

Abstract
Stress and/or antidepressants during pregnancy have been implicated in a wide range of long-term effects in the offspring. We investigated the long-term effects of prenatal stress and/or clinically relevant antidepressant exposure on male adult offspring in a model of the pharmacotherapy of maternal depression. Female Sprague-Dawley rats were implanted with osmotic minipumps that delivered clinically relevant exposure to the antidepressant escitalopram throughout gestation. Subsequently, pregnant females were exposed on gestational days 10–20 to a chronic unpredictable mild stress paradigm. The male offspring were analyzed in adulthood. Baseline physiological measurements were largely unaltered by prenatal manipulations. Behavioral characterization of the male offspring, with or without pre-exposure to an acute stressor, did not reveal any group differences. Prenatal stress exposure resulted in a faster return towards baseline following the peak response to an acute restraint stressor, but not an airpuff startle stressor, in adulthood. Microarray analysis of the hippocampus and hypothalamus comparing all treatment groups revealed no significantly-altered transcripts. Real time PCR of the hippocampus confirmed that several transcripts in the CRFergic, serotonergic, and neural plasticity pathways were unaffected by prenatal exposures. This stress model of maternal depression and its treatment indicate that escitalopram use and/or stress during pregnancy produced no alterations in our measures of male adult behavior or the transcriptome, however prenatal stress exposure resulted in some evidence for increased glucocorticoid negative feedback following an acute restraint stress. Study design should be carefully considered before implications for human health are ascribed to prenatal exposure to stress or antidepressant medication.
Keywords: behavior, gene expression, hypothalamic-pituitary-adrenal axis, microarray, neuropharmacology, prenatal antidepressant exposure, prenatal stress
1. Introduction
Depression affects 10–20% of pregnant women (Gavin et al., 2005). Although it is becoming increasingly well-known that untreated maternal depression during pregnancy is detrimental to infant outcome, the known and unknown risks associated with antidepressant treatment lead to increased discontinuation of antidepressant use during pregnancy (Bonari et al., 2005). Although the short-term effects of prenatal antidepressant exposures have been investigated, the long-term effects, if any, are relatively unknown. This study aimed to elucidate any long-term behavioral, endocrine, and gene expression alterations due to in utero manipulations in order to aid clinicians and their patients in treatment decisions.
Investigation of the long-term effects of prenatal stress, one possible proxy for maternal depression during pregnancy, and antidepressants concurrently has not been rigorously examined. In animal models, there are numerous studies that have examined types of prenatal stress (Mueller and Bale, 2006; Mueller and Bale, 2008; Newport et al., 2002) and several studies that have examined antidepressant-exposed pregnant rats from the perspective of the offspring (Cabrera-Vera and Battaglia, 1998; Cabrera-Vera et al., 1997; Cabrera and Battaglia, 1994; Forcelli and Heinrichs, 2008; Henderson and McMillen, 1993). However, recent health care data bases indicate that over 6% of pregnant women receive a prescription for an antidepressant (Andrade et al., 2008), therefore it can be argued that investigations of both stress and antidepressant exposure are potentially the most clinically relevant scenario. We hypothesized that, in our rat model, escitalopram intervention during the stress treatment may attenuate any long-term effects of the prenatal stress exposure on outcome measures in the offspring. Additionally, no published study to date has examined the transcriptome in the context of prenatal antidepressant exposure.
Translation of clinically relevant models of prenatal exposures is necessary to adequately evaluate the risk to the mother and infant. Previously, we have modeled stress and antidepressant exposure to the offspring (Bourke et al., 2013; Capello et al., 2011) with special emphasis on clinically relevant antidepressant exposure using the antidepressant escitalopram as representative of the class. These studies have demonstrated that chronic unpredictable mild stress during pregnancy produces an increase in basal plasma corticosterone concentrations indicative of a chronically activated hypothalamic-pituitary-adrenal (HPA) axis in response to this chronic stressor. Additionally, we have demonstrated that continuous delivery via osmotic minipumps results in serum escitalopram concentrations maintained within a clinically observed range and providing occupancy of the serotonin transporter in fetal rat brain >80% (Capello, et al., 2011). This methodology is utilized in this study to model antidepressant treatment of depression during pregnancy.
2. Materials and Methods
2.1. Animals
The experimental time line is depicted in Figure 1. Sprague-Dawley CD IGS strain #001 male, retired breeders and nulliparous females weighing 200–225 grams were purchased from Charles River Laboratories and bred (Charles River, Wilmington, MA). All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee (IACUC protocol number 079–2010) and carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Resources, 1996) as adopted and promulgated by the U.S. National Institutes of Health. All steps were taken to minimize animal suffering at each stage of the study.
Fig. 1.
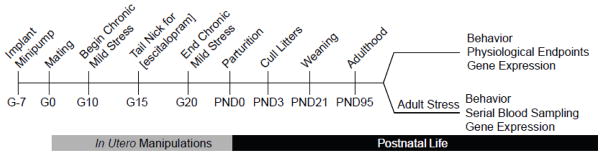
Timeline of chronic unpredictable mild stress and/or escitalopram administration during pregnancy and measurements in adulthood.
Rats were kept on a 12:12 light:dark cycle (lights on at 7:00 AM) in a humidity (60%) and temperature (20°C–23°C) controlled facility. Rodent diet 5001 chow (Purina Mills, Richmond, IN) and water were available ad libitum throughout the study. After two weeks at the Emory University animal facility, female rats were paired with male retired breeders in a breeding cage. Gestational day 0 (G0) was designated by the presence of a sperm plug and pregnant females were single-housed after breeding. Three days after birth, rat pups were sexed and litters were culled to six male and two female pups. Animals were weaned on PND21 and kept pair-housed until adulthood. Because pup growth rates and maternal licking and grooming behavior, a hallmark of good maternal care, were unaltered by either prenatal stress and/or escitalopram exposure, maternal care was performed by the biological dam rather than a foster dam (Bourke, et al., 2013). Escitalopram release from the minipumps ended between 0–2 days post parturition (Alzet model 2ML4 technical information).
For the adult offspring endpoints, in almost all circumstances only a single pup was used from each litter in order to prevent possible litter effects (Holson and Pearce, 1992). When that was not possible, no more than two pups per litter were assigned to a group and are noted at the appropriate point within the manuscript. Each group was assigned between 8 and 12 pups. Rats were killed via rapid decapitation on postnatal day (PND) 90–95 during the early phase of the light cycle. The brain was removed and immediately dissected on ice. Adrenals were removed and weighed. An additional manipulation of twelve days of chronic immobilization stress in adulthood was used in a separate cohort of animals to determine if a stress-susceptible phenotype existed. Animals were immobilized in DecapiCone® bags (Braintree Scientific, Braintree, MA) for two hours once a day for 12 days (Neigh et al., 2010). One day after the last stress session, control and immobilization stress animals were killed.
2.2 Behavioral Testing
2.2.1. Defensive Withdrawal Test
For the defensive withdrawal test, rats were placed in a black PVC withdrawal box (10cm diameter tube x 21cm in length, closed at one end) and the withdrawal box was then placed into the open field arena at a distance of 20cm from a corner with the open end facing the corner. Rats were recorded and scored based on time spent to exit the chamber with the Cleversys System.
2.2.2. Open Field Test
Rats were placed in the center of an open field arena (75 cm x 75 cm x 60 cm) in order to assess locomotor and exploratory behavior. Testing took place one hour after lights out and was conducted under red light. Rats were recorded for 15 minutes and scored based on time spent in the center versus time spent in the periphery by the Cleversys System.
2.2.3. Sucrose Consumption Test
Rats were given free access to one bottle of tap water and one bottle of a 0.8% sucrose solution in tap water. In order to prevent any effect due to side bias, bottle location was reversed after 24 hours. After 24 and 48 hours, the bottles were weighed to determine sucrose and water consumption. Data reported are the average consumption of day 1 and day 2 of the sucrose consumption test.
2.2.4. Elevated Plus Maze
As an index of anxiety-like behavior, animals were tested in the elevated plus maze. Rats were placed in the center of a plastic plus maze and recorded for five minutes. Testing took place two hours after lights out and was conducted under dim red light. Tapes were scored for number of entries and time spent in the closed or open arms by the CleverSys system. Percent of time spent in the open arm was calculated as time spent in the open arm divided by total testing time (300 seconds).
2.2.5. Acoustic Startle Response
During the light cycle, rats were placed in a ventilated startle chamber (San Diego Instruments, San Diego, CA) using an accelerometer to measure the startle reflex. After a white-noise (65 db) acclimatization period in the chamber lasting five minutes, individual rats were exposed to 16 trials consisting of a short pulse of noise (115 db). Between each trial, there was a period of white noise, which lasted 2, 5, 25, or 60 seconds.
2.2.6. Forced Swim Test
In order to assess motor activity in an inescapable environment, the forced swim test was administered. Rats were placed in a clear acrylic beaker (40 cm high×18 cm in diameter) filled with 30°C water for ten minutes during the light cycle. Following this training session, rats were tested the following day for five minutes. Behavior was scored by the CleverSys System.
2.3. Escitalopram Administration and Chronic Unpredictable Mild Stress Model
Nulliparous female rats were subcutaneously implanted with Alzet 28 day osmotic minipumps (model 2ML4, Alzet, Cupertino, CA) slightly posterior to the scapulae. Animals were anesthetized with isoflurane, checked for reaction to painful stimuli, and surgically implanted with pumps under aseptic conditions. Osmotic pumps delivered either 0.9% saline or 12.2 mg/kg/day escitalopram oxalate in 0.9% saline based upon a predicted weight of the pregnant dam on G21 of 400 grams (unpublished observations). After initial implantation (mean weight of 255 g), escitalopram oxalate dosage was initially 17.3 mg/kg/day. Escitalopram oxalate (gift of Lundbeck USA, Paramus, NJ 07652) was chosen as it offers the greatest aqueous solubility of the common SSRI antidepressants. Three days after minipump implantation, females were bred with retired breeder males. On G10, the chronic unpredictable mild stress model of depression began and consisted of restraint, cage tilt, damp bedding, noise, cage changes, and overnight illumination (Bourke, et al., 2013). G10 was selected to begin the stress paradigm because the fetal central nervous system begins substantial development at this point (Clancy et al., 2001) and to minimize premature termination of the pregnancy as a result of excessive stress. On G15, all pregnant dams, regardless of treatment group, were given a tail nick to determine the serum concentration of escitalopram. No pregnant dams with escitalopram minipumps had serum escitalopram concentrations less than 17 ng/mL. The stress model ended on G20 to prevent premature parturition.
2.4. Physiological Measures
Plasma corticosterone concentrations were assayed using the ImmuChem 125I Corticosterone RIA kit with a sensitivity of 1 ng/mL (MP Biomedicals, Orangeburg, NY). Plasma ACTH concentrations were assayed using the 125I ACTH RIA kit with a sensitivity of 1.5 pg/mL (Diasorin, Stillwater, MN). Non-fasting blood glucose concentrations were measured with the Freestyle Blood Glucose Meter with Freestyle Test Strips (TheraSense, Alameda, CA). Insulin concentrations were measured with a Mercodia Rat Insulin ELISA with a sensitivity of 0.15 μU/mL (ALPCO Diagnostics, Windham, NH). Insulin resistance was calculated by a product of insulin and glucose analogous to the homeostatic model assessment of insulin resistance (HOMA-IR) calculation: [glucose] x [insulin] / 405. Values were normalized to the control group median [glucose] x median [insulin] values such that control mean value would approximate 1. Glucose and insulin measures were not taken after 24 hours of fasting, which would have interfered with our main experimental interests and design. Heart tissues were placed in 0.9% sterile saline, dissected on ice, and weighed to determine total heart weight and right ventricle: left ventricle + septum weight as described previously (Fornaro et al., 2007).
2.5. Behavioral Testing
Behavioral testing was conducted as previously described (Bourke and Neigh, 2011). Briefly, animals were tested during the beginning of the dark cycle for the defensive withdrawal test (10 min), elevated plus maze (5 min), open field test (15 min), marble burying test (15 min) and acoustic startle response (15 min). The sucrose consumption test was conducted over 48 hours and fluid consumption amounts were adjusted for body weight. The forced swim training (10 min) and test (5 min) periods were both conducted in the light cycle. Although the test was administered, the animals showed extreme difficulty with maintaining an immobile floating position likely due to the heavy mass of the adult animals. While the test was conducted, it was not analyzed due to this confounding variable. Locomotion and exploratory behavior were assessed with the TopScan and ForcedSwimScan systems (Cleversys, Reston, VA). All tests were conducted between PND 90–120 and separated by seven days. A 15 minute restraint using plastic restrainers designed to minimize movement (Braintree Scientific, Braintree, MA) was used in a separate cohort of animals before the open field, marble burying test, acoustic startle response, and the forced swim test session. Other tests (elevated plus maze, defensive withdrawal, and sucrose consumption) did not incorporate the acute restraint in order to minimize habituation to a homotypic stressor.
2.6. Jugular Catheter Studies
Jugular catheters were implanted as described previously (Thrivikraman et al., 2002). Briefly, animals were anesthetized with a preparation of ketamine:xylazine:acepromazine and assessed for reaction to a painful stimuli prior to surgery. The jugular vein was implanted with a catheter to allow for repeated blood sampling and to prevent a stress response elicited by other sampling methods (i.e., tail nick). Animals were given four days to recover before initiation of an air puff startle stressor and serial blood sampling (Engelmann et al., 1996). Briefly, a baseline blood sample was collected at 9:00 AM. Animals were then exposed to three trials of three short air puffs (Air Duster, Office Max) on the head and given one minute to recover between trials. Blood samples (200 μL) were collected and an equal volume of sterile, 0.9% saline was injected to replace the blood volume lost. Catheters were flushed with 150 μL of sterile gentamicin (120 μg/mL) solution after sampling to prevent infection. The restraint stress serial blood sampling was conducted four days after air puff serial blood sampling. A baseline blood sample was collected at 9:00 AM and the animals were immobilized for 5 minutes in DecapiCone® bags (Braintree Scientific, Braintree, MA). Blood samples were collected as described above.
2.7. Gene Expression Studies
Brains were immediately dissected on ice with a Jacobowitz 2 mm Brain Slicer (Zivic Instruments, Pittsburgh, PA). The amygdala (bilateral) was removed with a 2 mm sterile biopsy punch (Miltex Inc, York, PA). The hippocampus and hypothalamus were dissected from the same single slice. RNA extractions were performed the day of the dissections. RNA was extracted with the TRIzol method (Invitrogen, Carlsbad, CA) and RNA integrity was assessed using a BioRad Spectrophotometer and an Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). Samples with RNA integrity numbers under 5.5 units were excluded. RNA was amplified using the GeneChip® WT cDNA Synthesis and Amplification Kit (Affymetrix, Santa Clara, CA). RNA was labeled using the GeneChip WT Sense Target Labeling Kit (Affymetrix, Santa Clara, CA), hybridized to a GeneChip Rat Exon 1.0 ST Array (Affymetrix, Santa Clara, CA) and analyzed on an Affymetrix GeneChip System (Affymetrix, Santa Clara, CA). Expression Console was used to normalize chip data by the RMA-Sketch method (Affymetrix, Santa Clara, CA). The data were filtered to remove duplicate transcript probe sets and analyzed with hierarchical clustering with TM4 Microarray Suite using a Pearson Correlation with Average Linkage Clustering (Saeed et al., 2003). To find differentially regulated genes, Significance Analysis of Microarrays (SAM) was used in R to analyze the normalized data (R Development Core Team, 2010; Tusher et al., 2001). SAM was set to examine each two-class unpaired comparison with 1,000 permutations, a 1.5 fold-change cut-off, and a False Discovery Rate (FDR) cut off of 10%. Power analysis was conducted with SAM. In a parallel analysis, data was filtered after SAM analysis with Ingenuity Pathway Analysis 8.7 (Ingenuity Systems, Redwood City, CA) to remove any transcripts that were not expressed in the regions of interest. Individual p-values from the t-tests performed in SAM were adjusted using a Bonferroni correction. To combine the different rounds of microarrays, a meta-analysis was performed using Fisher’s method of combining p-values from the MADAM package in R (Kugler et al., 2010).
For real time PCR studies, RNA from the dorsal hippocampus was extracted with TRIzol (Invitrogen, Carlsbad, CA) and to eliminate genomic DNA contamination, a DNAse step was performed using the Turbo DNA-free kit (Ambion, Austin, TX). Reverse transcription was carried out with the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). cDNA was quantified with the PicoGreen Assay (Invitrogen, Carlsbad, CA). A rat endogenous control plate (Applied Biosystems, Foster City, CA) was run in order to determine the ideal endogenous control (Hmbs). Primers were designed with Primer3Plus (Untergasser et al., 2007) (Table S1) and designed with Ensembl to overlap exon-exon junctions to eliminate amplification of genomic DNA (Flicek et al., 2011). Primer concentrations were optimized to eliminate primer dimers with a dissociation curve. ABsolute SYBR Green (Fisher Scientific, Pittsburgh, PA) was used to detect double stranded cDNA and real time PCR was run on the Applied Biosystems 7900HT system (Applied Biosystems, Foster City, CA). Gene expression changes were assessed with the following formula: Fold change in gene expression = (2−ΔΔCt) (Livak and Schmittgen, 2001).
Power analysis of the Affymetrix GeneChip Rat Exon 1.0 ST Array was conducted with R (R Development Core Team, 2010). After RMA-Sketch normalization, data was imported into R and analyzed with the samr package (Tusher, et al., 2001). Power analysis was determined with the samr package (Tibshirani, 2006).
2.8. Statistical Analyses
In all figures, values are expressed as mean ± SEM. GraphPad Prism 4.0 (GraphPad Software, La Jolla, CA) and SPSS 17.0 (IBM, Armonk, NY) were used to conduct non-microarray statistical analysis. Experimental differences were analyzed with a 2-way ANOVA (prenatal stress x prenatal escitalopram) followed by a Dunnett’s post hoc test where appropriate. A 3-way ANOVA was used to analyze prenatal escitalopram x prenatal stress x adult stress effects. Jugular catheter sampling measures were analyzed with a 3-way ANOVA with repeated measures on time using a Huynh-Feldt correction. Differences were considered significant if p < 0.05. R (R Development Core Team, 2010) was used to conduct statistical analysis of microarray data.
3. Results
3.1. Offspring Endpoints after Chronic Unpredictable Mild Stress
The experimental time line is depicted in Figure 1. Juvenile and adolescent weight gain of the offspring was not adversely affected by either treatment, indicating that maternal stress and/or escitalopram exposure does not interfere with normal growth (Table 1, top). Basal measures of pituitary and adrenal output were assessed in adulthood. ACTH concentrations in the prenatal stress alone group were somewhat lower than the other groups (Bonferroni post hoc test, p < 0.01). Plasma corticosterone concentrations were increased due to prenatal escitalopram treatment (F(1,63) = 7.1; p < 0.01); however, these increases, as well as the observed change in ACTH concentrations (vide supra), were well within the ranges routinely observed at trough baseline in nonstressed rats and the physiological significance of these changes are unclear. Adrenal weights were not significantly different.
Table 1.
Physiologic measures in adulthood.
| SAL | ESCIT | S-SAL | S-ESCIT | |
|---|---|---|---|---|
| Maternal Drug Conc on G15 (ng/mL) | <0.5 ± 0.07 (9) | 40.9 ± 3.38 (8) | <0.5 ± 0.23 (10) | 35.1 ± 2.70 (10) |
| PND 25–81 Rate of Weight Gain (g/day) | 6.9 ± 0.1 (27) | 7.0 ± 0.1 (18) | 7.1 ± 0.1 (26) | 6.7 ± 0.3 (29) |
| ACTH (pg/mL) | 28.2 ± 1.9 (26) | 24.5 ± 1.1 (16) | 20.4 ± 1.1 (16)* | 29.2 ± 2.1 (26) |
| Corticosterone (ng/mL) | 11.6 ± 1.1 (22) | 19.8 ± 4.6 (12)# | 10.8 ± 1.4 (15) | 15.0 ± 2.0 (18)# |
| Adrenal Weight (mg) | 57.4 ± 3.0 (26) | 61.4 ± 3.1 (18) | 60.8 ± 2.4 (18) | 61.7 ± 2.6 (26) |
| Glucose (mg/dL) | 102 ± 1.4 (26) | 101 ± 2.0 (18) | 101 ± 2.0 (18) | 102 ± 2.4 (25) |
| Insulin (μU/mL) | 3.4 ± 0.3 (26) | 2.8 ± 0.2 (18) | 2.7 ± 0.2 (18) | 2.4 ± 0.2 (25) ** |
| Insulin Resistance (IR) | 0.99 ± 0.08 (26) | 0.80 ± 0.05 (18) | 0.80 ± 0.06 (18) | 0.69 ± 0.060 (25) ** |
| Heart Weight (g) | 1.83 ± 0.05 (7) | 1.80 ± 0.08 (10) | 1.70 ± 0.03 (10) | 1.75 ± 0.04 (10) |
| RV:LV + Septum | 0.25 ± 0.01 (7) | 0.25 ± 0.01 (10) | 0.26 ± 0.01 (10) | 0.25 ± 0.01 (10) |
Groups were based on treatment to the pregnant dam throughout gestation: SAL (saline treatment during gestation), ESCIT (escitalopram exposure during gestation), S-SAL (chronic unpredictable mild stress + saline during gestation), S-ESCIT (chronic unpredictable mild stress + escitalopram during gestation). Individual males were analyzed for all other endpoints on PND90–95. All endpoints in Table 1 utilized two pups per litter. Data are mean ± SEM (N).
main effect due to prenatal stress,
p < 0.05,
p < 0.01 with a Dunnett’s post hoc Test.
Previous studies have shown a link between metabolic syndrome and prenatal stress (Li et al., 2010; Tamashiro et al., 2009); therefore, we examined glucose, insulin, and insulin resistance in adulthood. At baseline and after ad libitum food availability overnight, blood glucose was not different between groups. Plasma insulin was decreased due to prenatal stress (F(1,77) = 4.3; p < 0.05) and there was a trend of a decrease in plasma insulin due to prenatal escitalopram (F(1,77) = 3.4; p = 0.07). Insulin resistance, assessed using the HOMA-IR calculation, was decreased by both prenatal stress (F(1,75) = 4.1; p < 0.05) or prenatal escitalopram (F(1,75) = 4.3; p < 0.05) (Table 1, bottom). As with the basal ACTH and corticosterone measures, these were not considered to be physiologically significant.
An increased risk of offspring pulmonary hypertension following antidepressant exposure during pregnancy has been reported in human and animal studies (Chambers et al., 2006; Fornaro, et al., 2007). An assessment of pulmonary hypertension was conducted by measuring the right ventricle: left ventricle + septum weight ratio as previously described (Fornaro, et al., 2007). Neither total heart weight nor RV:LV + septum ratio was affected by prenatal treatments (Table 1, bottom).
3.2. Behavioral Characterization of Male Adult Offspring
Behavioral characterization was carried out to determine any phenotypic behavioral differences that arise from prenatal exposure to stress and/or escitalopram. Where denoted, an additional manipulation of chronic adult stress was used to investigate susceptibility or resistance to stress in adulthood. Several behavioral endpoints associated with locomotion, exploratory behavior, and risk-taking behavior were measured and analyzed but none reached statistical significance. The most representative measurement for each test is described in this section and displayed in Figure 2. In the elevated plus maze, the defensive withdrawal test, the sucrose consumption test, and the open field test, prenatal treatments had no effect on behavioral measures (Fig. 2A–D). In the marble burying test, the number of three quarter or fully buried marbles were unaltered due to prenatal stress (F(1,57) = 0.2; p > 0.05) or prenatal escitalopram (F(1,57) = 0.9; p > 0.05), however acute restraint stress challenge before the test decreased marble burying behavior for all treatment groups compared to rats not exposed to acute restraint (F(1,57) = 8.1; p < 0.01) (Fig. 2E). The magnitude of the responses to acute restraint stress challenge was not different amongst the various prenatal exposures. Neither prenatal stress, prenatal escitalopram, nor the response to acute restraint stress challenge altered the acoustic startle response in terms of percent habituation or initial startle amplitude (Fig. 2F).
Fig. 2.

Behavioral characterization of adult male offspring prenatally exposed to stress and/or escitalopram. Males were tested in adulthood in several tests for anxiety-like and depressive-like behavior (A–C). A separate cohort was challenged by exposure to 15 minutes of restraint before the test (white bars; D–F). Data is presented for defensive withdrawal (A), elevated plus maze (B), sucrose consumption test (C), open field test (D), marble burying test (E), and acoustic startle response (F). Data are mean ± SEM, N = 6–12 per group. SAL (saline treatment during gestation), ESCIT (escitalopram exposure during gestation), S-SAL (chronic unpredictable mild stress + saline during gestation), S-ESCIT (chronic unpredictable mild stress + escitalopram during gestation).
3.3. Serial Blood Sampling after Acute Stress
An air puff startle was used to assess pituitary (ACTH) and adrenal (corticosterone) responsiveness to a mild stressor in adulthood following the various prenatal exposures. The air puff startle increased plasma ACTH in all groups (F(1.52,36.39) = 25.3; p < 0.001) with no significant response differences due to prenatal escitalopram. Prenatal stress showed a trend towards increased ACTH responsiveness compared to the nonstressed groups (2-way ANOVA comparison of stress x drug, F(1,24) = 4.1; p = 0.055) (Fig. 3A). Cumulative output over 90 minutes was assessed using area under the curve calculations. Prenatal escitalopram did not alter cumulative plasma ACTH (F(1,28) = 0.03; p > 0.05), however prenatal stress produced a trend of increased total ACTH secretion (F(1,28) = 3.0; p = 0.095).
Fig. 3.

HPA axis endocrine response following acute stress in adult male offspring prenatally exposed to stress and/or escitalopram. Jugular catheters were implanted in adult males and the HPA response to an acute air puff startle (A, B) or restraint stress (C–E) was assessed. Data are mean ± SEM, N = 9–16 per group. SAL (saline treatment during gestation), ESCIT (escitalopram exposure during gestation), S-SAL (chronic, unpredictable, mild stress + saline during gestation), S-ESCIT (chronic, unpredictable, mild stress + escitalopram during gestation).
Acute air puff startle increased plasma corticosterone in all groups (F(4.36,104.64) = 60.2; p < 0.001) (Fig. 3B). The response was not different between any of the prenatal treatments, however there was a trend over time for prenatal stress to increase plasma corticosterone concentrations via a more prolonged response (F(4.36,104.64) = 2.3; p = 0.062) (Fig. 3B). Cumulative adrenal output was assessed by calculating the plasma corticosterone area under the curve. The corticosterone area under the curve over 90 minutes was unaffected by prenatal stress (F(1,27) = 2.1; p > 0.05) or prenatal escitalopram (F(1,27) = 0.1; p > 0.05). An acute restraint stress was used to evaluate the adrenal response to a heterotypic stressor. Acute restraint increased plasma corticosterone in all groups regardless of a history of prenatal stress (F(4.54,81.78) = 40.2; p < 0.001) (Fig. 3C). There was a trend of prenatal stress exposure to return corticosterone concentrations towards baseline more quickly following acute restraint (F(1,18) = 4.0; p = 0.06). This trend was also reflected by the area under the curve calculations: prenatal stress exposed rats had decreased cumulative plasma corticosterone over the 90 minute sampling period (F(1,21) = 7.6; p < 0.05). ACTH sampling was not possible in this experiment.
3.4. Microarray Analysis of the Amygdala, Hippocampus, and Hypothalamus
Microarray analysis was used to examine differentially regulated transcripts in response to prenatal exposures. Post-normalization analysis revealed that mean intensity of each chip was unaffected by chip number or chip run in the robust multiarray average normalization (F(17, 2045062) = 0.2; p = 1.0). Hierarchical clustering from hippocampus, hypothalamus and amygdala showed grouping of samples based on brain region as expected (Fig. 4). An initial microarray SAM analysis identified several differentially regulated transcripts due to prenatal treatments (Fig. S1). A post-hoc power analysis was performed to ensure adequate sample size. A SAM power analysis with an effect size of 1.5 fold, a false discovery rate of 10%, and an N equal to three revealed that approximately 100–500 differentially-regulated transcripts could be reliably reported (Fig. S2). Because all comparisons revealed less than 500 differentially regulated transcripts, the initial microarray was underpowered. Subsequent power analysis determined that an N of six or greater could reliably detect at least ten differentially regulated transcripts while keeping the Type I error below 0.01. To satisfy the power requirements, a second microarray was run using a separate cohort of animals having undergone the same prenatal exposures, but the amygdala was excluded from further iterations (Fig. 5A). Hierarchical clustering revealed that the first and second microarrays clustered separately (Fig. 5B and 5C), precluding direct comparison. Meta-analysis was run with Fisher’s Method of combining p-values using the p-values obtained in the SAM analysis. When multiple comparisons were adjusted for using a Bonferroni correction, no genes reached statistical significance. To relax the Bonferroni correction, regions of interest were filtered with IPA software to only analyze highly expressed transcripts in the hippocampus or hypothalamus (6,840 transcripts in the hippocampus, 7,339 transcripts in the hypothalamus). Using this relaxed Bonferroni correction, no transcripts reached statistical significance.
Fig. 4.

Heat map and hierarchical clustering of microarray results clustered by brain region. Data from the first microarray was used to generate the heat map because the second microarray did not cluster with the first array (Fig. 5). Each lane within each brain region represents an individual animal. As expected, analysis showed distinct clustering by brain region, indicating tissue-specific differential transcription.
Fig. 5.

Hierarchical clustering of microarray results by specific brain region. Hierarchical clustering in the amygdala (A), hippocampus (B), and hypothalamus (C) was performed to determine clustering of treatment groups and initial versus follow-up microarrays. For the top “Escit” row, a (−) is indicative of a minipump delivering saline during gestation (SAL) while a (+) is indicative of a minipump delivering escitalopram oxalate (ESCIT). For the lower “Stress” row, a (−) is indicative of the offspring’s mother exposure to regular handling (S-SAL) while a (+) is indicative of the offspring’s mother exposed to chronic, mild stress (S-ESCIT). Clustering analysis showed separate clustering of the initial and follow-up microarray likely due to differing chip lot numbers, time between runs, and/or environmental factors. While these factors precluded direct comparison in a SAM analysis, a meta-analysis was used to determine differential expression that minimizes the influence of extraneous variables.
3.5. Real-Time PCR Analysis of the Hippocampus
Real time PCR was used to complement the hippocampal microarray data and determine if more subtle differentially regulated transcripts that would not be detected using the 1.5 fold threshold were present. Target genes were selected based on the corticotropin-releasing factor (CRFergic) pathway (Fig. 6A), the serotonergic pathway (Fig. 6B), and markers of neural plasticity (Fig. 6C), all pathways promulgated to play plausible roles in mediating stress and/or antidepressant medication neurobiology. An additional manipulation of twelve days of chronic immobilization stress in adulthood was used in one half of the animals to determine if a stress-susceptible phenotype existed compared to basal (i.e., no stress) expression. Using a Bonferroni correction to account for multiple hypothesis testing (p < 0.003), no genes reached statistical significance for differential expression using a 2-way ANOVA (prenatal exposure x adult stress; p > 0.003).
Fig. 6.

Real time PCR of hippocampal tissue from adult male offspring with or without subchronic stress in adulthood. Following prenatal treatments, half of the male offspring were exposed to twelve days of restraint stress in adulthood in order to investigate stress-activated pathways. Targets related to the CRFergic pathway such as corticotropin-releasing factor (Crf), CRF binding protein (Crfbp), CRF1 receptor (Crfr1), glucocorticoid receptor (Nr3c1), mineralocorticoid receptor (Nr3c2), proopiomelanocortin (Pomc), and FKBP51 (Fkbp5) are depicted in panel (A). Targets related to the serotonergic pathway such as 5-HT1A receptor (5ht1a), 5-HT1B receptor (5ht1b), 5-HT2A receptor (5ht2a), p11 (S100a10), and the serotonin transporter (Slc6a4) are depicted in panel (B). Markers of neural plasticity such as brain-derived neurotrophic factor (Bdnf), TrkB receptor (Ntrk2), S100 calcium binding protein B (S100b), and AKT1 (Akt1) are depicted in panel (C). No significant effects of prenatal stress, prenatal escitalopram, or the combination were identified (p > 0.05). Data are mean ± SEM, N = 8–10 per group.
4. Discussion
The use of psychotropic medications may be clinically necessary in some patients and discontinuation during pregnancy is unrealistic and significantly increases the risk for recurrent depression during pregnancy (Cohen et al., 2006). The majority of epidemiological studies report that the risks to the infant are relatively low (Wisner et al., 1999). These studies have largely focused on birth defects, especially septal heart defects. Others have shown that the risk to the infant is typically overestimated and no higher than antibiotic or gastric medication use during pregnancy: approximately 1–3% (Bonari, et al., 2005). In our study, we found no immediate or long-term gross teratological effects from prenatal exposures.
Physiological measures of the male offspring showed almost no changes in growth, development, baseline endocrine measures, or gross histological differences compared to animals born to dams unexposed to stress or escitalopram (Table 1). While measures of ACTH and insulin were statistically significant, we believe these small differences are not physiologically meaningful. Stress-induced plasma levels of ACTH are typically observed above 75 pg/mL (Fig. 3A) and elevated plasma insulin levels are usually documented above four ng/mL (Tamashiro, et al., 2009).
A shown in Figure 2, when assessed in adulthood in males, we observed no changes in behaviors routinely utilized in rodents to study effects on measures of anxiety or affect. Additionally, a stress-challenge in these animals did not reveal any increased susceptibility and/or resiliency associated with any of the prenatal exposures. In the defensive withdrawal test, a high amount of variance may have contributed to a lack of statistical differences. Behavioral testing may have also been affected by the testing experience as seen in the high amount of time in the open arm of the elevated plus maze. A small difference in marble-burying behavior was found but there is no likely biological significance when compared to other behavioral tests run in this study. While we expected an increase in anxiety-like measures from the stress-challenge, these behavioral tests were originally developed for assessing efficacy of antidepressants and anxiolytics. Chronic stress would likely have a robust effect but the present study sought to determine the effect of in utero exposures on acute stress reactivity.
Gingrich and colleagues have investigated the use of serotonin reuptake inhibitors during postnatal development and find long-term behavioral alterations in mice (Ansorge et al., 2008; Ansorge et al., 2004). Our results do not replicate their findings, however there are several key differences, which may account for this discrepancy. Perhaps most importantly, these studies employed a dosing regimen during the postnatal period (PND4–21) that has no temporal overlap with the present studies. Concise ontological data on human serotonergic function in the CNS is lacking and definitive mapping of our in vivo exposure and their postnatal exposure onto human fetal/infant serotonergic development is not possible at present (Avishai-Eliner et al., 2002). Importantly, the distribution of serotonergic neurons in rats is similar on G19 to adult groupings (Lidov and Molliver, 1982). While the dosing regimens utilized by Gingrich and colleagues postnatally lead to “therapeutic” levels of SERT transporter occupancy and that drug exposure is maintained throughout their studies, we argue that this may not be fully relevant to humans because, following birth, exposure via lactation is low and infants are unlikely to receive any type of therapeutic dose of antidepressants for medical reasons for the first few years of life. Additionally, we observe that peak escitalopram concentrations following subcutaneous injection are up to 12-fold higher than that observed during continuous exposure (Bourke, et al., 2013). Similar peaks, of unknown magnitude, should also be expected following subcutaneous or intraperitoneal drug administration. These transient peaks would clearly be suprapharmacological, possibly toxic and may be responsible for their findings but there is no data to support this possibility at present.
Maternal depression poses significant risks to the developing fetus and can produce dysregulation of the child’s stress response measured as behavioral and salivary cortisol alterations (Brennan et al., 2008; Oberlander et al., 2008). Previously, we have shown that chronic unpredictable mild stress produces an increase in baseline and peak corticosterone in pregnant animals (Bourke, et al., 2013). In the adult male offspring, we observed no major differences in baseline plasma ACTH or corticosterone concentrations. An air-puff startle stress challenge revealed a nonsignificant trend towards increased ACTH and corticosterone responses in animals exposed to prenatal stress with no protective effects of escitalopram coexposure (Fig. 3A, B). In a similar cohort of rats, a restraint stress challenge did not reveal any increased plasma corticosterone responsivity in animals exposed to prenatal stress (Fig. 3C). Indeed, prenatal stress was associated with decreased total corticosterone responses (area under the curve) over the 90 minute time period with the curves showing similar peak responses but a faster return towards baseline in the prenatally stressed rats possibly indicative of more efficient negative feedback (no changes were observed in hippocampal glucocorticoid receptor or chaperone protein mRNA that might explain this effect; Fig. 6A). The failure of this study to replicate other studies investigating the long-term effects of prenatal stress may be due to developmental timing of the stress exposure. For example, data suggest that early gestational stress may be the most detrimental to the infant (Mueller and Bale, 2008). Alternatively, other groups have shown that the last trimester in pregnancy leads to the greatest difference in HPA responsivity (Vallee et al., 1997). A last possibility for the deviation of our results from others could be the nature of the stressor. Other groups have shown that repeated exposure to the same stressor elicits a greater difference from controls than varied stress (Richardson et al., 2006).
Although stress and antidepressants can plausibly target, and alter, parts of the HPA axis, the serotonergic system and/or neurotrophic factors, downstream targets affected by prenatal exposure cannot adequately be determined. Therefore, microarray analysis was initially performed to examine predictable and unpredictable targets for further study. Microarray analysis of the hippocampus and hypothalamus did not detect any differences in gene expression (Figures 4–5). The amygdala was not repeated so this region may still represent an area to explore with a suitably powered study. It should be noted that our two studies clustered separately and produced a significant “batch effect” as others have observed (Yang et al., 2008). Steps were taken to minimize this effect (concurrent array normalization) but the individual arrays still clustered separately. Fisher’s method was used to circumvent this confounding variable and these results are still a reliable measure of the transcriptome. While microarray analysis of the entire transcriptome is indeed a powerful tool, it is limited by certain constraints. Notably, microarray analyses employ a 1.5- or 2-fold change cut-off as a convention. Gene expression differences that are slightly below this cut-off may still have profound effects on the offspring. While other studies examining prenatal stress exposure using RT-PCR have found changes in gene expression (Mueller and Bale, 2008; Nyirenda et al., 2009; Pankevich et al., 2009), our study using a transcriptome-wide and target-specific approach (Figure 6) failed to reproduce these findings. However, some have found that these expression changes are dependent on developmental timing of the stressors (Mueller and Bale, 2008) and our stress model extended from mid to late pregnancy.
Serotonin plays a well-established role in neurodevelopment by affecting neuronal migration and recent data is clarifying the underlying mechanism(s) (Riccio et al., 2009). In addition to endogenous fetal serotonin, the delicate balance of maternal-placental-fetal interactions of the serotonin system supports a role of serotonin in fetal development (Bonnin et al., 2011). In animals, postnatal exposure to SSRIs utilizing dosing strategies different from our study alters behavior and cortical development (Ansorge, et al., 2008; Pawluski et al., 2012; Pawluski et al., 2012; Rayen et al., 2011; Simpson et al., 2011). To our knowledge, no studies have examined long-term changes in gene expression due to prenatal antidepressant exposure. Other studies have found differences in monoamine receptor density, transporter binding, monoamine concentrations and turnover (Cabrera-Vera and Battaglia, 1998; Cabrera-Vera, et al., 1997; Cabrera and Battaglia, 1994; Forcelli and Heinrichs, 2008; Henderson and McMillen, 1993). Except in the case of Forcelli 2008, these studies have not carefully controlled for the clinical relevance of the antidepressant exposure as is the case here. These studies have found subtle differences, which are mostly below 50% and would not be detectable with a microarray if the change were due to altered transcription. Additionally, these autoradiography and biochemical studies have found variable effects on the serotonergic system but the neurobiological consequences are unclear. Using real time PCR to investigate these discrete pathways and to determine changes that would not be detectable with the fold change threshold used in the microarray, we did not find that prenatal escitalopram exposure had any effect on targets in the hippocampus using our model of clinically relevant exposure to escitalopram during pregnancy. Using an epigenome-wide sequencing approach, Schroeder and colleagues found that neither maternal psychiatric illness in a clinical population nor prenatal antidepressant exposure are associated with methylation differences in umbilical cord blood (Schroeder et al., 2012) that is primarily of fetal origin.
The goal of the present study was to evaluate the long-term effects of prenatal stress and antidepressant exposure on adult male offspring. It should be noted that these results only apply to our particular developmental time frame for both exposure and testing. Testing of the exposed offspring in adulthood was chosen based on the fact that mammals, both humans and laboratory rodents, spend the majority of their lifespan as adults and we may have missed changes that may have occurred earlier in life and dissipate by adulthood or changes that do not occur until late life. Alterations due to prenatal exposures may be detected during these time frames but testing in adulthood allows determination of the persistent changes that may be present at earlier time points but do not normalize by adulthood. The present study also focused on male offspring. A study of sex differences after prenatal exposures using the model described in this study may reveal sex-specific differences based on the in utero environment.
5. Conclusion
In conclusion, we employed a novel stress model of maternal depression and its clinically relevant treatment with the antidepressant escitalopram to examine the long-term effects in the offspring. Prenatal exposures produced no alterations in behavior, transcriptome-wide gene expression, or gene expression of several neuropsychiatric targets in the hippocampus of male rats. Subtle differences were observed in the pituitary and adrenal function of prenatally stressed rats, indicating possible changes in HPA axis function. For the endpoints examined here, prenatal escitalopram exposure produced no physiologically relevant alterations in the adult male offspring.
Supplementary Material
Highlights.
We used a model of prenatal stress and/or antidepressant exposure in rats
Prenatally-exposed adult males were examined for long-term changes
No substantial groups differences were observed in several endpoints
Acknowledgments
Escitalopram oxalate was generously provided by Lundbeck Research USA (Paramus, NJ). We would like to thank Alicia Smith, Ph.D. for her advice during analysis of the microarray data and Catherine Capello and Faketa Zeljenovik for their assistance in breeding, treating and raising the animals.
Role of Funding Source
This work was supported by the National Institutes of Health National Institute of Mental Health [Grant 77928] (ZNS and MJO), the National Institute of Environmental Health Sciences [Grant 12870] (CHB), the National Center for Research Resources [Grant 012870] (CHB), the Howard Hughes Medical Institute [Grant 5600672] (CHB), and in part by the Emory Biomarker Service Center. Funding sources had no role in study design, data collection, data analysis, interpretation of data, manuscript preparation, or the decision to submit the manuscript for publication.
Abbreviations
- ACTH
adrenocorticotropic hormone
- CRF
corticotropin-releasing factor
- GN
gestational day N
- HPA
hypothalamic-pituitary-adrenal
- PNDN
postnatal day N
- SAM
significance analysis of microarray
Footnotes
Conflict of Interest Statement
CHB and DEO have no conflicts to disclose. ZNS has received research support from NIH, GSK, Pfizer and Wyeth, has served on speakers or advisory boards for Pfizer, Eli Lilly, Wyeth, BMS, and GSK, and has received honoraria from Eli Lilly, GSK, Pfizer, and Wyeth. GNN receives grant funding from NIMH, AHA, NARSAD, GSK, and Emory University. MJO has research grants from NIH, Eli Lilly, Lundbeck A/S, Cyberonics, Ortho-McNeil Janssen, AstraZeneca, Dainippon Sumitomo Pharma, Sunovion, and SK Life Sciences. He is a consultant for Takeda and RJ Reynolds. He receives consulting payments from H. Lundbeck A/S >$5,000 annually. This financial interest has been reviewed and is managed. He has a patent entitled: “Method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters” (US 7,148,027 B2).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade SE, Raebel MA, Brown J, Lane K, Livingston J, Boudreau D, et al. Use of antidepressant medications during pregnancy: a multisite study. Am J Obstet Gynecol. 2008;198:1–5. doi: 10.1016/j.ajog.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Morelli E, Gingrich JA. Inhibition of serotonin but not norepinephrine transport during development produces delayed, persistent perturbations of emotional behaviors in mice. J Neurosci. 2008;28:199–207. doi: 10.1523/JNEUROSCI.3973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram T. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonari L, Koren G, Einarson TR, Jasper JD, Taddio A, Einarson A. Use of antidepressants by pregnant women: evaluation of perception of risk, efficacy of evidence based counseling and determinants of decision making. Arch Womens Ment Health. 2005;8:214–20. doi: 10.1007/s00737-005-0094-8. [DOI] [PubMed] [Google Scholar]
- Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472:347–350. doi: 10.1038/nature09972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Capello CF, Rogers SM, Yu ML, Boss-Williams KA, Weiss JM, et al. Prenatal exposure to escitalopram and/or stress in rats: a prenatal stress model of maternal depression and its treatment. Psychopharmacology (Berl) 2013 doi: 10.1007/s00213-013-3030-z. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Neigh GN. Behavioral effects of chronic adolescent stress are sustained and sexually dimorphic. Horm Behav. 2011;60:112–120. doi: 10.1016/j.yhbeh.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Pargas R, Walker EF, Green P, Newport DJ, Stowe Z. Maternal depression and infant cortisol: influences of timing, comorbidity and treatment. J Child Psychol Psychiatry. 2008;49:1099–107. doi: 10.1111/j.1469-7610.2008.01914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Battaglia G. Prenatal exposure to fluoxetine (Prozac) produces site-specific and age-dependent alterations in brain serotonin transporters in rat progeny: evidence from autoradiographic studies. J Pharmacol Exp Ther. 1998;286:1474–1481. [PubMed] [Google Scholar]
- Cabrera-Vera TM, Garcia F, Pinto W, Battaglia G. Effect of prenatal fluoxetine (Prozac) exposure on brain serotonin neurons in prepubescent and adult male rat offspring. J Pharmacol Exp Ther. 1997;280:138–145. [PubMed] [Google Scholar]
- Cabrera TM, Battaglia G. Delayed decreases in brain 5-hydroxytryptamine2A/2C receptor density and function in male rat progeny following prenatal fluoxetine. J Pharmacol Exp Ther. 1994;269:637–645. [PubMed] [Google Scholar]
- Capello CF, Bourke CH, Ritchie JC, Stowe ZN, Newport DJ, Nemeroff A, et al. Serotonin transporter occupancy in rats exposed to serotonin reuptake inhibitors in utero or via breast milk. J Pharmacol Exp Ther. 2011;339:275–85. doi: 10.1124/jpet.111.183855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579–87. doi: 10.1056/NEJMoa052744. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Altshuler LL, Harlow BL, Nonacs R, Newport DJ, Viguera AC, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295:499–507. doi: 10.1001/jama.295.5.499. [DOI] [PubMed] [Google Scholar]
- Engelmann M, Thrivikraman KV, Su Y, Nemeroff CB, Montkowski A, Landgraf R, et al. Endocrine and behavioral effects of airpuff-startle in rats. Psychoneuroendocrinology. 1996;21:391–400. doi: 10.1016/0306-4530(96)00006-6. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, et al. Ensembl 2011. Nucleic Acids Res. 2011;39:800–806. doi: 10.1093/nar/gkq1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Heinrichs SC. Teratogenic effects of maternal antidepressant exposure on neural substrates of drug-seeking behavior in offspring. Addict Biol. 2008;13:52–62. doi: 10.1111/j.1369-1600.2007.00078.x. [DOI] [PubMed] [Google Scholar]
- Fornaro E, Li D, Pan J, Belik J. Prenatal exposure to fluoxetine induces fetal pulmonary hypertension in the rat. Am J Respir Crit Care Med. 2007;176:1035–40. doi: 10.1164/rccm.200701-163OC. [DOI] [PubMed] [Google Scholar]
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005;106:1071–83. doi: 10.1097/01.AOG.0000183597.31630.db. [DOI] [PubMed] [Google Scholar]
- Henderson MG, McMillen BA. Changes in dopamine, serotonin and their metabolites in discrete brain areas of rat offspring after in utero exposure to cocaine or related drugs. Teratology. 1993;48:421–30. doi: 10.1002/tera.1420480506. [DOI] [PubMed] [Google Scholar]
- Holson RR, Pearce B. Principles and pitfalls in the analysis of prenatal treatment effects in multiparous species. Neurotoxicol Teratol. 1992;14:221–228. doi: 10.1016/0892-0362(92)90020-b. [DOI] [PubMed] [Google Scholar]
- Institute for Laboratory Animal Resources, editor. Guide for the Care and Use of Laboratory Animals. National Research Council; Washington DC: 1996. [Google Scholar]
- Kugler K, Mueller L, Graber A. MADAM - An open source meta-analysis toolbox for R and Bioconductor. Source Code Biol Med. 2010:5. doi: 10.1186/1751-0473-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Olsen J, Vestergaard M, Obel C, Baker JL, Sorensen TIA. Prenatal stress exposure related to maternal bereavement and risk of childhood overweight. PLoS ONE. 2010:5. doi: 10.1371/journal.pone.0011896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidov HG, Molliver ME. Immunohistochemical study of the development of serotonergic neurons in the rat CNS. Brain Res Bull. 1982;9:559–604. doi: 10.1016/0361-9230(82)90164-2. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 2006;88:605–614. doi: 10.1016/j.physbeh.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigh GN, Owens MJ, Taylor WR, Nemeroff CB. Changes in the vascular area fraction of the hippocampus and amygdala are induced by prenatal dexamethasone and/or adult stress. J Cereb Blood Flow Metab. 2010;30:1100–1104. doi: 10.1038/jcbfm.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport DJ, Stowe ZN, Nemeroff CB. Parental depression: animal models of an adverse life event. Am J Psychiatry. 2002;159:1265–1283. doi: 10.1176/appi.ajp.159.8.1265. [DOI] [PubMed] [Google Scholar]
- Nyirenda MJ, Carter R, Tang JI, de Vries A, Schlumbohm C, Hillier SG, et al. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11beta-hydroxysteroid dehydrogenase type 1. Diabetes. 2009;58:2873–2879. doi: 10.2337/db09-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–9106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- Pankevich DE, Mueller BR, Brockel B, Bale TL. Prenatal stress programming of offspring feeding behavior and energy balance begins early in pregnancy. Physiol Behav. 2009;98:94–9102. doi: 10.1016/j.physbeh.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluski JL, Charlier TD, Fillet M, Houbart V, Crispin HT, Steinbusch HW, et al. Chronic fluoxetine treatment and maternal adversity differentially alter neurobehavioral outcomes in the rat dam. Behav Brain Res. 2012;228:159–68. doi: 10.1016/j.bbr.2011.11.043. [DOI] [PubMed] [Google Scholar]
- Pawluski JL, Rayen I, Niessen NA, Kristensen S, van Donkelaar EL, Balthazart J, et al. Developmental fluoxetine exposure differentially alters central and peripheral measures of the HPA system in adolescent male and female offspring. Neuroscience. 2012;220:131–41. doi: 10.1016/j.neuroscience.2012.06.034. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, R. A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2010. [Google Scholar]
- Rayen I, van den Hove DL, Prickaerts J, Steinbusch HW, Pawluski JL. Fluoxetine during development reverses the effects of prenatal stress on depressive-like behavior and hippocampal neurogenesis in adolescence. PLoS ONE. 2011;6:e24003. doi: 10.1371/journal.pone.0024003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio O, Potter G, Walzer C, Vallet P, Szabo G, Vutskits L, et al. Excess of serotonin affects embryonic interneuron migration through activation of the serotonin receptor 6. Mol Psychiatry. 2009;14:280–290. doi: 10.1038/mp.2008.89. [DOI] [PubMed] [Google Scholar]
- Richardson HN, Zorrilla EP, Mandyam CD, Rivier CL. Exposure to repetitive versus varied stress during prenatal development generates two distinct anxiogenic and neuroendocrine profiles in adulthood. Endocrinology. 2006;147:2506–17. doi: 10.1210/en.2005-1054. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Schroeder JW, Smith AK, Brennan PA, Conneely KN, Kilaru V, Knight BT, et al. DNA methylation in neonates born to women receiving psychiatric care. Epigenetics. 2012;7:409–14. doi: 10.4161/epi.19551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson KL, Weaver KJ, de Villers-Sidani E, Lu JY-F, Cai Z, Pang Y, et al. Perinatal antidepressant exposure alters cortical network function in rodents. Proc Natl Acad Sci U S A. 2011;108:18465–70. doi: 10.1073/pnas.1109353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashiro KLK, Terrillion CE, Hyun J, Koenig JI, Moran TH. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes. 2009;58:1116–1125. doi: 10.2337/db08-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrivikraman KV, Huot RL, Plotsky PM. Jugular vein catheterization for repeated blood sampling in the unrestrained conscious rat. Brain Res Brain Res Protoc. 2002;10:84–94. doi: 10.1016/s1385-299x(02)00185-x. [DOI] [PubMed] [Google Scholar]
- Tibshirani R. A simple method for assessing sample sizes in microarray experiments. BMC Bioinformatics. 2006;7:106. doi: 10.1186/1471-2105-7-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:71–74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone secretion. J Neurosci. 1997;17:2626–36. doi: 10.1523/JNEUROSCI.17-07-02626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisner KL, Gelenberg AJ, Leonard H, Zarin D, Frank E. Pharmacologic treatment of depression during pregnancy. JAMA. 1999;282:1264–1269. doi: 10.1001/jama.282.13.1264. [DOI] [PubMed] [Google Scholar]
- Yang H, Harrington CA, Vartanian K, Coldren CD, Hall R, Churchill GA. Randomization in laboratory procedure is key to obtaining reproducible microarray results. PLoS ONE. 2008:3. doi: 10.1371/journal.pone.0003724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.