

Abstract
We previously reported that tumor-evoked regulatory B cells (tBregs) play an essential role in breast cancer lung metastasis by inducing TGFβ-dependent conversion of metastasis-promoting FoxP3+ Tregs. Here we show that resveratrol (RSV), a plant-derived polyphenol, at low and non-cytotoxic doses for immune cells can efficiently inhibit lung metastasis in mice. The mechanism of this process is that RSV inactivates Stat3 preventing the generation and function of tBregs, including expression of TGFβ. As a result, it frees antitumor effector immune responses by disabling tBreg-induced conversion of FoxP3+ Tregs. We propose that RSV at low doses may also benefit humans to control cancer escape-promoting tBreg/Tregs without non-specific inactivation of effector immune cells.
Keywords: tBregs, Tregs, lung metastasis, resveratrol, Stat3, TGFβ
INTRODUCTION
Successful metastasis is an active process controlled by primary tumors to employ regulatory immune cells to suppress antitumor effector immune responses. For example, murine mammary 4T1 adenocarcinoma (which represents a highly aggressive model of human breast carcinoma (1)) produce GM-CSF, IL-1β, and TGFβ to expand and activate various myeloid-derived suppressor cells (MSC and MDSC) and M2 macrophages (2–4). They impair antitumor immune responses and promote metastases, acting either directly or indirectly via induction/ expansion of Tregs (5–7). As such, the increase in MDSCs and Tregs is often associated with a poor disease outcome in mice and humans with cancer (8–10). However, our recent attempts to link myeloid cells with the induction of immune suppression needed for the successful lung metastasis in mice with 4T1 cancer (4T1 adenocarcinoma, such as 4T1 or 4T1.2 cancer cells, established in mammary gland) failed (11–14). Instead we found that cancer metastasis requires an additional player, a unique subset of TGFβ-producing regulatory B cells designated tumor-evoked Bregs (tBregs) (13, 14). The role of tBregs is to induce TGFβ-dependent conversion of metastasis-promoting FoxP3+Tregs from non-Treg CD4+ T cells (13) to inactivate antitumor NK cells and effector CD8+ T cells and thereby to protect metastasizing cancer cells (11, 14). This process is actively controlled by the primary tumor, in particular, by non-metastatic cancer cell subsets that induce the generation of tBregs from normal B cells utilizing metabolites of 5-lypoxigenase (15). The clinical implication of this is that, as long as cancer persists, tBregs will continue to be induced, initiating the chain of suppressive events. In the absence of B cells, such as mice with B-cell deficiency, 4T1 cancer can only progress at the primary site (mammary gland) but will fail to metastasize into the lungs, unless replenished with WT B cells or tBregs (11, 13, 14). Thus, tBregs need to be inactivated to block cancer metastasis. However, the lack of available tBreg-specific markers makes this task difficult. tBregs substantially differ from the immune tolerance-inducing IL-10-producing B cells and Bregs (16–18) and even from a handful of B cells/Bregs that induce carcinogenesis-supporting inflammation (19, 20) or promote cancer by disabling CD4+ T cell help in mice (21). Although tBregs express CD25 and IL-10 as recently found human granzyme B-expressing human CD19+CD38+CD1d+ IgM+CD147+ Bregs (22), phenotypically they resemble poorly proliferative B2-like cells (IgDHigh) that express constitutively active Stat3 and surface markers CD25High, B7–H1High, CD81High, CD86High, CCR6High, and CD62LLow, IgMInt/Low and CD20Low (13, 14). Although lung metastasis can be abrogated injecting anti-B220 antibody (Ab; targets B cells and pDCs) and PC61 Ab (depletes CD25+ cells such as Tregs and tBregs), but they are not specific and may also deplete lymphocytes required for the elimination of cancer cells. Similarly, Rituximab (anti-CD20 Ab widely used in humans with B-cell malignancies) cannot be used, as it worsens metastasis in mice with 4T1 cancer (14), enhances tumor burden in mice intravenously injected with B16–F10 melanoma (23) and, importantly, failed to benefit patients with renal cell carcinoma and melanoma (24). At least in mice with 4T1 cancer, we linked these effects with the anti-CD20 Ab-induced enrichment of tBregs expressing low levels of CD20 due to the depletion of beneficial B cells (14).
Resveratrol (3,5,4″-trihydroxystilbene, RSV) is a phytoalexin found in grapes, mulberries and peanuts. As a potential anticancer therapeutic drug and inhibitor of cancer angiogenesis, RSV controls mammalian cell apoptosis via multiple molecular pathways, such as by targeting p53, Rb and cell cycle kinases (25). This probably explains why RSV differentially acts on various cells depending on their activation and differentiation state and exhibits both estrogenic and anti-estrogenic properties on mammary cancer cells (26). Although it can inhibit initiation and progression of tumors in mice (26–29), it fails to suppress breast cancer metastasis in mice (30) or instead worsens survival of mice with prostate cancer xenografts (31) and human patients with refractory multiple myeloma (32). RSV can also either inhibit or induce expression of TGFβ and thereby affects downstream functions mediated by TGFβ (33–36). This probably explains why only Foxp3+ Tregs were inhibited in mice treated with RSV, whereas MDSCs were instead expanded (29, 37). RSV is also a potent inhibitor of Stat3 phospshorylation and acetylation (38, 39) and as such it specifically induces apoptosis of malignant cells expressing activated Stat3 (40). Since tBregs also express activated Stat3 and induce conversion of metastasis-promoting Foxp3+ Tregs in a TGFβ-dependent way (13), RSV may also inactivate tBregs and thereby block breast cancer lung metastasis.
Here we show that low and non-cytotoxic doses of RSV can indeed preferentially inhibit tBregs and concurrently block lung metastasis in mice with highly metastatic 4T1.2 cancer. The mechanism of this process is that RSV blocks phosphorylation of Stat3 (i.e. inhibiting its activity), such as production of TGFβ in tBregs. Since TGFβ is a key factor used by tBregs to convert metastasis-promoting FoxP3+ Tregs (13), its RSV-induced inactivation in tBregs is sufficient to almost completely block lung metastasis by releasing the suppressed state of effector cellular immune responses. Hence, this is a first mechanistic proof of principle that underscores the therapeutic relevance of non-cytotoxic low-doses of RSV to combat cancer escape mediated by tBregs.
MATERIALS AND METHODS
Reagents, cells and mice
Resveratrol (ResVida, > 98% pure) was gift from DSM-nutraceuticals (Aurangabad, India). Stat3 inhibitor V (Stattic) and VI (S31–201) were purchased from Calbiochem (EMD Millipore, San Diego, USA). 4T1 adenocarcinoma cells and B16F10 melanoma cells were purchased from American Type Culture Collection. 4T1.2 cells, a subset of 4T1 cells, were a gift from Dr. Robin L. Anderson (Peter McCallum Cancer Center, Australia). Non-metastatic 4T1.2-PE cells were generated from 4T1.2 cells by using TARC-PE38 chemotoxin (11). Female BALB/C and C57Bl/6 mice were from the Jackson Laboratory (Bar Harbor, ME). μMT mice in BALB/c background were a gift from professor Dr. Thomas Blankenstein (MDC, Berlin, Germany). Pmel mice were a gift from Dr. Nicolas P. Restifo (National Cancer Institute, Bethesda, MD) and have been described elsewhere (41).
Separation of RSV and its O-sulfated metabolite by HPLC
The extraction of trans-RSV and its metabolites were carried out as described elsewhere (42). Briefly, 70 μl of methanol and 10 μl of hexestrol (internal standard) were added to 20 μl of serum samples collected at different time points, which were vortex-mixed and centrifuged at 20,800 rcf at 4°C for 10 min. The supernatant was analyzed in Shimadzu Prominence HPLC system (Shimadzu, Columbia, MD) using Shimadzu SIL-20A autosampler at 4°C with 20 ml injections. The separation of RSV and RSV-3-O-sulfate was accomplished using an Eclipse XDB-C18 guard column (4.6 mm ×12.5 mm) and an analytical column Discovery C18 (150x 4.6 mm ID, 5 mm; Supelco). The mobile phase consisted of water containing 0.1% acetic acid and 0.07% triethylamine as component A and acetonitrile as component B. A linear gradient was run as follows: 0–3 min 20% B; 3–25 min 20–60% B; 25–30 min 60–20% B at a flow rate of 1.0 ml/min. The total run time was 30 min per sample.
Mass spectrometry analysis was performed using a triple quadrupole mass spectrometer model API 4000 system from Applied Biosystems/MDS Sciex equipped with Turbo Ion Spray® (TIS) (Applied Biosystems, Foster City, CA, USA). The data was acquired and analyzed using Analyst version 1.4.2 (Applied Biosystems). Negative electrospray ionization data were acquired using multiple reaction monitoring (MRM), the standards were characterized using the following MRM transitions: RSV (227–185); RSV-Sulf (307–227) and Hexestrol (269–134). The TIS instrumental source settings for temperature, curtain gas, ion source gas 1 (nebulizer), ion source gas 2 (turbo ion spray), entrance potential and ion spray voltage were 500 °C, 10 psi, 60 psi, 70 psi, -10V and -4500 V, respectively. The TIS compound parameter settings for declustering potential, collision energy, and collision cell exit potential were -70V, -25V, -7V for RSV; -50V, -28V and -9V for RSV-3-O-sulfate and -82V, -20V and -8V for hexesterol.
In vitro immune cell manipulations
tBreg generation, T cell suppression and Treg conversion assays were performed following our previously described methods (11, 13). In brief, tBregs were generated from murine splenic B cells (>95% purity, isolated by negative selection using the RoboSep system, StemCell Technologies, Vancouver, Canada) by incubating for two days in 50% conditioned medium of 4T1-PE cells (CM-PE) in complete RPMI (cRPMI: RPMI 1640 with 10% heat-inactivated fetal bovine serum, 10 mM HEPES-Sodium Pyruvate, 1 mM sodium pyruvate, 0.01% 2-Mercaptoethanol, 2mM L-glutamine, 100U/ml penicillin and 100 μg/ml streptomycin, all from Invitrogen) at a 37°C in humidified atmosphere with 5% CO2. Control B cells were treated with 100 ng/ml of recombinant mouse BAFF/BlyS (R&D) in cRPMI (B+BAFF). Both tBregs and B+BAFF were generated in the presence or absence of different concentrations of resveratrol (stock solution of 200mM in EtOH). tBregs were characterized as CD81hi CD25+ cells gated within CD19+ cells using respective mAbs (CD19-PerCP/Cy5.5, CD81-PE, CD25-Pacific blue purchased from Biolegend). To test the suppressive activity of B cells, splenic CD3+ T cells were isolated using the mouse T-cell enrichment columns (R&D Systems) and labeled with the cell proliferation dye eFluor670 (eBioscience) before incubating with B cells at 1:1 ratio for 4–5 days in the presence of 1.5μg/ml of anti-mouse CD3 Ab (BD Biosciences). Cells were stained with anti-mouse CD4-PerCP and CD8-FITC (Biolegend). Decrease in eFluor670 expression of CD4+ and CD8+ T cells correlates with the proportion of cells that underwent divisions. To test the ability of tBregs to convert Tregs, non-Treg CD25−CD4+ T cells [isolated with CD3+ T cell enrichment column (R&D Systems) and CD4+ T cell isolation kit II (Miltenyi Biotec) and depleted of CD25+ cells (Dynabeads, Invitrogen)] were mixed with B cells at a 2:1 ratio and cultured for 5 days in the presence of bead-conjugated anti-CD3/CD28 Abs (Invitrogen) and 500 U/ml mouse recombinant IL-2. 4T1.2 cell proliferation was assessed using the cell WST-1 proliferation reagent (Roche) following manufacturers instructions. Briefly, cells were cultured in 96-wells plate with cRPMI for 24h prior addition of RSV. After 72h, supernatants were replaced with fresh cRPMI+10%WST and incubated for 30 minutes at 37°C. The formazan dye conversion/viable cells were quantified using ELISA plate reader (BioRad) at 450 nm.
To generate Tregs, in vitro generated tBregs or B cells isolated from LN of BALB/c mice with 4T1.2 cancer were incubated with non-Treg cells (CD25−CD4+ from BALB/C mice or GFP−CD4+ cells from FoxP3-GFP mice, purity >99.5%) at a 1:1 ratio and cultured for 5 days in the presence of bead-conjugated anti-CD3/CD28 Abs and 500 U/ml IL-2 in the absence or presence of DMSO or SB431542 (Tocris bioscience, Ellisville, MO).
In vivo manipulations
Animal care was provided in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 86–23, 1985). The experiments were performed using 4–8 weeks old female mice in a pathogen-free environment at the National Institute on Aging Animal Facility, Baltimore, MD. Female BALB/c mice or C57BL/6 and Pmel were subcutaneously (s.c.) challenged with respective syngeneic tumor cells, such as 5 ×104 4T1 cells (in the fourth mammary gland) or 1 ×105 B16F10 melanoma cells, and euthanized at 28 and 20 days, respectively, to assess tumor weight. For the euthanized 4T1 cancer-bearing mice, lungs were analyzed for metastasis by ex vivo injecting India ink through the trachea, which was distained in Fekete’s solution to count tumor nodules. For in vivo tBreg and T-cell generation, mice were euthanized at 13 – 20 days post tumor challenge.
To assess the effect of systemic administration of RSV, 4T1-tumor bearing BALB/c mice were intraperitoneally (i.p.) injected with RSV (20 or 50 μg/mouse) or mock (EtOH) every other day from day 3 to 21 post tumor challenge. C57Bl/6 mice with B16F10 melanoma were i.p. injected with 50 or 500 μg/mouse RSV or mock every other day from day 3 to 16 post-tumor challenge. To evaluate in vivo effects of RSV to block the tBreg generation, 4T1 tumor-bearing BALB/C mice were i.p. injected with RSV (50μg/mouse) or mock ever other day from days 3 to 11. Then, at day 14, mice were randomized and i.v. injected with tBregs (5×106) treated with either RSV (12.5μM) or mock. Alternatively, untreated Pmel mice were i.v. injected with tBregs (5×106) treated with either RSV (12.5μM) or mock one day before and 5 days after B16F10 melanoma challenge. To assess effects of RSV on in vivo generation of tBregs, tumor-bearing BALB/c mice were adoptively transferred with B cells from donor tumor-bearing BALB/c mice treated with RSV or mock. Specifically, the donor mice with 106 4T1.2 cells were i.p injected with RSV (50μg/mouse) or mock every other day from day 3 to 11. Then, their B cells (4×106) were isolated and i.v. transferred into host BALB/c mice at 13 and 15 day post challenge with 5×104 4T1.2 cells. The host mice were i.p. pretreated with RSV (50μg/mouse) every other day from day 3 to 11. Donor mouse lymph node B cells were isolated at day 13 and 15 using magnetic separation with anti-mouse CD19-FITC (Biolegend) and anti-FITC MicroBeads (Miltenyi Biotec). All B-cell samples were tested in parallel for the ability to suppress T cell proliferation and to convert naïve T cell into Foxp3+ T cells as described above.
TGFβ production and pStat3 expression
Production of TGFβ (active form) was quantified in supernatants using ELISA Ready-SET Go kit for human/mouse TGFβ1 (eBiosciences) following manufacturers instructions. To evaluate intracellular expression, cells were pretreated with 1/1000 monensin (eBioscience) for two hours before permebilization and staining with Foxp3 and IFNγ (eBioscience) or TGFβ-PE (Biolegend). TGFβ expression from splenocytes, LN and tumor B cells from tumor-bearing mice (± i.p RSV treatment) were analyzed by stimulating cells for 4h with phorbol 12-myristate 13-acetate (PMA, 5ng/ml) and ionomycin (50 ng/ml), both from Tocris, R&D. Stat3 expression was assessed in 10 μg whole cell lysate with western blotting using anti-Stat3 (9132) and anti-phosphorylated Stat3 (Tyr705, 9138) mAbs (Cell Signaling). For intracellular expression, cells were first fixed with 2% paraformaldehyde in PBS for 10 min at 37°C, and chilled on ice for 1 min. Cells were then spined down, resuspended in pre-chilled 90% methanol (in water) and incubated for 30min on ice. The cells were stained with anti-mouse CD19-PerCP/Cy5.5 (Biolegend) and rabbit anti-mouse pStat3-Alexa Fluor 647 (Tyr705, Cell Signaling) at 1/200 dilution.
Statistical Analysis
The results are presented as the mean ± SEM. Differences were tested using Student’s t test and a 2-sided p-value less than 0.05 was considered statistically significant.
RESULTS
RSV blocks cancer progression and metastasis
High doses of RSV (>100 mg/Kg) suppress cancer progression in mice (37, 43) via direct induction of apoptosis of phosphorylated Stat3-expressing malignant cells (40) and, indirectly, blocking the generation of cancer escape-promoting FoxP3+ Tregs (29, 37). Since tBregs also express activated Stat3 and induce conversion of FoxP3+ Tregs (13), we hypothesized that some anticancer benefits of RSV could be through the inactivation of tBregs. To test this idea, we screened for lower doses of RSV to avoid the involvement of its non-specific cytotoxicity. As others reported (30, 40), we also found that RSV at doses above 12 μM was cytotoxic for most cells tested, such as on 4T1.2 cancer cells (Suppl. Fig. 1A), naïve mouse B cells and T cells (B+ BAFF and CD4+CD25−, respectively, Suppl. Fig. 1B,C), although consistently yielding a higher cytotoxicity for Tregs (CD4+CD25+) and tBregs (Suppl. Fig. 1B,C). In vivo, RSV is usually quickly metabolized (44, 45) and its plasma levels were reported to drastically decrease from 95 μM to 1 μM within 480 minutes post intravenous (i.v.) injection of 20 mg/Kg RSV (46). In concordance, we did not detect RSV in plasma of mice (detection threshold 39 ng/ml, about 170 nM) intraperitoneally injected with lower dose RSV (5 mg/Kg, 100 μg/mouse), although its metabolite resveratrol 3-O-sulfate was only transiently present at maximum of 450 nM by 60 minutes after injection (140±48 ng/ml, Table 1). After 60 minutes or in mice injected with 50 μg RSV (Table 1) we failed to detect RSV or its metabolite. Thus, RSV injected at doses below 100 μg probably will not generate systemic levels of RSV in mice sufficient to elicit non-specific cytotoxicity to cancer cells or T cells and B cells in mice. Despite this, progression of subcutaneously challenged B16–F10 melanoma was substantially decreased in C57BL/6 mice treated with 50 μg RSV (Suppl. Fig. 2A). Similarly, it also reduced 4T1.2 breast cancer growth in the mammary gland (Fig. 1A) and its lung metastasis (Fig. 1B) in BALB/c mice. Hence, considering the importance of tBregs inducing metastasis-promoting Tregs in 4T1 cancer lung metastasis (11, 13), the low dose of RSV could probably disable cancer escape by inactivating tBregs/Tregs. In concordance, RSV-treated mice also had significantly reduced tBregs (CD25+CD81High cells within CD19+ B cells, Fig. 1C,D) and FoxP3+ Tregs (Fig. 1E and Suppl. Fig. 2B,C). In contrast and in support with a recent report from others (29), RSV did not affect MDSCs, as they were comparably expanded in both RSV and mock-treated tumor-bearing mice (Suppl. Fig. 2D).
Table 1.
Detection of resveratrol and its metabolites, resveratrol 3-O-Sulfate, in the plasma of mice.
| Time point | Groups | Resveratrol 3-O-Sulfate(Mean ng/ml) | ± SEM |
|---|---|---|---|
| 1h | RSV 50μg | 45.5 | 7.5 |
| RSV 100μg | 141.1 | 48.4 | |
| 3h | RSV 50μg | BQ | - |
| RSV 100μg | BQ | - | |
| 6h | RSV 50μg | BQ | - |
| RSV 100μg | BQ | - |
Mice were i.p. injected with RSV 50 or 100μg in 200μl sterile PBS. Serum samples were collected at different time points (1, 3 and 6h post-RSV injection). Neither RSV nor resveratrol-3-O-glucoronides were detected in samples. The only RSV metabolite that presented measurable levels was the resveratrol 3-O-Sulfate (data are shown as mean ± SEM from two mice per group experiment). BQ, below the detection sensitivity (≤39 ng/ml)
Figure 1. RSV attenuates the development of 4T1 breast cancer and lung metastasis.
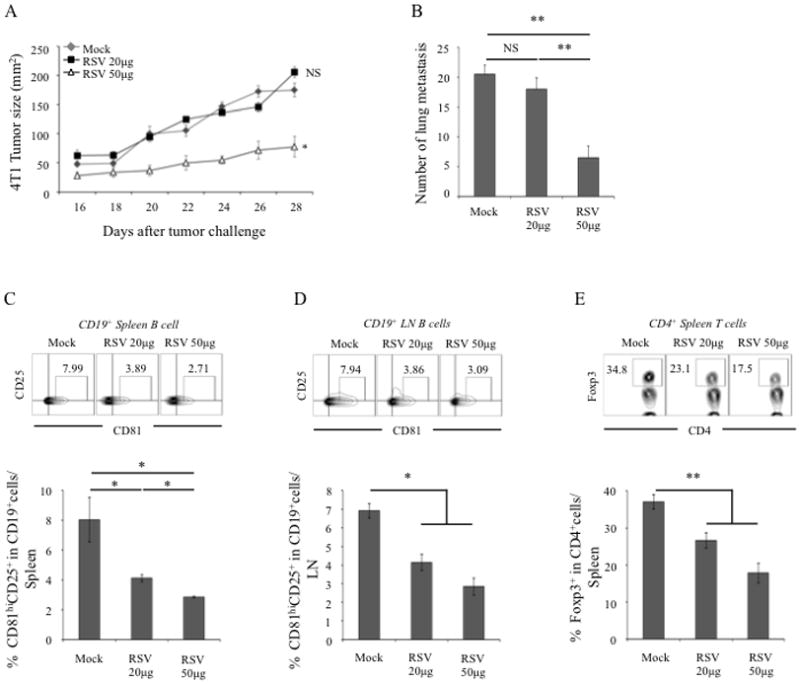
Female BALB/c mice were i.p. injected with either 20 μg RSV or 50 μg RSV every other day starting from day 3 post s.c. challenge with 4T1 cancer cells (5×104). Control tumor-bearing mice (mock) were injected with empty vehicle. Y-axis in A shows tumor size 2 ± SEM measured at indicated times (X-axis) and in B depicts number of lung metastatic foci ± SEM at day 28 post tumor challenge in 4–5 mice per group experiment reproduced three times. The proportion (%) of tBregs (CD81HiCD25+ gated on CD19+ cells in spleen (C) and lymph node (D), respectively) and splenic Tregs (Foxp3+ gated on CD4+ cells, E) was measured in mice shown in A and B. Dot blots in C–E are representative data of the experiments shown in lower panels (C–E). From hereon, NS: statistically no significant; *p<0.05; ** p<0.01; ***p<0.001.
RSV blocks metastasis by inactivating tBregs
To confirm the role of RSV on tBregs, we tested whether it affects in vitro generation of tBregs from naïve B cells treated with conditioned media (CM) from breast cancer cells (13). RSV significantly blocked the generation of tBregs (Fig. 2) even at non-cytotoxic doses (Suppl. Fig. 1B). For example, it reduced expression of tBreg-associated surface markers (Fig. 2A) and disabled tBregs to suppress T-cell proliferation stimulated with anti-CD3 Ab (both CD4+ and CD8+ T cells, respectively, Fig. 2B,C). Importantly, 1–3 μM RSV (non-cytotoxic dose, Suppl. Fig. 1B) was sufficient to almost completely block a second feature of tBregs, the induction of FoxP3+ Tregs from naïve CD4+CD25− T cells (p<0.05, Fig. 2D). In contrast, RSV did not affect control B cells (incubated with BAFF, B+BAFF), which neither inhibited T cell proliferation (Fig. 2A–D) nor converted FoxP3+ Tregs (Fig. 2D). Hence, low doses of RSV can indeed block in vitro generation of tBregs, suggesting that it could also do so in vivo to abrogate metastasis.
Figure 2. RSV abrogates tBreg generation.
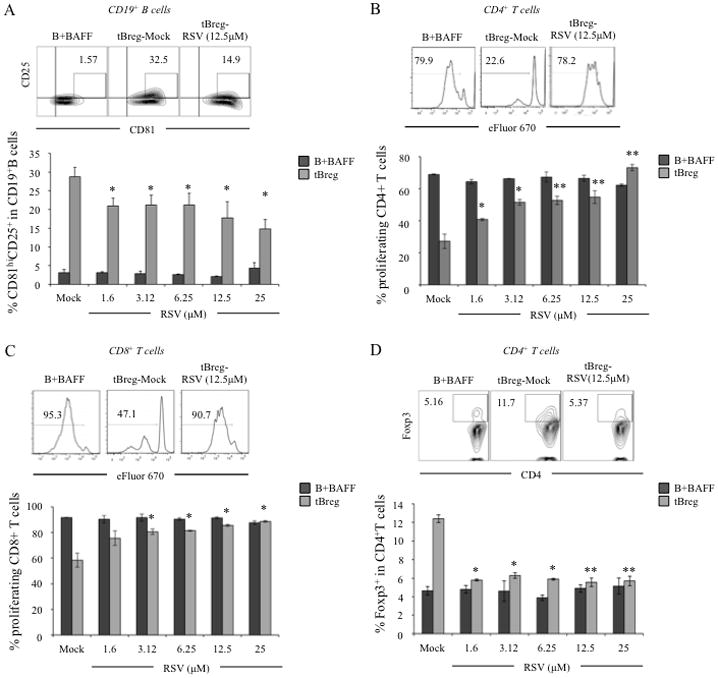
To test effects of RSV on in vitro generation of tBregs, titrated doses of RSV (X-axis) were incubated from the beginning of tBreg generation. Controls were BAFF-treated B cells (B+BAFF) cultured with indicated doses of RSV. Y-axis shows expression of tBreg-like surface markers on B cells (%± SEM of CD81HiCD25+ gated on CD19+ cells, A), or % ± SEM of proliferated (eFlour 670 diluted) CD4+ cells (B) and CD8+ (C) T cells, or % ± SEM of Treg conversion from non-Treg CD4+ T cells (FoxP3+ gated on CD4+ cells, D). In B–D, T cells were stimulated with 3 μg/ml anti-CD3 mAb for 5 days in the presence of tBregs and B cells from A. Dot plot in A and histograms (B–D) show representative flow cytometry data of the experiments shown in A–D. All data were reproduced 4 times in triplicate experiments. P-values are for comparisons of Mock vs RSV of the same type of cells.
To test this idea, we performed adoptive transfer experiments in 4T1.2 cancer-bearing mice pretreated with 50 μg RSV every other day from day 3 through 11 post tumor challenge to eliminate endogenous tBregs and Tregs (Fig. 3A, see also Fig. 1C,D). At day 13 when circulating RSV presumably fully disappeared, mice were randomized and adoptively transferred with in vitro-generated tBregs either treated with RSV (tBreg-RSV) or ethanol (tBreg-mock, Fig. 3A). As expected for tBregs (13), mice replenished with tBreg-mock cells significantly increased lung metastasis as compared with PBS injected RSV-pretreated mice (p<0.05, PBS vs. tBregs-Mock, Fig. 3A). In contrast, transfer of tBregs-RSV completely failed to augment metastasis (p<0.05, tBregs-Mock vs. tBreg-RSV, Fig. 3A), confirming our in vitro conclusion that RSV inactivates tBregs. To further confirm these results and to rule out artifacts of in vitro manipulations, we performed similar experiments using B cells isolated from 4T1 cancer-bearing mice treated with RSV (Fig. 3B). As we previously reported (13, 14), purified B cells from mice with 4T1.2 cancer (Mock B) contained cancer-induced tBregs, as they readily suppressed proliferation of T cells (Fig. 3C) and converted FoxP3+ Tregs from non-Treg CD4+ T cells (Fig. 3D) ex vivo. Importantly, adoptive transfer of these Mock B cells (isolated from untreated cancer-bearing mice) significantly increased cancer progression (Fig. 3E) and lung metastasis (Fig. 3F) in RSV-pretreated mice with 4T1.2 cancer. In contrast, B cells from RSV-treated cancer-bearing donor mice did not contain tBregs, as they failed to suppress T cells and induce Treg conversion (Fig. 3C,D) nor enhance cancer progression and metastasis (RSV B, Fig. 3E,F) when transferred into RSV-pretreated mice. Thus, low doses of RSV blocks tBregs and thereby inhibits 4T1 breast cancer escape and metastasis. The mechanism of this process could be the inhibition of metastasis-promoting FoxP3+Tregs induced by tBregs (11, 13). Although high doses of RSV are cytotoxic for Tregs (both in vitro, suppl. Fig. 1C, and in vivo (29, 37)), Tregs were also significantly reduced in tumor-bearing mice treated with low dose of RSV (Fig. 1E). Of note: RSV appears to only affect its generation but not function, as Tregs surviving RSV treatment readily suppressed target T cells (Suppl. Fig. 1D).
Figure 3. RSV inhibits tBreg function in vivo.
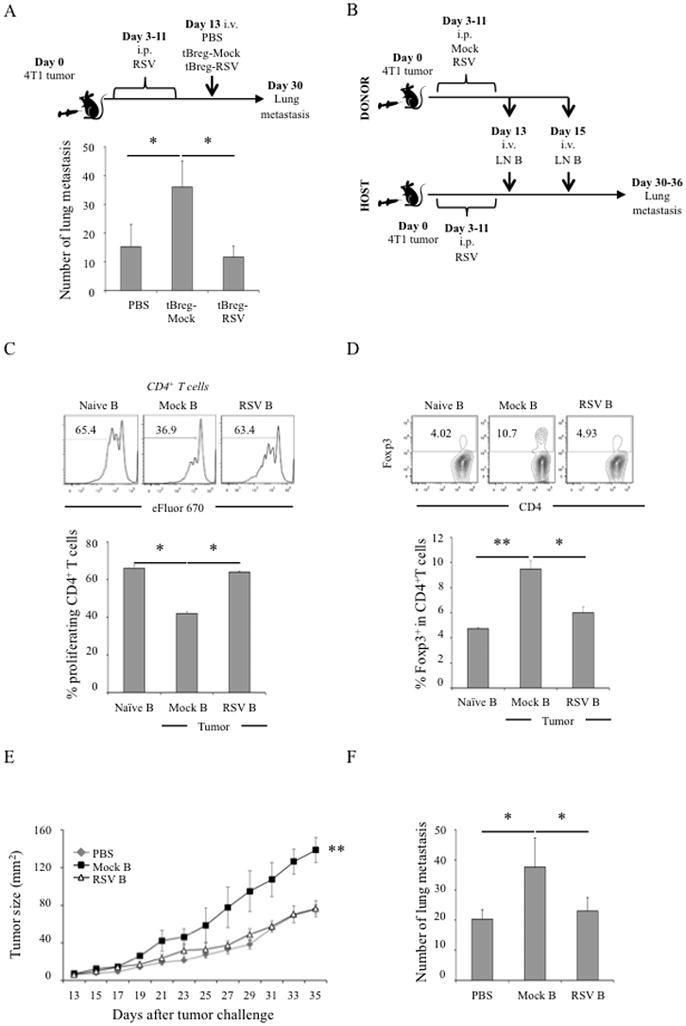
To confirm tBreg inactivation by RSV, we performed series of adoptive transfer experiments with either in vitro (A) or in vivo (B–F) inactivated donor tBregs/B cells (see schemas in A and B). Host female BALB/c mice with 4T1 cancer were “depleted” of tBregs by treating every other day with RSV (50 μg) from day 3 through 11 after tumor challenge. At D13, mice were randomized and were adoptively transferred with ex vivo-generated tBregs in the presence of 12.5 μM RSV or mock-treated tBregs (A). In B–F, B cells were isolated from LN of donor tumor-bearing mice treated with RSV (50 μg) or mock (see schema in B) and used for adoptive transfer. These B cells were also tested for the ability to suppress CD4+ T cell proliferation (C) and convert FoxP3+Tregs (D) as in Fig.2B,D. The results were also compared with activity of naïve mouse B cells (naïve, C and D). B cells/tBregs from mock, but not RSV, treated mice augment 4T1.2 cancer growth (E) and lung metastasis (F) when adoptively transferred into host RSV-pretreated mice, as compared with control mice injected with PBS (PBS, E and F). Histograms in C and D are representative flow cytometry data of the graphs (mean ± SEM) shown in lower panels. Every result was reproduced at least twice in 4–5 mice per group experiments.
RSV inhibits tBreg-Treg axis
To test the indirect effect of low dose of RSV on Tregs, we quantified Treg numbers in mice adoptively transferred with tBregs (Fig. 3B). Whereas FoxP3+ Treg numbers infiltrating in the secondary lymphoid organs and tumor (Fig. 4A–D) were significantly increased in tumor-bearing mice adoptively transferred with Mock B cells (isolated from untreated tumor bearing mice), we failed to detect any enhancement of Tregs when mice replenished with B cells from RSV-treated donor tumor-bearing mice (RSV B, Fig. 4A–D). In fact, the numbers of Tregs in mice transferred with RSV-B cells and RSV-pretreated control (PBS) mice were almost indistinguishable (Fig. 4A–D). Note: compared with naïve mice, Tregs were still increased in these two groups of mice (p<0.05, naïve vs PBS, Fig. 4A–D), suggesting the tumor-induced restoration of Tregs and tBregs after two weeks of cessation of RSV treatment. Thus, the low dose of RSV reduces FoxP3+Tregs by inhibiting tBregs and thereby may release the immunosuppressed state of antitumor effector immune cells.
Figure 4. Modulation of Foxp3+ Tregs and effector CD8+ T cells in tumor-bearing mice adoptively transferred with tBregs.
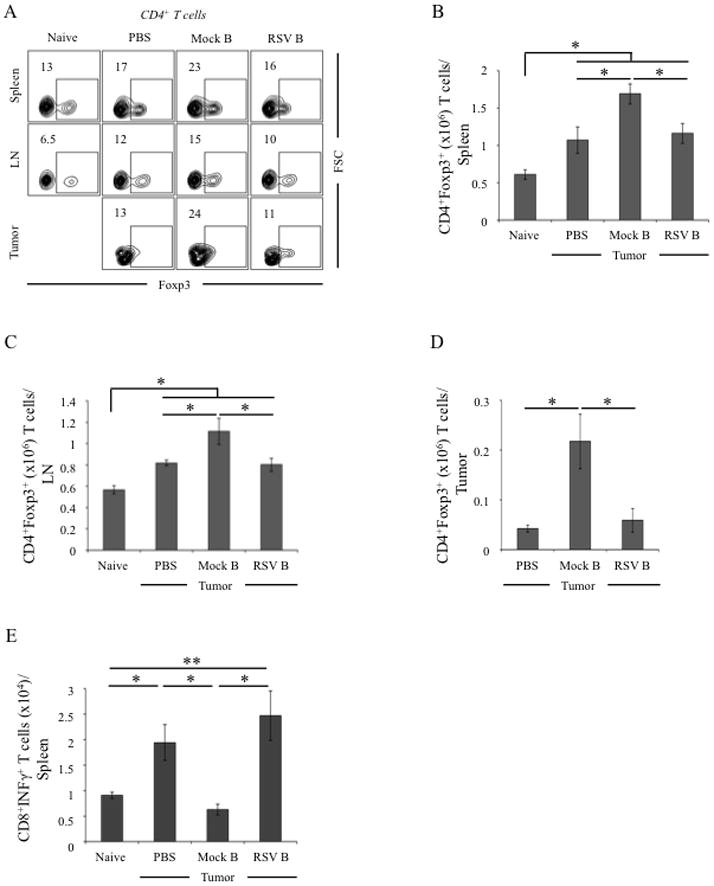
Mice were treated and adoptively transferred with B cells as in Fig.3B and the presence of FoxP3+ Tregs and IFNγ+ CD8+ T cells were evaluated in the host mice. As expected, B cells from mock-treated tumor-bearing mice (mock B) increased proportion (%, A) and numbers of FoxP3+ Tregs in spleen (B), draining LN (C) and tumor (D), as compared with naïve and tumor-bearing PBS-treated host mice. The tBreg transfer also reduced numbers of splenic IFNγ+ CD8+ T cells (E). Conversely, B cells/tBregs from RSV-treated tumor-bearing mice (RSV B) did not increase Tregs (A–D) and failed to reduce IFNγ+ CD8+ T cells (E). Shown, representative data of 4 mice per group experiments repeated twice.
To test this possibility, we evaluated CD8+ T cells in RSV pretreated tumor-bearing mice adoptively transferred with tBregs (see Fig. 3B). Although the role of CD8+ T cells in 4T1.2 cancer-bearing mice is not known, RSV significantly increased numbers of IFNγ-producing CD8+ T cells in mice with 4T1.2 cancer (p<0.05, Naïve vs. PBS, Fig. 4E). This increase was almost completely reduced to the levels of naïve mice, if RSV-treated mice were adoptively transferred with B cells/tBregs from 4T1.2 cancer-bearing mice (Fig. 4E). In contrast, transfer of B cells/tBregs from RSV treated cancer-bearing donor mice failed to affect or inhibit CD8+ T cells (p<0.05, Mock B vs. RSV B, Fig. 4E). Thus, RSV releases suppression of antitumor effector immune responses by inhibiting tBregs. To further support this conclusion, we used a different tumor model with defined CD8+ T cell responses, pmel mice transgenic with CD8+ T cells specific for B16 melanoma-expressed gp10025–32 epitope (41). In these mice adoptive transfer of tBregs (generated from congenic WT C57BL/6 mice) also significantly enhanced progression of B16–F10 melanoma (Fig. 5A). Importantly, the enhanced tumor growth was also accompanied with a significant increase in FoxP3+Tregs (Fig. 5B–D) and substantial reduction of IFNγ-expressing CD8+ T cells specific to gp10025–32 peptide (Fig. 5E,F). Conversely, these effects (the increase in tumor growth and Tregs and the decrease in CD8+ T cells) were completely reversed, if mice were transferred with tBregs pretreated with RSV (Fig. 5A–F). Taken together, the low dose of RSV inactivates tBregs thereby abrogating the generation of cancer escape-promoting Tregs and concurrently activating antitumor effector CD8+ T cells.
Figure 5. RSV reverses tBreg-induced suppression of tumor-specific CD8+ T cells.
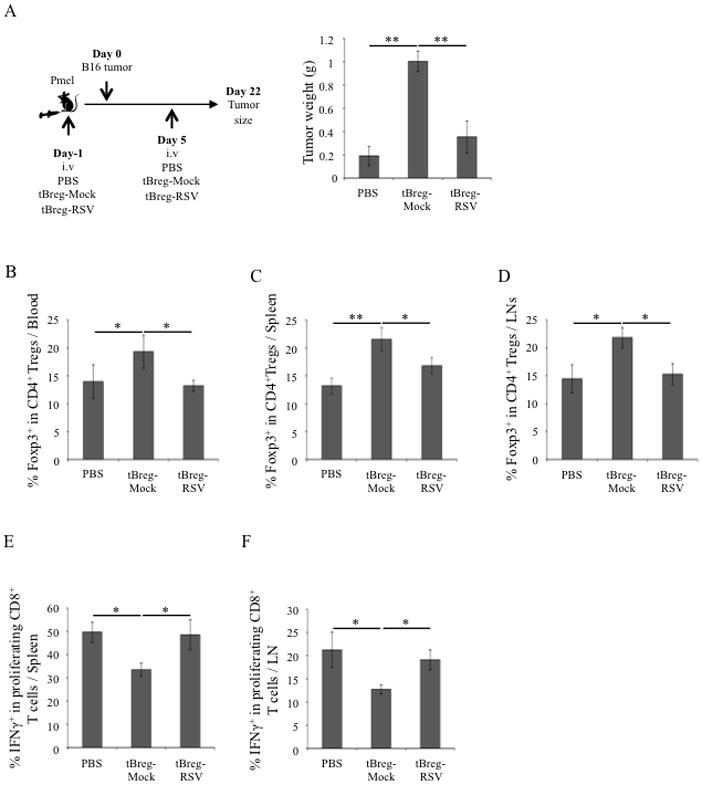
(A) TCR-transgenic Pmel mice were adoptively transferred with ex vivo-generated tBregs (5×106) either mock (tBreg-mock) or RSV treated (12.5 μM, tBreg-RSV) 1 day prior and 5 days after challenge with B16F10 melanoma. Control tumor-bearing mice were injected with PBS. Y-axis shows mean ± SEM of tumor weight (B), proportion of FoxP3-expressing CD4+ T cells (B–D) and proliferating IFNγ+ CD8+ T cells (E–F) in the blood (B), spleen (C, E) and draining LN (D, F). In E and F, T cell proliferation was assessed using eFluor 670-labeled splenocytes and LN cells from tumor-bearing mice after incubation with 5 μg/ml hgp10025–32 peptide in the presence of mouse IL2 (20 U) for 5 days. Shown, representative data of 4–5 mice per group experiments repeated three times.
RSV inhibits tBreg-mediated Treg conversion by blocking TGFβ1
A unique feature of tBregs is that they constitutively express activated (phosphorylated) Stat3 (13). Since RSV is a potent inhibitor of phosphorylation and acetylation of Stat3 (38, 39), it may affect function of tBregs by blocking Stat3 and thereby disabling TGFβ-induced FoxP3+ Treg conversion (13). To test this possibility, first we confirmed the importance of TGFβ in tBreg-mediated conversion of FoxP3+ Tregs (13, 14) by incubating naïve mouse CD25−CD4+ T cells (non-Tregs) with in vitro-generated syngeneic tBregs or B cells (tumor B) isolated from BALB/c mice with 4T1.2 cancer for 5 days in the presence or absence of ALK5 inhibitor SB431542, a selective inhibitor of the TGFβ type I receptor activity. Unlike control B cells from naïve BALB/c mice, which did not induce FoxP3 in non-Tregs regardless of the presence or absence of the inhibitor (Fig. 6A), tBregs or tumor B cells only converted FoxP3+ Tregs in the absence of SB431542 (Fig. 6A). Next, we tested whether RSV affects TGFβ production from already established (ex vivo generated) tBregs (Fig. 6B). As shown by ELISA (Fig. 6B) and intracellular staining (Fig. 6C), TGFβ1 expression in tBregs was significantly and in dose-dependent manner reduced by treatment with low non-cytotoxic dose of RSV (<12 μM). Similarly, in vivo, B cells from tumor-bearing mice treated with 50 μg RSV also failed to secrete significant amounts of TGFβ1 (RSV, Fig. 6D), whereas B cells from mock-treated mice with 4T1.2 cancer abundantly produced TGFβ (p<0.01, Mock vs RSV, Fig. 6D). Confirming this, we found significant loss of TGFβ expression in tBregs (Fig. 6E–G) and B cells (Suppl. Fig. 3) infiltrating the secondary lymphoid organs (Fig. 6E,F and suppl. Fig. 3A–C) and in the tumor (Fig. 6G and suppl. Fig. 3D).
Figure 6. RSV inhibits TGFβ production from tBregs.
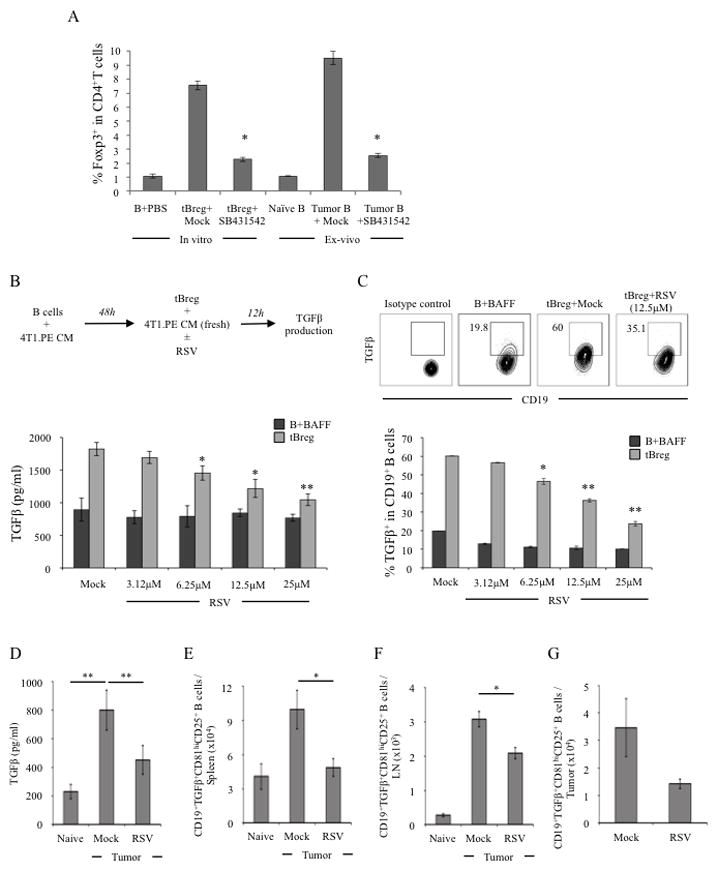
For in vitro conversion assay of Tregs (A), CD25−CD4+ T cells from naïve BALB/c mice were incubated with B cells treated with PBS, or in vitro generated tBregs, or B cells isolated from naïve or tumor (4T1.2)-bearing BALB/c mice in the presence or absence of 10 μM SB431542. T cells were stimulated with anti-CD3 Ab and 500 U/ml IL-2 for 5 days and FoxP3 expression was detected by intracellular stating (Y-axis, % of FoxP3 in CD4+ T cells ± SEM of a triplicate experiment). As shown in schema (upper panel, B), RSV was added after the generation of tBregs. Control B cells were BAFF-treated B cells (B+BAFF) incubated with RSV (lower panel, B, C). X-axis shows concentration of RSV used, and Y-axis is for levels of secreted TGFβ1 ± SEM (pg/ml, lower panel, B) and % ± SEM of intracellular TGFβ1 expression in B cells (lower panel, C) after 12 h incubation. Dot plot in C (upper panel) is representative data of triplicate experiments shown in lower panel (C) repeated three times. To assess in vivo effects of RSV, BALB/c mice were treated with RSV (50 μg) or mock every other day staring 3 days after challenge with 4T1.2 cancer cells (D). At day 10, B cells were isolated from LN tested for ability to secrete TGFβ by ELISA after 48 h stimulation with PMA/ionomycin (D). Similarly, numbers of TGFβ-expressing tBregs (CD81High CD25+ CD19+ B cells) were assessed via intracellular staining in freshly isolated B cells from spleen (E), draining LN (F) and tumor (G). Shown, representative data of 4 mice per group experiments repeated three times.
Next, to prove this reduction is a result of Stat3 inhibition, we compared phosphorylated state of Stat3 (pStat3) in tBregs after treatment with RSV and specific Stat3 inhibitors, S31–201 and stattic. As shown by western blotting of total cell lysates (Fig. 7A) and confirmed by intracellular staining (Fig. 7B), pStat3 was significantly reduced in tBregs after treatment with RSV and Stat3 inhibitors, respectively. Importantly, like RSV, Stat3 inhibitors also inhibited TGFβ1 expression in tBregs (Fig. 7C), a key factor required for tBreg-mediated conversion of FoxP3+ Tregs from non-Treg cells (13). In support and similarly with RSV, Stat3 inhibitor-treated tBregs also failed to in vitro convert (Fig. 7D) and in vivo expand FoxP3+ Tregs (Suppl. Fig. 4) after adoptive transfer into B-cell deficient mice with 4T1 cancer. As a result, unlike adoptive transfer of mock-treated tBregs, which significantly increased lung metastasis in these mice (tBreg-Mock, Fig. 7E), we failed to increase metastasis if mice were replenished with tBregs treated with Stattic or RSV (Fig. 7E). Taken together, the mechanism of anti-metastatic activity of a non-cytotoxic low dose of RSV is in the inactivation of Stat3 in tBregs, which blocks TGFβ production thereby disabling conversion of metastasis-promoting FoxP3+ Tregs.
Figure 7. RSV inactivates Stat3 in tBregs and disables conversion of Tregs via down regulation of TGFβ.
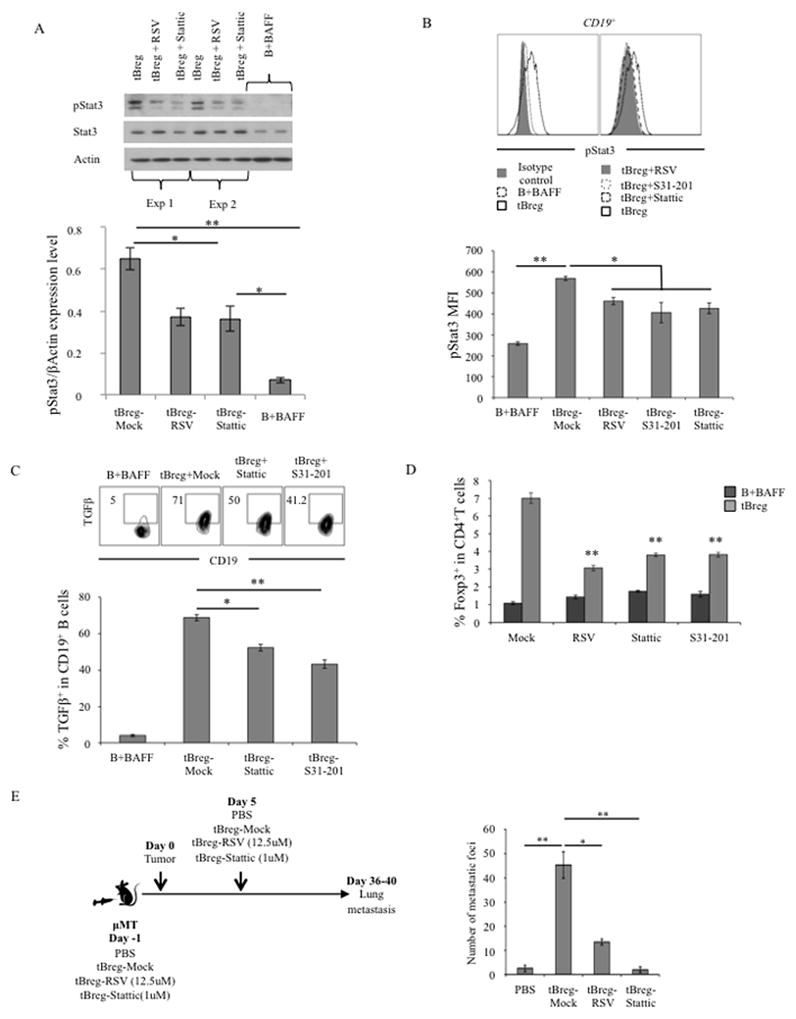
tBregs were overnight treated with RSV (12.5 μM, A–D), or specific Stat3 inhibitor Stattic (1 μM, A–D) and S31–201 (5 μM, B–D) to test phosphorylation of Stat3 (A, B) and TGFβ expression (C) in tBregs using western blotting (A) and intracellular staining (B, C). Upper panels in A–C are representative data of mean ± SEM of triplicate experiments shown in lower panels (A – C) independently reproduced at least 5 times. In A, upper panel, shown data of two independent tBreg lysates (Exp 1 and Exp 2, respectively). The loss of Stat3 disables tBregs to convert FoxP3+ Tregs from non-Treg CD4+ T cells in vitro (D). Shown are representative data of triplicate experiment repeated four times. P-values are for comparisons of Mock vs treated tBregs (D). To confirm the inability of Stat3-inactivated tBregs to promote lung metastasis, 4T1.2 cancer-bearing μMT mice genetically deficient in B cells (5 per group) were adoptively transferred with congenic BALB/c tBregs pretreated with RSV, static or mock (E) and compared with untreated tumor-bearing mice (PBS). Lung metastasis (Y-axis, number of metastatic foci) was assessed at day 30 post tumor challenge.
DISCUSSION
We previously reported that cancer metastasis requires active involvement of regulatory immune cells, such as FoxP3+CD4+ Tregs and TGFβ-expressing tBregs (11, 13, 14). Breast cancer, by producing metabolites of 5-lipoxygenase, induces the generation of tBregs from normal B cells (13, 15) to convert FoxP3+ CD4+ Tregs from non-Tregs CD25− CD4+ T cells utilizing TGFβ (13). The Tregs in turn protect metastasizing cancer cells by inactivating antitumor NK cells using galectin-1/β-galactoside-binding protein (11) and down regulating effector CD8+ T cells (14). Thus, Tregs and tBregs need to be controlled to successfully combat lung metastasis. Although the inactivation of tBregs alone appears to be sufficient (13–15), there are no simple and specific strategies that inactivate tBregs. They cannot be depleted with anti-CD20 antibody/rituximab, a current clinical strategy to combat B cell malignancies, as it instead worsen cancer escape by enriching tBregs expressing low levels of CD20 (14). Here we demonstrate that, unlike the majority of reports which used relatively high doses of RSV (up to 100 mg/Kg) that are cytotoxic both for cancer cells and potentially beneficial effector immune cells in vitro and in vivo (26–29, 37, 39, 43), low doses and non-cytotoxic doses of RSV (1–5 mg/Kg) can inhibit progression of B16 melanoma and 4T1.2 breast cancer cells and abrogates lung metastasis by inactivating tBregs. Depending on a high and low dose RSV use, it differentially acts on target cells. High amounts of RSV (300 μM) reduce cellular energy and activate AMPK (47), while at less than 10 μM concentrations it activates AMPK without decreasing energy (48). In rats and humans, a high-dose of RSV (>3000 mg/Kg), also induces renal toxicity (32, 49), limiting its potential clinical use.
Our data shown here indicate that the immunological mechanism of a low-dose RSV use is that it inactivates tBregs thereby disables their ability to convert non-Treg cells into FoxP3+Tregs. For example, in vitro a low dose RSV-induced inactivation of tBregs almost completely disabled Treg conversion, and in vivo inactivation tBregs reduced endogenous pool of FoxP3+ Tregs in tumor-bearing mice. As a result, while control tBregs (either ex vivo generated or isolated from donor tumor-bearing mice) increased Tregs and concurrently lung metastasis upon adoptive transfer into tumor-bearing mice, RSV-treated Bregs failed to affect this process. In concordance with our recent finding that the loss of tBregs (due to the inhibition of cancer-produced metabolites of 5-lypoxigenase) also leads to disappearance of cancer-associated Foxp3+ Tregs (15), these data further underscore the sufficiency of the inactivation of tBregs alone to abrogate breast cancer lung metastasis. Thus, although high doses of RSV are cytotoxic for normal immune cells and other regulatory immune cells, such as Foxp3+ Tregs, as reported by others (29, 37), we show that cancer escape-supporting Tregs can also be reduced by inactivating their key inducers, tBregs. In contrast, RSV appears does not affect MDSCs, although they are reported to also promote 4T1 cancer escape (2–4) and expand Tregs (5–7). In support and confirming the ability of RSV to expand MDSCs to protect endothelial cells from a high-dose IL2-induced injury (29), we did not detect any change in MDSCs in tumor bearing mice after treatment with a low dose RSV. Instead, to underscore the primary role of tBregs in 4T1 cancer escape, we consistently observed that proportion of MDSCs was increased by adoptive transfer of mock, but not RSV-treated, tBregs in tumor-bearing B cell deficient mice (A.B., personal communication, and see ref. (13–15)). Moreover, despite the presence and expansion of MDSCs, only the loss or conversely the restoration of tBregs abrogated or augmented metastasis in mice with 4T1 cancers, respectively (11, 13–15). Thus, it is tempting to speculate that, although MDCSs play important regulatory and tumorigenic role, they may be required for other steps in cancer progression, such as induction of Tregs via arginase-dependent but TGFβ-independent way, promoting Th2-skewed responses, survival and cancer angiogenesis (50–52).
Here we also show that the mechanism of this process is that, as a potent inhibitor of Stat3 phosphorylation and acetylation (38, 39), RSV at a low and non-cytotoxic dose (3–10 μM) inhibits the generation and function of tBregs by inactivating Stat3. Although the molecular mechanism of this process is not fully understood and is a focus of a different study, this inactivation of Stat3 in tBregs presumably leads to the inhibition of TGFβ expression, a down-stream target of Stat3 (53). Confirming this, the specific inhibitors of Stat3 also blocked tBregs and inhibited production of TGFβ, thereby disabling tBregs ability to convert FoxP3+ Tregs, a process that we reported to require TGFβ (13). As a result and as we reported (11, 13, 14), the loss of tBregs/Tregs in turn “releases” the suppressed state of effector immune cells, such as anti-tumor IFNγ-producing CD8+ T cells and NK cells (11), thus protecting the lungs from metastasizing cancer cells. Using two different tumor models we show that, unlike its high and cytotoxic dose (>10 μM) for cancer (39, 40) and immune cells, RSV at low and non-cytotoxic concentrations does not prevent activation of effector IFNγ-expressing CD8+ T cells. Importantly, the low dose RSV-induced inactivation of tBregs was sufficient to reduce Tregs and abrogate lung metastasis in mice with highly aggressive breast cancer. Taken together, we propose that low doses of RSV can be safely used to provide therapeutic benefit in cancer patients via blockage of tBreg/Treg axis and concurrent induction of antitumor effector responses.
Supplementary Material
Acknowledgments
We are grateful to Cheryl Sherman-Baust (NIA/NIH) for helpful comments, Drs. Thomas Blankenstein and Thomas Kammertons (MDC, Berlin, Germany) for a gift of μMT mice in BALB/c background; and Drs. Jorge M. Sztein (NIAID/NIH, Rockville, MD) and Dr. Kathy Perdue (NIA/NIH) for the help with re-derivation of μMT mice at NIA.
Footnotes
This research was supported by the Intramural Research Program of the National Institute on Aging, NIH.
Conflict of Interest: The authors do not have any conflict of interest. The entire work was funded and performed at the National Institute on Aging
References
- 1.Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, Lowen D, Javni J, Miller FR, Slavin J, Anderson RL. A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis. 1999;17:163–170. doi: 10.1023/a:1006689719505. [DOI] [PubMed] [Google Scholar]
- 2.Sinha P, V, Clements K, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 3.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 5.Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer research. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH. De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+CD25- cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4103–4108. doi: 10.1073/pnas.0407691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos RN, Chin LS, Dos Santos AP, Bergami-Santos PC, Laginha F, Barbuto JA. Monocyte-derived dendritic cells from breast cancer patients are biased to induce CD4+CD25+Foxp3+ regulatory T cells. J Leukoc Biol. 2012;92:673–682. doi: 10.1189/jlb.0112048. [DOI] [PubMed] [Google Scholar]
- 8.Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, Kaiser LR, June CH. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 9.Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, Knolle PA, Thomas RK, Bergwelt-Baildon M, Debey S, Hallek M, Schultze JL. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–2025. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 10.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 11.Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, Deng J, Xu M, Briest S, Biragyn A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olkhanud PB, Rochman Y, Bodogai M, Malchinkhuu E, Wejksza K, Xu M, Gress RE, Hesdorffer C, Leonard WJ, Biragyn A. Thymic stromal lymphopoietin is a key mediator of breast cancer progression. Journal of immunology. 2011;186:5656–5662. doi: 10.4049/jimmunol.1100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4 T cells to T-regulatory cells. Cancer research. 2011;71:3505–3515. doi: 10.1158/0008-5472.CAN-10-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bodogai M, Lee-Chang C, Wejksza K, Lai JP, Merino M, Wersto RP, Gress RE, Chan AC, Hesdorffer C, Biragyn A. Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer research. 2013 doi: 10.1158/0008-5472.CAN-12-4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wejksza K, Lee-Chang C, Bodogai B, Bonzo B, Gonzalez FJ, Lehrmann E, Becker K, Biragyn A. Cancer-produced metabolites of 5-lipoxygenase induce tumor-evoked Bregs via peroxisome proliferator-activated receptor alpha. J Immunol. 2013 doi: 10.4049/jimmunol.1201920. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, Chalasani G, Sayegh MH, Najafian N, Rothstein DM. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. The Journal of clinical investigation. 2011;121:3645–3656. doi: 10.1172/JCI46274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne SN, Halliday GM. B cells activated in lymph nodes in response to ultraviolet irradiation or by interleukin-10 inhibit dendritic cell induction of immunity. J Invest Dermatol. 2005;124:570–578. doi: 10.1111/j.0022-202X.2005.23615.x. [DOI] [PubMed] [Google Scholar]
- 18.Sun JB, Flach CF, Czerkinsky C, Holmgren J. B lymphocytes promote expansion of regulatory T cells in oral tolerance: powerful induction by antigen coupled to cholera toxin B subunit. Journal of immunology. 2008;181:8278–8287. doi: 10.4049/jimmunol.181.12.8278. [DOI] [PubMed] [Google Scholar]
- 19.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 20.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat Med. 1998;4:627–630. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- 22.Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, Beyer T, Reister F, Fabricius D, Lotfi R, Lunov O, Nienhaus GU, Simmet T, Kreienberg R, Moller P, Schrezenmeier H, Jahrsdorfer B. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer research. 2013;73:2468–2479. doi: 10.1158/0008-5472.CAN-12-3450. [DOI] [PubMed] [Google Scholar]
- 23.Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano A, Palma G, Arra C, Maiolino P, Adcock IM, Pinto A. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am J Respir Crit Care Med. 2011;183:1369–1379. doi: 10.1164/rccm.201010-1738OC. [DOI] [PubMed] [Google Scholar]
- 24.Aklilu M, Stadler WM, Markiewicz M, Vogelzang NJ, Mahowald M, Johnson M, Gajewski TF. Depletion of normal B cells with rituximab as an adjunct to IL-2 therapy for renal cell carcinoma and melanoma. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2004;15:1109–1114. doi: 10.1093/annonc/mdh280. [DOI] [PubMed] [Google Scholar]
- 25.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhat KP, Lantvit D, Christov K, Mehta RG, Moon RC, Pezzuto JM. Estrogenic and antiestrogenic properties of resveratrol in mammary tumor models. Cancer research. 2001;61:7456–7463. [PubMed] [Google Scholar]
- 27.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 28.Jeong MH, Yang KM, Choi YJ, Kim SD, Yoo YH, Seo SY, Lee SH, Ryu SR, Lee CM, Suh H, Jo WS. Resveratrol analog, HS-1793 enhance anti-tumor immunity by reducing the CD4+CD25+ regulatory T cells in FM3A tumor bearing mice. Int Immunopharmacol. 2012;14:328–333. doi: 10.1016/j.intimp.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Guan H, Singh NP, Singh UP, Nagarkatti PS, Nagarkatti M. Resveratrol prevents endothelial cells injury in high-dose interleukin-2 therapy against melanoma. PLoS One. 2012;7:e35650. doi: 10.1371/journal.pone.0035650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bove K, Lincoln DW, Tsan MF. Effect of resveratrol on growth of 4T1 breast cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2002;291:1001–1005. doi: 10.1006/bbrc.2002.6554. [DOI] [PubMed] [Google Scholar]
- 31.Klink JC, Tewari AK, Masko EM, Antonelli J, Febbo PG, Cohen P, Dewhirst MW, Pizzo SV, Freedland SJ. Resveratrol worsens survival in SCID mice with prostate cancer xenografts in a cell-line specific manner, through paradoxical effects on oncogenic pathways. Prostate. 2012 doi: 10.1002/pros.22619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popat R, Plesner T, Davies F, Cook G, Cook M, Elliott P, Jacobson E, Gumbleton T, Oakervee H, Cavenagh J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br J Haematol. 2012 doi: 10.1111/bjh.12154. [DOI] [PubMed] [Google Scholar]
- 33.Tiwari V, Chopra K. Resveratrol prevents alcohol-induced cognitive deficits and brain damage by blocking inflammatory signaling and cell death cascade in neonatal rat brain. J Neurochem. 2011;117:678–690. doi: 10.1111/j.1471-4159.2011.07236.x. [DOI] [PubMed] [Google Scholar]
- 34.Rahal K, Schmiedlin-Ren P, Adler J, Dhanani M, Sultani V, Rittershaus AC, Reingold L, Zhu J, McKenna BJ, Christman GM, Zimmermann EM. Resveratrol has antiinflammatory and antifibrotic effects in the peptidoglycan-polysaccharide rat model of Crohn’s disease. Inflamm Bowel Dis. 2012;18:613–623. doi: 10.1002/ibd.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suenaga F, Hatsushika K, Takano S, Ando T, Ohnuma Y, Ogawa H, Nakao A. A possible link between resveratrol and TGF-beta: resveratrol induction of TGF-beta expression and signaling. FEBS Lett. 2008;582:586–590. doi: 10.1016/j.febslet.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 36.Serrero G, Lu R. Effect of resveratrol on the expression of autocrine growth modulators in human breast cancer cells. Antioxid Redox Signal. 2001;3:969–979. doi: 10.1089/152308601317203512. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y, Paik JH, Cho D, Cho JA, Kim CW. Resveratrol induces the suppression of tumor-derived CD4+CD25+ regulatory T cells. Int Immunopharmacol. 2008;8:542–547. doi: 10.1016/j.intimp.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S, Aggarwal BB. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007;109:2293–2302. doi: 10.1182/blood-2006-02-003988. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, Yu H. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7765–7769. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5:621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 41.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boocock DJ, Patel KR, Faust GE, Normolle DP, Marczylo TH, Crowell JA, Brenner DE, Booth TD, Gescher A, Steward WP. Quantitation of trans-resveratrol and detection of its metabolites in human plasma and urine by high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:182–187. doi: 10.1016/j.jchromb.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2012 doi: 10.1038/onc.2012.519. [DOI] [PubMed] [Google Scholar]
- 44.Walle T, Hsieh F, DeLegge MH, Oatis JE, Jr, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 45.Poulsen MM, Vestergaard PF, Clasen BF, Radko Y, Christensen LP, Stodkilde-Jorgensen H, Moller N, Jessen N, Pedersen SB, Jorgensen JO. High-Dose Resveratrol Supplementation in Obese Men: An Investigator-Initiated, Randomized, Placebo-Controlled Clinical Trial of Substrate Metabolism, Insulin Sensitivity, and Body Composition. Diabetes. 2012 doi: 10.2337/db12-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrer P, Asensi M, Segarra R, Ortega A, Benlloch M, Obrador E, Varea MT, Asensio G, Jorda L, Estrela JM. Association between pterostilbene and quercetin inhibits metastatic activity of B16 melanoma. Neoplasia. 2005;7:37–47. doi: 10.1593/neo.04337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crowell JA, Korytko PJ, Morrissey RL, Booth TD, Levine BS. Resveratrol-associated renal toxicity. Toxicol Sci. 2004;82:614–619. doi: 10.1093/toxsci/kfh263. [DOI] [PubMed] [Google Scholar]
- 50.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer research. 2008;68:5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serafini P, De SC, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 53.Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M, Kobayashi T, Yoshimura A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25:2520–2530. doi: 10.1038/sj.onc.1209281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.