

Abstract
SIRT1 and PPARγ, host defenses regulating inflammation and metabolic functions, are suppressed under chronic high oxidant stress and inflammation (OS/ Infl) conditions. In diabetes, dietary advanced glycation end products (dAGEs) cause OS/Infl and suppress SIRT1. Herein, we ask whether dAGEs also suppress host defense in adults without diabetes. The relationships between dAGEs and basal SIRT1 mRNA, PPARγ protein levels in mononuclear cells (MNC) and circulating inflammatory/metabolic markers were examined in 67 healthy adults aged >60 years and in 18 subjects, before and after random assignment to either a standard diet (regular>15 AGE Eq/ day) or an isocaloric AGE-restricted diet (<10 AGE Eq/ day) for 4 months. Also, the interactions of AGEs and anti-AGE receptor-1 (AGER1) with SIRT1 and PPARγ were assessed in wild type (WT) and AGER1-transduced (AGER1+) MNC-like THP-1 cells. We found that dAGE, but not caloric intake, correlated negatively with MNC SIRT1 mRNA levels and positively with circulating AGEs (sAGEs), OS/infl, MNC TNFα and RAGE. Basal MNC PPARγ protein was also lower in consumers of regular vs. AGE-restricted diet. AGE restriction restored MNC SIRT1 and PPARγ, and significantly decreased sAGEs, 8-iso-prostanes, VCAM-1, MNC TNFα and RAGE. Model AGEs suppressed SIRT1 protein and activity, and PPARγ protein in WT, but not in AGER1+ cells in vitro. In conclusion, chronic consumption of high-AGE diets depletes defenses such as SIRT1 and PPARγ, independent of calories, predisposing to OS/Infl and chronic metabolic disease. Restricted entry of oral AGEs may offer a disease-prevention alternative for healthy adults.
Keywords: Nutrition, Oxidative stress, Insulin resistance, Host defense mechanisms, Glycation
Introduction
Chronic diseases often affecting the older population, such as diabetes, cardiovascular disease (CVD) and autoimmune conditions are characterized by elevated oxidant stress (OS) and inflammation (Infl) (Dandona et al. 2005; Libby 2003). Food-derived pro-oxidant advanced glycation end products (dAGEs), largely generated by heat processing, are absorbed as AGE-peptides and AGE-lipids (Koschinsky et al. 1997; Cai et al. 2002). Over-consumption of dAGEs promotes high basal OS/Infl, as shown in studies in mice fed a diet with AGEs or a synthetic methylglyoxal-BSA derivative (MG) (Cai et al. 2008). Dietary AGE restriction in mice reduced OS and inflammation, prevented age-related diabetes and its complications (Hofmann et al. 2002; Peppa et al. 2003; Lin et al. 2002; Zheng et al. 2002). These data were confirmed in studies of healthy persons and patients with diabetes or kidney disease (Vlassara et al. 2002, 2009; Uribarri et al. 2003), while recently insulin resistance (IR) was found substantially improved in patients with insulin-resistant diabetes type 2 fed an AGE-restricted diet (Uribarri et al. 2011). Further evidence has implicated high levels of MG with declining cognitive function in elderly subjects (Beeri et al. 2011).
SIRT1, a member of the sirtuin (silent mating type information regulation 2 homolog) 1 family of NAD+ deacetylases, a survival-associated factor (Guarente 2011), is a key regulator of signaling and transcription factors, including the insulin receptor and its substrates, NF-κB, forkhead box class O (FOXO), PGC-1a or α and adiponectin (Liang et al. 2009). The role of SIRT1 extends to insulin non-sensitive cells, i.e. peripheral mononuclear cells (MNC) and macrophages, where it regulates inflammatory responses (Olefsky and Glass 2010). SIRT1 and its activity are suppressed in high OS conditions, such as diabetes, metabolic syndrome, CVD and aging (De Kreutzenberg et al. 2010; Cardellini et al. 2009).
We recently reported that SIRT1 expression and activity in MNC are suppressed in connection with elevated circulating AGEs in T2D patients, possibly contributing to persistent inflammation and IR in these patients (Uribarri et al. 2011).
PPARγ, a member of the nuclear receptor superfamily of transcription factors, also plays a major role in metabolic and inflammatory processes (Kersten et al. 2000). It negatively regulates inflammatory mediators that promote IR in tissues, such as fat and skeletal muscle, as well as in MNC-derived macrophages (Kersten et al. 2000; Olefsky and Glass 2010; Chinetti et al. 2001; Chawla et al. 2001; Hevener et al. 2007). PPARγ is shown to interact with SIRT1 and inhibit its activity via a negative feedback loop that plays a role in cell senescence (Chinetti et al. 2001). As with SIRT1, PPARγ gene expression and activity are also suppressed under high OS both in vitro and in vivo (Han et al. 2010; Blanquicett et al. 2010; El Midaoui et al. 2006; Cai et al. 2012). There have been many studies on the effects of PPARγ activation by fatty acids and their derivatives, as well as of thiazolidinediones (TZDs) in the treatment of T2D (Kersten et al. 2000; Chawla et al. 2001). However, the effects of high OS on constitutive PPARγ have not been assessed in older persons even though they often display an excessive oxidant load.
In the current proof-of-concept study, we show an inverse relationship between diet-derived AGEs and SIRT1 and PPARγ in MNC from older adults, free of diabetes. This relationship, which was independent of caloric intake, was restored by AGE restriction and further supported by in vitro mechanistic studies.
Subjects and methods
Observational study
Healthy adult volunteers (NL) over the age of 60 years were recruited from the New York City urban community (n = 67). Exclusion criteria were major medical conditions including diabetes, cardiovascular or kidney disease, and cancer. Information was collected on medical history, medications and dietary caloric and AGE intake.
All participants consented to participate in the studies and the study protocol was approved by the Mount Sinai School of Medicine Institutional Review Board.
Interventional study
A group of 18 healthy participants over the age of 60, whose usual diet was rich in AGEs (dietary AGE intake >15 AGE Eq/day), were randomized to either a low-AGE diet (n = 10) or their usual diet (n = 8) for a period of 4 months. Gender ratio (6/2 F/M vs. 7/3 F/M), age (63.5 ± 5 vs. 65 ± 2 years) and BMI (29 ± 2 vs. 26 ± 3) were not significantly different between the low-AGE diet vs. the usual AGE diet, respectively. Fasting blood samples and 3-day food records were obtained at the beginning and at the end of the intervention (Vlassara et al. 2009). Participants prepared their own food at home after being individually instructed on how to reduce dietary AGE intake by modifying the cooking method (time and temperature) without changing the quantity of food. Specifically, they were instructed to use moist heat methods such as boil, poach, stew or steam, and to avoid dry heat methods such as frying, baking, roasting or grilling. We previously found that switching to these suggested methods of cooking limits new AGE formation, particularly in animal food products (Vlassara et al. 2002, 2009).
This clinical trial was registered in www.clinicaltrials.gov under number NCT00579774.
Methods
Human subjects
After an evaluation of medical history and a physical examination, participants provided a fasting blood sample at baseline and 4 months later. Routine blood tests were performed in the hospital clinical laboratory.
Dietary intake
Daily dietary AGE content from 3-day food records that emphasized cooking methods was estimated from a database of ~560 foods which listed AGE values (Uribarri et al. 2010) expressed as AGE Equivalents (Eq/day) (1 AGE equivalent = 1000 kilounits) (Uribarri et al. 2010). The 3-day food record estimates the amount of food and beverages consumed based on established guidelines developed to assist in estimating portions. A dietitian reviewed the record with the subject, probing for additional details on portions and preparation methods, to enhance accuracy and completeness. In addition, all subjects were administered a 15 min food frequency questionnaire on dietary habits and methods of food preparation, as described (Uribarri et al. 2003b). The information obtained evaluates the frequency at which certain foods were consumed (meat, fish, poultry, cheese, egg yolk, fats, fast foods, and convenience breakfast and snack foods), the portion sizes (in household measures), and methods of cooking (boiled, roasted, broiling/grilling, frying, or canned). Each item was assigned a numerical value, multiplied by the number of entries; the sum of entries per day provided an AGE score (Uribarri et al. 2003b). Additional estimates on intakes (based on a 24-h recall record) were obtained from subjects by twice weekly phone calls intended to verify and/or emphasize the subject’s compliance. The data from the 3-day records significantly correlated with the 24-h recall data, as well as with the previously described 7-day AGE-food frequency test, obtained at baseline and end of study (Uribarri et al. 2003b).
Nutrient calculations were estimated from food records using a nutrient software program (Food Processor version 10.1; ESHA Research, Salem, Oregon).
Measurement of AGEs, insulin resistance and inflammation
εN-carboxymethyl-lysine (CML) and methylglyoxal derivatives (MG) in serum were quantified by ELISAs, using two non-cross-reactive monoclonal antibodies (4G9 and MG3D11 mabs) raised against synthetic standards, CML-BSA and MG-BSA, respectively (Cai et al. 2008, 2002). In our laboratory, the test sensitivity for CML and MG derivatives is 0.1 u/ml and 0.004 nmol/ml, respectively; the intra-assay variation is ±2.6 % (for CML) and ±2.8 % (for MG) and the inter-assay variation is ±4.1 % (CML) and ±5.2 % (MG). Insulin was measured by an ELISA kit (ALPCO Diagnostics, Salem, NH). IR was estimated according to the Homeostasis Model Assessment (HOMA) index as: FI x Fasting glucose/22.5, where FI is insulin in μU/mL and fasting glucose is expressed in mmol/L. Leptin and adiponectin were determined with human leptin or adiponectin ELISA kits (Millipore, Billerica, MA, USA). TNFα was measured in PMNC lysates by an ELISA kit (Biosource International, Camarillo, CA, USA).
Peripheral blood mononuclear cells (MNCs)
MNCs were separated from fasting, EDTA anti-coagulated blood by Ficoll-Hypaque Plus gradient (American Biosciences, Uppsala, Sweden) and used to isolate mRNA and protein (Vlassara et al. 2002, 2009). Total RNA was extracted by Trizol (Molecular Probes, Inc.). The extracted RNA had an A260/280 ratio between 1.8 and 2.0. Total RNA was reverse-transcribed using Superscript III RT (Invitrogen).
Quantitative RT-PCR assay
AGER1, RAGE, and SIRT1 mRNA expression was analyzed by quantitative SYBR Green real-time PCR. Amplification was performed with 40 cycles of denaturation at 95 °C for 15 s, annealing at 55 °C for 20 s and elongation at 72 °C for 30 s. Primer sequences were: for AGER1, forward primer 5′-CTGGGGCTCTTCATCTT CAG-3′, reverse primer 5′-GTTGCATCTCCCACA GAGGT-3′, for RAGE, forward primer 5′-AGGAGCGT GCAGAACTGAAT-3′, reverse primer 5′-TTGGCAA GGTGGGGTTATAC-3′, and for SIRT1, forward primer 5′-CGGAAACAATACCTCCACCT-3′, reverse primer 5′-CACCCCAGCTCCAGTTAGAA. During thermal cycling, emission from each sample was recorded and the raw fluorescence data was processed using SDS software to produce threshold cycle (Ct) values for each sample. β-actin and GAPDH housekeeping genes were used for internal normalization. The transcript copy number of target genes was determined based on their Ct values (Vlassara et al. 2009).
Western analysis
Cell lysates were prepared by sonication in 500 μl lysis buffer (New England Biolabs) and cell proteins were separated on 8 % SDS-PAGE gels and transferred onto nitrocellulose (NT) membranes for probing with primary and secondary antibodies for SIRT1 (Millipore Corporation, Billerica, MA, USA), PPARγ and AGER1 (Santa Cruz Biotechnology) and secondary antibodies and then visualized by an enhanced chemiluminescence system (Roche) (Uribarri et al. 2011; Olefsky and Glass 2010).
Model AGEs used for in vitro studies
Synthetic preparations of MG-BSA (containing 22 MG-Arg/mol), by GC–MS (Cai et al. 2008) and, together with BSA (Fraction V, Sigma), were rendered LPS-free prior to use (using a Detoxigel column, Pierce, Rockford, IL, USA). The pass-through tested negative for Lipopolysaccharide (LPS), (Limulus assay, Bio Whittaker, Walkerville, MD, USA), and contained MG-BSA: 0.12 nmol MG/μg and BSA: 0.00001 nmol MG/μg.
Statistical analysis
Data analyses were performed using SPSS 17.0 software (SPSS, Chicago, IL, USA). Data in the tables and figures are presented as mean ± SEM. The Kolmogorov–Smirnov goodness-of-fit test was used to test for normal distribution. Variables not normally distributed were logarithmically converted for analyses. Differences of means between groups were analyzed by the Student t test or ANOVA, followed by the Bonferroni correction for multiple comparisons, depending on the number of groups. Correlation analyses were also examined by the Pearson correlation coefficient.
Significance of changes during the interventional study was assessed by comparing deltas (differences) between end of study and baseline values for both the AGE-restricted and the regular AGE diet groups by unpaired t test. Significant differences were defined as a value of p < 0.05 and are based on two-sided tests.
Results
Observational study
Selected demographics, clinical and biochemical parameters of the study population are described in Table 1. While within the range expected for subjects over 60 years of age, there were significant gender differences with respect to BMI, waist circumference, diastolic blood pressure, total cholesterol and HDL cholesterol. There was also a significant gender difference in MNC SIRT1 mRNA levels.
Table 1.
Baseline characteristics of the cross-section population
| Parameters | All | Women | Men | p* |
|---|---|---|---|---|
| N | 67 | 40 | 27 | |
| Age (years) | 70 ± 1 | 71 ± 1 | 68 ± 2 | 0.173 |
| BMI (kg/m2) | 26 ± 0.6 | 25 ± 0.7 | 28 ± 1.0 | 0.012 |
| Weight (kg) | 72 ± 2 | 65 ± 2 | 87 ± 3 | 0.000 |
| Waist circumference (cm) | 94 ± 2 | 88 ± 2 | 108 ± 4 | 0.000 |
| Systolic blood pressure (mmHg) | 127 ± 2 | 132 ± 3 | 125 ± 3 | 0.127 |
| Diastolic blood pressure (mmHg) | 69 ± 1 | 75 ± 2 | 67 ± 1 | 0.003 |
| Creatinine Clearance (ml/min) | 91 ± 3 | 88 ± 4 | 96 ± 6 | 0.262 |
| BUN (mg/dl) | 18 ± 0.6 | 18 ± 0.7 | 18 ± 1.0 | 0.718 |
| Serum albumin (g/dl) | 4.2 ± 0.04 | 4.2 ± 0.06 | 4.2 ± 0.04 | 0.560 |
| Triglycerides (mg/dl) | 96 ± 6 | 95 ± 7 | 99 ± 8 | 0.682 |
| Cholesterol (mg/dl) | 198 ± 4 | 205 ± 4 | 179 ± 6 | 0.001 |
| HDL cholesterol (mg/dl) | 66 ± 2 | 71 ± 2 | 54 ± 3 | 0.000 |
| LDL cholesterol (mg/dl) | 113 ± 3 | 116 ± 4 | 106 ± 5 | 0.121 |
| sCML (U/ml) | 9.2 ± 0.4 | 9.0 ± 0.5 | 9.5 ± 0.7 | 0.600 |
| Fasting blood glucose (mg/dl) | 81.5 ± 1 | 81 ± 1 | 83 ± 3 | 0.458 |
| Insulin (μU/ml) | 6.6 ± 0.4 | 6.4 ± 0.5 | 6.9 ± 0.9 | 0.640 |
| HOMA-IR | 1.30 ± 0.09 | 1.28 ± 0.09 | 1.38 ± 0.18 | 0.626 |
| Leptin (ng/ml) | 20 ± 2 | 21 ± 3 | 18 ± 3 | 0.554 |
| Adiponectin (μg/ml) | 8.7 ± 0.7 | 8.5 ± 0.8 | 9.1 ± 1.2 | 0.656 |
| Leptin/adiponectin | 4.2 ± 1 | 4.6 ± 1 | 3.4 ± 1 | 0.473 |
| sMG (nmol/ml) | 0.89 ± 0.03 | 0.89 ± 0.03 | 0.89 ± 0.05 | 0.927 |
| 8-isoprostane (pg/ml) | 152 ± 8 | 154 ± 11 | 147 ± 13 | 0.655 |
| VCAM1 (ng/ml) | 797 ± 34 | 810 ± 43 | 767 ± 57 | 0.556 |
| TNFα (pg/mg protein) | 8.5 ± 0.4 | 8.4 ± 0.5 | 8.7 ± 0.7 | 0.714 |
| RAGE (mRNA) | 356 ± 24 | 371 ± 31 | 322 ± 34 | 0.296 |
| AGER1 (mRNA) | 188 ± 13 | 192 ± 16 | 180 ± 24 | 0.678 |
| SIRT1 (mRNA) | 342 ± 20 | 364 ± 27 | 294 ± 21 | 0.047 |
| Caloric intake (kcal/d) | 1834 ± 95 | 1821 ± 106 | 1861 ± 198 | 0.861 |
| Protein intake (g/day) | 83 ± 6 | 77 ± 5 | 98 ± 14 | 0.165 |
| Carbohydrate intake (g/day) | 235 ± 15 | 225 ± 18 | 256 ± 28 | 0.345 |
| Fat intake (g/day) | 68 ± 4 | 68 ± 5 | 66 ± 7 | 0.760 |
| Saturated fat intake (g/day) | 21 ± 1 | 21 ± 2 | 22 ± 3 | 0.623 |
| AGE intake (Eq/day) | 12.4 ± 1.0 | 12.8 ± 1.0 | 11.5 ± 1.0 | 0.498 |
All values are expressed as mean ± SEM
p value reflects the difference between men and women as estimated by unpaired t test
The daily dietary AGE intake correlated directly with sCML (r = 0.650, p = 0.001), sMG (p = 0.495, p = 0.001), and with markers of inflammation and OS, including 8-isoprostanes (r = 0.442, p = 0.001), TNFα (r = 0.490, p = 0.001), VCAM-1 (r = 0.469, p = 0.001) and full length RAGE mRNA (p = 0.457, p = 0.001) levels and inversely with the anti-inflammatory protein, adiponectin (r = −0.304, p = 0.023). Thus, persons consuming a high AGE diet had elevated basal OS and inflammation. There was also a positive association between circulating AGEs, sCML or sMG and, MNC AGER1 mRNA (r = 0.651, p = 0.001 for sCML and r = 0.384, p = 0.001 for sMG) (Vlassara et al. 2009).
MNC SIRT1 mRNA levels inversely correlated with levels of sCML (r = −0.597, p = 0.001), sMG (r = −0.406, p = 0.001), VCAM1 (r = −0.424, p = 0.001) as well as with plasma 8-isoprostanes (r = −0.384, p = 0.002), MNC TNFα protein (r = −0.424, p = 0.001) and RAGE mRNA (r = −0.461, p = 0.001). Highly noteworthy was the strong inverse correlation of SIRT1 mRNA with dietary AGE intake (r = −0.510, p = 0.001) (Fig. 1a), which remained unchanged after adjusting for caloric intake (r = −0.496, p = 0.001) or for intake of protein, carbohydrate and fat (r = −0.557, p = 0.001). It should be noted that SIRT1 mRNA levels did not correlate with dietary intake of calories, protein, carbohydrates, total fat or saturated fats (data not shown).
Fig. 1.
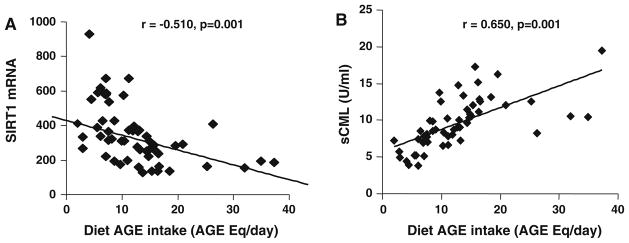
a Dietary AGE intake (dAGE) correlates negatively with MNC SIRT1 mRNA in older healthy subjects (n = 67). Baseline dietary AGE intake was estimated from 3-day food records and is expressed as AGE Eq/day. MNC SIRT1 mRNA levels were measured by RT-PCR. b Dietary AGE intake (dAGE) correlates positively with fasting serum CML (sCML) levels, based on CML ELISA. Analyses were based on the Pearson correlation coefficient
Interventional study
Serum, plasma and MNC changes
Dietary AGE restriction (by 50 %) for a period of 4 months, without caloric restriction, led to markedly lower levels of sCML (13.7 ± 1.0 down to 9.2 ± 0.8 U/ml) and sMG (1.19 ± 0.05 down to 0.79 ± 0.05 nmol/ml), as well as of 8-isoprostanes (170 ± 23 down to 85 ± 6 pg/ml) (Table 2; Fig. 2).
Table 2.
Differences for selected biochemical parameters and BMI before and after intervention in each dietary group
| Parameter | Low-AGE diet
|
Regular AGE diet
|
p* | ||
|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | ||
| sCML (U/ml) | −3.71 | 1.03 | 1.87 | 1.05 | 0.002 |
| sMG (nmol/ml) | −0.28 | 0.07 | 0.18 | 0.10 | 0.002 |
| SIRT1 (mRNA, AU) | 92 | 52 | −189 | 62 | 0.003 |
| AGER1 (mRNA, AU) | −70 | 24 | 69 | 27 | 0.001 |
| RAGE (mRNA, AU) | −127 | 41 | 253 | 137 | 0.021 |
| p66shc (mRNA, AU) | −27 | 10 | 26 | 11 | 0.003 |
| 8-Isoprostane (pg/ml) | −48 | 11 | 48 | 20 | 0.002 |
| TNFα (pg/mg cell protein) | −2.1 | 1.6 | 3.2 | 0.8 | 0.013 |
| VCAM1 (pg/ml) | −270 | 92 | 182 | 46 | 0.002 |
| hsCRP (mg/l) | 1.3 | 1.4 | 0.4 | 0.4 | 0.582 |
| Leptin (ng/ml) | −4.2 | 3.5 | 4.5 | 4.4 | 0.162 |
| Adiponectin (ug/ml) | −0.96 | 1.4 | −0.34 | 1.1 | 0.744 |
| HOMA-IR | 0.04 | 0.44 | 0.14 | 0.15 | 0.834 |
| Triglycerides (mg/dl) | −10 | 7 | −6 | 7 | 0.683 |
| HDL (mg/dl) | −7 | 2 | 2 | 4 | 0.098 |
| LDL (mg/dl) | −5 | 5 | 0.1 | 4 | 0.479 |
| BMI | −0.43 | 0.27 | −0.29 | 0.25 | 0.722 |
Differences or deltas were calculated as last minus first visit under the index treatment
p statistical significant differences between deltas
Fig. 2.

Dietary AGE restriction restores SIRT1 and improves inflammatory and oxidative stress markers in healthy subjects. After 4 months of AGE-restricted diet (closed bars) or regular diet (open bars), changes in circulating CML, MG and 8-isoprostane, and in MNC TNFα protein and SIRT1 mRNA (by RT-PCR) levels are shown as percentage from baseline (mean ± SEM). *p < 0.050 refers to statistical significance in percent change between baseline and end of study between both groups
In addition, there were reductions in baseline levels of MNC TNFα protein, full length RAGE and AGER1 mRNA in the AGE-restricted group. Interestingly, there was a significant increase in SIRT1 mRNA levels in this group (Table 2; Fig. 2), compared to the control group (Table 2; Fig. 2). As previously reported, the changes in mRNA correlated with changes in SIRT1 protein and deacetylase activity, namely, levels of NF-κB acetylated p65 (Uribarri et al. 2011; Cai et al. 2012).
Since high OS can also influence PPARγ, protein levels of this nuclear receptor were assessed. Baseline MNC PPARγ protein levels inversely correlated both with the corresponding dAGE intake as well as serum AGE levels (Fig. 3a). Specifically, PPARγ was significantly lower in persons reporting a dietary AGE intake>15 Eq/day than in those reporting 10 Eq/day or below, (Fig. 3a). After AGE restriction, MNC PPARγ protein levels were significantly increased (by ~50 %), while they decreased further in those encouraged to maintain their customary high-AGE diet (Fig. 3b). These data were consistent with the findings on SIRT1 mRNA, shown above.
Fig. 3.

a Dietary AGE intake correlates inversely with PPARγ protein expression in MNC of healthy older subjects. Upper panel Western blot analysis of PPARγ protein in MNC of a subgroup of randomly selected study participants (n = 8) with widely varying levels of dAGE consumption, as indicated. Middle panel Densitometric analysis of western blot data shown in upper panel and expressed as PPARγ/β-actin ratio. Individual density data on PPARγ protein are juxtaposed with the corresponding fasting serum CML, based on CML ELISA, expressed in U/ml. Lower panel PPARγ/β-actin ratios are grouped in those from persons with high dietary AGE intake (>15 Eq/day, open bar) and those with low-AGE intake (<10 Eq of AGE/day, closed bar), *p = 0.01. b AGE restriction restores MNC PPARγ expression. Representative western blot analysis and densitometric analysis data from study participants exposed to their regular diet (open bar) or a low-AGE diet (closed bar) for 4 months, indicating entry (1) and end of study (2). Data, based on PPARγ/β-actin ratio, are shown as the percentage (mean ± SEM) of change from baseline. *p = 0.001 indicates the significance between low-AGE and regular diet
In vitro effects of AGEs on SIRT1 and PPARγ expression
Using monocyte-like THP-1 cells, we explored the mechanisms of the changes observed in circulating MNCs. Chronic exposure to the synthetic AGE, MG-BSA (MG) led to the suppression of SIRT1, as well as of anti-AGE receptor AGER1 protein (Fig. 4a). MG-stimulation also reduced SIRT1-dependent deacetylase activity, as shown by increased levels of acetylated NF-κB p65 (Fig. 4b), and a reduction in intracellular NAD+/NADH ratio (Fig. 4c), which directly regulates SIRT1 via ROS. These MG-mediated effects were blocked in cells over-expressing the AGER1 gene (AGER1+), consistent with the anti-AGE, anti-ROS and pro-SIRT1 properties of AGER1 (Uribarri et al. 2011; Cai et al. 2012).
Fig. 4.

Specific AGEs, MG-BSA (MG), modulate SIRT1 and PPARγ expression in THP-1 in vitro. a Time-dependent suppression of SIRT1 protein by MG in vitro parallels that of AGER1. Western blots (upper panels) and densitometry (lower panels) are shown for SIRT1 (black squares) and AGER1 (open squares) in THP-1 cells treated with MG-BSA (60 μg/mL) for up to 96 h. Data on test/β-actin ratios are shown as mean ± SEM (n = 3 tests). *p < 0.001 vs. non-stimulated cells (CL). b SIRT1 expression and Nf-kB p56 acetylation (Ac-p65) are AGER1-dependent. Wild-type THP-1 cells (WT) or THP-1 cells transfected with AGER1 (AGER1+) were stimulated by MG (60 μg/ mL) for 72 h before Western blots (upper panels) and densitometry plots (lower panels; AGER1+, black bars; WT, open bars). Data (n = 3–5 tests) are shown as mean ± SEM *p <0.001 vs. WT. c MG-induced effects on SIRT1 and ac-p56 are NAD+-dependent and regulated by AGER1. WT or AGER1+ THP-1 cells exposed to MG-BSA (60 μg/mL) for up to 72 h. [NAD+/NADH] ratio is shown as fold (mean ± SEM) above control (n = 3, each in triplicate). *p < 0.001 vs. WT control. §p <0.001 vs. WT treated by MG. d MG suppresses PPARγ protein, but not in cells over-expressing AGER1 (AGER1+). Western blots and density plots, derived from PPARγ/β-actin ratio, are shown as mean ± SEM (n = 4 tests)
Furthermore, PPARγ protein levels were suppressed, in a time-dependent manner in MG-stimulated THP-1 cells, but remained enhanced in AGER1+ cells (Fig. 4d). These data were in agreement with the clinical findings in healthy subjects with intact MNC AGER1.
Discussion
The current study demonstrates that gene expression of SIRT1, a key regulator of inflammation and insulin function, can be suppressed in MNC of older adults as a function of diet-derived oxidant AGEs (dAGEs), and independent of caloric or nutrient intake, or presence of known high OS-related conditions such as diabetes. Furthermore, high dAGEs appeared associated with reduced levels of the anti-inflammatory nuclear receptor PPARγ. Notably, brief dietary restriction of AGEs, without altering caloric intake, largely restored both these host defense mechanisms in MNC and improved systemic markers of OS and inflammation in non-diabetic older subjects. Thus, chronic exposure to AGE-rich diets can deplete key defense mechanisms in a manner, which may raise susceptibility to chronic metabolic conditions. The clinical findings, which confirm extensive multi-generational animal studies focused on a single class of higly reactive AGEs, MG derivatives (Cai et al. 2012), are herein supported by mechanistic evidence in genetically manipulated cells underscoring the relevance of AGE burden, and of the anti-AGE receptor AGER1, in the regulation of SIRT1 and PPARγ.
Since MNC SIRT1 levels have been found suppressed in persons with pre-diabetes (De Kreutzenberg et al. 2010), we reasoned that high dAGEs could act as modulators of SIRT1 expression even prior to clinical disease, especially in older persons, a population that is prone to OS/Infl and thus to cardio-metabolic disorders. The cross-sectional data showing a strongly negative association of dAGEs with SIRT1 in MNC lend support to this hypothesis, and are further supported by the interventional data. Together they introduce a potential mechanistic pathway by which diet-derived AGEs can over time down-regulate SIRT1 gene and activity levels, initiating pro-inflammatory events. In this respect, there was also a strong negative correlation of SIRT1 with TNFα, a key inflammatory cytokine, as well as pro-OS/Infl AGE receptor RAGE and VCAM-1, further supporting this view.
The present study utilized MNC, which can reflect systemic changes in the inflammatory/IR axis (De Kreutzenberg et al. 2010). Inflammation is promoted via a transition of monocyte-derived macrophages from M2 to M1 phenotype, with decreased anti-inflammatory mediators (IL-10, IL-4), and higher TNFα, IL-6 or IL-1β levels, changes that are linked to suppressed SIRT1 (Olefsky and Glass 2010; Cai et al. 2012). Thus, in T2D decreased MNC SIRT1, hyper-acetylated NF-κB p65 and increased TNFα levels were consistent with such a switch (Uribarri et al. 2011; Cai et al. 2012).
In the current study of non-diabetic subjects, the findings, including MNC SIRT1 or TNFα levels, reveal a pro-inflammatory pattern albeit in the absence of diabetes. Moreover, dietary AGEs appeared to play a central role, since along with systemic AGEs and OS, SIRT1, a key regulator of inflammatory events, was reversed by AGE restriction, as demonstrated in mice (Cai et al. 2012). A chronic AGE overload may, thus, explain the surreptitious presence of activated MNC in circulation. Tissue infiltration by these cells in concert with local macrophages, secreting TNFα and other inflammatory cytokines, may set the stage for vascular or metabolic disease in due time (Olefsky and Glass 2010; De Kreutzenberg et al. 2010; Cai et al. 2012).
Furthermore, the study reveals for the first time that along with SIRT1, nuclear receptor PPARγ expression is markedly reduced in MNC of persons under increased OS, lending support to animal studies on targeted macrophage PPARγ deficiency (Chawla et al. 2001). Particularly noteworthy here was the co-recovery in PPARγ together with SIRT1, following AGE restriction. A similar coordinate regulation of PPARγ and SIRT1 was observed in THP-1 cells pre-exposed to AGEs (MG). Of note, AGER1 transduction blocked AGE-mediated down-regulation of PPARγ as well as of SIRT1 (Uribarri et al. 2003a), consistent with the protective role of AGER1 against AGEs. Of note, MNC AGER1 mRNA levels, although found reduced in diabetes or CKD (Vlassara et al. 2009; Uribarri et al. 2011), did not differ from normal in these subjects. While the size of this pilot clinical study limits definitive answers, SIRT1 and PPARγ could have a lower in vivo threshold to elevated OS relative to AGER1. Larger studies are required to unravel the molecular underpinnings of these findings. However, suppressed MNC SIRT1 or PPARγ gene levels may serve as sensitive “rheostats” of homeostatic imbalance in older subjects.
Calories and nutrients have long been thought to modulate SIRT1 in man and animals (Guarente 2011). The current intervention, which allowed equal caloric intake to both groups, provides first clinical evidence that AGEs, rather than calories, play a primary role in the regulation of SIRT1 and related pathways, thus confirming in humans similar evidence obtained in mice (Cai et al. 2012). Since calories closely correlate with dAGE content, a significant implication of this finding is that food glycoxidants are partly responsible for the down-regulation of SIRT1 attributed to high calorie diets. A related advantage that emerges from these data is that normal SIRT1, as well as PPARγ, levels can be restored by adopting a regimen of reduced AGE consumption, without drastically altering calories/nutrients.
Conclusions
The main implication of this human study is that the modern lifestyle by encouraging consumption of AGE-enriched food can prematurely deplete key host defense mechanisms, such as SIRT1 and PPARy, thus impeding normal metabolism long before disease onset. A diet nutritionally optimal but low in AGEs can preserve or restore such defenses, thus averting chronic diseases.
Acknowledgments
AG-23188 and AG-09453 (to H. Vlassara) from the National Institute of Health and National Institute of Research Resources, MO1-RR-00071, awarded to the General Clinical Research Center at Mount Sinai School of Medicine, for clinical and statistical support.
Footnotes
Conflict of interest No conflicts of interested declared by any of the authors.
Contributor Information
Jaime Uribarri, Email: jaime.uribarri@mssm.edu, Department of Medicine, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Weijing Cai, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Renata Pyzik, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Susan Goodman, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Xue Chen, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Li Zhu, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Maya Ramdas, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
Gary E. Striker, Department of Medicine, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA. Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA
Helen Vlassara, Email: Helen.vlassara@mssm.edu, Department of Geriatrics, The Mount Sinai School of Medicine, One Gustave Levy Place, New York, NY 10029, USA.
References
- Beeri MS, Moshier E, Schmeidler J, Gobold J, Uribarri J, Reddy S, Sano M, Grossman HT, Cai W, Vlassara H, Silverman JM. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech Ageing Dev. 2011;132:583–587. doi: 10.1016/j.mad.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPARγ in vascular endothelial cells. Free Radic Biol Med. 2010;48:1618–1625. doi: 10.1016/j.freeradbiomed.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Gao QD, Zhu L, Peppa M, He C, Vlassara H. Oxidative stress-induced carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol Med. 2002;8:337–346. [PMC free article] [PubMed] [Google Scholar]
- Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlassara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173(2):327–336. doi: 10.2353/ajpath.2008.080152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA. 2012;109:15888–15893. doi: 10.1073/pnas.1205847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardellini M, Menghini R, Martelli E, Casagrande V, Marino A, Rizza S, Porzio O, Mauriello A, Solini A, Ippoliti A, Lauro R, Folli F, Federici M. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SIRT1. Diabetes. 2009;58:2396–2401. doi: 10.2337/db09-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Barrak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Chinetti G, Lestavel S, Bocher V, Remaley AT, Never B, Torra IP, Treiussier E, Minnich A, Jaye M, Duverger N, Brewer HB, Fruchart JC, Clavey V, Staels B. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- Dandona P, Alijada A, Chaudhuri A, Morahanty P, Grag R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes and inflammation. Circulation. 2005;111:1448–1454. doi: 10.1161/01.CIR.0000158483.13093.9D. [DOI] [PubMed] [Google Scholar]
- De Kreutzenberg SV, Ceolotto G, Papparella I, Bortoluzzi A, Semplicini A, Dalla Man C, Cobelli C, Fadini GP, Avogaro A. Downregulation of the longevity-associated protein SIRT1 in insulin resistance and metabolic syndrome. Potential biochemical mechanisms. Diabetes. 2010;59:1006–1015. doi: 10.2337/db09-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Midaoui A, Wu L, Wang R, de Champlain J. Modulation of cardiac and aortic peroxisome proliferator-activated receptor-gamma expression by oxidative stress in chronically glucose-fed rats. Am J Hypertens. 2006;19:407–412. doi: 10.1016/j.amjhyper.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Guarente L. Sirtuins, aging and medicine. New Eng J Med. 2011;364:2235–2243. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010;38:7458–7468. doi: 10.1093/nar/gkq609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, Olefsky JM, Reichart D, Nguyen MTA, Bandyopadyhay G, Leung HY, Watt MJ, Bebber C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass C, Ricote M. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thaizolidinediones. J Clin Invest. 2007;117:1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann S, Dong HJ, Cai W, Altomonte J, Thung SN, Zeng F, Fisher EA, Vlassara H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes. 2002;51:2082–2089. doi: 10.2337/diabetes.51.7.2082. [DOI] [PubMed] [Google Scholar]
- Kersten S, Desverfgne B, Wahil W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Koschinsky T, He JC, Liu C, Buenting C, Heitmann K, Vlassara H. Orally absorbed reactive advanced glycation end products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci. 1997;94:6474–6479. doi: 10.1073/pnas.94.12.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol. 2009;5:367–373. doi: 10.1038/nrendo.2009.101. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2003;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lin RY, Reis ED, Dore AT, Lu M, Ghodsi N, Fallon JT, Fisher EA, Vlassara H. Lowering of dietary advanced glycation endproducts (AGE) reduces neointimal formation after arterial injury in genetically hypercholesterolemic mice. Atherosclerosis. 2002;163:303–311. doi: 10.1016/s0021-9150(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation and insulin resistance. Ann Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Peppa M, He C, Hattori M, McEvoy R, Zheng F, Vlassara H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes. 2003;52:1441–1448. doi: 10.2337/diabetes.52.6.1441. [DOI] [PubMed] [Google Scholar]
- Uribarri J, Peppa M, Cai W, Goldberg T, Lu M, He C, Vlassara H. Restriction of dietary glycotoxin reduces excessive advanced glycation end products in renal failure patients. J Am Soc Nephrol. 2003a;14(3):728–731. doi: 10.1097/01.asn.0000051593.41395.b9. [DOI] [PubMed] [Google Scholar]
- Uribarri J, Peppa M, Cai W, Goldberg T, Lu M, Baliga S, Vassalotti JA, Vlassara H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am J Kid Dis. 2003b;43:532–538. doi: 10.1016/s0272-6386(03)00779-0. [DOI] [PubMed] [Google Scholar]
- Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, Yong A, Striker GE, Vlassara H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–916. doi: 10.1016/j.jada.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribarri J, Cai W, Ramdas M, Goodman S, Pyzick R, Chen X, Zhu L, Striker GE, Vlassara H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34:1610–1616. doi: 10.2337/dc11-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, Peppa M, Rayfield EJ. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci. 2002;99:15596–15601. doi: 10.1073/pnas.242407999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassara H, Cai W, Goodman S, Pyzik R, Young A, Chen X, Zhu L, Neade T, Beeri M, Silverman JM, Ferrucci L, Tansman L, Striker GE, Uribarri J. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: role of the antiinflammatory AGEreceptor-1. J Clin Endocrinol Metab. 2009;94:4483–4491. doi: 10.1210/jc.2009-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, He C, Cai W, Hattori M, Steffes M, Vlassara H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diab Metab Res Rev. 2002;18:224–237. doi: 10.1002/dmrr.283. [DOI] [PubMed] [Google Scholar]