17-tert-butyl diether. Tetrahedron. 1991;47:7583–7592. [Google Scholar]
- [35].Tietze LF, Wolfling J, Schneider G, Noltemeyer M. Synthesis of new 16-spirosteroids. Steroids. 1994;59:305–309. doi: 10.1016/0039-128x(94)90118-x. [DOI] [PubMed] [Google Scholar]
- [36].Collins DJ. The structure and function of estrogens. V. Synthesis of (9,12,12-2H3)- and (11ξ12,12-2H3)-estradiol. Aust J Chem. 1983;36:403–407. [Google Scholar]
- [37].Perez E, Liu R, Yang S-H, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005;1038:216–222. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- [38].Mahoney LR. Antioxidants. Angew Chem Int Ed. 1969;8:547–555. [Google Scholar]
- [39].Prokai L, Oon S-M, Prokai-Tatrai K, Abboud KA, Simpkins JW. Synthesis and biological evaluation of 17β-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants. J Med Chem. 2001;44:110–114. doi: 10.1021/jm000280t. [DOI] [PubMed] [Google Scholar]
- [40].Dodge JA, Lugar CW., III Alcohol inversion of 17β-steroids. Bioorg Med Chem Lett. 1996;6:1–2. [Google Scholar]
- [41].Loibner H, Zbiral E. Reactions using triphenyhlphosphane/azodicarboxylate. 2. Reactions with organophosphorus compounds. XLII. Nucleophilic substitution reactions of hydroxysteroids using triphenylphosphane/diethylazodicarboxylate. Helv Chim Acta. 1977;60:417–425. [Google Scholar]
- [42].Posner GH, Lee JK, White MC, Hutchings RH, Dai H, Kachinski JL, Dolan P, Kensler TW. Antiproliferative hybrid analogs of the hormone 1β,25-dihydroxyvitamin D3: design, synthesis, and preliminary biological evaluation. J Org Chem. 1997;62:3299–3314. doi: 10.1021/jo970049w. [DOI] [PubMed] [Google Scholar]
- [43].The designation of compounds in this paper (compound numbers) is different than that used in reference 18 (abbreviations and code numbers) which reports the details of their biological evaluation. For the reader’s convenience, the designations for the compounds in the two publications are as follows: 3 (ent-E2), 12a (ZYC34), 12b (ZYC33), 14a (ZYC10), 14b (ZYC27), 17b (ZYC12), 20 (ZYC13), 22 (ZYC9).