

Abstract
We report the characterization of a new rapid-onset model of Huntington’s disease (HD) generated by adeno-associated virus (AAV) vector-mediated gene transfer of N-terminal huntingtin constructs into the rat striatum. Expression of exon 1 of mutant huntingtin containing 70 CAG repeats rapidly led to neuropathological features associated with HD. In addition, we report novel data relating to neuronal transduction of AAV vectors that modulated the phenotype observed in this model. Quantitative RT-PCR revealed AAV vector-mediated expression in the striatum to over 100-fold the endogenous huntingtin level. Moreover, AAV vectors exhibited non-uniform transduction patterns in striatal neuronal populations and also axonal transport that led to transduction and neuronal cell death in the globus pallidus and substantia nigra. These findings may inform future studies utilizing AAV vectors for neurodegenerative disease modeling. Furthermore, RNA interference (RNAi) of mutant huntingtin expression mediated by virus vector delivery of short hairpin RNAs (shRNAs) ameliorates early-stage disease phenotypes in transgenic mouse models of HD, however whether shRNA-mediated knockdown of mutant huntingtin expression is neuroprotective has not been reported. AAV-shRNA mediated dramatic knockdown of HD70 expression, preventing striatal neurodegeneration and concomitant motor behavioral impairment. These results provide further support for AAV vector-mediated RNAi as a therapeutic strategy for HD.
Keywords: Huntington’s disease, animal model, adeno-associated virus vector, neurodegeneration, behavior
INTRODUCTION
Huntington’s disease (HD) is caused by a CAG repeat expansion within exon 1 of the HD gene [1]. This mutation confers a toxic gain of function to the protein huntingtin (htt) containing an expanded polyglutamine (polyQ) tract and leads to the dysfunction and death of GABAergic medium spiny neurons in the striatum [2]. Symptomatically, HD patients suffer from a progressive loss of motor control, cognitive function and psychiatric disturbances [3]. There is currently no effective treatment for the disease, which progresses towards death within twenty years of onset. A number of transgenic or knock-in mouse models of HD have been developed [4]. Phenotypes reminiscent of early-stage HD were reported to varying degrees in some of these models, including neuronal dysfunction, reduced brain weight, striatal atrophy, and motor deficits. However, none of these models recapitulate the substantial striatal neuronal cell loss that is characteristic of HD. Also, it is currently not possible to generate transgenic models in higher organisms such as non-human primates, limiting translational research.
A complementary approach to modeling genetic disorders of the central nervous system utilizes recombinant viral vectors to deliver expression cassettes into the brain of experimental animals [5], which has potential advantages over transgenic approaches [6]. HD models have been developed based on viral vector-mediated gene transfer of N-terminal fragments of mutant htt to the striatum. Initial studies using AAV serotype 2 [7] or lentiviral vectors [8] recapitulated some key elements of the disease and resulted in a limited degree of neurodegeneration 5–8 weeks post-injection in the striatum of rats. However, a behavioral phenotype was not characterized in these studies. More recently, a non-human primate model of HD generated by lentiviral gene transfer to the macaque putamen has been reported that exhibits striatal neuronal cell loss and a progressive motor phenotype [9]. DiFiglia et al. also recently reported a robust, rapid-onset phenotype including substantial striatal neurodegeneration and behavioral impairment in mice as early as 2 weeks post-injection of an AAV serotype1/8 mutant htt construct [10]. These studies support the use of viral vector-mediated gene transfer for the further elucidation of the molecular mechanisms underlying HD, and as a platform for testing neuroprotective therapeutic strategies that can be translated to non-human primates.
However, further characterization of viral vector-based models is required. Using a similar approach, we have developed a rapid-onset rat model of HD using AAV vector-mediated gene transfer of an N-terminal mutant htt construct into the striatum. We used AAV serotype 1/2 vectors to achieve robust neuronal transduction leading to a rapidly progressive neuropathological phenotype. Moreover, we report novel findings relating to AAV vector transduction in the brain that influenced the phenotype observed in this model including characterization of a non-uniform transduction pattern of neuron populations and relative quantification of the high level of AAV-mediated transgene expression in the striatum, axonal vector transport to and toxicity in other associated areas of the basal ganglia, and a comprehensive analysis of mutant huntingtin-mediated neuronal toxicity. These novel findings may inform future studies utilizing AAV vectors for neurodegenerative disease modeling.
Finally, we assessed whether our model would have utility in screening new therapeutic treatments. Virus vector-mediated delivery of short hairpin RNAs (shRNAs) has been reported to ameliorate early-stage disease phenotypes in HD transgenic mice [11–13], but whether shRNA-mediated inhibition of mutant htt expression is neuroprotective has not been demonstrated. Here we report that AAV vector-mediated RNAi resulted in protection from N-terminal mutant htt-mediated neurodegeneration and motor behavioral impairment in our model. These results further support the use of AAV vector-mediated RNAi as a therapeutic strategy for HD.
RESULTS
AAV1/2 vector-mediated expression of N-terminal htt constructs in rat striatum
Using a similar approach to previous studies [7–10], AAV1/2 vector-mediated gene transfer of N-terminal htt constructs (AAV-HD70, -HD20, -Hdh8, or Empty vector, Fig. S1) to the rat striatum was used to establish a genetic animal model of HD. In line with previous findings, neuronal intranuclear inclusions, a feature of polyQ expansions diseases such as HD, were apparent by 2 weeks in the AAV-HD70 injected brains, with more diffuse anti-htt staining for AAV-HD20 or -Hdh8 (Fig. 1). HD70 immunostaining was diminished by 5–8 weeks, due to neuronal cell death within the striatum (see below). The spread of expression throughout the rat striatum achieved using AAV1/2 vectors (Fig. S2) appeared to be substantially greater than that achieved using AAV2 [7] and lentiviral [8] vectors, and similar to that reported recently using AAV1/8 in mice [10]. However, expression appeared most rapidly in large interneurons, with more extensive immunoreactivity throughout the striatum between 2–8 weeks (Fig. 1).
Fig. 1. Temporal expression of AAV vector N-terminal htt constructs in rat striatum.
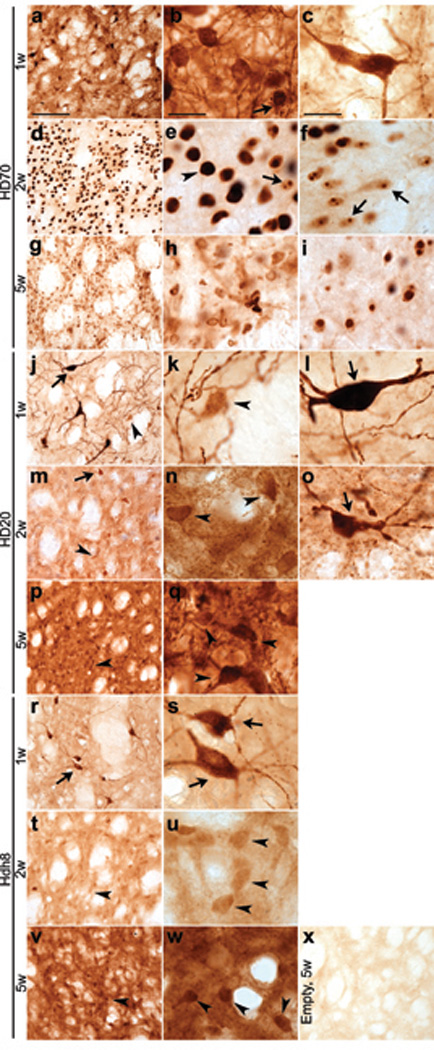
(a–c) Diffuse anti-htt immunostaining in cell bodies and fibers at 1 week post-injection of AAV-HD70. Robust anti-htt staining in medium-sized neurons was observed towards the center of the expression area (b), whereas at the periphery, immunostaining of large cells resembling interneurons was more prevalent (c). (d–f) Immunostaining of single, large HD70 inclusion bodies (arrowheads, (e) at 2 weeks whereas smaller single or multiple HD70 protein inclusion bodies (arrows) were observed towards the periphery of the expression area (f), (g–i) By 5 weeks, anti-htt immunoreactivity was diminished towards the centre of the expression area (h). A greater level of immunostaining of inclusion bodies was preserved towards the periphery of the expression area (i). (j–l) Robust anti-htt immunostaining expression in large interneurons (arrow) and weaker immunoreactivity of medium-sized neurons (arrowhead) at 1 week post-injection of AAV-HD20. (m–o) Immunostaining of medium-sized neurons was stronger at 2 weeks, with diffuse and punctate staining of cell bodies and fibers. Interneurons expressing HD20 appeared dystrophic (o). (p–q) HD20 immunostaining of cell bodies and fibers of medium-sized neurons was stronger and more punctate at 5 weeks, while immunostaining of large interneurons was absent, except at the periphery of the expression area (not shown). (r–w) The pattern of anti-htt immunoreactivity of Hdh8 was similar to that observed for HD20 at 1 week (r–s), 2 weeks (t–u) and 5 weeks (v–w) and appeared to be more diffuse and less punctate than that of HD20. (x) Weak anti-htt immunostaining in striatum of Empty vector injected rats at 5 week. Scale bars, 100 µm (low power, left column) 20 µm (high power, middle and right columns).
AAV1/2-HD70-mediated striatal neuropathology
Striatal atrophy and neuronal cell loss is the hallmark of HD neuropathology [14] and AAV1/2-HD70 expression led to striatal neurodegeneration and neuropathological features associated with HD, consistent with previous studies [7–10]. Substantial loss of neuronal immunoreactivity (NeuN, calbindin D28k or DARPP-32) was already evident at 2 weeks (not shown) and by 5 weeks immunostaining was almost completely absent in the AAV-HD70 expression area (Figs. 2a,b,f,g,k), comprising ~35% of the ipsilateral striatum (Figs. 2e,j). HD70-induced cell loss contributed to marked striatal atrophy as indicated by increased density of Nissl staining, pyknotic, condensed nuclei and enlargement of the ipsilateral lateral ventricle (Figs. 2q–s), Interestingly, this contrasts with reduced striatal Nissl staining reported by DiFiglia et al. 2 weeks post-injection of AAV1/8Htt100Q [10]. Cell death was confirmed by extensive striatal Fluoro-Jade B staining and reactive astrogliosis within the lesion area (Figs. 2m,o). Striatal HD70–mediated neurodegeneration also led to decreased innervation of the globus pallidus (GP), indicated by decreased enkephalin immunoreactivity (Fig. S3). In addition to the substantial loss of projection neurons, we also investigated the impact on striatal interneuron populations. In human HD brain, there is a relative sparing of striatal interneuron populations that are immunopositive for choline acetyltransferase (ChAT), parvalbumin (parv) or neuropeptide Y (NPY) [15–17]. However, there was almost complete loss of NPY, parv, and ChAT immunoreactivity within the AAV-HD70 striatal expression area (Figs. 3j–m). Surprisingly, ChAT immunostaining was also lost in the striatum of rats injected with AAV-HD20 or -Hdh8 vectors (Figs. 3f,i,m), whereas NPY and parv immunoreactivity was preserved (Figs. 3d,e,g,h,m). These results contrast with those reported by de Almeida et al., where 47% of cholinergic interneurons and 80% of NADPH-d interneurons within the DARPP-32-depleted region were spared in animals injected with LV-htt171-82Q vector [8]. Interneuron survival was not assessed in other studies [7, 9, 10].
Fig. 2. Striatal neurodegeneration in rats injected with AAV-HD70.
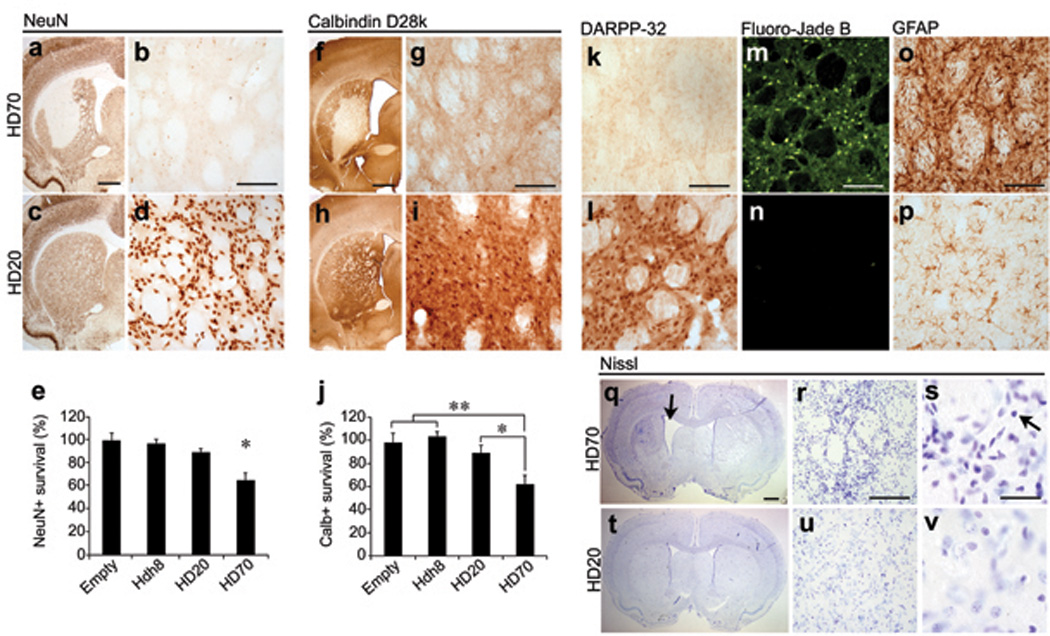
Loss of NeuN (a,b), calbindin D28k (f,g) and DARPP-32 (k) immunoreactivity within the expression area in HD70 rats at 5 weeks. Immunoreactivity for these markers was preserved in rats injected with AAV-HD20 (c,d,h,i,l), or Hdh8 or Empty vectors. (e,j) Stereological quantification of neuronal cell death. Data are presented as the mean±SEM (n=6) percentage of the number of cells counted in the ipsilateral compared to the contralateral hemisphere. (One-way ANOVA with post-hoc analysis and Bonferroni correction, *p<0.05, **p<0.01). Increased Fluoro-Jade B staining (m) and GFAP immunoreactivity (o) in the striatum of HD70 animals at 5 weeks compared to HD20 (n,p) or Hdh8 or Empty vector rats (not shown). Altered Nissl staining in the striatum of HD70 rats (q–s) and marked striatal atrophy in the ipsilateral hemisphere (arrow) compared to HD20 (t–v) or other control vector-injected rats (not shown). At higher magnification, pyknotic, condensed nuclei and increased cell density was observed (s, arrow). Scale bars: 1mm (low power, a,f,q), 100 µm (b,g,k,m,o,r), 20 µm (s).
Fig. 3. Loss of striatal interneurons in rats injected with AAV htt vectors.
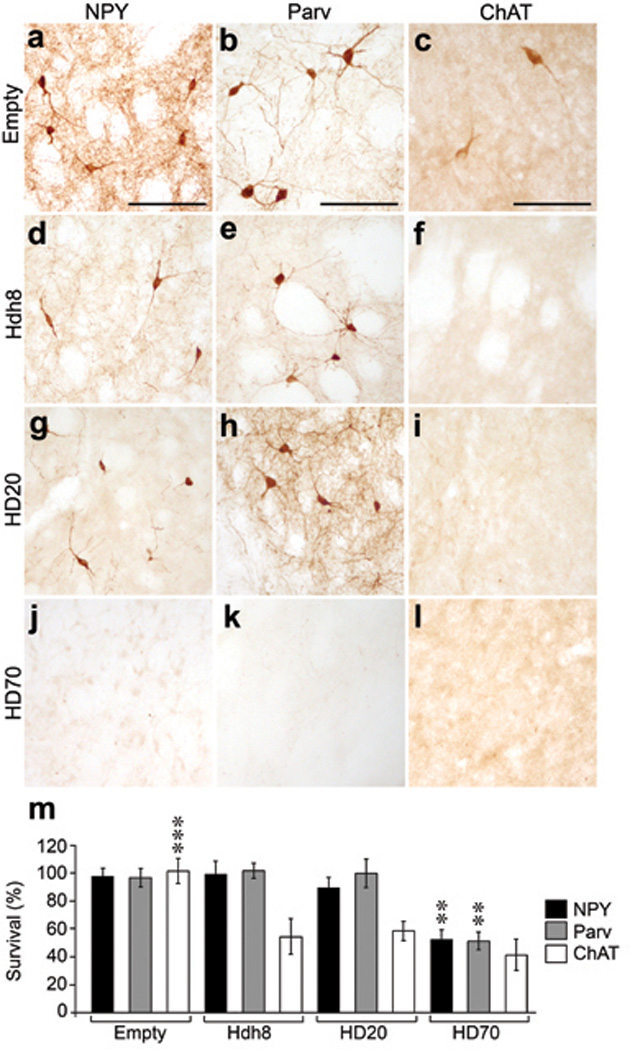
NPY, Parv and ChAT immunoreactivity in the striatum of rats 5 weeks post-injection of Empty vector (a–c), Hdh8 (d–f), HD20 (g–i), HD70 (j–l). (m) Stereological quantification of striatal interneuron cell death. Each bar represents mean±SEM (n=6) of the number of cells counted in the ipsilateral injected hemisphere as a percentage of the cells counted in the contralateral hemisphere. AAV-HD70 expression led to the loss of 47.7±7.0% of NPY-positive interneurons and 48.7±6.4% of parv-positive interneurons in the ipsilateral striatum. Expression of AAV-HD70, -HD20, or -Hdh8 also led to loss of ChAT-positive interneurons (HD70, 58.8±11.4% cell loss; HD20, 41.6±6.9% cell loss; Hdh8 45.5±12.7% cell loss; one-way ANOVA with post-hoc analysis and Bonferroni correction, **p<0.01, ***p<0.001). Scale bars, 50 µm.
Non-uniform transduction of striatal neuronal populations by AAV1/2 vectors
We hypothesized that the pattern of striatal neuron loss was partly related to non-uniformity in AAV1/2 vector transduction of striatal neuronal populations. Transduction of projection neurons (DARPP-32) and interneuron populations (ChAT, parv, NPY) was investigated at 1, 2 and 5 weeks post-injection of AAV-HD70, -HD20, or -Hdh8. An AAV-EGFP treatment group was included as an additional non-disease-related reporter gene control. The findings of this experiment are depicted in Fig. 4 and summarized in Table S1. DARPP-32 containing projection neurons were transduced more rapidly by AAV-HD70 and -EGFP than AAV-HD20 or -Hdh8 and NPY-positive interneurons were far more readily transduced by AAV-HD70 than the other vectors. ChAT-positive interneurons were strongly transduced at 1 week by all vectors, with ChAT immunostaining absent by 2 weeks, indicating death of cholinergic interneurons. These results confirmed that the large interneurons with strong anti-htt immunoreactivity at 1 week, which appeared dystrophic or absent at 2–5 weeks in Fig. 1 were cholinergic interneurons. We propose that the rapid, high-level AAV vector transduction of cholinergic interneurons led to toxicity and neuronal cell death, irrespective of the transgene expressed, with wild-type or mutant N-terminal htt constructs or EGFP all causing cholinergic neuronal cell death. Neuronal toxicity resulting from high transduction levels by high-titer AAV8-EGFP has been previously reported [18]. Interestingly, the differences between our findings and those reported previously for AAV2 [7] or lentivirus [8] indicate that the phenotype in viral vector-based models of neurodegenerative disease can be significantly influenced by cell-specific targeting depending on vector system employed.
Fig. 4. Differential transduction of striatal neuron populations by AAV1/2 vectors.
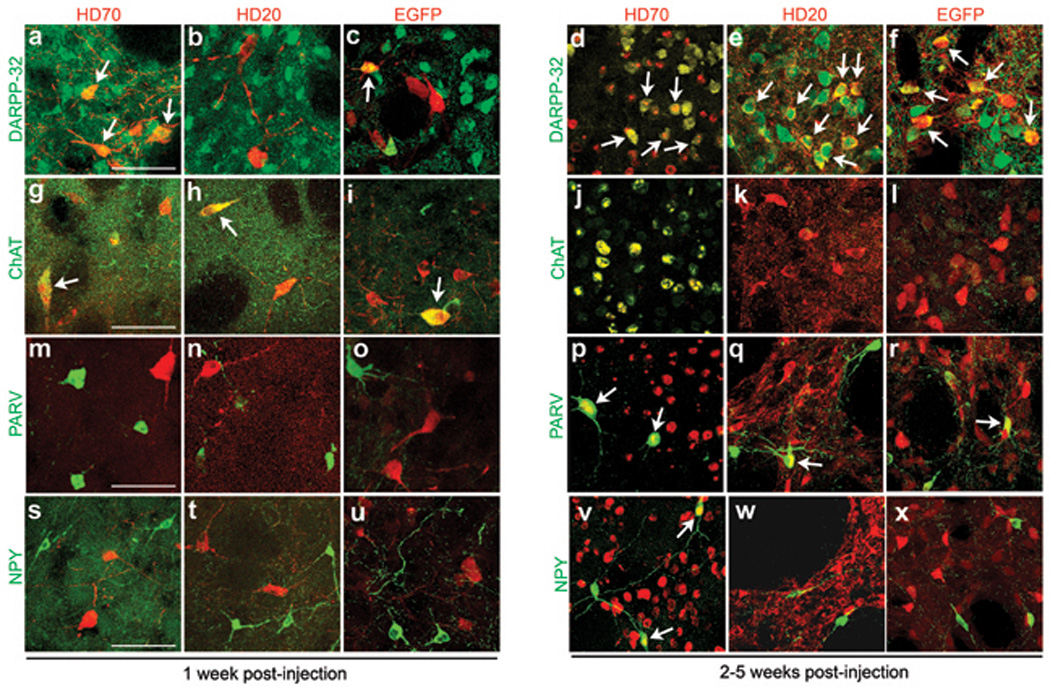
Merged confocal microscopy images of double immunofluorescent labeling with antibodies to striatal neuronal populations and AAV vector-mediated transgenes. Arrows indicate co-localization of immunofluorescence. Robust transduction of DARPP-32-immunoreactive projection neurons at 1 week in AAV-HD70 or -EGFP-injected brains (a,c), which was increased at 2 weeks post-injection (d,f). In contrast, projection neurons were very weakly transduced by AAV-HD20 at 1 week (b), with increased transduction observed at 2–5 weeks post-injection (e). Robust transduction of cholinergic interneurons (ChAT) by all AAV vectors at 1 week (g–i). Parvalbumin- (parv) or neuropeptide-Y- (NPY) positive interneurons lacked immunoreactivity to AAV transgenes at 1 week (m–o, s–u). At 2–5 weeks, ChAT immunoreactivity was diminished due to cell death (j–l) but there was artifactual cross-reactivity of ChAT staining with HD70 inclusions (j). At 2–5 weeks, parv-positive interneurons were transduced by all AAV transgenes (p–r). Transduction of NPY-positive interneurons at 2–5 weeks was only observed in AAV-HD70-injected rats (v–x). Results for AAV-Hdh8 transduction (not shown) were similar to those for AAV-HD20. Scale bars, 50 µm.
Axonal transport of AAV1/2-HD70 led to toxicity in distal regions of the basal ganglia
In line with previous reports of axonal transport of AAV virions or transgene products [19, 20], 5 weeks after striatal infusion of AAV1/2 vectors robust Ab1 anti-htt immunoreactivity was detected in the GP and substantia nigra (SN) (Figs. 5a–d,i–p). Anterograde transport led to staining of fibers of striatopallidal or striatonigral projections in the GP and substantia nigra pars reticulata (SNpr), respectively. In addition, retrograde transport led to immunoreactivity in cell bodies of the GP and substantia nigra pars compacta (SNpc). Axonal transport of vector genomes to the SN was confirmed by PCR (Fig. 5y). Neuronal cell loss and atrophy of the GP was visualized by loss of NeuN immunoreactivity and increased Nissl staining density (Figs. 5e–h). Similarly, loss of immunostaining for the neuronal protein HuC/D and tyrosine hydroxylase (TH) was observed, indicating toxicity in dopaminergic neurons of the SNpc (Figs. 5q–x). In contrast, Senut et al. observed AAV2-97Q-GFP expression in the SNpc 5 weeks after striatal injection, however no toxicity in the SNpc was reported [7].
Fig. 5. AAV vector axonal transport and neurodegeneration in the globus pallidus and substantia nigra.
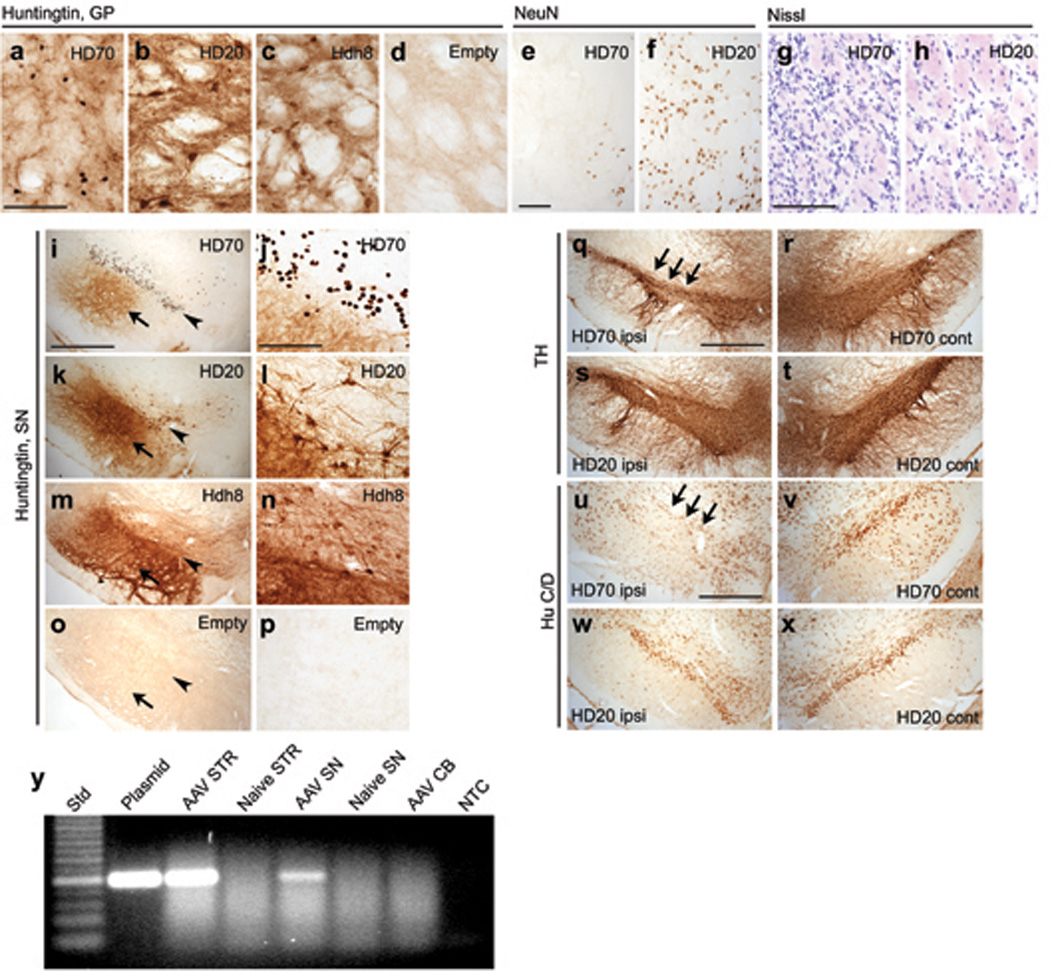
(a–d) Ab1 anti-htt immunostaining in the globus pallidus (GP) at 5 weeks. (e–h) Neuronal cell loss in the GP 5 weeks after striatal injection of AAV-HD70, visualized by loss of NeuN immunoreactivity (e) and increased Nissl staining density (g). NeuN immunoreactivity and Nissl staining in the GP was unchanged in animals injected with AAV-HD20 (f,h) and other control vectors (not shown). (i–p) Ab1 anti-htt immunostaining of robust HD70, HD20 and Hdh8 expression in the substantia nigra (SN) of rats 5 weeks post-injection into the striatum. Diffuse fiber staining was observed in the substantia nigra pars reticulata (SNPr, arrow), whereas neuronal cell bodies and fibers were stained in the pars compacta (SNPc, arrowhead). The pattern of staining in the SNPc resembled that observed in the striatum and GP, with HD70 inclusions (j) and more diffuse staining for HD20 (l) and Hdh8 (n). (q–x) Expression of HD70 in the substantia nigra caused loss of (q) TH and (u) HuC/D immunostaining compared to the contralateral hemisphere (r,t). No loss of TH or HuC/D immunoreactivity was observed in brains injected with HD20 (s,t,w,x) or other control vectors (not shown). (y) Agarose gel of PCR to detect AAV vector genome in striatum (STR), substantia nigra (SN) or cerebellum (CB) 5 weeks post-injection into the striatum or in naïve animals. Std = 25bp ladder, plasmid = AAV plasmid positive control, NTC = no template control. Scale bars, 50 µm (a,e,g,j), 1mm (i,q,u).
Quantitative RT-PCR analysis of expression levels of N-terminal htt constructs
Quantitative analysis of transgene expression levels has not been determined in previous viral vector-mediated HD models, therefore RT-PCR was used to analyze the level of AAV1/2 vector-mediated over-expression of N-terminal htt constructs in the striatum (Fig. 6). AAV-HD70 mRNA expression rose to ~150-fold that of endogenous Hdh mRNA in the rat striatum by 2 weeks and dropped to near-endogenous level by 5 weeks. This pattern of expression corroborated observations using anti-htt immunohistochemistry of HD70 expression (Fig. 1), and was consistent with immunohistochemical and histological evidence for neurodegeneration between 2–5 week time-points (Fig. 2). In contrast, both AAV-HD20 and -Hdh8 mRNA levels rose more gradually, to ~150-fold by 5 weeks. The level of endogenous rat Hdh mRNA in AAV-Empty vector rats was relatively unchanged over 5 weeks. The expression levels of mutant N-terminal htt achieved using AAV1/2 vector-mediated gene transfer were significantly higher than has been reported in transgenic or knock-in mouse models of HD, which were typically close to that of endogenous mouse Hdh expression levels [4]. Interestingly, AAV-HD70-mediated transgene levels determined by RT-PCR were higher at 1–2 weeks post-injection than that observed for AAV-HD20 or -Hdh8, despite the fact that vectors were titer-matched and the expression cassettes contained the same promoter and post-transcriptional regulatory elements. This finding reflects the broader neuronal transduction of AAV-HD70 at these time points.
Fig. 6. Quantitative real-time RT-PCR analysis of htt construct over-expression levels.
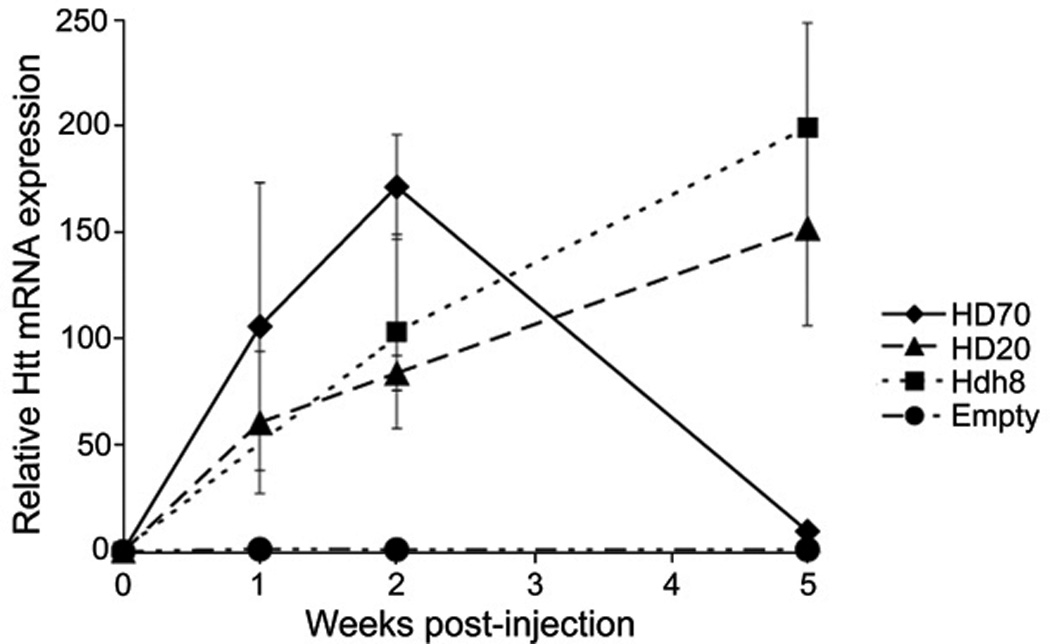
AAV vector-mediated N-terminal htt mRNA expression levels relative to endogenous rat Hdh mRNA expression levels in rat striatae injected with AAV-HD70, -HD20, -Hdh8 or -Empty vectors, quantified by real-time RT-PCR. Each data point represents mean±SEM (n=4).
AAV vector-mediated RNAi prevents HD70-induced neurodegeneration and behavioral impairment
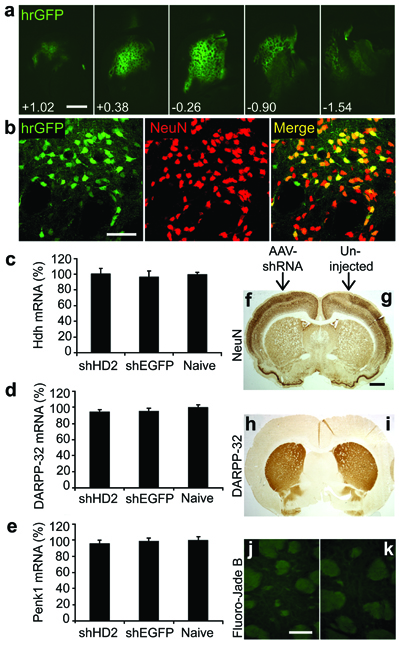
Finally, we used the AAV-HD70 model to assess the neuroprotective efficacy of shRNA-mediated knockdown of htt expression. We used AAV vectors expressing shRNA constructs(Fig.S4) targeting a similar site of the human htt mRNA as siHUNT-2 previously reported [13] (AAV-shHD2) or a control shRNA targeting EGFP (AAV-shEGFP) [21]. As a prelude to the neuroprotection study, we first injected naive rats with AAV-shRNA vectors alone. In contrast with Rodriguez-Lebron et al. [13] we observed no ‘off-target’ knock-down of proenkephalin (Penk1) and DARPP-32 mRNA, assessed by RT-PCR (Fig. S5), however it is possible that microarray analysis would reveal additional unintended gene expression inhibition [22]. In agreement with our findings, Wang et al. used in vivo transfected siRNAs targeting a similar site encoded in exon 1 of the HD gene and reported no deleterious side effects in mice [23]. Consistent with previous reports [11, 13], we found no difference in the level of endogenous rat Hdh mRNA expression in striatae injected with AAV-shHD2 or -shEGFP vectors compared with uninjected striatae (Fig. S5), likely due to two mismatches between human and rat htt mRNAs in the target sequence. In addition, toxicity due to shRNA expression in vivo has been reported [24], however, we observed no toxicity in the striatae of rats 5 weeks after AAV-shHD2 or -shEGFP vector infusion (Fig. S5). Next, we injected AAV-shHD2 or -shEGFP into the rat striatum followed 2 weeks later by AAV-HD70. Rats were euthanized 2 weeks after AAV-HD70 infusion, at the time of predicted maximal transgene expression (Fig. 6). HD70 mRNA and protein expression was dramatically reduced in rats that were injected with shHD2, in contrast with robust HD70 expression in rats injected with shEGFP, determined by immunofluorescence and confocal microscopy, RT-PCR and Western blot analyses (Fig. 7). Moreover, RNAi-mediated inhibition of HD70 expression by AAV-shHD2 led to substantial neuroprotection. Stereological cell counts revealed survival of 91.7±2.9% of NeuN-positive cells (Figs. 8a,b,g) and 98.1±6.8% of calbindin-positive neurons (Fig. 8h). In addition, Fluoro-Jade B staining was rare in AAV-shHD2 plus -HD70 vector injected rats (Fig. 8e). The cylinder test [25] was used to analyze spontaneous exploratory fore paw use in the rats (Fig. 8i). No significant difference in forepaw use between groups was observed in naïve animals or at 13 days post-injection of AAV-shRNA vectors. However, at 13 days after infusion of AAV-HD70, rats that had received AAV-shEGFP plus -HD70 exhibited a 10.1±2.1% impairment of the contralateral forepaw. In contrast, AAV-shHD2 plus -HD70-treated rats showed no impairment of the contralateral forepaw, exhibiting symmetrical use of forepaws in this task. These results suggest that AAV-shHD2 mediated functional protection from motor impairment by blocking striatal expression of a neurotoxic mutant htt construct.
Fig. 7. AAV vector-mediated knockdown of HD70 expression in rat striatum.
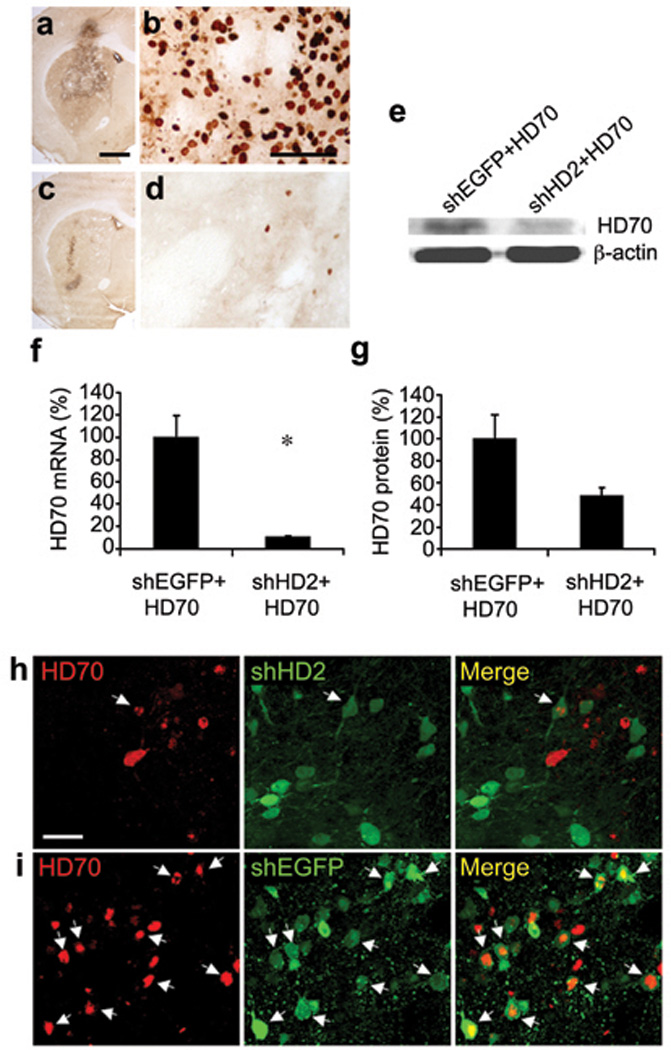
(a–d) Ab1 anti-htt immunohistochemistry on striatal sections from animals injected with AAV-shEGFP plus AAV-HD70 (a,b) or AAV-shHD2 plus AAV-HD70 (c,d).
(e) Autoradiograph images of bands visualized in western blot of rat striatal lysate. Molecular weights of the bands presented here were: HD70 ~35kDa, β-actin ~42kDa.
(f) Mean±SEM (n=4) HD70 mRNA expression quantified by real-time RT-PCR analysis. Student’s t-test: *p=0.012. (g) Mean±SEM (n=4) HD70 protein expression quantified by western blot. Student’s t-test: p=0.064 (not statistically significant). (h,i) Confocal microscopy images of striatal sections showing immunofluorescent Ab1 anti-htt staining (red) and native hrGFP fluorescence (green). Arrows indicate co-localization of expression of HD70 and shHD2 (h) or shEGFP (i). Scale bars (a) 1mm, (b) 50µm, (h) 25µm.
Fig. 8. AAV vector-mediated knockdown of HD70 expression prevented degeneration of striatal neurons and motor behavioral impairment.
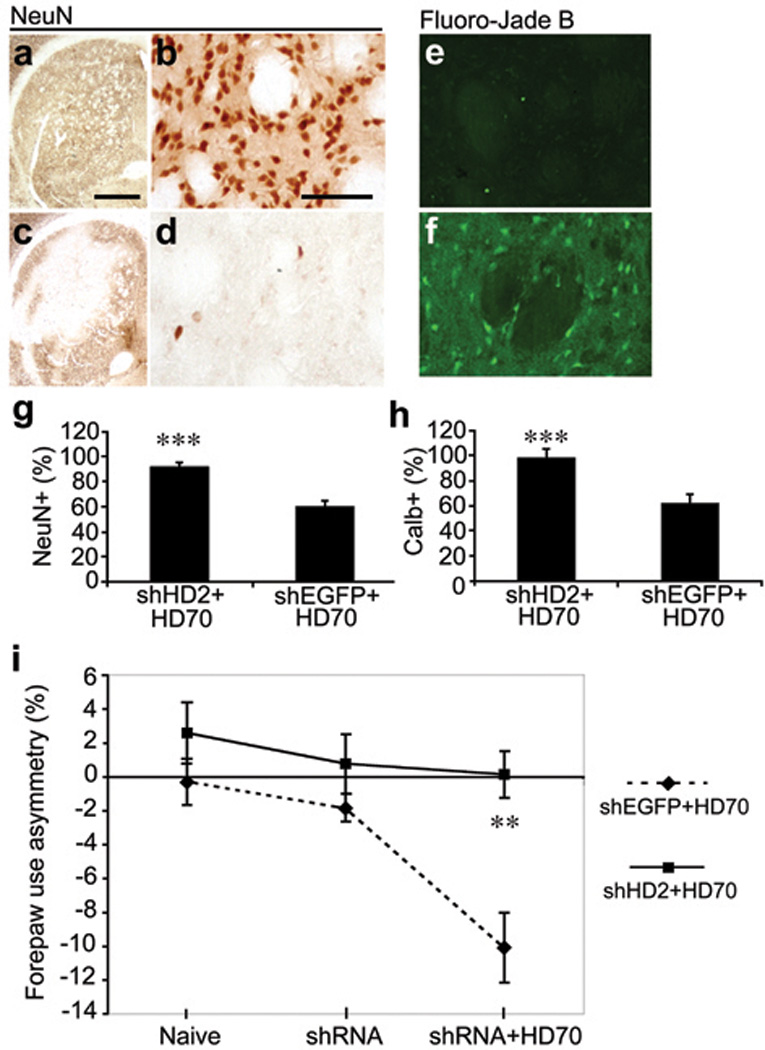
Striatal sections from rats injected with AAV-shHD2 or -shEGFP followed by AAV-HD70, immunostained with antibodies to NeuN (a–d) or stained with Fluoro-Jade B (e,f). High power images were taken in the middle of the transduced areas of striatae. Stereological cell counts of NeuN-positive cells (g) or calbindin d28k-positive projection neurons (h) of the rat striatum transduced by AAV-shHD2 or -shEGFP followed by AAV-HD70. Mean±SEM (n=6) ipsilateral data are presented as a percentage of the number of cells counted in the contralateral hemisphere. NeuN-positive striatal cell survival: shHD2+HD70 = 91.7 ± 2.9%; shEGFP+HD70 = 60.1±4.2%. Calbindin-positive projection neuron survival: shHD2+HD70 = 98.1±6.8%; shEGFP+HD70 = 61.7±7.5%. Student’s t-test, ***p<0.001. (i) Spontaneous exploratory forepaw use in the cylinder test in rats that were injected with AAV-shHD2 or -shEGFP, followed by AAV-HD70. Mean±SEM (n=14) percentage of fore-paw use asymmetry, where negative scores indicate contralateral fore-paw impairment. For each group, results are presented for tests conducted prior to shRNA vector injection (Naïve), 13 days post-injection of shRNA vectors (shRNA) and 13 days post-injection of HD70 vector (shRNA+HD70). ANOVA: shEGFP+HD70 v other groups, all time points ** p < 0.01. Scale bars (a) 1mm, (b) 50µm, applies to all high magnification images.
DISCUSSION
Here we report a comprehensive characterization of a new model of HD generated by AAV1/2 vector-mediated transfer of N-terminal htt constructs to the rat striatum. Recombinant AAV has emerged as a vector of choice for gene transfer to the CNS, due to its strong neuronal tropism and lack of pathogenicity in mammals [5]. The packaging capacity of 4.7kb precludes AAV vector-mediated over-expression of full-length htt, however N-terminal truncated htt constructs have been successfully used previously to recapitulate elements of HD in experimental animals [8–10, 26–28]. Moreover, there is growing evidence to support a ‘toxic fragment hypothesis’, whereby neuropathological events in HD are triggered by proteolytic cleavage of an N-terminal htt fragment [2]. The AAV-HD70 model recapitulates some critical elements of HD and bears both similarities and differences to several previously reported genetic models of HD. The rapidly progressive phenotype most closely resembles previous models generated by virus vector-mediated gene transfer [7–10]. The extent of striatal neuronal cell loss observed was greater than previously reported in earlier viral vector-based models of HD in rats [7, 8] and similar to that recently reported using AAV1/8 in mice by DiFiglia et al. [10] as a consequence of the increased expression achieved using AAV1/2 vectors, both in terms of expression level and the volume of striatal transduction. Moreover, the striatal neuronal cell loss observed as early as 5 weeks post-injection of AAV-HD70 was over twice that reported for 12 month old YAC128 mice that exhibit the greatest extent of striatal neuronal cell loss of all the transgenic HD mouse models [29]. Differences in the rapidity of disease progression correlate to some degree with mutant htt expression level in genetic models of HD. Indeed, analysis of the temporal expression profile by RT-PCR revealed a dramatic level of N-terminal htt expression resulting from AAV vector-mediated gene transfer. This may prove to be advantageous in that it may facilitate more rapid in vivo screening of therapeutics than is currently available using transgenic models.
Interestingly, we found that AAV1/2 vector-mediated transduction patterns influenced the neuropathological phenotype in our model. First, it was found that AAV1/2 transduction of striatal neurons is non-uniform, most notably with increased transduction of cholinergic interneurons compared to other neuronal populations. The reason for this is unclear, however it may be that cholinergic interneurons express different cell-surface receptors that facilitate enhanced tropism through more rapid internalization of AAV1/2 virions. Entry of AAV into cells is dependent upon binding to cell surface glycosaminoglycan receptors and other co-receptors, although the mechanisms underlying selective tropisms of AAV serotypes remain largely unknown [30]. Moreover, differences in intra-cellular processing of AAV vectors may play a role in transduction of various neuronal populations [30]. Over 100 serotypes of AAV have been isolated to date, and characterization of novel AAV serotypes revealed different patterns of transduction in a diverse array of tissues [31]. Moreover, it has been shown that transduction patterns in the nigro-striatal system can vary greatly between different AAV serotypes [19, 32–34]. Further investigation to elucidate differences in AAV tropism and transduction among neuronal populations in the striatum and other brain regions may lead to improved modeling of neurodegenerative diseases in experimental animals. Secondly, we found that neuronal transduction by AAV1/2 vectors was altered by changing the transgene alone. It has been proposed that changes in secondary structure of AAV vector genomes containing different constructs may alter tropism by changing the conformation of capsid proteins on the virion surface, leading to altered interactions with cell surface receptors [35]. This hypothesis awaits verification by structural studies of recombinant AAV vectors.
Axonal vector transport also played a role in the neuropathological phenotype in our model. Robust expression of AAV-HD70 was observed in the GP and SN following striatal injection of AAV vectors, leading to toxicity in distal structures of the basal ganglia. These results confirm previous reports that retrograde transport can lead to transduction of neurons in brain regions distal to the site of injection [20], but also indicate that neuronal cell loss in distal brain regions can result from expression of neuro-toxic gene constructs such as HD70. Although the primary neuropathology in HD occurs in the striatum, neurodegeneration and atrophy of the GP and SNpr is also observed in HD brains, however neuronal cell loss in the SNpc is less typical of HD [14]. Taken together, these results raise some issues regarding the use of viral vectors for modeling neurodegenerative diseases that warrant caution and require to be addressed. For example, these findings indicate that the use of viral vector systems such as AAV could potentially introduce artifacts into functional genomics and disease modeling studies.
Nevertheless, the robust striatal neurodegenerative phenotype observed in the AAV-HD70 model suggests that it is well-suited for the rapid screening of neuroprotective strategies for HD. Here, we demonstrated for the first time that AAV vector-mediated RNAi was highly effective as a neuroprotective therapy in an animal model of HD. Our findings are in agreement with previous reports supporting the utility of RNAi as a therapeutic approach for HD and other polyQ expansion diseases in cell culture [36, 37] and transgenic mouse models [11–13, 23, 38, 39]. Recently, DiFiglia et al. also reported that a cholesterol-conjugated siRNA targeting human htt mRNA inhibited expression of the AAV1/8Htt100Q vector and led to increased survival of striatal neurons and improved motor behaviors in mice [10]. Importantly, however, siRNA-mediated silencing is transient and would require frequent repeat interventions for treating HD patients. In contrast, AAV vector-mediated delivery of shRNAs offers a long-term approach for therapeutic disease gene silencing following a single injection [40].
Despite the clear therapeutic potential of RNAi-based gene therapy for HD, several issues are challenging translation towards the clinic, including allele-specific silencing, ‘off-target effects’ and potential toxicity [41]. Despite these caveats, RNAi may prove to be an effective therapeutic strategy for HD and other neurodegenerative diseases. Our results support AAV vector-mediated knockdown of mutant htt expression as a direct approach to preventing striatal neurodegeneration and associated behavioral impairment. Translation of viral vector-mediated RNAi to a non-human primate genetic model of HD [9] would provide further opportunity for pre-clinical assessment of the safety and efficacy of this therapeutic approach.
MATERIALS AND METHODS
Recombinant AAV vector expression constructs
An AAV vector expression cassette (Fig. S1), consisting of rat neuron-specific enolase (NSE) promoter, woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and bovine growth hormone polyadenylation sequence (BGHpA) flanked by AAV2 inverted terminal repeats (ITR) was used to regulate expression of all transgenes used in this study [42]. An exon 1 mutant HD transgene (HD70) was obtained by PCR amplification of genomic DNA from the YAC HD72 mouse [43]. As a control, a wild-type exon 1 HD transgene (HD20) was PCR amplified from a Hybrid Hunter cDNA library (Invitrogen, Carlsbad, CA). The primers used to amplify both transgenes were: GCCATGGCGACCGTCGAAAAGCT (forward), CTATCGGTGCAGCGGCTCCTCAGC (reverse). Amplicons were cloned into pCR2.1-TOPO (Invitrogen). DNA sequencing of pCR2.1-TOPO-HD70 revealed deletion of two CAG repeats during PCR amplification of the original YAC HD72 template. Transgenes were cloned into the polylinker site (pl) of the AAV expression cassette plasmid pAM/NSE-pl-WPRE-BGHpA. As an additional control, cDNA from exon 1 of the rat homologue, Hdh, containing 8 CAG repeats (Hdh8) was also cloned into the AAV expression cassette plasmid. A 534bp amplicon (<www.ncbi.nlm.nih.gov> Entrez Nucleotide GI: 39983010) was amplified from rat brain cDNA using the following primers: CCTTGCTGCTAAGTGGCG (forward), CTCAGCAAACCTCCACAG (reverse) and cloned into pCR2.1-TOPO (Invitrogen). A 198bp amplicon corresponding to Hdh exon 1 was amplified from this plasmid template using the following primers: CCGATGGCAACCCTGGAAAAACT (forward), CTATCGGTGCAGCGGCTCCTCGGC (reverse) and cloned into the AAV expression plasmid. DNA sequencing confirmed stability of CAG repeats in HD70, HD20 and Hdh8 constructs following propagation of plasmids in bacterial culture. A construct containing regulatory elements but lacking a transgene (AAV-Empty) was also used as a control. Finally, cDNA encoding enhanced green fluorescent protein (EGFP) was excised from pIRES-EGFP (Clontech, Mountain View, CA) and also cloned into the AAV expression cassette plasmid for inclusion as a non-disease related gene control in some of the experiments in this study.
A second AAV vector cassette (Fig. S4) was used to regulate expression of short hairpin RNA (shRNA) constructs, under control of the human U6 promoter (U6). The U6 promoter and the shRNA hairpin cloning site were amplified from pSilencer™ 2.1-U6 hygro (Ambion, Inc., Austin, TX) by PCR using the following primers: CGGGGTACCCTCGAGGAGGAGAAGCATGAATTC (forward), CGGGGTACCACTAGTACGGCCAGTGCCAAGCTT (reverse). The PCR product was cloned into pAM/CBA-hrGFP-WPRE-BGHpA to generate pAM/U6-pl-CBA-hrGFP-WPRE-BGHpA. This expression cassette plasmid also contained a hrGFP reporter gene, under regulatory control of a 1.1kb CMV enhancer/chicken β-actin promoter (CBA), WPRE and BGHpA. Ambion’s Insert design tool for the pSilencer™ Vectors (http://www.ambion.com/techlib/misc/psilencer_converter.html) was used to generate short hairpin siRNA-encoding, complementary forward and reverse DNA oligonucleotide insert sequences. These were based on previously published siRNA targets siHUNT-2 [44] and siRNA targeting EGFP [21] (termed shHD2 and shEGFP, respectively). Complementary sense/anti-sense DNA oligonucleotides (40ng/µl) were annealed at room temperature for 1 hour in 10mM Tris-HCl, pH7.5, 100mM NaCl, 1mM EDTA. Annealed oligonucleotides were cloned into the polylinker site (pl) of the AAV expression cassette plasmid pAM/U6-pl-CBA-hrGFP-WPRE-BGHpA.
AAV vectors
Chimeric AAV vectors containing equal numbers of AAV serotype 1 and 2 capsid proteins [45] were generated as described previously [46]. Briefly, HEK293 cells were transfected using calcium phosphate with an AAV expression plasmid (HD70, HD20, Hdh8, EGFP, Empty, shHD2 or shEGFP), the adenovirus helper plasmid (pFΔ6) and both the AAV1 (pH21) and AAV2 (pNLrep) helper plasmids mixed at a 1:1 ratio to form chimeric capsids. Sixty hours after transfection, cells were harvested and vectors were purified using heparin affinity columns (HiTrap Heparin HP, Amersham Pharmacia Biotech, Uppsala, Sweden). Genomic titers of virus vector stocks were determined by real time PCR using an ABI 7900HT Sequence Detection Sytem (Applied Biosystems, Foster City, CA) with primers designed to WPRE and were matched to 1×1012 viral vector genomes/mL by appropriate dilution in sterile 1x phosphate-buffered saline (PBS).
Sterotaxic surgery
Adult male Wistar rats (250–300g) (Animal Resources Unit, The University of Auckland) were used in this study. Approvals for all animal experiments were gained from The University of Auckland Animal Ethics Committee. The rats were housed in a humidity- and temperature-controlled containment facility that was kept on a 12 hour light/dark cycle, with food and water available ad libitum. Rats were anaesthetized with sodium pentobarbitone (Nembutal, Virbac Laboratories, Auckland, New Zealand; 80mg/kg i.p) and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) before receiving a 3µL intra-striatal infusion (coordinates: RC+0.4mm, ML-3.0, DV-5.5, bregma =0; [47]) of AAV vectors, at an infusion rate of 100nL/min.
Experimental groups
Groups of animals used in this study were as follows. (1) HD model characterization - Immunohistochemistry and Histology: AAV-HD70, -HD20, -Hdh8, or -Empty vector (n=6 rats per time point) were killed at 1, 2, 5 and 8 weeks post-injection; AAV-EGFP (n=4 rats per time point) were killed at 1, 2, and 5 weeks post-injection. (2) HD model characterization - quantitative real-time PCR: AAV-HD70, -HD20 or -Empty vector (n=4 per time point) were euthanized at 1, 2 and 5 weeks post-injection; AAV-Hdh8 vector, n=4 per time point were killed at 2 and 5 weeks post-injection. (3) HD model characterization – axonal transport of vectors PCR: AAV vector-injected (n=3) or naïve (n=1) rats were euthanized at 5 weeks post-injection and brains were used for PCR-based detection of vector genomes. (4) Expression of AAV-shRNA vectors: Two groups of rats received unilateral intra-striatal injections of AAV-shHD2 or -shEGFP (n=12 rats per group) and were killed two weeks post-injection (n=3 per group) or five weeks post-injection (n=9 per group). Brains were analyzed using immunohistochemistry, Fluoro-Jade B staining or fluorescence microscopy (n=3 rats per group at 2w, n=6 rats per group at 5w). Rats were also killed at 5 weeks post-injection and brains were analyzed by quantitative real-time RT-PCR (n=3 striatae per shRNA vector and n=3 uninjected striatae). (5) HD70 knockdown: Two groups of rats received a unilateral intra-striatal injection of either AAV-shHD2 (n=14) or -shEGFP (n=14). Two weeks later, all rats (n=28) were also injected unilaterally into the same hemisphere with AAV-HD70. Rats were killed two weeks post-injection of AAV-HD70 and brains were analyzed using immunohistochemistry, Fluoro-Jade B staining or fluorescence microscopy (n=6 rats per group), western blot (n=4 rats per group), or quantitative real-time RT-PCR (n=4 rats per group). (6) Behavioral testing: The cylinder test was used to assess impairment in spontaneous exploratory forelimb use, as described previously [25]. Behavioral testing was conducted by an investigator blinded to experimental groups. Testing was conducted (n=14 per group) in naïve animals prior to injection of AAV-shHD2 or -shEGFP, 13 days post-injection of AAV-shRNA vectors and finally at 13 days post-injection of AAV-HD70. Briefly, forelimb use during exploratory activity of rats in a transparent cylinder (20cm diameter × 30cm height) was recorded by video camera for 5 minutes. Two mirrors were placed behind the cylinder at an angle to enable recording of forelimb movements when the rat was facing away from the camera. The forelimb used for push-off and landing of vertical movements within the cylinder was counted by reviewing the video footage in slow motion. Net ipsilateral forelimb use values were determined as a percentage of total forelimb use.
Stereological cell counts
Cell counting was performed using NeuN, calbindin d28k, ChAT, NPY or parvalbumin immuno-stained striatal sections. For each neuronal marker, 10 sections per brain were selected 320µm apart throughout the rostral-caudal extent of the striatum for immunohistochemistry. Quantification of the number of immunopositive cells throughout the extent of the striatum was estimated using unbiased stereological techniques. Stereological cell counting was performed using the optical fractionator method, Stereo Investigator 7 (Microbrightfield, Inc. Williston, VT), attached to an Olympus AX70 microscope (Center Valley, PA), using a 40x objective, by an investigator blinded to experimental groups. An estimate of the fraction of surviving cells in the injected striatum was calculated by dividing the number of cells counted in the ipsilateral hemisphere by the number of cells counted in the contralateral hemisphere.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Vicky Tsang and Alexandre Mouravlev (The University of Auckland) for generating various plasmid constructs, Claudia Leichtlein (The University of Auckland) for technical support, Anassuya Ramachandran, Russell Snell, Henry Waldvogel and Richard Faull (The University of Auckland) for helpful discussions and Marian DiFiglia (Mass. General Hospital) for anti-htt antibodies. This work was supported by a University of Auckland doctoral scholarship to NRF and grants from the NZ Health Research Council and Foundation for Science Research & Technology to DY and MJD.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Orr HT, Zoghbi HY. Trinucleotide Repeat Disorders. Annual review of neuroscience. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 3.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Menalled LB, Chesselet MF. Mouse models of Huntington's disease. Trends Pharmacol Sci. 2002;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- 5.Janson CG, McPhee SW, Leone P, Freese A, During MJ. Viral-based gene transfer to the mammalian CNS for functional genomic studies. Trends in neurosciences. 2001;24:706–712. doi: 10.1016/s0166-2236(00)01954-8. [DOI] [PubMed] [Google Scholar]
- 6.Kirik D, Bjorklund A. Modeling CNS neurodegeneration by overexpression of disease-causing proteins using viral vectors. Trends in neurosciences. 2003;26:386–392. doi: 10.1016/S0166-2236(03)00164-4. [DOI] [PubMed] [Google Scholar]
- 7.Senut MC, Suhr ST, Kaspar B, Gage FH. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J Neurosci. 2000;20:219–229. doi: 10.1523/JNEUROSCI.20-01-00219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palfi S, et al. Expression of Mutated Huntingtin Fragment in the Putamen Is Sufficient to Produce Abnormal Movement in Non-human Primates. Mol Ther. 2007 doi: 10.1038/sj.mt.6300185. [DOI] [PubMed] [Google Scholar]
- 10.DiFiglia M, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harper SQ, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machida Y, Okada T, Kurosawa M, Oyama F, Ozawa K, Nukina N. rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem Biophys Res Commun. 2006;343:190–197. doi: 10.1016/j.bbrc.2006.02.141. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntingtin's disease transgenic mice. Molecular Therapy. 2005;12 doi: 10.1016/j.ymthe.2005.05.006. 6-8-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Jr, Martin JB. Sparing of acetylcholinesterase-containing striatal neurons in Huntington's disease. Brain research. 1987;411:162–166. doi: 10.1016/0006-8993(87)90694-9. [DOI] [PubMed] [Google Scholar]
- 16.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science (New York, N.Y. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 17.Harrington KM, Kowall NW. Parvalbumin immunoreactive neurons resist degeneration in Huntington's disease striatum. J Neuropathol Exp Neurol. 1991;50:309. doi: 10.1097/00005072-199107000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger C, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 20.Kaspar BK, Erickson D, Schaffer D, Hinh L, Gage FH, Peterson DA. Targeted retrograde gene delivery for neuronal protection. Mol Ther. 2002;5:50–56. doi: 10.1006/mthe.2001.0520. [DOI] [PubMed] [Google Scholar]
- 21.Donze O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic acids research. 2002;30:e46. doi: 10.1093/nar/30.10.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson AL, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA (New York, N.Y. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YL, Liu W, Wada E, Murata M, Wada K, Kanazawa I. Clinico-pathological rescue of a model mouse of Huntington's disease by siRNA. Neuroscience research. 2005;53:241–249. doi: 10.1016/j.neures.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 24.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 25.Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 26.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 27.Schilling G, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Human molecular genetics. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 29.Slow EJ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Human molecular genetics. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 30.Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Current gene therapy. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 32.Davidson BL, et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paterna JC, Feldon J, Bueler H. Transduction profiles of recombinant adeno-associated virus vectors derived from serotypes 2 and 5 in the nigrostriatal system of rats. Journal of virology. 2004;78:6808–6817. doi: 10.1128/JVI.78.13.6808-6817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taymans JM, et al. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Human gene therapy. 2007;18:195–206. doi: 10.1089/hum.2006.178. [DOI] [PubMed] [Google Scholar]
- 35.Mastakov MY, Baer K, Symes CW, Leichtlein CB, Kotin RM, During MJ. Immunological aspects of recombinant adeno-associated virus delivery to the mammalian brain. Journal of virology. 2002;76:8446–8454. doi: 10.1128/JVI.76.16.8446-8454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia H, Mao Q, Paulson HL, Davidson BL. siRNA-mediated gene silencing in vitro and in vivo. Nature biotechnology. 2002;20:1006–1010. doi: 10.1038/nbt739. [DOI] [PubMed] [Google Scholar]
- 38.Huang B, Schiefer J, Sass C, Landwehrmeyer GB, Kosinski CM, Kochanek S. High-capacity adenoviral vector-mediated reduction of huntingtin aggregate load in vitro and in vivo. Human gene therapy. 2007;18:303–311. doi: 10.1089/hum.2006.160. [DOI] [PubMed] [Google Scholar]
- 39.Xia H, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nature medicine. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 40.Grieger JC, Samulski RJ. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Advances in biochemical engineering/biotechnology. 2005;99:119–145. [PubMed] [Google Scholar]
- 41.Raoul C, Barker SD, Aebischer P. Viral-based modelling and correction of neurodegenerative diseases by RNA interference. Gene therapy. 2006;13:487–495. doi: 10.1038/sj.gt.3302690. [DOI] [PubMed] [Google Scholar]
- 42.Fitzsimons HL, Bland RJ, During MJ. Promoters and regulatory elements that improve adeno-associated virus transgene expression in the brain. Methods (San Diego, Calif. 2002;28:227–236. doi: 10.1016/s1046-2023(02)00227-x. [DOI] [PubMed] [Google Scholar]
- 43.Hodgson JG, et al. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauck B, Chen L, Xiao W. Generation and characterization of chimeric recombinant AAV vectors. Mol Ther. 2003;7:419–425. doi: 10.1016/s1525-0016(03)00012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.During MJ, Young D, Baer K, Lawlor P, Klugmann M. Development and optimization of adeno-associated virus vector transfer into the central nervous system. Methods Mol Med. 2003;76:221–236. doi: 10.1385/1-59259-304-6:221. [DOI] [PubMed] [Google Scholar]
- 47.Paxinos G, Watson C. The Rat Brain In Stereotaxic Coordinates. Orlando: Academic Press; 1986. [Google Scholar]
- 48.Young D, Lawlor PA, Leone P, Dragunow M, During MJ. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nature medicine. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- 49.Sharp AH, et al. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 50.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002 doi: 10.1186/gb-2002-3-7-research0034. 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.