

Abstract
We describe an engineered fluorescent optogenetic sensor, SuperClomeleon, that robustly detects inhibitory synaptic activity in single, cultured mouse neurons by reporting intracellular chloride changes produced by exogenous GABA or inhibitory synaptic activity. Using a cell-free protein engineering automation methodology that bypasses gene cloning, we iteratively constructed, produced, and assayed hundreds of mutations in binding-site residues to identify improvements in Clomeleon, a first-generation, suboptimal sensor. Structural analysis revealed that these improvements involve halide contacts and distant side chain rearrangements. The development of optogenetic sensors that respond to neural activity enables cellular tracking of neural activity using optical, rather than electrophysiological, signals. Construction of such sensors using in vitro protein engineering establishes a powerful approach for developing new probes for brain imaging.
Introduction
Optogenetic sensors (OSs) based on the green fluorescent protein (GFP), combined with advances in optical imaging methods, have enabled quantitative monitoring of cellular processes with high spatiotemporal resolution (Okumoto, 2010). Many such OSs have been developed to track subcellular localization (Flach et al., 1994) or monitor protein–protein interactions (Xia et al., 2001), and have great potential for measurement of neuronal activity by enabling precise, cellular-level reporting of neural circuit activity (Zhang et al., 2010; Mancuso et al., 2011) based on optical rather than electrophysiological signals (Siegel and Isacoff, 1997; Kuner and Augustine, 2000; Hires et al., 2008). However, engineering OSs that monitor the small and rapid changes in the concentrations of signaling molecules typically associated with neuronal activity has been challenging (Miyawaki et al., 1997; Kuner and Augustine, 2000; Okumoto et al., 2005; Hires et al., 2008; Arosio et al., 2010).
Presynaptic release of GABA opens postsynaptic GABA receptors, resulting in Cl− fluxes that underlie synaptic inhibition. Fluorescent OSs that monitor intracellular Cl− concentration, [Cl−]i, therefore could report on this important class of synaptic action. A first-generation Cl− sensor, Clomeleon, has been used to monitor such dynamic changes in [Cl−]i in cultured neurons (Kuner and Augustine, 2000) and in brain tissue (Berglund et al., 2006). Clomeleon is a fusion of the yellow fluorescent protein (YFP), which contains a serendipitous Cl− binding site adjacent to its chromophore (Wachter et al., 2000), and the chloride-insensitive cyan fluorescent protein (CFP). Halide binding to YFP quenches fluorescence emission (Wachter and Remington, 1999), altering fluorescence resonance energy transfer (FRET) between the CFP donor and the YFP acceptor (Kuner and Augustine, 2000). The use of FRET enables ratiometric determination of [Cl−]i, which is unaffected by variations in fluorescence emission intensity associated with differences in indicator concentration, optical path length, or excitation intensity (Bright et al., 1989; Miyawaki, 2005). However, it remains extremely challenging to detect GABA-induced [Cl−]i changes with Clomeleon because the Cl− affinity of Clomeleon (∼100 mm) is well beyond the physiological range of [Cl−]i (∼5–6 mm in most adult neurons; Berglund et al., 2006). This limits the signal-to-noise ratio (s/n), requiring averaging of multiple trials before a response can be detected via fluorescence imaging.
Here we report the use of recently developed protein engineering automation techniques (Cox et al., 2007; Allert et al., 2010) to improve the fluorescence response of Clomeleon by manipulating its halide affinity and fluorophore characteristics. The resulting sensor, SuperClomeleon, has a significantly improved s/n in the relevant range of postsynaptic [Cl−]i and greatly enhances imaging of synaptic inhibition in neurons.
Materials and Methods
Protein engineering.
Oligonucleotides (79–80 bases) were synthesized by solid phase oligonucleotide synthesis (MerMade 192; BioAutomation) and assembled into full-length open reading frames (ORFs) by automation (Cox et al., 2007), which were reamplified by PCR using 5′-biotinylated primers.
Protein was produced by in vitro-coupled transcription and translation (TnT) reactions (Allert et al., 2010): 2.4 μg of linear, biotinylated dsDNA was added to 30 μl of a BL21 Star (DE3; Invitrogen) cell lysate and a solution of amino acids, nucleotide triphosphates, and cofactors and water (final volume 120 μl) in 96-well PCR plates, which were covered with a breathable seal for optimum aerobic expression and chromophore maturation (Heim et al., 1994), and incubated with shaking for 8 h at 30°C, followed by 6 h at 4°C.
All proteins had combined C-terminal Flag and His6 peptides for affinity purification. Proteins were purified at 4°C from TnT reactions using anti-Flag M2 agarose (Sigma) and Cl−-free buffer: four (15 min) washes of agarose-bound FPs with 20 mm HEPES, pH 7.1, removed Cl− before elution with 120 μl of 3×Flag (Sigma). Protein expression and purity relative to a sample of the Clomeleon chloride-sensitive YFP Topaz domain (CT) was determined by densitometry of the YFP band in protein gels stained with GelCode Blue (Thermo Scientific) using ImageJ (release 1.42q) software (good category of expression >90% pure; poorly expressed proteins were less pure). Protein expression levels neither indicated nor predicted fluorescence brightness, although high fluorescence was correlated with good protein expression.
The linear ORF of CT was optimized for improved expression in Escherichia coli (Allert et al., 2010) is as follows: 5′-CGGCGTAGAGGATCGAGATCTCGATCCCGCGAAATTAATACGACTCACTATAGGGGAATTGTGAGCGGATAACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGGTTAGCAAAGGAGAAGAATTATTTACAGGCGTTGTTCCTATATTAGTAGAATTAGATGGAGACGTAAATGGTCATAAATTCAGCGTATCGGGTGAAGGTGAAGGTGACGCAACCTATGGCAAACTGACCCTGAAATTCATCTGCACCACCGGCAAATTACCAGTACCATGGCCGACCTTAGTAACCACCTTCGGATACGGCGTCCAATGCTTCGCTCGTTATCCGGATCACATGCGTCAGCACGACTTCTTTAAAAGCGCCATGCCGGAAGGCTATGTACAGGAACGCACGATCTTCTTCAAAGACGACGGCAACTACAAAACCCGTGCGGAAGTGAAATTCGAAGGCGATACCCTGGTAAACCGCATCGAACTGAAAGGCATAGACTTCAAAGAAGACGGCAACATCCTGGGCCACAAACTGGAATACAACTACAACAGCCACAACGTCTACATCATGGCGGACAAACAGAAAAACGGCATCAAGGTGAACTTCAAAATCCGCCACAACATCGAAGATGGCAGCGTACAACTGGCGGATCACTATCAGCAGAATACCCCAATCGGCGATGGTCCAGTTCTGCTTCCGGATAACCACTACCTGAGCTATCAGAGCGCACTGAGCAAAGATCCGAACGAAAAACGCGATCACATGGTGCTCCTGGAATTCGTAACCGCAGCAGGCATTACCTTAGGCATGGACGAACTGTACAAAGGCGGCAGCAACGATTACAAAGACGACGACGATAAAGGCGGTAGCCATCATCACCACCACCACTAATAAGAGATCCGGCTGCTAACAAAGCCCGAAAGGAAGCTGAGTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGG-3′, where the italicized portions highlight the regulatory elements before and after the gene. CT is the FRET acceptor in Clomeleon (Kuner and Augustine, 2000) and includes Val1a, L68V, K79R, R80Q, Q148H, and H231L mutations relative to the YFP used to identify halide-binding Site I (Wachter et al., 2000).
The protein sequences of CFP and YFP from Clomeleon were 95% identical. To favor unique sites for primer annealing, the CFP DNA sequence was diverged from YFP to only 79% identity. CFP ORFs, assembled in the manner described above, encoded the C-terminal 24 aa linker and the AgeI restriction site of Clomeleon (Kuner and Augustine, 2000), but with a scrambled rTEV sequence of FQENLY. The diverged DNA sequence of Clomeleon CFP is as follows: 5′-ATGGTAAGCAAAGGTGAAGAACTGTTCACGGGCGTTGTCCCGATTTTGGTTGAACTCGATGGTGATGTTAATGGTCACCGTTTTTCTGTCAGCGGCGAGGGTGAAGGTGATGCCACCTACGGTAAACTGACCTTGAAATTTATTTGCACGACCGGCAAACTGCCGGTTCCATGGCCGACGTTGGTCACCACGCTGACCTGGGGTGTTCAATGCTTTTCTCGCTACCCGGATCACATGAAACAGCATGACTTTTTCAAGTCCGCCATGCCGGAAGGTTATGTGCAGGAACGCACCATCTTTTTCAAAGATGACGGCAACTACAAGACCCGTGCTGAAGTCAAGTTTGAAGGTGATACCTTGGTTAATCGTATCGAGCTGAAAGGTATTGATTTTAAAGAAGATGGCAACATTCTGGGTCACAAACTGGAATACAACTATATCAGCCACAATGTTTACATCACCGCAGACAAACAAAAGAATGGCATCAAAGCGCATTTCAAAATTCGCCACAACATTGAAGATGGTAGCGTTCAACTGGCGGACCATTATCAACAAAATACGCCAATTGGCGATGGCCCGGTCTTGCTGCCGGACAACCATTACCTGTCCACCCAATCTGCCCTCTCGAAAGATCCGAACGAAAAGCGTGACCAvCATGGTCTTGCTGGAGTTTGTGACCGCAGCGGGCATTACACATGGCATGGATGAACTGTACAAAAAGCTCACCGGTTCGGGCTCGGGTTTTCAGAACGAACTCTATGGTGGCGGCTCCGGCGGTACTAGCTCGACCGGCGGCAGCAACGATTACAAAGACGACGACGATAAAGGCGGTAGCCATCATCACCACCACCAC-3′, where the 72 italicized bases correspond to the 24 aa linker.
The linear DNA ORFs of select variants were amplified with primers to clone each ORF into the BamHI (5′-CGCGGATCCCGGCGTAGAGGATCGAGATCTCGATCCC-3′) and HindIII (5′-CCCAAGCTTCCTCAAGACCCGTTTAGAGGCCCCAAGG-3′) restriction sites of pUC19 (Invitrogen). For the gene fusions encoding the 24 aa linker, the CFP vector was digested with AgeI and HindIII. The YFP genes were amplified from their vectors with the 3′ HindIII primer and a new 5′ primer (5′-AAGCTCACCGGTTCGGGCTCGGGTTTTCAGAACGAACTCTATGGTGGCGGCTCCGGCGGTACTAGCTCGACCATGGTTAGCAAAGGAGAAGAATTATTTACAG-3′) encoding the linker and AgeI site.
Truncated fusions connected by the two-residue linker were accessed from either the 24 aa-linked fusions or the individual FP clones. Site-directed mutagenesis (5′-GAGTTTGTGACCGCAGCGCTCGAGGAAGAATTATTTACAGGC-3′ and its reverse complement) provided direct replacement of 40 aa (11 C-terminal donor, 24 linker, and 5 N-terminal acceptor) with the two-residue Leu-Glu (XhoI site) linker (Shimozono et al., 2006).
CT variants were titrated with halide under ambient conditions in 384-well microtiter plates (Corning 3821) using a fluorescence plate reader (Tecan Genios) with appropriate excitation (485 ± 10 nm) and emission (535 ± 12.5 nm) filters. Constant ionic strength (screen, 520 mm; conventionally prepared, 146 mm) was maintained with potassium d-gluconate (Wachter and Remington, 1999)containing the following: 20 mm buffer (KOAc, pH 4.0, 5.0; MES, pH 5.5, 6.0, 6.5; HEPES, pH 7.1, 7.5; TAPS pH 8.0; CHES, pH 9.0; CAPS, pH 10.0; pH adjusted with KOH and HOAc). The in vitro screen used proteins purified from TnT reactions, KCl buffers (0–500 mm; pH 7.1, 0.1% bovine serum albumin), and 150 mm KF buffer for endpoint determination of fully quenched fluorescence emission intensity. Titrations of conventionally prepared CT variants (5 μm dialyzed protein diluted to 1 μm with 20 mm buffer, 150 mm ionic strength) used 14 potassium halide buffers (0–108 mm) and a 108 mm KF endpoint buffer (four replicates per data point).
For conventional protein expression and purification, the pUC19 FP recombinants were transformed into KRX cells (Promega) and expressed in ZYM-5052 auto-induction media (Studier, 2005) with the T7 RNA polymerase under the control of the rhamnose promoter. Cultures were grown in shaker flasks at 25°C for up to 24 h. Proteins were purified (>95% pure) using nickel-charged immobilized ion affinity chromatographic (IMAC) beaded agarose and eluted into 50 mm sodium phosphate, 300 mm NaCl, and 300 mm imidazole at pH 7.8. Bound Cl− was removed by five rounds of 1:1000 dialysis with 10 kDa cutoff membranes in 20 mm HEPES, pH 7.1, at 4°C. The single domain YFP variants were stable at 4°C for months. The CFP–YFP fusions were stored stably at 4°C for up to 1 week or flash frozen in liquid nitrogen for prolonged storage at −80°C. Fusion proteins were titrated with Cl− in triplicate in a spectrofluorometer (SLM Aminco-Bowman series 2; Spectronic Instruments): 25°C, 4 ml quartz cuvettes, exciting at 450 ± 1 nm, collecting emission spectra (460–550 nm), and 4 nm slit widths.
Thermal stabilities were measured following fluorescence emission intensities in a real-time PCR instrument (LightCycler 480 II; Roche), 384-well microtiter plates, heating at 1.1°C/min (20−95°C), 10 acquisitions/°C, 1 s integration time, and a 498 ± 40 nm excitation filter and 580 ± 20 nm emission filter. The denaturation transition midpoint temperature (Tm) values were obtained from first-derivative transforms of the data. Conventionally cloned and expressed single-domain Q69T/V163A variant (79.7 μm in 20 mm HEPES, pH 7.1) was diluted to 3 μm in 2 ml of 20 mm MES, pH 6.0, in a quartz cuvette with a Teflon-coated stir bar and stopper. Coincidence of thermal melts monitored by fluorescence and by circular dichroism (CD) was determined using an Aviv model 202 CD spectrophotometer, the sample was heated 20−98°C while simultaneously monitoring the CD (222 nm) and fluorescence (513 nm excitation monochromator and 530 nm Schott high-pass filter) signals.
Changes in error, δS/S (Marvin et al., 1997), of variant i relative to a reference species, ref, at the midpoint of analyte fluctuations, SM, can be defined as follows:
 |
where 32 ρM > 1 for improved variants (for [Cl−]i sensing, the midpoint, M, is 5.5 mm).
X-ray crystallography.
The Q69T/V163A variant was purified by IMAC (see above), followed by gel filtration (Superdex200; GE Healthcare), eluting with 50 mm Tris and 150 mm NaCl, pH 7.5. The protein was concentrated, dialyzed, spin concentrated (10 kDa cutoff; Sartorius) to 16.4 mg/ml, and crystallized in hanging drops: 2 μl of protein, 2 μl of mother liquor (either 21% (w/v) PEG 2K, 50 mm NaOAcat, pH 4.6, 90 mm MgCl2; or 10% PEG 3K, 150 mm NH4OAc at pH 5.4) for 3 d at 17°C. The crystals were transferred to cryoprotectant (22% PEG 2K, 50 mm NaOAc at pH 4.6) with 20% ethylene glycol and either water (apo) or 100 mm KI (Wachter et al., 2000), equilibrated at room temperature (2 h), and flash frozen in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source SIBYLS beam line at 1.1109 Å (apo, 3ST0) and 1.1169 Å (iodide, 3SV5). The diffraction images were processed using HKL2000 (Otwinowski and Minor, 1997). The crystal structure was solved by molecular replacement using Phaser (McCoy et al., 2007) and the YFP structure 2O24 (Arosio et al., 2007) as the search model. Initial model building and refinement was done in REFMAC (Murshudov et al., 1997), further refinement including ligand occupancy was performed in PHENIX (Adams et al., 2010), model rebuilding was done in Coot (Emsley and Cowtan, 2004), and figures were prepared using PyMOL (DeLano Scientific). Data collection at the SIBYLS beam line, Advanced Light Source, Lawrence Berkeley National Laboratory, was supported in part by the U.S. Department of Energy programs for Integrated Diffraction Analysis Technologies, Molecular Assemblies Genes, and Genomics Integrated Efficiently (Contract DE-AC02-05CH11231).
SuperClomeleon.
The protein sequence of SuperClomeleon with the C-terminal Flag and His6 epitope tags (italicized) used in the in vitro assays was as follows:
MVSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTWGVQCFARYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNAISDNVYITADKQKNGIKANFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAALEEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFGYGVTCFARYPDHMRQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKANFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSYQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYKGGSNDYKDDDDKGGSHHHHHH.
Fusion constructs for eukaryotic expression.
The C-terminal truncated CFP of Clomeleon was subcloned from pECFP (R26K, H164N, and H231L relative to Clomeleon CFP; Clontech) by a sense primer containing a NheI site (5′-GTCAGATCCGCTAGCGCTAC-3′) and an antisense primer containing an XhoI site (5′-AATAACTCGAGGGCGGCGGTCACGAAC-3′). The N-terminal truncated YFP was subcloned from the pRK5-YFP vector by using a sense primer containing an XhoI site (5′-CCACTCGAGGAGGAGCTGTTCACC-3′) and an antisense primer containing an EcoRI site (5′-CGCGAATTCTATTAAAGCTTCTTGTAC-3′). The original Clomeleon (Kuner and Augustine, 2000) was constructed in the mammalian expression vector pRK5. Removing the CFP-containing portion by cutting and resealing at the SpeI site gave the pRK5-YFP vector. pECFP vector was cut open at the NheI and EcoRI site. The fragments encoding CFP and YFP were ligated into the NheI and EcoRI sites of pECFP. Mutations on YFP and CFP were performed by site-directed mutagenesis (Stratagene).
Cellular imaging.
Neurons were obtained from cortex or hippocampus of postnatal day 0–2 mice (Mus musculus) of either sex and cultured according to standard procedures (Kuner and Augustine, 2000). Neurons were transfected 3–4 d after culturing, via Lipofectamine 2000 (Invitrogen; Gitler et al., 2004a).
OS FRET was imaged in the cell body of transfected neurons by using an upright two-photon microscope (Prairie Technology) equipped with a 60× water-immersion objective (Olympus), collecting fluorescence emission of CFP (485 ± 20 nm) and YFP (535 ± 15 nm) in parallel. The CFP donor was excited by 840 nm light from a Ti: sapphire laser (Chameleon; Coherent) with a Pockels cell to attenuate laser power. Images were acquired and analyzed with Prairie View software. Illumination and detection conditions were fixed for all measurements to facilitate comparisons between measurements made on different OSs. The experiments shown in Figure 9, D–F, were done similarly, but used an Olympus FV-1000 two-photon microscope with a 25× objective. It is important to note that the absolute values of FRET ratios depend on the acquisition conditions, such as photomultiplier gain and filter properties, and will therefore differ between microscopes.
Figure 9.

Imaging synaptic inhibition with SuperClomeleon. A, A presynaptic inhibitory neuron was stimulated by an extracellular electrode (stimulate), while a postsynaptic neuron expressing SuperClomeleon was patch clamped via a recording pipette (record). B, A brief train of electrical stimuli (10 Hz, 2 s) applied to the inhibitory neuron produced IPSCs (lower) and changes in FRET ratio (535/485 nm emission, upper) in voltage clamped (−50 mV) postsynaptic neurons expressing either Clomeleon (black) or SuperClomeleon (red). Note different timescales for upper and lower traces. Four FRET signal trials were averaged in an effort to extract a detectable signal from the Clomeleon-expressing neuron; for comparison purposes, the same was done for the SuperClomeleon responses. These examples were chosen because they had similar synaptic charge (integral of IPSCs) in response to the stimuli. C, s/n characteristics for FRET ratio changes resulting from inhibitory synaptic activity. Response s/n characteristics were quantified by integrating the stimulus-induced change in FRET signal (signal) produced by either Clomeleon (black) or SuperClomeleon (red) and dividing this by the baseline FRET noise. These responses were then normalized by dividing them by the synaptic charge produced by the stimulus, accounting for cell-to-cell variations in IPSCs. Individual measurements (n = 16 for each indicator) were then binned into groups to form the cumulative distributions, which were fit by Gaussian functions (smooth curves). Dashed line indicates s/n = 0; i.e., no signal. D, IPSPs (lower) and changes in FRET ratio (535/485 nm emission, upper) measured in response to a brief train of electrical stimuli (10 Hz, 2 s) in neurons not under voltage clamp. Black traces come from a neuron expressing Clomeleon (black), while red traces are from a neuron expressing SuperClomeleon. E, Mean values for integrated change in FRET signal, or s/n (F), measured in response to IPSPs in neurons expressing Clomeleon or SuperClomeleon (n = 9 cells for each). Error bars indicate SEM.
To vary [Cl−]i, neurons (3–4 days in vitro) were transfected with OS variants, placed in a perfusion chamber, and treated with ionophores (10 μm nigericin and 5 μm tributyltin acetate) to clamp [Cl−]i and intracellular pH to extracellular levels. Physiological salines containing various [Cl−] were perfused through the chamber at a rate of 1 ml/min. High-[Cl−] saline contained 161 mm Cl−, 10 mm HEPES, 20 mm d(+)-glucose, 2 mm Na-EGTA, and 4 mm MgCl2. Cl−-free saline consisted of 10 mm HEPES, 20 mm d(+)-glucose, 48 mm Na-gluconate, 105 mm K-gluconate, 2 mm Na-EGTA, and 4 mm Mg (gluconate)2. Intermediate [Cl−] solutions were prepared by mixing the two salines. The KF saline used to fully quench YFP fluorescence contained 10 mm HEPES, 20 mm d(+)-glucose, 48 mm NaF, 105 mm KF, 2 mm Na-EGTA, and 4 mm Mg (gluconate)2. All salines were adjusted to pH 7.1.
Quantifying FRET measurements.
FRET-based Cl− indicators such as Clomeleon and SuperClomeleon have emission spectra that change shape as a function of [Cl−]. These changes in the shape of the emission spectrum are quantified by measuring fluorescence emission at two wavelengths, λ1 and λ2, corresponding to the emission maxima for the CFP donor and YFP acceptor, respectively. An emission ratio, R, is then calculated from the fluorescence emission (F) measured at these two wavelengths, F1 and F2 as follows:
 |
Cl− decreases the fluorescence of the YFP acceptor, F2. As a result, this ratio declines from a maximum, Rmax, in the absence of Cl− to a minimum, Rmin, in saturating [Cl−]. To convert the fluorescence emission ratio into [Cl−]i, the calibration scheme presented in Grynkiewicz et al. (1985) must be modified to take into account the fact that the titration curves decrease as a function of [Cl−]. Here we recapitulate the scheme of Grynkiewicz et al. (1985), in terms appropriate for measurement of [Cl−]i by indicators such as Clomeleon and SuperClomeleon.
The starting point in this scheme is to state the fluorescence intensity at the two emission wavelengths in terms of four proportionality constants, s, representing the relative fluorescence of the Cl− bound (subscript b) and Cl− free (subscript f) forms of indicator at emission wavelengths λ1 and λ2. The total fluorescence of the indicator at each wavelength is then:
where c indicates the concentrations of the Cl− bound (subscript b) and Cl−-free (subscript f) species. sf1 and sb1 represent the fluorescence of the CFP donor in Cl−-free and saturating conditions, respectively. The relative fractions of the Cl− bound and Cl−-free forms of the indicator vary with [Cl−] according to the Cl− dissociation constant, Kd, of the indicator:
 |
Combining Equations 3a and b allows R to be expressed in terms of s and c:
 |
Combining Equations 4 and 5 then yields:
 |
This can be rearranged into the following expressions:
 |
The limiting values for the titration curve, Rmax and Rmin, can be restated in terms of their component s constants:
 |
 |
Substituting these into Equation 8 yields:
 |
Note that although the form of this equation closely resembles Equation 5 of Grynkiewicz et al. (1985), many of the terms are different.
It is worth emphasizing that while Kd is independent of the specific instrument used to measure R, all other variables on the right side of Equation 11 are instrument-specific. Thus, Rmax, Rmin, sf1, and sb1 must be determined empirically for each instrument used to measure R for Clomeleon, SuperClomeleon, or other FRET-based Cl− indicators. This can be done by varying [Cl−]i using the procedures described above.
Electrophysiology.
Neurons were patch clamped via glass pipettes (5–6 MΩ resistance) filled with an internal solution containing 5 mm Cl−: 140 mm K-gluconate, 10 mm HEPES, 4 mm Na-ATP, 0.4 mm Na-GTP, 5 mm EGTA, 0.5 mm CaCl2, and 2 mm MgCl2. Extracellular saline contained 150 mm NaCl, 3 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 20 mm d(+)-glucose, and 10 mm HEPES, pH 7.4. Electrophysiological signals were recorded using a Multiclamp 700B amplifier (Molecular Devices) and digitized with a Digidata 1440 (Molecular Devices) A-D converter. Data were acquired and analyzed with pClamp10.1 software (Molecular Devices). GABA was locally applied onto neurons via pressure ejection. For this purpose, a glass pipette containing GABA (1 mm) was connected to a Picospritzer II (General Valve) to provide 100 ms pulses. IPSCs were evoked by stimulating presynaptic neurons using a concentric bipolar electrode (FHC). A train of extracellular current pulses (75 μA, 10 Hz, 2 s) were applied near the neuron being recorded from, while blocking EPSCs by adding 20 μm CNQX and 50 μm APV to the extracellular solution.
Throughout this paper, error determinations represent the SEM, unless indicated otherwise. Statistical comparisons were made with the Student's t test (Press et al., 2007, p. 729).
Results
Matching sensor affinity with [Cl−]i
Structural studies of YFP (Wachter et al., 2000; Griesbeck et al., 2001; Fig. 1A) have shown that its halide-binding site comprises the chromophore and nine nearby residues (Fig. 1B). The principles of molecular recognition determining halide binding are not well defined. Consequently, it is impossible to predict a priori how mutations in the halide-binding site contribute to binding affinity. We therefore used recently developed cell-free protein engineering methods (Cox et al., 2007; Allert et al., 2010) that bypass cloning steps (Fig. 2) to rapidly explore the Cl− binding and fluorescence properties of single, double, and triple mutants in the crystallographically defined halide-binding site. We mutated seven of these residues, avoiding Arg96, which catalyzes chromophore maturation (Barondeau et al., 2003; Sniegowski et al., 2005) and Tyr203, which tunes the spectral characteristics of YFP (Dickson et al., 1997). Starting with all 133 single mutants at these positions, double and triple mutations were iteratively constructed in CT in the absence of the CFP FRET donor (Kuner and Augustine, 2000). Another iterative set of mutants beginning with the 133 single mutations were also tested in CT H148Q, a mutation previously used to improve Cl− affinity (Jayaraman et al., 2000; Galietta et al., 2001; Markova et al., 2008) but with diminished fluorescence (Elsliger et al., 1999).
Figure 1.

Iodide-binding site in YFP. A, The structure of YFP (Wachter et al., 2000) with I− (purple) in relation to the chromophore (yellow). B, The iodide-binding site including residues selected for mutagenesis (underlined).
Figure 2.

Protein engineering method.
In noncooperative binding to a single site, changes in fluorescence emission intensity are directly proportional to the fraction of binding sites occupied by Cl− (ȳ). Sensor responses are maximal when the dissociation constant, Kd, matches the midpoint of the relevant [Cl−]i range (Marvin et al., 1997); in a mature neuron, [Cl−]i rises from ∼5 to ∼6 mm during synaptic inhibition (Berglund et al., 2006) with a midpoint of 5.5 mm. For a single-site hyperbolic binding isotherm, over such a range the theoretical maximal signal is 4.6% (Kd = 5.5 mm, Δȳ5,6 = ȳ6 − ȳ5 = 0.046) of the difference in fluorescence intensities observed in the absence and saturating presence of Cl− (ΔFmax). For Clomeleon (Kd = 119 mm), the maximal signal is 0.8% of ΔFmax; matching Cl− affinity therefore could achieve a 5.9-fold improvement in signal, if ΔFmax remained constant.
Clomeleon variants were rapidly screened using a cell-free protein engineering methodology that obviates cloning steps (Fig. 2). Linear DNA fragments encoded ORFs flanked by T7 RNA polymerase promoter and terminator sites were assembled robotically from synthetic oligonucleotides (Cox et al., 2007). The ORF mRNA sequence of CT was optimized computationally for E. coli expression (Allert et al., 2010). The resulting DNA fragments were then used to produce proteins by combined in vitro transcription and translation in 120 μl aliquots of E. coli extracts, purified by small-scale affinity purification using beads, and assayed for Cl− binding in microtiter plates.
A total of 424 variants, encoded in genes constructed from 344 unique oligonucleotides, were tested in this manner. The genes encoding the initial 266 single-point mutants were assembled from 269 oligonucleotides. Following an initial triage of these single-point variants based on fluorescence and expression levels, the Cl− affinities of 131 variants were determined by Cl− titration using a fluorescence plate reader. Their response to Cl− was evaluated using a metric that takes into account both fluorescence and affinity matching, ρ5.5. (see Materials and Methods) Twelve single mutants showed performance improvements (ρ5.5 > 1) with Q69T the only one having ρ5.5 > 2 (Fig. 3A,B).
Figure 3.
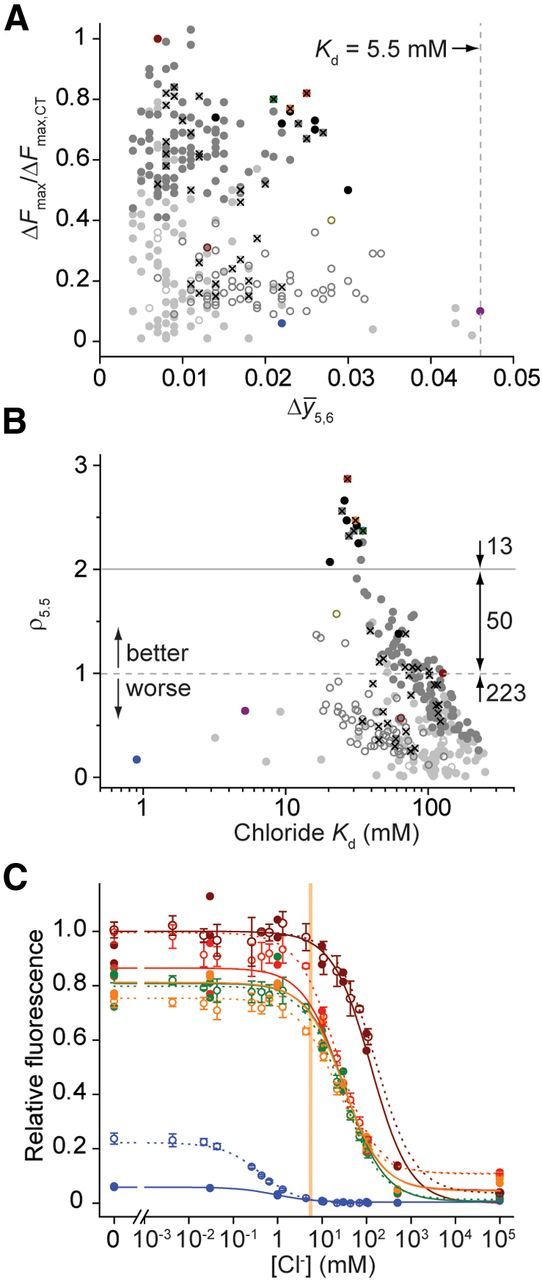
Chloride-binding properties of YFP variants. A, Performance assessment, correlating Cl− affinity matching (Δȳ5,6) and ΔFmax relative to CT (ΔFmax,CT): His 148 variants, open circles; His148Q variants, closed circles; single mutants, light grey; double mutants, medium grey; triple mutants, black; CT, dark red; H148Q, red open circle; Q69T/V163A, light red; Q69T, yellow; V150A/V163A, green; Q183A, blue; Q183G, magenta; H138Q/V163A/L201I, green open circle; single or double mutants that were subsequently included in higher order constructs, X. Vertical dashed line indicates a Kd of 5.5 mm, at the midpoint of the [Cl−]i range of relevance for imaging synaptic inhibition in mature neurons. B, Performance assessment, correlating values for the performance metric, ρ5.5, with Cl− affinity. Distribution of mutants with decreased (ρ5.5 < 1), improved (1 < ρ5.5 < 2), and superior (ρ5.5 < 2) performance is indicated. Colors and symbols as in A. C, Cl− titration of selected variants produced in vitro (filled circles, solid lines; no replicates) or by conventional expression (open circles, dashed lines; four replicates). Error bars indicate 1 SD. Fluorescence values are scaled to Clomeleon. Colors as in A. Vertical bar indicates [Cl−]i between 5 and 6 mm, the range of relevance for imaging synaptic inhibition in mature neurons.
In subsequent assembly rounds, we constructed 150 double mutants from a subset of 29 single mutants and included single-point variant with ρ5.5 < 1 to test whether improvements can arise by combining suboptimal intermediates. Out of these 150 double mutants, the five best (ρ5.5 ≥ 2.32) were combinations of only six single mutations (Q69T, V150A/I, and V163A/G/T), from which the set of all six possible triple mutants was constructed. With this automated gene assembly method, only 75 additional oligonucleotides were needed to construct the genes encoding the 156 higher-order mutants.
The average Kd values for Cl− binding of the 285 characterized CT variants improved from 101 mm (single) to 77.5 mm (double) to 33.3 mm (triple mutants), with a concomitant increase in average ρ5.5 values from 0.43 to 0.83 to 2.21. Based on their ρ5.5 values, 63 variants (22%) were improvements over CT (Fig. 3B) and had mutations in at least one of six binding site residues. The five highest affinity variants (Kd < 10 mm) were single mutations at Gln183; these exhibited diminished fluorescence brightness and a reduced ΔFmax response, precluding their use in cellular measurements (Fig. 3A,B). The 13 best variants had ρ5.5 > 2 and included mutations at only three positions (Gln69, Val150, and Val163). The best CT mutant, Q69T/V163A, was composed of individual single mutants each with ρ5.5 > 1 and was almost three times more sensitive than CT in vitro (Table 1).
Table 1.
Chloride binding to single domain YFP variants
| YFP mutation | Cell-free Cl− screen ρ5.5 | Cl− |
Conventionally expressed protein |
||||||
|---|---|---|---|---|---|---|---|---|---|
| F− Kd (mm) | Br− Kd (mm) | I− Kd (mm) | pKa | Tm (°C) | |||||
| ΔFmaxa | Kd (mm) | ρ5.5 | |||||||
| CT | 1.00 | 1.00 | 163 ± 4 | 1.00 | 2.77 ± 0.07b | 89.6 ± 4.8 | 35.4 ± 2.0 | 5.7 ± 0.1 | 68.6 ± 0.1 |
| Q69T/V163A | 2.87 | 0.92 | 21.2 ± 2.2 | 4.77 | 11.2 ± 0.8b | 33.3 ± 1.7 | 24.1 ± 1.7 | 6.4 ± 0.1 | 72.5 ± 0.1 |
| Q69T | 2.47 | 0.67 | 21.0 ± 1.2 | 3.48 | 16.0 ± 1.2b | 53.2 ± 3.2 | 13.3 ± 0.9b | 6.3 ± 0.1 | 64.6 ± 0.1 |
| V150A/V163A | 2.37 | 0.81 | 28.6 ± 1.0 | 3.48 | 1.09 ± 0.02b | 19.2 ± 0.5 | 12.4 ± 0.3 | 6.4 ± 0.1 | 67.3 ± 0.1 |
| H148Q/V163A/L201I | 1.57 | 0.40 | 20.1 ± 0.4 | 2.14 | |||||
| H148Q/I152L/V163S | 0.17 | 29.8 ± 1.5 | 0.71 | ||||||
| H148Q | 0.57 | 0.27 | 58.9 ± 3.8 | 0.67 | 7.0 ± 0.1 | ||||
| H148Q/V163S | 0.36 | 0.17 | 35.2 ± 1.6 | 0.62 | |||||
| Q183Ab | 0.17 | 0.23 | 0.417 ± 0.030 | 0.48 | 3.67 ± 0.29 | 0.272 ± 0.014 | 0.482 ± 0.034 | 6.7 ± 0.1 | 57.1 ± 0.3 |
| Q69H | { | No response to Cl− | } | 5.5 ± 0.1 | 73.4 ± 0.1 | ||||
| Q69M | { | No response to Cl− | } | 5.7 ± 0.1 | 73.7 ± 0.1 | ||||
Chloride binding to single domain YFP variants. All halide titration and thermal stability data determined at pH 7.1.
aΔFmax values are scaled relative to Clomeleon Topaz.
bThe F− endpoint was not included in the halide titration fits of conventionally prepared protein.
Using conventionally cloned and expressed protein, we assayed the binding and stability of 3 of the top 13 variants, as well as the original CT, the highest affinity variant Q183A, chloride-insensitive mutants Q69H and Q69M (Griesbeck et al., 2001), and four H148Q variants. Cl− affinities were determined by titration in a fluorescence plate reader. For most variants, the results were similar to the in vitro screening observations (Fig. 3C; Table 1). The Cl− dependence of thermal stabilities, which is thermodynamically linked to binding (Isom et al., 2010; Layton and Hellinga, 2010), was used to confirm the binding properties determined by titration. To determine thermal stabilities, the temperature dependence of the chromophore fluorescence was followed, which was coincident with the thermal stability determined by circular dichroism (Fig. 4A). Cl− increased the stability of chloride-responsive proteins, whereas the nonresponsive variants Q69H and Q69M showed minimal changes (Fig. 4B), consistent with the presence or absence of Cl−-binding sites inferred from fluorescence observations. No consistent trend in relative affinities for F−, Br−, and I− was observed (Table 1), indicating that molecular recognition of halides is quite complex (Wachter and Remington, 1999; Jayaraman et al., 2000; Kuner and Augustine, 2000). The pH dependence of fluorescence response and halide affinity (Fig. 5A,B) reveals linkage between chromophore ionization and binding affinity (see Discussion).
Figure 4.

Thermal stabilities and their dependence on chloride binding. A, Q69T/V163A CD (black) and fluorescence (red) data acquired simultaneously at pH 6.0 and fit to a two-state unfolding mechanism. Tm values of 67.0 ± 0.1°C (CD) and 67.2 ± 0.1°C (fluorescence) differ with the entry in Table 1 due to differences in pH, buffer, heating rate, and equipment. B, The effect of Cl− addition, pH 7.1, on thermal stability (four replicates; error bars indicate 1 SD) of YFP variants. Q69H and Q69M data points and error bars removed for clarity.
Figure 5.

The effect of pH on chloride affinity. A, Effect of pH on Cl− titrations illustrated for conventionally expressed Q69T/V163A. Fluorescence emission is normalized relative to the amount of fluorescence measured at pH 7.1 and in the absence of Cl−. B, The pH dependence of selected variants.
Structural analysis
High-resolution x-ray structures of the halide-free (apo) and I− complexes were determined for the Q69T/V163A variant that was used in cellular measurements (Fig. 6, Table 2). With the exception of some distant loops, the backbones of these two structures and YFP (Wachter et al., 2000) are essentially the same. Two I− sites were observed by anomalous scattering in the vicinity of the chromophore (Fig. 6A–E). One site (I) was observed previously (Wachter et al., 2000). The second site (II) is located 9 Å away, where I− replaces an interior water observed in the apo-protein. Small rearrangements of Thr63 and Thr108 side chains in the complex and apo-protein (Fig. 6F), relative to YFP, enable I− binding at Site II. The two side chain hydroxyls interact with the bound I−. Ethylene glycol (EG, used as cryoprotectant at 3.6 m in structure determination) also bound to Site I (Fig. 6C,E). Both I− and EG were observed at less than full occupancy in the complex, suggesting that these two ligands compete. EG binding neither was detectable by fluorescence quenching nor affected Cl− affinity and therefore EG is a low-affinity ligand. Formate was also observed to bind in the vicinity of the chromophore (Fig. 6G); it is likely to originate from ultrafiltration membranes used in protein purification.
Figure 6.

Iodide-binding sites in Q69T/V163A. A, Locations of iodide-binding Sites I (bottom) and II (top). B, Anomalous difference map (4.0 σ) of the iodide-bound structure with the Site I EG removed for visual clarity. The occupancy values were 0.2 (Site I iodide), 0.4 (Site II iodide), and 0.8 (Site I EG). Composite omit maps of the I−(purple) complex (C) and apo structure (D), both at 1.2σ. Residues surrounding Sites I (E) and II (F), superimposing the I− complex (blue), apo protein (magenta), and YFP (light brown; Wachter et al., 2000). Coloring of the bound I− matches its scaffold. EG (yellow) at Site I is shown for the apo protein. G, Composite omit map (1.5 σ) of formate (yellow) in the apo structure with the chromophore and Tyr203.
Table 2.
Crystallographic data collection and refinement statistics
| Q69T/V163A apo | Q69T/V163A iodide-bound | |
|---|---|---|
| Space group | P212121 | P212121 |
| Cell parameters | ||
| a, b, c (Å) | 51.026, 62.964, 69.078 | 51.488, 63.025, 66.070 |
| Data collection | ||
| Wavelength (Å) | 1.1109 | 1.1169 |
| Resolution (Å) | 50.0–1.19 (1.21–1.19) | 50.0–1.53 (1.56–1.53) |
| Completeness (%) | 92.7 (55.5) | 97.4 (94.6) |
| Redundancy | 7.4 (4.1) | 6.8 (6.7) |
| Rsym (%) | 4.4 (43.2) | 5.0 (47.2) |
| I/σI | 27.6 (2.7) | 31.3 (4.3) |
| Refinement | ||
| Resolution (Å) | 46.5–1.19 | 45.6–1.53 |
| No. reflections | 66756 | 32217 |
| Rwork/Rfree (%) | 14.1/16.6 | 16.0/18.3 |
| No. atoms | ||
| Total | 2303 | 2137 |
| Water | 400 | 245 |
| B-factors | 20.3 | 19.8 |
| RMS deviations | ||
| Bond lengths (Å) | 0.008 | 0.012 |
| Bond angles (°) | 1.292 | 1.491 |
| PDB code | 3ST0 | 3SV5 |
Crystallographic data collection and refinement statistics. Values in parentheses refer to the highest resolution shell. All atoms are included in the B-factor calculations.
FRET performance
We fused the affinity-tuned CT variants with a CFP FRET donor to reconstruct improved versions of Clomeleon. The performance of these OSs was evaluated in vitro (Table 3) and in a series of cellular FRET measurements. The Cl− affinities of these sensors were determined by Cl− titration experiments (Fig. 7), using two-photon imaging to measure sensor FRET in the cell bodies of individual cultured hippocampal neurons transfected with the OS of interest (Fig. 7A). [Cl−]i was varied by permeabilizing the cell membrane to external Cl− via ionophores (Berglund et al., 2006). The resulting changes in FRET emission (Fig. 7B) yielded titration curves such as those shown in Figure 7C. These curves were fit using Equation 11 to determine the Kd for Cl− binding. When imaging synaptic inhibition in neurons, the most important parameter is s/n. We therefore also estimated the s/n for the novel OSs, using the approach described above: “signal” corresponded to the change in FRET as [Cl−]i was raised from 5 to 6 mm, and “noise” was the SD of the FRET ratio determined at 5 mm [Cl−]i for each cell.
Table 3.
In vitro chloride binding to fusion proteins
| Fusion protein components |
In vitro |
||||
|---|---|---|---|---|---|
| Donora | Acceptor | Linkerb | S30Rc | ΔRmax | Kd (mm) |
| CFPd | CT | Long | − | 2.68 | 119 ± 3 |
| CFP | Q69T/V163A | Long | − | 1.89 | 30.4 ± 1.0 |
| Cerulean | Q69T/V163A | Long | − | 1.20 | 23.9 ± 1.7 |
| CFP | CT | Long | + | 2.77 | 143 ± 3 |
| CFP | Q69T/V163A | Long | + | 1.95 | 28.0 ± 2.7 |
| Cerulean | Q69T/V163A | Long | + | 1.26 | 22.6 ± 2.4 |
| CFP | CT | Short | + | 6.48 | 77.9 ± 1.1 |
| CFP | Q69T/V163A | Short | + | 4.84 | 23.5 ± 1.6 |
| Ceruleane | Q69T/V163A | Short | + | 2.75 | 22.6 ± 2.2 |
In vitro chloride binding to fusion proteins.
aCFP corresponds to the FRET donor in Clomeleon (Kuner and Augustine, 2000).
bConstructs with a long linker tested in vitro contained a scrambled protease cleavage site.
cThe S30R mutation was in both FPs.
dThis is the original Clomeleon (Kuner and Augustine, 2000).
eSuperClomeleon.
Figure 7.

Titration of SuperClomeleon in neurons. A, Imaging of Cl−-dependent changes in the FRET ratio (535/485 nm emission) in a single hippocampal neuron expressing SuperClomeleon. Ionophore treatment was used to clamp [Cl−]i to the values indicated. B, Time course of the changes in FRET emission ratio produced in response to changes in [Cl−]i in the cell shown in A. C, Cl− titration curves for indicated Clomeleon variants (23 replicates for Clomeleon and 4–21 replicates for its variants, as described in the text). Curves show fits to Equation. 11, with SEM indicated by error bars. F−indicates FRET values measured in the presence of 161 mm F−. Inset illustrates data normalized by Rmin and Rmax values, to allow comparison of the relative Cl− affinities of the Clomeleon variants. D, s/n estimated from the data shown in C. The transformation of Clomeleon into SuperClomeleon consists of the following additive improvements: Clomeleon (black), shorten interdomain linker (red), S30R point mutation in both domains (blue), intermediate affinity-tuning mutations (white), Q69T/V163A double mutant in YFP (teal), and replacement of the CFP domain with Cerulean (magenta). Also shown for comparison is the H148Q/I152L/V163S variant (white) described by Markova et al. (2008).
FRET depends upon both the distance between, and the orientation of, the fluorescent donor and acceptor. As a result, the linker between the donor and acceptor is an important determinant of FRET efficiency in GFP-based OSs (Evers et al., 2006). Therefore, to improve FRET efficiency, we used the strategy of Shimozono et al. (2006) to truncate the linker between the CFP and YFP of Clomeleon-based OSs: 40 residues (11 residues from the C terminus of CFP, the 24-residue linker, and 5 residues from the N terminus of YFP) were replaced with two residues. This truncation (short linker) did increase FRET efficiency, evident as an increase in the difference in FRET ratios (ΔRmax) observed in the absence of Cl−and in the presence of saturating concentrations of Cl− (or F−; Fig. 7C). This version of Clomeleon exhibited enhanced Cl− binding affinity (Fig. 7C, inset) and improved s/n (Fig. 7D; n = 4). The mean s/n measured for this version was significantly better than that of Clomeleon (Student's t = 52.1, p = 0.000).
Because donor and acceptor fluorescence also play important roles in determining s/n, we next introduced into both the donor and acceptor fluorophores a mutation (S30R) that increases brightness and stability (Pédelacq et al., 2006). This pair of mutations produced the largest improvement in ΔRmax (Fig. 7C) and further improved s/n (Fig. 7D; n = 18), but had no effect on Cl− affinity (Fig. 7C, inset).
After making these improvements to the FRET properties of Clomeleon, we next replaced the YFP FRET acceptor with several affinity-tuned CT variants. All of these variants increased the Cl− affinity of Clomeleon: V163S (6.4 ± 0.4 mm; n = 8), I152L/V163A (4.8 ± 0.2 mm; n = 4), V150A/V163A (4.6 ± 0.2 mm; n = 5), and V163A/L201I (2.5 ± 0.6 mm; n = 5). All of these also improved s/n above that produced by the Clomeleon variant with the short linker and S30R mutations (Fig. 7D). We also tested an OS with YFP H148Q/I152L/V163S, a variant previously reported to have improved properties (Markova et al., 2008). However, for FRET-based measurements, this OS had s/n properties no better than those of Clomeleon (Fig. 7D). This is due to its relatively poor Cl− affinity (27.0 ± 5.0 mm; n = 4) and the diminished fluorescence of the FRET acceptor (Table 1).
The largest improvement in s/n was observed when the affinity-tuning mutations in the best CT variant from the cell-free screen, Q69T/V163A, were included in the OS FRET acceptor. This sensor had a Cl− affinity of 8.1 ± 0.5 mm. ΔRmax for this version was smaller than for the short linker/S30R version of Clomeleon, but was still approximately twice as large as the value measured for Clomeleon (Fig. 7C). Most importantly, s/n for this variant was substantially larger than that measured for any of the previous versions of Clomeleon (Fig. 7D; n = 21).
In the last step, Clomeleon was further improved by replacing CFP with a brighter donor, Cerulean (Rizzo et al., 2004). This substitution decreased ΔRmax somewhat (Fig. 7C) but did not affect Cl− affinity. s/n for this construct was the best of any of the Clomeleon variants that we produced (Fig. 7D) and is significantly better than that of Clomeleon. We therefore called it SuperClomeleon and next characterized its performance for measuring transient changes in [Cl−]i associated with neuronal inhibition.
Detecting GABA-mediated changes in [Cl−]i
We determined the ability of SuperClomeleon to detect transient [Cl−]i changes produced in neurons by brief applications of the inhibitory neurotransmitter GABA. Cultured hippocampal neurons were transfected with SuperClomeleon and GABA was locally applied from a pipette positioned near the neuron (Fig. 8A). The neurons were voltage clamped to monitor Cl− flux electrically and to control neuronal membrane potential; introduction of a 5 mm Cl− solution into the cells from the patch pipette maintained resting [Cl−]i near physiological levels (Kuner and Augustine, 2000; Berglund et al., 2006).
Figure 8.

SuperClomeleon monitoring of GABA-induced chloride fluxes. A, GABA was locally applied from a pipette (GABA) onto individual neurons that were patch clamped via a second recording pipette (record). B, Simultaneous measurement of GABA-activated Cl− currents (lower) and changes in SuperClomeleon FRET ratio (535/485 nm emission; upper) from a single neuron held at various membrane potentials (underlined). GABA application (100 ms) indicated by arrows. Note different timescales for upper and lower traces. C, Comparison of time course of changes in integrated Cl− current (Charge) and 535/485 nm emission ratio (FRET) in response to GABA application (at arrow), measured at a holding potential of −50 mV. Traces have been normalized (and inverted, for the case of charge) to allow a comparison of their kinetics. D, Relationship between GABA-induced changes in integrated Cl−currents (Charge) and FRET signals for B. E, Aggregate data for cells expressing either SuperClomeleon (red, 10 cells) or Clomeleon (black, 7 cells). Lines indicate linear regression fit to the data, with slopes of 10.1 ± 0.5 (SEM) s · nC−1 for SuperClomeleon and 2.1 ± 0.2 s · nC−1 for Clomeleon. Units for y-axes (integrated FRET signals) in D and E are seconds.
A brief (100 ms) application of GABA generated a transient Cl− flux that was observed both as a transmembrane electrical current and as a change in SuperClomeleon FRET (Fig. 8B,C). The magnitude and direction of both current and FRET responses varied with membrane potential according to the direction of Cl− flux. At a holding potential of −40 mV, Cl− influx was observed as a large, outward current and a decrease in SuperClomeleon FRET (Fig. 8B, left). At more negative potentials, where the electrochemical driving force on Cl− influx was reduced, the currents and FRET responses became smaller and reversed their polarities at −90 mV (Fig. 8B, right). The reversal potential of approximately −80 mV was consistent with the Cl− equilibrium potential of −89 mV predicted by the Nernst equation.
To define the time course and magnitude of the GABA-induced Cl− flux, we integrated the GABA-induced current over time (transmembrane charge). Comparison of the rising phases of the transmembrane charge and the change in SuperClomeleon FRET ratio indicated that the FRET response lags behind the Cl− flux by 1–2 s (Fig. 8C). This is consistent with previous observations made with Clomeleon (Kuner and Augustine, 2000). For observations made on the single neuron shown in Figure 8B, there was a linear relationship between the Cl− flux, as measured by the charge, and the integrated FRET signal (Fig. 8D). This correlation was also evident in 49 measurements from 10 neurons (Fig. 8E, red) and indicates that SuperClomeleon reliably reports the Cl− fluxes and resulting changes in [Cl−]i produced by activation of GABA receptors. Much smaller FRET responses were observed in neurons expressing Clomeleon (Fig. 8E, black). The slope of the relationship between transmembrane charge and integrated FRET signal indicated that SuperClomeleon responses were 4.8-fold greater than those of Clomeleon for a given amount of Cl− flux (Fig. 8E). This indicates that SuperClomeleon improves FRET signal amplitude ∼5-fold compared with Clomeleon. FRET signals declined with time constants of 6.9 ± 0.6 s (n = 38) for SuperClomeleon and 4.9 ± 0.7 s (n = 18) for Clomeleon, reflecting slow Cl− efflux from neurons (Kuner and Augustine, 2000; Berglund et al., 2006). By analogy with fluorescent calcium indicators (Regehr and Atluri, 1995), the slower time course of decay observed by SuperClomeleon is probably due to the improved ability of this indicator to detect small and persistent changes in [Cl−]i.
Imaging GABA-mediated synaptic inhibition with SuperClomeleon
One of the potentially most important applications of OSs is imaging the spatiotemporal dynamics of synaptic inhibitory circuits (Mancuso et al., 2011). While Clomeleon is capable of reporting the small changes in [Cl−]i associated with synaptic inhibition (Berglund et al., 2006, 2008), its low Cl− sensitivity and very low s/n in the desired concentration range has prevented routine imaging of synaptic inhibition. We therefore evaluated the ability of SuperClomeleon to perform this demanding task in cultured hippocampal neurons by activating inhibitory synapses via electrical stimulation of nearby interneurons (Fig. 9A).
Postsynaptic neurons expressing Clomeleon or SuperClomeleon were voltage clamped (−50 mV) to measure the postsynaptic Cl− currents associated with IPSCs. Under these conditions, brief trains of electrical stimuli produced IPSCs, which decreased during the train (Fig. 9B, bottom) due to synaptic depression (Gitler et al., 2004b). In neurons expressing Clomeleon, inhibitory synaptic activity rarely produced detectable FRET changes (Fig. 9B, left). However, similar stimuli reliably produced transient reductions in SuperClomeleon FRET (Fig. 9B, right), revealing that this indicator is capable of detecting the relatively small changes in [Cl−]i associated with synaptic inhibition. To compare the performance of the two OSs, we measured synaptic responses in eight cells expressing SuperClomeleon and in five cells expressing Clomeleon. While 100% (16/16) of the trials yielded detectable SuperClomeleon responses, Clomeleon reported responses only 50% (8/16) of the time.
Responses to inhibitory synaptic activity were quantified to provide a direct comparison of s/n for inhibitory synaptic responses reported by SuperClomeleon and Clomeleon. For the examples shown in Figure 9B, the s/n ratio was ∼5.8 for the neuron expressing SuperClomeleon and was too small to be determined reliably in the neuron expressing Clomeleon. This was further quantified by first calculating the s/n by integrating the stimulus-induced change in FRET signal and dividing this value by the baseline FRET noise (as done in Fig. 8D). Because of substantial cell-to-cell variations in IPSC amplitude, these responses were then normalized for the amount of synaptic Cl− flux by dividing their values by the synaptic charge produced by the stimulus. The cumulative probability plots shown in Figure 9C show the results of this analysis, with the dashed vertical line indicating values where there was no measurable response (i.e., a s/n of 0). This analysis revealed that, on average, the s/n of SuperClomeleon responses (median = 0.15 ± 0.01 s · pC−1) was 5.4-fold larger than those of Clomeleon (median = 0.028 ± 0.008 s · pC−1).
For many potential applications of SuperClomeleon, it is likely that the postsynaptic neuron would not be voltage clamped. Indeed, in many applications it is possible that there would be no electrophysiological measurements of postsynaptic activity at all. FRET responses should be smaller when the neurons are not voltage clamped, because the electrochemical driving force on Cl− will change as the membrane potential changes during the IPSP. Thus, we completed our analysis by comparing the ability of SuperClomeleon and Clomeleon to detect inhibitory activity in postsynaptic neurons that were not voltage clamped.
Under current-clamp conditions, the same brief trains of inhibitory synaptic activity produced a series of IPSPs (Fig. 9D, bottom). In neurons expressing Clomeleon, detectable FRET signals were observed in only three of nine experiments (Fig. 9D, left). In contrast, a similar amount of inhibitory synaptic activity produced detectable changes in FRET in every case (nine of nine experiments) for neurons expressing SuperClomeleon (Fig. 9D, right). The mean integrated change in FRET signal measured with SuperClomeleon was 5.4-fold larger than for the integrated change in FRET signal measured in neurons expressing Clomeleon (Fig. 9E). The difference in the performance of Clomeleon and SuperClomeleon for detecting changes in [Cl−]i associated with IPSPs was even greater when considering s/n, rather than integrated change in FRET signal: the s/n for SuperClomeleon was >6 times larger than that measured for Clomeleon (Fig. 9F). In summary, our measurements under both voltage-clamp and current-clamp conditions indicate that the s/n was ∼6-fold better for SuperClomeleon than for Clomeleon. We therefore conclude that SuperClomeleon is superior for measurements of changes in [Cl−]i associated with synaptic inhibition.
Discussion
We used a multistage strategy to improve Cl− sensing by Clomeleon. First, using a cell-free automation methodology to rapidly prototype variants in vitro, we improved YFP by more closely matching its Cl− affinity to postsynaptic [Cl−]i. Next, we incorporated this affinity-tuned single domain into Clomeleon for FRET-based sensing, enhancing s/n in cellular imaging experiments. Improvements in affinity matching, while retaining fluorescence, resulted in the largest enhancement of cellular imaging performance. Shortening the interdomain linker and introducing brightening mutations significantly improved the FRET response. This process culminated in identification of SuperClomeleon, a variant that greatly enhanced observation of synaptic inhibition in neurons.
Choosing single-point mutants with clearly improved ρ5.5 values was a critical step for success; incremental improvements also arose from combinations of initially suboptimal variants. Nevertheless, given that the number of variants that need to be examined in an enumerative strategy can increase approximately exponentially with each iteration that includes more positions, carrying forward only clearly improved variants balances experimental load and likelihood of success. We observed good agreement between the affinities determined in the initial screen and the proteins produced conventionally. Cell-free protein engineering therefore is a powerful tool for generating and prototyping mutant proteins.
Our structural analysis revealed that the effects of the mutations are complex. The halide-binding site of SuperClomeleon resides within the internal cavity that accommodates the conformational change required for chromophore cyclization (Barondeau et al., 2003). This internal cavity accommodates two halide-binding sites, as well as a molecule of EG and a molecule of formate. The appearance of a second halide-binding site accompanied by motions of the hydrogen-bonding side chains in the otherwise rigid structure shows that the lining of this cavity is mobile and can accommodate binding sites through subtle rearrangements. The presence of small organics, such as ethylene glycol and formate, suggests that this cavity can be further diversified to bind and detect small molecules.
The YFP chromophore exists in four possible charge states (anionic, cationic, zwitterionic, and neutral), of which only the anionic form is fluorescent (Elsliger et al., 1999). Bound Cl− and the anionic chromophore interact unfavorably by electrostatic repulsion, so that Cl− binding is favored in the protonated, neutral, nonfluorescent state. These interactions link [Cl−] and affinity with pH and chromophore pKa, setting up a trade-off between Cl− affinity and sensor brightness. Improvements in Cl− affinity correlate with raised chromophore pKa values relative to CT, which decreases the concentration of anionic chromophore in the halide-free protein at a given pH, thereby diminishing sensor brightness. This decreased fluorescence reduces ΔFmax because Cl− binding quenches emission. The highest affinity variant Q183A has a raised pKa value and diminished fluorescence brightness. Similarly, other mutations, including H148Q, increase affinity at the expense of brightness (Jayaraman et al., 2000; Wachter et al., 2000; Galietta et al., 2001; Markova et al., 2008;Fig. 3A,B). In constructing SuperClomeleon these trade-offs could be balanced sufficiently to improve sensing substantially; that is, SuperClomeleon offers improved sensitivity to physiological Cl− fluxes despite a higher interference by protons.
Affinity matching together with other enhancements improved the s/n characteristics of SuperClomeleon in cellular imaging. As a result, SuperClomeleon represents a significant improvement over Clomeleon and consistently outperformed Clomeleon in all of our neuronal imaging experiments: Cl− titrations, activation of GABA receptors via exogenous GABA, and electrically evoked synaptic inhibition. Quantitatively, our GABA application experiments indicate a 4.8-fold improvement in signal for SuperClomeleon relative to Clomeleon, while the synaptic inhibition data indicate a s/n that is 5- to 6-fold larger than that measured for Clomeleon. The key qualitative improvement is that SuperClomeleon robustly detects inhibitory synaptic activity in single neurons even under the relatively noisy conditions of two-photon imaging; this was not the case for Clomeleon (Berglund et al., 2006, 2008). Of particular note is that SuperClomeleon can detect responses to brief IPSP trains, an advantage that will prove useful for many future applications and will enable optogenetic imaging of the spatiotemporal dynamics of synaptic inhibitory circuits at the cellular level of resolution (Mancuso et al., 2011).
In conclusion, we have partnered automated in vitro protein engineering technology with cellular imaging to develop an OS with improved ability to image inhibitory synaptic activity. This development not only enables novel brain imaging experiments but also establishes a general approach for developing and improving many other types of OSs.
Footnotes
This work was supported by the Duke Institute for Brain Science and the National Institutes of Health Director's Pioneer Award (5DPI OD000122), by a Competitive Research Program grant from the National Research Foundation (NRF) of Singapore, and by the World Class Institute Program of the NRF of Korea funded by the Ministry of Education, Science, and Technology of Korea (NRF Grant number WCI 2009-003). We thank Ken Berglund for many helpful discussions.
The authors declare no competing financial interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allert M, Cox JC, Hellinga HW. Multifactorial determinants of protein expression in prokaryotic open reading frames. J Mol Biol. 2010;402:905–918. doi: 10.1016/j.jmb.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio D, Garau G, Ricci F, Marchetti L, Bizzarri R, Nifosì R, Beltram F. Spectroscopic and structural study of proton and halide ion cooperative binding to GFP. Biophys J. 2007;93:232–244. doi: 10.1529/biophysj.106.102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F. Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods. 2010;7:516–518. doi: 10.1038/nmeth.1471. [DOI] [PubMed] [Google Scholar]
- Barondeau DP, Putnam CD, Kassmann CJ, Tainer JA, Getzoff ED. Mechanism and energetics of green fluorescent protein chromophore synthesis revealed by trapped intermediate structures. Proc Natl Acad Sci U S A. 2003;100:12111–12116. doi: 10.1073/pnas.2133463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Krieger P, Loo LS, Wang D, Cant NB, Feng G, Augustine GJ, Kuner T. Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 2006;35:207–228. doi: 10.1007/s11068-008-9019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Wang H, Feng G, Hall WC, Kuner T, Augustine GJ. Imaging synaptic inhibition through the brain via genetically targeted Clomeleon. Brain Cell Biol. 2008;36:101–118. doi: 10.1007/s11068-008-9031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright GR, Fisher GW, Rogowska J, Taylor DL. Fluorescence imaging microscopy. Methods Cell Biol. 1989;30:157–192. doi: 10.1016/S0091-679X(08)60979-6. [DOI] [PubMed] [Google Scholar]
- Cox JC, Lape J, Sayed MA, Hellinga HW. Protein fabrication automation. Protein Sci. 2007;16:379–390. doi: 10.1110/ps.062591607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- Elsliger MA, Wachter RM, Hanson GT, Kallio K, Remington SJ. Structural and spectral response of green fluorescent protein variants to changes in pH. Biochemistry. 1999;38:5296–5301. doi: 10.1021/bi9902182. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evers TH, van Dongen EM, Faesen AC, Meijer EW, Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–13192. doi: 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- Flach J, Bossie M, Vogel J, Corbett A, Jinks T, Willins DA, Silver PA. A yeast RNA-binding protein shuttles between the nucleus and the cytoplasm. Mol Cell Biol. 1994;14:8399–8407. doi: 10.1128/mcb.14.12.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galietta LJ, Haggie PM, Verkman AS. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–224. doi: 10.1016/S0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004a;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004b;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires SA, Zhu Y, Tsien RY. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc Natl Acad Sci U S A. 2008;105:4411–4416. doi: 10.1073/pnas.0712008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Vardy E, Oas TG, Hellinga HW. Picomole-scale characterization of protein stability and function by quantitative cysteine reactivity. Proc Natl Acad Sci U S A. 2010;107:4908–4913. doi: 10.1073/pnas.0910421107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Haggie P, Wachter RM, Remington SJ, Verkman AS. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J Biol Chem. 2000;275:6047–6050. doi: 10.1074/jbc.275.9.6047. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/S0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Layton CJ, Hellinga HW. Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand-affinities with extrinsic fluorescent dyes. Biochemistry. 2010;49:10831–10841. doi: 10.1021/bi101414z. [DOI] [PubMed] [Google Scholar]
- Mancuso JJ, Kim J, Lee S, Tsuda S, Chow NB, Augustine GJ. Optogenetic probing of functional brain circuitry. Exp Physiol. 2011;96:26–33. doi: 10.1113/expphysiol.2010.055731. [DOI] [PubMed] [Google Scholar]
- Markova O, Mukhtarov M, Real E, Jacob Y, Bregestovski P. Genetically encoded chloride indicator with improved sensitivity. J Neurosci Methods. 2008;170:67–76. doi: 10.1016/j.jneumeth.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Corcoran EE, Hattangadi NA, Zhang JV, Gere SA, Hellinga HW. The rational design of allosteric interactions in a monomeric protein and its application to the construction of biosensors. Proc Natl Acad Sci U S A. 1997;94:4366–4371. doi: 10.1073/pnas.94.9.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A. Innovations in the imaging of brain functions using fluorescent proteins. Neuron. 2005;48:189–199. doi: 10.1016/j.neuron.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Okumoto S. Imaging approach for monitoring cellular metabolites and ions using genetically encoded biosensors. Curr Opin Biotechnol. 2010;21:45–54. doi: 10.1016/j.copbio.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumoto S, Looger LL, Micheva KD, Reimer RJ, Smith SJ, Frommer WB. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc Natl Acad Sci U S A. 2005;102:8740–8745. doi: 10.1073/pnas.0503274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical recipes: the art of scientific computing. Ed 3. New York: Cambridge UP; 2007. [Google Scholar]
- Regehr WG, Atluri PP. Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variants useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Shimozono S, Hosoi H, Mizuno H, Fukano T, Tahara T, Miyawaki A. Concatenation of cyan and yellow fluorescent proteins for efficient resonance energy transfer. Biochemistry. 2006;45:6267–6271. doi: 10.1021/bi060093i. [DOI] [PubMed] [Google Scholar]
- Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19:735–741. doi: 10.1016/S0896-6273(00)80955-1. [DOI] [PubMed] [Google Scholar]
- Sniegowski JA, Lappe JW, Patel HN, Huffman HA, Wachter RM. Base catalysis of chromophore formation in Arg96 and Glu222 variants of green fluorescent protein. J Biol Chem. 2005;280:26248–26255. doi: 10.1074/jbc.M412327200. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Wachter RM, Remington SJ. Sensitivity of the yellow fluorescent protein to halides and nitrate. Curr Biol. 1999;9:R628–R629. doi: 10.1016/S0960-9822(99)80408-4. [DOI] [PubMed] [Google Scholar]
- Wachter RM, Yarbrough D, Kallio K, Remington SJ. Crystallographic and energetic analysis of binding of selected anions to the yellow variants of green fluorescent protein. J Mol Biol. 2000;301:157–171. doi: 10.1006/jmbi.2000.3905. [DOI] [PubMed] [Google Scholar]
- Xia Z, Zhou Q, Lin J, Liu Y. Stable SNARE complex prior to evoked synaptic vesicle fusion revealed by fluorescence resonance energy transfer. J Biol Chem. 2001;276:1766–1771. doi: 10.1074/jbc.M008741200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]