

Abstract
Multiple genetic alterations are associated with prostate carcinogenesis. Tumor-suppressor genes phosphatase and tensin homolog deleted on chromosome 10 (Pten) and androgen upregulated gene 19 (U19), which encodes ELL-associated factor 2 (EAF2), are frequently inactivated or downregulated in advanced prostate cancers. Previous studies showed that EAF2 knockout caused tumors in multiple organs and prostatic intraepithelial neoplasia (PIN) in mice. However, EAF2-knockout mice did not develop prostate cancer even at 2 years of age. To further define the roles of EAF2 in prostate carcinogenesis, we crossed the Pten +/− and EAF2 +/− mice in the C57/BL6 background to generate EAF2 −/− Pten +/−, Pten +/−, EAF2 −/− and wild-type mice. The prostates from virgin male mice with the above four genotypes were analyzed at 7 weeks, 19 weeks and 12 months of age. Concomitant loss of EAF2 function and inactivation of one Pten allele induced spontaneous prostate cancer in 33% of the mice. Prostatic tissues from intact EAF2 −/− Pten +/− mice exhibited higher levels of phospho-Akt, -p44/42 and microvessel density. Moreover, phospho-Akt remained high after castration. Consistently, there was a synergistic increase in prostate epithelial proliferation in both intact and castrated EAF2 −/−Pten +/− mice. Using laser-capture microdissection coupled with real-time reverse transcription–PCR, we confirmed that co-downregulation of EAF2 and Pten occurred in >50% clinical prostate cancer specimens with Gleason scores of 8–9 (n = 11), which is associated with poor prognosis. The above findings together demonstrated synergistic functional interactions and clinical relevance of concurrent EAF2 and Pten downregulation in prostate carcinogenesis.
Keywords: prostate cancer, EAF2/U19, Pten, knockout
INTRODUCTION
Prostate cancer is the second most common malignancy in males worldwide and the second leading cause of cancer deaths in the US males.1 Despite significant research progress in recent decades, prostate cancer remains a major lethal disease. Elucidating the mechanisms of prostate carcinogenesis may lead to new approaches for the prevention, diagnosis, prognosis and/or treatment of prostate cancer. Prostate carcinogenesis involves multiple genetic and molecular abnormalities.2 Defining the functional significance of these abnormalities will provide new insights into the mechanisms of prostate carcinogenesis. Genetically engineered mouse models provide a powerful tool to define the importance of various genes and molecules in prostate carcinogenesis.3
One frequent molecular change in prostate carcinogenesis is the downregulation of a novel prostate tumor suppressor, ELL-associated factor 2 (EAF2), which is encoded by an androgen upregulated gene 19 (U19).4 Our previous studies showed EAF2 downregulation in ~80% of advanced human prostate cancer specimens, and that EAF2 downregulation was associated with allelic loss, promoter hypermethylation and/or potential homozygous deletion. Ectopic expression of EAF2 induced apoptosis in prostate cancer cells both in the culture and xenografts, and also inhibited the growth of tumor xenografts.4 Consistent with its potential tumor suppressive role, EAF2-knockout mice with mixed genetic background developed late-onset lung adenocarcinoma, hepatocellular carcinoma, B-cell lymphoma and high-grade prostatic intraepithelial neoplasia (PIN), which is the putative precursor of prostate cancer.5 This raised the possibility that EAF2 inactivation may drive prostate cancer progression in the presence of other molecular abnormalities.
Another frequent molecular change in prostate carcinogenesis is the loss of tumor suppressor gene phosphatase and tensin homolog deleted on chromosome 10 (Pten). Pten is one of the best characterized tumor suppressors and is one of the most frequently mutated/deleted genes in multiple human cancers, including prostate cancer.6,7 Pten counteracts phosphoinositide-3-kinase signaling by balancing phosphatidylinositol 4, 5-phosphate and phosphatidylinositol 3, 4, 5-phosphate levels.8–10 Phosphatidylinositol 3, 4, 5-phosphate accumulation leads to the phosphorylation of downstream targets, including Akt, and subsequently modulates the activities of further downstream effectors and cellular biology functions, including proliferation, apoptosis, cell size, polarity, metabolism, adhesion, migration and angiogenesis.9,11,12 Complete Pten inactivation has been reported in about 20% of primary prostate tumors and up to 60% of prostate cancer metastases.8 Homozygous Pten deletion is embryonically lethal in the mouse model, indicating its essential role in early animal development. Pten heterozygous deletion mice developed severe hyperplastic and dysplastic lesions in different organs, including the prostate.13,14 Prostate-specific Pten homozygous knockout mice developed prostatic hyperplasia, murine PIN (mPIN) and ultimately prostate cancer, demonstrating its role as a tumor suppressor in the prostate.15–18 Because Pten is frequently mutated/inactivated in human prostate cancer along with other molecular and genetic abnormalities, Pten-knockout mice have been frequently used to generate compound gene knockout mice to evaluate the role of other genes in prostate cancer progression.3,15,19,20
In the present study, we have generated EAF2−/−Pten +/− mice and demonstrated synergism between EAF2 homozygous deletion and Pten heterozygous deletion in promoting prostate carcinogenesis. In addition, we have provided evidence for concurrent downregulation of EAF2 and Pten in advanced human prostate cancer specimens. These findings demonstrate a clinical relevance for the murine prostate cancer induced by concurrent EAF2 homozygous deletion and Pten heterozygous deletion.
RESULTS
EAF2−/− Pten +/− prostate was significantly enlarged in the mouse model
To determine whether EAF2 homozygous deletion and Pten heterozygous deletion can have synergistic effect on the prostate in vivo, we crossed EAF2 +/− mice5 with Pten +/− mice21 and generated wild-type, EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice. Each animal was genotyped by PCR analysis of tail genomic DNA and confirmed by repeating PCR analysis of muscle DNA after animal euthanasia (Figure 1a). At the beginning of the study, each experimental group consisted of nine virgin male mice. Animals were euthanized at 7 weeks, 19 weeks and 12 months of age to assess the impact of EAF2 homozygous deletion, Pten heterozygous deletion or both on prostate development, homeostasis and pathogenesis. One of each pair of prostate lobes was isolated and weighed. There was no significant difference in prostatic wet weight at 7 weeks of age between the four different experimental cohorts (Supplementary Figure S1A). At the age of 19 weeks, EAF2−/− Pten +/− prostate exhibited wet weight increase as compared with the wild-type prostate, with a trend in wet weight increase in the order of the anterior (AP), dorsal (DP) and ventral (VP) prostate, but no increase observed in the lateral prostate (LP) (Supplementary Figure S1B). At the age of 12 months, EAF2−/− and Pten +/− mice exhibited wet weight increase in the AP and VP as compared with the wild-type mice. EAF2−/− Pten +/− mice had significant wet weight increase in all the four prostate lobes at the age of 12 months (Figures 1b and c), suggesting that simultaneous deficiency in EAF2 and Pten could act synergistically to promote prostate growth.
Figure 1.

Increased prostate growth in EAF2−/− Pten +/− mice. (a) Representative genotyping PCR results for each genotype. W, wild-type. K, knockout. (b) Representative morphology of 12-month-old mouse anterior prostate with indicated genotypes. (c) Wet weight of 12-month-old mouse prostates with indicated genotypes (nine mice per group). *P<0.05 compared with other groups.
EAF2−/− Pten +/− mice developed prostate cancer
Histological analysis did not identify hyperplasia in the prostate at the age of 7 weeks in any of the mice (Supplementary Figure S2). At the age of 19 weeks, wild-type and EAF2−/− mice prostates did not exhibit any morphological change. Prostate hyperplasia was diagnosed in for out of nine (44%) Pten +/− mice. In contrast, nine out of nine EAF2−/− Pten +/− mice exhibited hyperplasia, but no mPIN was observed (Supplementary Figure S3). However, as shown in Figure 2a, mice at the age of 12 months exhibited obvious morphological changes in the prostate of EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice. EAF2 knockout prostate displayed prostate epithelial hyperplasia. Pten +/− prostate developed a cribiform phenotype within a pre-existing glandular lumen. Hyperplasia was much more significant in the EAF2−/− Pten +/− prostate. Most of the hyperplasia changes were focal. Overall, EAF2−/− prostate presented less extensive morphological changes than Pten +/− prostate, while morphologic changes were the most dramatic in EAF2−/− Pten +/− prostate.
Figure 2.

Histological changes of prostates in EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice. (a) Hematoxylin and eosin (H&E) staining of AP, DP, LP and VP prostates of wild-type, EAF2−/−, Pten +/− and EAF2−/− Pten +/− at 12 months. (b) H&E staining of epithelial hyperplasia in the LP, high-grade PIN in the VP, early carcinomas in the AP and VP in the EAF2−/− Pten +/− mice. (c) Incidence rate of the indicated phenotype in different prostate lobes from mice with indicated genotypes (nine mice per group). (d) Percentage of mice that displayed the indicated histological phenotype in prostates with indicated genotypes (nine mice per group). Original magnification × 20. Scale bars represent 100 μm.
Further histological analysis showed that three out nine (33%) EAF2−/− Pten +/− mice developed prostate cancer at 12 months of age, in addition to high-grade mPIN and extensive hyperplasia (Figure 2b). Prostate cancer was found in the AP (1/9), LP (2/9) and VP (2/9) of EAF2−/− Pten +/− mice, and extensive mPIN lesions were presented in all the four lobes of the same cohort. In contrast, no prostate cancer was found in Pten +/− or EAF2−/− mice. Pten +/− mice developed mPIN lesions frequently (8/9) (Figures 2b and c), with both high-and low-grade mPIN being found in the LP (8/9) and VP (3/9), but not in the AP or DP. EAF2−/− mice exhibited the same pattern but to a lesser degree, with two out of nine EAF2−/− mice being diagnosed with mPIN. Prostatic hyperplasia was found in all the four lobes of EAF2−/−, Pten +/− or EAF2−/− Pten +/− mice at the age of 12 months (Figure 2). As expected, some wild-type mice developed mild prostatic hyperplasia at 12-months of age (Figures 2c and d), which was much less severe and extensive than those observed in the knockout prostate (Figure 2a). No mPIN was found in the wild-type mice. The hyperplasia lesions, which developed in the LP exhibited epithelial cells piled up greater than nine cell layers deep with a still intact basement membrane. High-grade mPIN in the VP showed cellular pleomorphism, but the basement membrane remained intact. Cancer lesions were characterized by cellular and nuclear pleomorphism, loss of normal glandular architecture, neoplastic cells effacing the normal glandular architecture, particularly in the AP, loss of integrity of the basement membrane with small foci of necrosis and cellular debris in the VP.
A hallmark of prostate carcinogenesis is the loss of basal epithelial cells.22,23 To further characterize the lesions in the EAF2−/− Pten +/− prostate, we performed immunostaining of p63, a basal cell marker. All lobes of the wild-type prostate displayed the expected p63 staining pattern beneath the glandular epithelial layer (Figure 3). In all the lobes of EAF2−/− Pten +/− mice, p63 staining revealed no apparent basal cell loss in the mPIN lesions (Figure 3). As expected, loss of p63 staining was observed in a cancer lesion in the LP, which was identified by our pathologist LHR (Figure 3).
Figure 3.
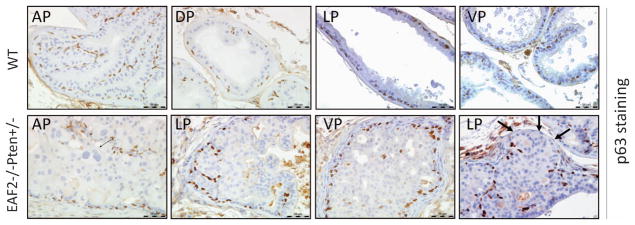
p63 staining of high-grade PIN/early carcinoma lesions in EAF2−/− Pten +/− mouse prostate at 12 months. p63 staining of high-grade PIN in AP and LP, VP and cancer in LP in wild-type and EAF2−/− Pten +/− mouse prostates. Original magnification × 40. Scale bars represent 50 μm. Arrows indicate loss of p63 staining.
EAF2−/− Pten +/− prostate displayed significant increases in epithelial proliferation in both testes-intact and castrated mice
As significantly increased prostate wet weight, hyperplasia, mPIN, and prostate cancer were observed in EAF2−/− Pten +/− mice, we investigated whether these phenotypic changes were associated with enhanced cellular proliferation. In the wild-type mice, proliferative indicator, Ki-67-positive luminal epithelial cells were rare and less than 1% in the prostate at 12 months of age (Figures 4a and b). EAF2 knockout alone slightly increased luminal epithelial cell proliferation, and Pten +/− prostate exhibited a more dramatic increase in the Ki-67-positive cells (Figures 4a and b). Combination of EAF2−/− and Pten +/− significantly increased Ki-67-positive cells in the hyperplastic regions (Figures 4a and b), suggesting that EAF2 and Pten double deficiencies had a synergistic effect in promoting prostate epithelial cell proliferation. The different staining patterns between basal epithelial cell markers p63, CK-14 and luminal epithelial marker androgen receptor verified that those Ki-67-positive proliferating cells were luminal epithelial cells and not basal epithelial cells. Thus the prostate cancer cells in the EAF2−/− Pten +/− mice displayed a proliferative luminal epithelial cell phenotype, as is characteristic of a majority of human prostate cancer cells (Supplementary Figure S4). Moreover, 9.7% of the luminal epithelial cells displayed positive Ki-67 staining in the EAF2−/− Pten +/− prostate 2 weeks after castration (Figures 4a and c), which was more than 10-fold higher than that in the wild-type control, indicating that hyperplastic or neoplastic EAF2−/− Pten +/− prostatic cells were highly proliferative under castration conditions.
Figure 4.

Enhanced epithelial cell proliferation in both intact and castrated EAF2−/− Pten +/− mouse prostate. (a) Representative Ki67 staining of the DP from intact or castrated wild-type, EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice. Prostate tissues were collected 2 weeks after castration. (b) Quantitative analysis of Ki67-positive cells of the DP from intact mice. (c) Quantitative analysis of Ki67-positive cells of the DP from castrated mice. Results are presented as Ki67-positive cells per 1000 epithelial cells (mean±s.d.). Nine mice were used for intact mice for each genotype. Four mice were used for castrated mice for each genotype. *P<0.05, when compared with other groups. Original magnification ×40. Scale bars represent 50 μm.
EAF2−/− Pten +/− mouse prostate displayed elevated levels of phosphorylated Akt, p44/42 and loss of Pten expression
One important mechanism for Pten loss to promote carcinogenesis is the activation of the Akt pathway.14,24–29 In order to further explore the potential mechanism of prostate carcinogenesis in EAF2−/− Pten +/− mice, we examined the status of phosphorylated Akt (pAkt). As shown in Figure 5a, wild-type and EAF2−/− mice prostates were pAkt negative (Figure 5a), indicating that EAF2 knockout alone does not activate Akt. In Pten +/− mouse prostates, pAkt was focally expressed in the hyperplasia regions with 100% penetrance (Figure 5a). Compared with the findings in Pten +/−, pAkt staining in the EAF−/− Pten +/− prostate was widespread, with large sections of prostatic tissues exhibiting intense staining and a significantly higher H-score than that in Pten +/− prostate tissues (Figure 5a, Supplementary Figure S5A). Also, EAF2−/− Pten +/− mouse prostates sustained the similar widespread and intense pAkt staining pattern 2 weeks after castration (Figure 5b, Supplementary Figure S5B), indicating that the activation of Akt pathway in EAF2−/− Pten +/− prostate was castration resistant.
Figure 5.

Synergistic activation of Akt and p44/42 signaling pathways in prostate epithelial cells of EAF2−/− Pten +/− mice. (a) Representative pAkt staining in AP and dorsal-lateral prostate (DLP) from intact mice with indicated genotype. (b) Representative pAkt staining in the AP and DLP from castrated mice with indicated genotype. (c) Representative phosphorylated p44/42 staining in the AP and DLP from intact mice with indicated genotype. Orignal magnification ×10, inset ×40. Scale bars represent 200 μm in ×10 and 50 μm in ×40.
The p44/42 mitogen-activated protein kinase, which is also negatively regulated by Pten, is a key transducer of proliferation signaling.29–31 Only a few sporadic prostate epithelial cells were phospho-p44/42 (p-p44/42)-positive in the wild-type prostates with 78% (7/9) penetrance (Figure 5c). A slight but not statistically significant increase in p-p44/42 expression was observed in eight out of nine EAF2−/− mice (Figure 5c), suggesting that the loss of function of EAF2 could activate p44/42 in the context of wild-type Pten function. In Pten +/− mouse prostates (Figure 5c), we detected extensive p-p44/42 expression, particularly in the hyperplasia and PIN regions with 100% penetrance. However, EAF2−/− on a Pten heterozygous background resulted in extensive and intensive p-p44/42 staining as evidenced by the increased H-score (Figure 5c, Supplementary Figure S5C). Compared with the single mutants, the increase in p-p44/42 staining intensity and extensiveness in the EAF2−/− Pten +/− mice was synergistic, suggesting a potential molecular basis independent of Akt signal pathway for the functional interaction of EAF2 and Pten loss of function.
To a lesser extent, the similar patterns of phospho-Akt and-p44/42 staining were also observed in 7 weeks and 19 weeks old mice prostates (Supplementary Figures S6 and S7, Figure 5). The extensiveness of phospho-Akt and -p44/42 staining in the single mutants and the EAF2−/− Pten +/− animals increased with age. These above findings suggested these changes were related to both the genotype and size of the lesion.
One potential mechanism for the widespread and intense pAkt staining could be the inactivation of the wild-type Pten allele in the EAF2−/− Pten +/− prostate. To test this possibility, we performed Pten staining and found significant reduction or loss of Pten staining in a few high-grade PIN lesions (Figure 6). As shown in Supplementary Figure S8, local reduction or loss of Pten expression in the LP was observed in four of nine EAF2−/− Pten +/− mice only, but not in other genotypes (P = 0.0412). Expression of phospho-Akt and –p44/42 was minimal in the wild-type and EAF2−/− mice and significantly increased in Pten +/− and EAF2−/− Pten +/− mice (see supplementary Figure S5). This finding suggested that Pten inactivation, in part, contributed to the widespread increased phospho-Akt and -p44/42 staining.
Figure 6.

Loss of Pten expression in prostate epithelial cells of EAF2−/− Pten +/− mice. Immunostaining of Pten was performed for indicated prostatic lobes of wild-type or EAF2−/− Pten +/− mouse prostates. Arrows indicates reduced or diminished Pten staining. Original magnification × 20 and ×40. Scale bars represent 100 μm in upper panel and 50 μm in lower panel.
EAF2 and Pten gene deletion co-operate to increase vascular density in the prostate
Given that EAF2−/− mice displayed an increase in blood vessel formation in the prostate,32 and tumor angiogenesis is critical to tumor growth and survival,33 we investigated whether concomitant loss of EAF2 and Pten had co-operative effects on microvessel density in the prostate. We examined microvessel density in the various genotypes of mice by immunostaining (Figure 7). We found that at the age of 12 months, the number of CD31-positive vessels increased in the prostates of EAF2−/− and Pten +/− compared with wild-type animals, and double deletion of EAF2 and Pten further increased microvessel density compared with the single mutants. This study indicated that prostate carcinogenesis induced by the concomitant loss of EAF2 and Pten is associated with increased angiogenesis. Additional studies will be needed to determine whether the increased microvessel density in the double mutant is a cause or result of carcinogenesis.
Figure 7.
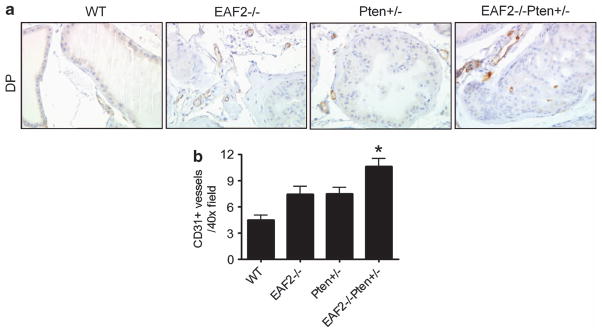
Elevated CD31 + blood vessel density in EAF2−/− Pten +/− mouse prostate. (a) CD31 immunostaining of vessels in prostate dorsal lobes from wild-type, EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice at age 12 months. Original magnification ×40. Scale bars represent 50 μm. (b) Quantification of CD31-positive vessels in the prostate. Data represent of nine mice per group (*P<0.05).
Phospho-JNK and WNT immunostaining were not altered in EAF2−/− Pten +/− mouse prostate
The c-Jun NH2-terminal kinases (JNK) signaling pathway has been identified as a functional target of the tumor suppressor Pten.34 To test whether the JNK signaling pathway was also synergistically activated in EAF2−/− Pten +/− mice, phospho-JNK expression was determined by immunohistochemistry in the prostates of 12 months old mice. We did not observe any obvious changes in phospho-JNK staining pattern among the different genotypes (Supplementary Figure S9). This suggests that the JNK signaling pathway is not involved in EAF2 and Pten functional interaction. As EAF2 has been implicated in the regulation of Wnt signaling, including Wnt4, Wnt5 and Wnt11,35–37 we chose one of the genes Wnt4 to evaluate whether there was any alteration in Wnt4 staining in EAF2−/− Pten +/− mouse prostate. As shown in Supplementary Figure S10, Wnt4 staining was comparable among the different genotypes. Enhanced Wnt4 expression was found in only one EAF2−/− Pten +/− mouse, suggesting that Wnt4 is unlikely a major mediator of functional interactions between EAF2 and Pten in the mouse prostate. However, Wnt signaling alteration through Wnt5 and/or Wnt11 cannot be ruled out.
EAF2/U19 and Pten are concurrently downregulated in human prostate cancer specimens with Gleason score 8 or higher
Both EAF2 and Pten are frequently downregulated in human prostate cancer specimens. Therefore, EAF2 and Pten are likely downregulated concurrently in some clinical prostate cancer specimens. To evaluate the expression levels of EAF2 and Pten in human prostate cancer specimens (Supplementary Table S1), we isolated prostate cancer cells using laser-capture microdissection and then performed real-time reverse transcription–PCR analysis of EAF2 and Pten gene expression. Using a twofold downregulation as a cut off, no co-downregulation (0/9) was observed in tumors with Gleason score of 7 or lower. EAF2 downregulation was observed in one out of nine, and no Pten downregulation (0/9) was observed in tumors with low Gleason scores (Figures 8a and b). In contrast, co-downregulation of EAF2 and Pten was detected in >50% (6/11) prostate cancer specimens with Gleason scores of 8–9. The difference of EAF2/Pten co-downregulation incidence between the two cohorts, one with Gleason 7 or lower (0/9) and the other with Gleason 8 or 9 (6/11), was statistically significant (P = 0.018) (Figure 8). These findings suggest that EAF2/Pten co-downregulation is associated with aggressive prostate cancer.
Figure 8.

Concurrent downregulation of EAF2 and Pten in human prostate cancer specimens with higher Gleason score. Treatment naïve fresh frozen human prostate samples were laser-capture microdissected and real-time PCR was performed on nine samples with Gleason grade 6–7, and 11 samples with Gleason grade 8–9. Gene expression of epithelial cells from the areas of cancer and normal adjacent tissue was analyzed by real-time reverse transcription–PCR. (a) EAF2 and PTEN are co-downregulated in prostate cancer epithelia Gleason grade 8 or higher (6/11). A change is defined as an increase or decrease of more than twofold. Fold change was determined by dividing tumor by normal Ct. Each point on the graph shows the fold change of one patient sample. The dotted line shows the cut off defined in our study as differentially regulated tumor compared with normal. (b) Percent of prostate cancer specimens with downregulation of EAF2 (1/9), but not Pten (0/9) in lower Gleason score samples. (c) Percent of prostate cancer specimens with co-downregulation (6/11) of EAF2 and Pten in higher lower Gleason score samples. A Fisher’s exact test was used to determine whether co-downregulation was significant in 6/11 high-Gleason-grade samples compared with lower-Gleason-grade samples (P = 0.018).
DISCUSSION
Using EAF2−/− Pten +/− mice and clinical prostate cancer specimens, this study provides strong evidence for EAF2 as a prostate tumor suppressor in the prostate, and demonstrates the importance of multiple genetic defects, specifically concurrent EAF2 and Pten downregulation, in prostate carcinogenesis. The findings described herein further substantiate the importance of exploring the mechanisms of EAF2 action, as well as its interactions with other important signaling pathways in prostate carcinogenesis.
Our studies argue that EAF2 is an important tumor suppressor in the prostate, particularly in the presence of other genetic abnormalities such as Pten downregulation. EAF2 inactivation increased hyperplasia and PIN but not prostate cancer in the mouse prostate (Figure 2).5 However, EAF2 deletion in Pten +/− mice induced an increased incidence of prostate cancer (3/9, 33%) compared with Pten deletion alone (0/9, 0%). These observations argue that EAF2 inactivation can exacerbate defects caused by Pten downregulation, leading to prostate cancer development and progression. It will be interesting to determine whether EAF2 inactivation can also synergistically enhance the defects caused by other genetic and/or molecular abnormalities in the prostate. Understanding the mechanisms by which EAF2 loss promotes prostate carcinogenesis, and the synergism between EAF2 inactivation and other genetic abnormalities may lead to new approaches to prevent and/or treat prostate cancer. As prostate cancers are associated with multiple genetic abnormalities,2 investigating the crosstalk between EAF2-regulated signaling and other pathways, such as Pten will be clinically relevant.
EAF2−/−, Pten +/− and EAF2−/− Pten +/− mice developed prostate, and no obvious growth defects or histological changes were observed in these mice as compared with the wild-type control prostate at 7 weeks of age. This observation suggested EAF2 and Pten defects did not affect early prostate development. The prostatic defect in the EAF2 and Pten-knockout mice was mild at 19 weeks of age, and became more severe at 12 months. Thus, EAF2 and/or Pten has an important role in the mature prostate. Individual EAF2 homozygous deletion or Pten heterozygous deletion could disrupt prostate homeostasis, but was not sufficient to drive the development of prostate cancer. The impact on prostate homeostasis by EAF2 homozygous deletion together with Pten heterozygous deletion was much more profound, leading to significant increases in prostate wet weight, elevated proliferation and eventually the development of prostate cancer.
The prostate cancers in EAF2−/− Pten +/− mice also appeared to resist castration. The prostate cancer in EAF2−/− Pten +/− was highly proliferative in the absence of androgens, as evidenced by a Ki-67 index, more than 10 times higher than that of castrated wild-type prostate. The Ki-67 index in EAF2−/− Pten +/− prostate was also significantly higher than that in EAF2−/− or Pten +/− prostate under castration conditions, corresponding to the increased cellular proliferation displayed by prostate cancer. Also, increased immunostaining of pAkt was widespread and intense in castrated EAF2−/− Pten +/− prostate, suggesting that Akt signaling remained active under castration conditions. Taken the above data together, prostate cancers induced by concomitant loss of EAF2 and Pten were resistant to castration in the mouse model, and loss of EAF2 could accelerate or enhance castration-resistance associated with Pten deletion.15,38 These observations also suggest that clinical prostate tumors with concurrent EAF2 and Pten downregulation are prone to castration-resistance.
We did not observe macroscopic tumors in other organs of the EAF2−/− Pten +/− mice on a C57/BL6 genetic background, suggesting that disruption of EAF2 and Pten pathways in these animals may not have synergism in organs other than the prostate. Genetic background appears to have an important role in carcinogenesis associated with EAF2 loss, because our previous studies showed that EAF2 knockout alone in a mixed genetic background was sufficient to cause lung adenocarcinoma, hepatocellular carcinoma and B-cell lymphoma.5 As these types of tumors were not detected in the EAF2−/− or EAF2−/− Pten +/− mice on a C57/BL6 background, the C57/BL6 genetic background appeared to be suppressive in EAF2 knockout-induced carcinogenesis.
EAF2−/− Pten +/− mice represent a clinically relevant prostate cancer model. EAF2 homozygous knockout mice developed prostatic hyperplasia and mPIN. Similarly, Pten heterozygous knockout mice developed prostatic hyperplasia and mPIN. Neither EAF2 knockout nor Pten heterozygous mice developed prostate cancer, even at an advanced age. However, 33% of mice with EAF2 homozygous deletion and Pten heterozygous deletion developed prostate cancer by the age of 12 months. These observations argue for a synergistic interaction between signaling pathways activated by EAF2 knockout and Pten heterozygous knockout, which subsequently drive the development of an invasive prostate carcinoma. The synergism is supported by the dramatic elevation of AKT and p44/42 phosphorylation (Figure 5), and increased vascularity (Figure 7) in the EAF2−/− Pten +/− mouse prostate as compared with the EAF2−/− or Pten +/− prostate. Microvessel density is considered to be highly correlated with prostate cancer progression.39 Elevated AKT and p44/42 phosphorylation are also common in advanced prostate cancer specimens, further substantiating the clinical relevance of the EAF2−/− Pten +/− prostate cancer model. EAF2−/− Pten +/− mice represent an excellent model for testing new therapeutics for a major subset of prostate cancers, which exhibit concurrent EAF2 and Pten downregulation.
In human prostate cancer specimens, co-downregulation of EAF2 and Pten was observed in >50% of prostate cancers with Gleason scores of 8–9 (n = 11), which is associated with poor prognosis. Moreover, the difference of EAF2/Pten co-downregulation between the two cohorts, one with Gleason 7 or lower and the other with Gleason 8 or 9, was statistically significant, suggesting that EAF2/Pten co-downregulation is associated with poor prognosis (Figure 8).
In summary, our findings argue that EAF2 is an important tumor suppressor in the prostate, and its inactivation can synergize with Pten heterozygous loss in driving prostate cancer development and progression. The EAF2−/− Pten +/− mouse model displayed an increased incidence in the prostate cancer, which was accompanied by activation of the AKT and p44/42 signaling pathways and enhanced angiogenesis. EAF2 and Pten were downregulated concurrently in more than half of the advanced human prostate cancer specimens examined, suggesting that this model represents a clinically relevant model of prostate cancer development and progression. This study provides a strong foundation to further elucidate the mechanisms of EAF2 action and its interaction with other important signaling pathways in prostate carcinogenesis.
MATERIALS AND METHODS
Generation of Pten +/− and EAF2−/− mice
Generation and characterization of Pten +/− (http://mouse.ncifcrf.gov/)21 and EAF2 +/−4,5 mice have been described previously. By crossing the Pten +/− mice on C57BL/6 genetic background and EAF2 +/− mice also on C57BL/6 genetic background, we produced EAF2−/−;Pten +/−, EAF2 +/+; Pten +/−, EAF2−/−;Pten +/+ and wild-type mice. Offspring were genotyped by PCR analysis of tail DNA. Genotyping was confirmed on muscle DNA after the animals were killed. Primers used for PCR analysis of EAF2 +/− mice were EAF2-F: 5′-CTGGATCTGGTTCTA ACTACCC-3′ (wild-type and mutated forward), EAF2-WR: 5′-CAAAGTTGATTTTGCTTCCTCTG-3′ (wild-type reverse), EAF2-KOR: 5′-GCC AGAGGCCACTTGTGTAG-3′ (mutated reverse). Primers used for Pten +/− mice genotyping were Pten-F: 5′-TTGCACAGTATCCTTTTGAAG-3′ (wild-type and mutated forward), Pten-WR: 5′-GTCTCTGGTCCTTACTTCC-3′ (wild-type reverse), Pten-KOR: 5′-ACGAGACTAGTGAGACGTGC-3′ (mutated reverse). The F:WR was used to detect the wild-type allele and F:KOR was used to detect the mutant allele. The PCR products are 508 bp (wild-type allele of EAF2), 231 bp (mutant allele of EAF2), 240 bp (wild-type allele of Pten) and 320 bp (mutant allele of Pten). All the animal studies outlined in this work were performed in accordance with the Institutional Animal Use and Care Committee.
Animal study design
Ten or nine virgin male mice with the above four different genotypes were killed at each time point of 7 weeks, 19 weeks and 12 months of age. For androgen ablation studies, five mice for each of the four genotypes at the age of 12 months were castrated and then killed 2 weeks later.
Histological and pathological analysis
Prostate tissues were formalin fixed and paraffin embedded. Sections were prepared for hematoxylin and eosin and antibody staining using routine procedures. The following antibodies were used for immunohistochemical staining: mouse monoclonal anti-p63 (sc-8431) and rabbit polyclonal anti-androgen receptor antibody (N-20, sc-816) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), rabbit monoclonal anti-Ki-67 (Epitomics Inc., Burlingame, CA, USA), rabbit monoclonal anti-Pten (138G6), rabbit monoclonal anti-phospho-Akt (Ser 473), rabbit polyclonal anti-phospho-p44/42, rabbit polyclonal anti-phospho-SAPA/JNK (Thr183/Tyr185) antibodies (Cell Signaling Inc., Danvers, MA, USA), rat monoclonal anti-CD31 (MEC 13.3, 550274, BD Biosciences, San Jose, CA, USA), rabbit polyclonal anti-WNT4 antibody (center) (Epitomics Inc). DBA (3,3′-Diaminobenzidine) (Sigma, St Louis, MO, USA) staining was performed according to manufacturer’s protocol (Zymed Laboratories Inc., San Francisco, CA, USA). Histological analysis was performed by an animal pathologist according to the standard described previously.40 For quantification of Ki-67 staining in the prostate tissues, the percentage of Ki-67-positive cells was calculated by counting totally 1000 epithelial cells in the field, which contained Ki-67-positive cells. Assessment of CD31-positive microvessel density was determined based on CD31-positive blood vessel count as previously described.32,41
LCM and real-time reverse transcription–PCR analysis of human prostate cancer specimens
Treatment naïve human prostate cancer specimens with Gleason score 6–7 or 8–9 were sectioned and evaluated by a board-certified pathologist. Prostate cancer cells and adjacent normal glandular epithelial cells were isolated by laser-capture microdissection using Leica LMD6000 Microsystems (Wetzlar, Germany). The expression of EAF2 and Pten genes was analyzed by ΔRT-qPCR. The oligos were used as follows: EAF2 sense 5′-CCAGGACTCCCAATCTTGTAAA-3′, antisense 5′-TAGCTTCTGCCTTCAGTTCTC TT-3′, hybridization oligo 5′FAM-CTCCATCTGAAGATAAGATGTCCCCAG CA-TAMRA3′, GAPDH sense 5′-CATGTTCGTCATGGGTGTGA-3′, antisense 5′-GGTGCTAAGCAGTTGGTGGT-3′, hybridization oligo 5′FAM-ACAGCCT CAAGATCATCAGCAATGCCTC-TAMRA3′ and Pten sense 5′-CGGAACTTGC AATCCTCAGT-3′, antisense 5′-AACTTGTCTTCCCGTCGTCT-3′, hybridization oligo 5′FAM-TGTGGTCTGCCAGCTAAAGGTGA-TAMRA3′. Reverse transcription and quantitative real-time PCR was done using Cells Direct One-Step qRT-PCR kit with ROX from Invitrogen (Carlsbad, CA, USA) and run on an ABI Step One Plus (Applied Biosystems Life Technologies, Grand Island, NY, USA). Data were analyzed using ABI Step One Plus software calculating ΔΔCt method comparing cancer to normal adjacent tissue.
Statistical analysis
All statistical analysis was performed using online GraphPad software (http://www.graphpad.com/). Statistical differences were determined by χ2 test without Yates correction in Figure 1c and Student’s t-test in Figures 4b and c, and Figure 7. A Fisher’s exact test was used to determine whether co-downregulation was significant in Figure 8. P<0.05 was considered significant.
Acknowledgments
This investigation was supported in part by the National Institutes of Health Grants R01 CA 120386, 5 R37 DK51193, 1 P50 CA90386 and T32 DK007774. This project used the UPCI Animal Facility and Tissue and Research Pathology Services (TARPS), and was supported in part by award P30CA047904. We thank Marie Acquafondata, Aiyuan Zhang, Katie Leschak and Dawn Everard (University of Pittsburgh, Pittsburgh, PA) for excellent technical assistance. We also thank all members of the Wang laboratory for insightful discussion and critical reading of the manuscript. LEP is a Tippins Foundation Scholar.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM, et al. Cooperativity of Nkx3. 1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci USA. 2002;99:2884–2889. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao W, Zhang Q, Jiang F, Pins M, Kozlowski JM, Wang Z. Suppression of prostate tumor growth by U19, a novel testosterone-regulated apoptosis inducer. Cancer Res. 2003;63:4698–4704. [PubMed] [Google Scholar]
- 5.Xiao W, Zhang Q, Habermacher G, Yang X, Zhang AY, Cai X, et al. U19/Eaf2 knockout causes lung adenocarcinoma, B-cell lymphoma, hepatocellular carcinoma and prostatic intraepithelial neoplasia. Oncogene. 2008;27:1536–1544. doi: 10.1038/sj.onc.1210786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 7.Bedolla R, Prihoda TJ, Kreisberg JI, Malik SN, Krishnegowda NK, Troyer DA, et al. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of PTEN expression and Akt activation. Clin Cancer Res. 2007;13:3860–3867. doi: 10.1158/1078-0432.CCR-07-0091. [DOI] [PubMed] [Google Scholar]
- 8.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 9.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382(Pt 1):1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–2065. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273:175–184. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 14.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 15.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 16.Backman SA, Ghazarian D, So K, Sanchez O, Wagner KU, Hennighausen L, et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc Natl Acad Sci USA. 2004;101:1725–1730. doi: 10.1073/pnas.0308217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005;65:5730–5739. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- 18.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad I, Sansom OJ, Leung HY. Advances in mouse models of prostate cancer. Expert Rev Mol Med. 2008;10:e16. doi: 10.1017/S1462399408000689. [DOI] [PubMed] [Google Scholar]
- 20.Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, et al. Nkx3. 1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63:3886–3890. [PubMed] [Google Scholar]
- 21.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bostwick DG, Brawer MK. Prostatic intra-epithelial neoplasia and early invasion in prostate cancer. Cancer. 1987;59:788–794. doi: 10.1002/1097-0142(19870215)59:4<788::aid-cncr2820590421>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 23.Bostwick DG. Prospective origins of prostate carcinoma. Prostatic intraepithelial neoplasia and atypical adenomatous hyperplasia. Cancer. 1996;78:330–336. doi: 10.1002/(SICI)1097-0142(19960715)78:2<330::AID-CNCR22>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene. 2008;27:5398–5415. doi: 10.1038/onc.2008.238. [DOI] [PubMed] [Google Scholar]
- 25.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 26.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335(Pt 1):1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 29.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. 2005;65:8096–8100. doi: 10.1158/0008-5472.CAN-05-1888. [DOI] [PubMed] [Google Scholar]
- 31.Koul HK, Maroni PD, Meacham RB, Crawford D, Koul S. p42/p44 Mitogen-activated protein kinase signal transduction pathway: a novel target for the treatment of hormone-resistant prostate cancer? Ann N Y Acad Sci. 2004;1030:243–252. doi: 10.1196/annals.1329.030. [DOI] [PubMed] [Google Scholar]
- 32.Pascal LE, Ai J, Rigatti LH, Lipton AK, Xiao W, Gnarra JR, et al. EAF2 loss enhances angiogenic effects of Von Hippel-Lindau heterozygosity on the murine liver and prostate. Angiogenesis. 2011;14:331–343. doi: 10.1007/s10456-011-9217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 34.Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer cell. 2007;11:555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 35.Maurus D, Heligon C, Burger-Schwarzler A, Brandli AW, Kuhl M. Noncanonical Wnt-4 signaling and EAF2 are required for eye development in Xenopus laevis. EMBO J. 2005;24:1181–1191. doi: 10.1038/sj.emboj.7600603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu JX, Hu B, Wang Y, Gui JF, Xiao W. Zebrafish eaf1 and eaf2/u19 mediate effective convergence and extension movements through the maintenance of wnt11 and wnt5 expression. J Biol Chem. 2009;284:16679–16692. doi: 10.1074/jbc.M109.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan X, Ji W, Mei X, Zhou J, Liu JX, Fang C, et al. Negative feedback regulation of Wnt4 signaling by EAF1 and EAF2/U19. PLOS One. 2010;5:e9118. doi: 10.1371/journal.pone.0009118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143:401–409. [PMC free article] [PubMed] [Google Scholar]
- 40.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 41.Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36:169–180. doi: 10.1007/BF00666038. [DOI] [PubMed] [Google Scholar]