

Abstract
Huntington disease (HD, OMIM #143100) is a dominantly inherited neurodegenerative disorder due to a CAG repeat expansion in the HTT gene, encoding a polyglutamine tract in the N-terminal part of the huntingtin protein. Most cases are inherited from an affected parent, but in about 10% of cases the condition appears to be de novo.1 De novo or sporadic cases are usually due to CAG repeat expansion of intermediate alleles. Intermediate alleles have 27–35 CAG repeats, and the higher the number of repeats, the higher the risk for expansion into disease range, usually upon paternal transmission.2 In most cases, the change in repeat size is minor, and gradual increases into the disease range over several generations is the basis of new genetic mutations and stable disease prevalence. So far, the largest single-step expansions reported were from 27 to 383 and from 35 to 582 CAG repeats. It has recently been shown that intermediate alleles and disease alleles share the same haplotypes, which is expected if intermediate alleles are the main source of new mutation cases. The high-risk haplotypes are called A1 and A2, and are both prevalent among Caucasians but rare in other ethnic groups.4
Huntington disease (HD, OMIM #143100) is a dominantly inherited neurodegenerative disorder due to a CAG repeat expansion in the HTT gene, encoding a polyglutamine tract in the N-terminal part of the huntingtin protein. Most cases are inherited from an affected parent, but in about 10% of cases the condition appears to be de novo.1 De novo or sporadic cases are usually due to CAG repeat expansion of intermediate alleles. Intermediate alleles have 27–35 CAG repeats, and the higher the number of repeats, the higher the risk for expansion into disease range, usually upon paternal transmission.2 In most cases, the change in repeat size is minor, and gradual increases into the disease range over several generations is the basis of new genetic mutations and stable disease prevalence. So far, the largest single-step expansions reported were from 27 to 383 and from 35 to 582 CAG repeats. It has recently been shown that intermediate alleles and disease alleles share the same haplotypes, which is expected if intermediate alleles are the main source of new mutation cases. The high-risk haplotypes are called A1 and A2, and are both prevalent among Caucasians but rare in other ethnic groups.4
We describe a de novo case of HD that defies this general rule. The patient is a 45-year-old white woman with advanced HD with extensive involuntary movements, unsteady gait, and dementia. Symptoms started around age 33 with swallowing problems, restlessness, asthenia, and poor concentration. Two years later, she was no longer able to work as an accountant or do activities she previously enjoyed. Four years later, increasing involuntary movements led to a diagnosis of HD, confirmed by finding 44 CAG repeats in the HTT gene. Family follow-up revealed allele sizes of 15/15 in the mother and 15/26 in the father (figure). This unexpected result led to uncertainty within the family concerning paternity, and the parents asked for paternity confirmation, which was verified. The HD alleles of the patient and her parents were subsequently subcloned, and an A > G polymorphism in the polyproline (CCG) tract following the polyglutamine (CAG) tract distinguished the father's 26-CAG allele from his 15-CAG allele; otherwise, the sequence surrounding the CAG repeat was identical to consensus, including a penultimate CAA following the CAG repeat. It turned out that the father's 26-CAG allele had expanded into his daughter's 44-CAG allele (figure), with no evidence of paternal mosaicism. Single nucleotide polymorphism haplotyping showed that the expansion had occurred on a low-risk B haplotype (subtype B44), not a high-risk A1 or A2 haplotype, as would be expected (figure).4
Figure. Family pedigree demonstrates paternal expansion of an allele with 26 CAG repeats into a pathologic allele with 44 CAG repeats.
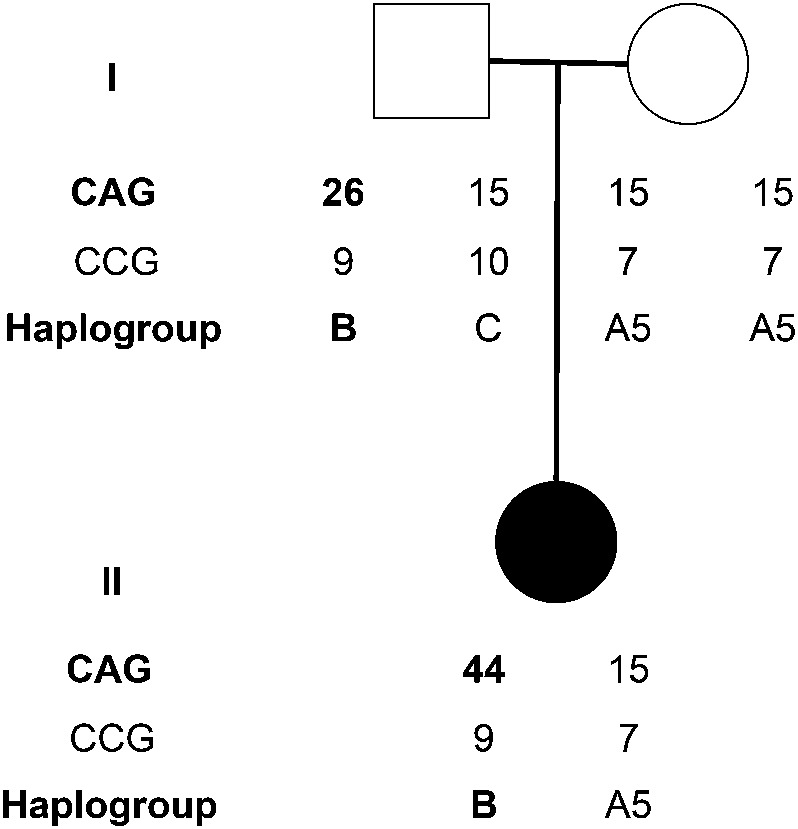
The CAG and CCG repeat lengths and haplogroups are defined for each family member. The father's alleles can also be distinguished by 9 uninterrupted CGG triplets due to a common A > G polymorphism turning a CCA into (the second) CCG. For details on the determination of HTT haplogroups, see Warby et al.1 Symbols: circle = female; square = male; black circle = patient.
Our case shows that an expansion of 18 CAG repeats may occur on a low-risk haplotype from an allele size that would usually be considered stable.5 This suggests that unknown predisposing factors, either genetic or environmental, may contribute to CAG repeat expansion in HD. The possibility that CAG repeats may unexpectedly expand into the disease range is important information when genetic counseling a family with a truly de novo case of HD.
Acknowledgments
Acknowledgment: The authors thank Dr. Harald Hovdal, the referring clinician, and the patient and her family for their interest in resolving this case.
Footnotes
Author contributions: Gunnar Houge: writing of manuscript, clinical work, interpretation of data. Ove Bruland: acquisition and analysis of data. Inga Bjørnevoll: acquisition of data, clinical contact. Michael R. Hayden: supervision of part of study. Alicia Semaka: acquisition and interpretation of data, revision of manuscript.
Disclosures: G. Houge, O. Bruland, and I. Bjørnevoll report no disclosures. M. Hayden is a Killam University Professor; holds a Canada Research Chair in Human Genetics and Molecular Medicine; and is supported by the Canadian Institutes of Health Research (CIHR). A. Semaka is supported by the Canadian Institutes of Health Research (CIHR) and is funded by a Doctoral Award from the CIHR and a Senior Trainee Award from the Michael Smith Foundation for Health Research. Go to Neurology.org for full disclosures.
References
- 1.Warby SC, Montpetit A, Hayden AR, et al. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet 2009;84:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Semaka A, Collins JA, Hayden MR. Unstable familial transmissions of Huntington disease alleles with 27-35 CAG repeats (intermediate alleles). Am J Med Genet B Neuropsychiatr Genet 2010;153B:314–320 [DOI] [PubMed] [Google Scholar]
- 3.Kelly TE, Allinson P, McGlennen RC, Baker J, Bao Y. Expansion of a 27 CAG repeat allele into a symptomatic Huntington disease-producing allele. Am J Med Genet 1999;87:91–92 [PubMed] [Google Scholar]
- 4.Warby SC, Visscher H, Collins JA, et al. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet 2011;19:561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maat-Kievit A, Losekoot M, Van Den Boer-Van Den Berg H, et al. New problems in testing for Huntington's disease: the issue of intermediate and reduced penetrance alleles. J Med Genet 2001;38:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]