

Abstract
Fluorescent proteins (FPs) are widely used in real-time single virus particle studies to visualize, track and quantify the spatial and temporal parameters of viral pathways. However, potential functional differences between the wild type and the FP-tagged virus may specifically affect particular stages in the virus life-cycle. In this work, we genetically modified the E2 spike protein of Sindbis virus (SINV) with two FPs. We inserted mApple, a red FP, or Venus, a yellow FP, at the N-terminus of the E2 protein of SINV to make SINV-Apple and SINV-Venus. Our results indicate that SINV-Apple and SINV-Venus have similar levels of infectivity and are morphologically similar to SINV-wild-type by negative stain transmission electron microscop. Both mutants are highly fluorescent and have excellent single-particle tracking properties. However, despite these similarities, when measuring cell entry at the single-particle level, we found that SINV-Apple and SINV-Venus are different in their interaction with the cell surface and FPs are not always interchangeable. We went on to determine that the FP changes the net surface charge on the virus particles, the folding of the spike proteins, and the conformation of the spikes on the virus particle surface, ultimately leading to different cell-binding properties between SINV-Apple and SINV-Venus. Our results are consistent with recent findings that FPs may alter the biological and cellular localization properties of bacterial proteins to which they are fused.
Keywords: Alphaviruses, fluorescent proteins, surface properties
1. INTRODUCTION
Alphaviruses are enveloped, positive-strand RNA viruses in the Togaviradae family. The alphavirus particle contains an internal nucleocapsid core which consists of the capsid protein surrounding the viral genome. On the surface of the particle are 80 trimeric spikes, anchored in the lipid membrane (Cheng et al., 1995; Jose, Snyder, and Kuhn, 2009; Zhang et al., 2011). Each spike in the mature virus is a trimer of two proteins, E2 and E1. The interactions between E2 and E1 are critical for particle entry. The E2 protein binds to the host cell receptor and the particle is endocytosed (Byrnes and Griffin, 1998; Davis et al., 1987; Marsh, Kielian, and Helenius, 1984). The E2-E1 heterodimer dissociates in response to the low pH environment of the endosome, and E1 mediates fusion between the host and viral membranes (Omar and Koblet, 1988; Wahlberg and Garoff, 1992). Both E2 and E1 undergo conformational and oligomeric changes following their dissociation from each other (Gibbons et al., 2003; Gibbons et al., 2004; Wahlberg et al., 1992). During particle assembly or when the spike is in the immature form, the E2 protein is attached to a smaller protein, E3 (Supplemental Figure 1). E3 is cleaved by the host protease furin in the trans-Golgi transport of the spikes to the plasma membrane (de Curtis and Simons, 1988; Jain, DeCandido, and Kielian, 1991). Although there are interactions between E3, E2, and E1 that are required for particles assembly and entry, there are a few positions within the E2 proteins where peptides and proteins can be inserted and viable virus is recovered (Mukhopadhyay et al., 2006; Navaratnarajah and Kuhn, 2007).
Fusing FPs to specific proteins of interest allows one to visualize, track, and quantify the spatial and temporal parameters of cellular processes in real-time at single particle resolution (Chudakov et al., 2010; Rizzo, Davidson, and Piston, 2009a). In the past two decades, new FPs have been isolated from several aquatic species to expand the spectral range that can be used during imaging. Although dimeric or tetrameric in nature, FPs have been engineered to be monomeric and minimize steric interferences, and thus thought to maintain the native structure and localization of the protein being tagged (Rizzo, Davidson, and Piston, 2009b).
The overall structure of the FPs is remarkably conserved regardless of its source. FPs are embodied by a rigid beta barrel consisting of 11 β-strands, linked through proline-rich loops that surround a central alpha helix and a chromophore consisting of three amino acids located in the center of the β-barrel (Kremers et al., 2011). The entire folded protein molecule is required for fluorescence. Because of their conserved structural properties, FPs with different spectral properties are often interchanged under the tacit assumption of equivalence in their physical and chemical properties.
In order to prepare Sindbis virus (SINV) (a species of the Alphavirus genus) for single-particle tracking measurements during interaction with the host-cell, we genetically fused the mApple FP and Venus FP between the E3 and E2 proteins making red-fluorescent SINV-Apple and yellow-fluorescent SINV-Venus, respectively. This strategy allowed for uniform expression of the FP on the viral surface at a known position selected to avoid disruption of virus assembly or altering the receptor-binding site of the virus to the host cell (Davis et al., 1987; Meyer and Johnston, 1993; Strauss et al., 1991). However, during single-particle tracking measurements, we found that despite the similarity in FP structure and the identical FP tag location within the genome, the initial binding event between SINV-Apple and SINV-Venus to the host cell was different. Nevertheless, once SINV-Apple or SINV-Venus entered the cell, all intracellular trafficking was statistically indistinguishable between the two viruses. Thus, difference in interaction only concerned the cell binding step suggesting possible variation between the spikes of SINV-Apple and SINV-Venus.
2. MATERIALS AND METHODS
2.1 Cells and viruses
BHK-21 cells (American Type Tissue Culture, Rockville, MD) were grown in minimal essential medium (MEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville, GA), non-essential amino acids, glutamine, and penicillin/streptomycin. Cells were grown at 37 °C in the presence of 5% CO2.
Sindbis virus strain TE12 (Lustig et al., 1988) was the parental virus in these studies. Wildtype and mutant virus cDNA clones were linearized with SacI and in vitro transcribed with SP6 polymerase (Owen and Kuhn, 1996). Virus stocks were generated by transfecting in vitro transcribed RNA into BHK-21 cells using Lipofectamine 2000 (Parrott et al., 2009). Media containing the virus particles was harvested 24–36 hours post-transfection and purified using two separate methods (Zhang et al., 2002). Briefly, media was collected and pelleted through a 5 ml 27% sucrose cushion in PBS at 130,000 ×g for 2.5 h at 4 °C. The pellet was resuspended in PBS and applied to a 15–60% linear sucrose gradient. The samples were centrifuged at 180,000 × g for 2.5 hours at 4 °C. Virus banded at ~40% sucrose and was isolated, buffer exchanged in PBS, and concentrated if needed. Alternatively, four hours post infection, cells were washed with PBS and Serum-free media (Invitrogen) was added. Fifteen hours later the media was collected and concentrated using an 100 kDa MWCO centrifugal concentrator (EMD Millipore Corporation, Billerica, Massachusetts).
2.2 Cloning and expression of Sindbis-Apple and Sindbis-Venus
mApple FP and Venus FP were each cloned into the TE12 strain of Sindbis virus using overlapping, fusion PCR as described previously (Sokoloski et al., 2012). FPs were inserted between the E3 and E2 proteins of Sindbis virus and were flanked by two linkers, Gly-Ala-Pro-Gly-Ser-Ala at the N-terminus (primer 5′-GGCGCGCCAGGATCAGCA-3′) and Ala-Gly-Pro-Gly-Ser-Gly at the C-terminus (primer 5′-GCCGGCCCAGGAAGCGGA-3′) of the fluorescent protein (Waldo et al., 1999). The furin cleavage site between E3 and E2 was not altered. The entire structural region of the Sindbis virus (~4500 bp) was sequenced to confirm no additional mutations were present. SINV-Apple and SINV-Venus refer to Apple and Venus fluorescent proteins incorporated into Sindbis particles respectively. SINV-WT refers to wild-type SINV.
2.3 Analysis of virus growth
BHK-21 cells were infected with the indicated virus at a multiplicity of infection of 5. The cell media was harvested and replaced at 3, 6, 9, 12, and 24 hours post-infection. The collected supernatant was plaqued on BHK-21 cells to quantify the number of infectious particles. At 48 hours post-infection, the cells were fixed with 10% formaldehyde for 45 minutes, and stained with 0.5% crystal violet.
2.4 Negative stain transmission electron microscopy
Samples of SINV-WT, SINV-Apple, and SINV-Venus (5 μl) were applied to 400-mesh carbon-coated formvar copper grids and stained with 1% uranyl acetate. The grids were examined on a JEOL 1010 transmission electron microscope (Tokyo, Japan) at 80 kV. Images were recorded using a Gatan UltraScan 4000 CCD camera (Pleasanton, CA). TEM diameters were measured with ImageJ software using at least two grids.
2.5 Particle determination
The number of genome-containing virus particles was determined by quantitative PCR (qPCR)(Sokoloski et al., 2012). Briefly, 4 μl of virus sample, or ~105 total particles, was transferred to PCR tubes containing 500 ng of both nsP1 and E2 reverse transcription primers: nsP1 5′-AACATGAACTGGGTGGTG -3′; E2 5′-ATTGACCTTCGCGGTCGGATTCAT -3′. The sample was heated to 94 °C for five minutes prior to 70 °C for five minutes. The sample was then moved to ice and processed using the Improm-II Reverse Transcriptase (Promega, Madison, WI) according to the manufacturer’s instructions. The sample was then either used immediately in the qPCR or stored at −20 °C for later use. Detection of the SINV nsP1 and E2 regions was performed according to the SYBR Brilliant Green III Supermix instructions (Agilent Technologies, Santa Clara, CA) with the following primer sets: SINV nsP1 Forward 5′-AAGGATCTCCGGACCGTA -3′, SINV nsP1 Reverse 5′-AACATGAACTGGGTGGTGTCGAAG -3′; SINV E2 Forward 5′-TCAGATGCACCACTGGTCTCAACA -3′, SINV E2 Reverse 5′-ATTGACCTTCGCGGTCGGATTCAT -3′. For qPCR, cDNA samples were mixed with either nsP1 or E2 primer sets, 2 x SYBR Green QPCR Mater Mix, and reference dye according to the manufacturer’s protocol. DNA was amplified for 40 cycles (5 seconds at 95 °C and 10 seconds at 60 °C) on StepOnePlus System (Applied Biosystems, Carlsbad, CA). Determination of the number of genomic RNA copies was performed with a standard curve (correlation coefficient of >0.995) of a quantified cDNA plasmid containing the SINV nsP1 and E2 sequence.
2.6 Single-particle data collection
For virus-cell binding imaging, between 3.6–7.2 × 105 BHK-21 cells were grown in 35mm glass bottom dishes (MatTek, Ashland, MA) in phenol red-free minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS. Virus particles (in 50 μl) were added to cells at a multiplicity of infection of 1–5.
For single-particle virus imaging a Revolution XD microscope system (Andor Technology, South Windsor, CT) with an inverted Nikon Ti microscope and Yokogawa confocal scanning unit with Nipkow disk was used. Samples were excited through a high-numerical aperture 60x oil-immersed objective (CFI APO TIRF, NA 1.49, Nikon) with a 488 nm laser (25 mW) or 640 nm laser (40 mW). The optimal excitation for Venus is 515 nm but fluorescence can be measured and quantified at 488 nm. The resulting fluorescence was collected back through the objective, passed through an emission filter (525/30 or 685/40) to eliminate residual laser light and recorded on CCD camera iXon DU-897-BV (Andor™ Technology, South Windsor, CT). Images were processed and analyzed using Andor iQ and ImageJ (National Institutes of Health, Bethesda, MD) software. Photobleaching decay of fluorescence from single viruses was obtained from time-lapsed images of single particles attached to the surface of a coverslip. Curve fitting and histograms were done with IgorPro software (WaveMetrics, Inc).
2.7 Isolation of viral glycoprotein spikes by detergent extraction
To isolate viral glycoprotein spikes, virus was treated with Nonidet-P40, as described previously (Wengler and Rey, 1999). Briefly, purified SINV-WT, SINV-Apple, and SINV-Venus was suspended in TNE buffer containing 10 mM Tris (pH 7.5), 10 mM NaCl, and 20 mM EDTA, and incubated in the presence of 1% Nonidet-P40 for 10 min at 25 °C. The cores were separated from the glycoprotein spikes by sucrose density gradient centrifugation. The top of the gradients, containing the membrane proteins was analyzed by SDS-PAGE and probed for E2 protein using polyclonal antibody. For native gel electrophoresis, protein samples were loaded onto a 0.5% agarose gel and run in 1x TAE buffer for an hour at 80V. Efficiency of fluorescence protein incorporation in the spikes was detected using a Typhoon 9200 imaging system (GE Healthcare, Piscataway, NJ) at the appropriate wavelengths corresponding to Apple and Venus fluorescent proteins and the images were overlaid.
2.8 Coverslip binding assay
Glass coverslips were cleaned by sequential sonication in acetone, rinsing with milliQ water, sonication in 1M KOH, and another rinse with milliQ water. Clean coverslips were stored in water and dried by burning with a propane torch before coating. Coverslip silanisation was performed by immersing dried coverslips in 3% aminopropyltriethoxisilane (APTES) solution in acetone for 15 minutes, then rinsed one time in acetone and two times with water and incubated overnight at 60°C to dry. To make poly-L-lysine coated coverslips, dried coverslips were immersed in 0.01% of poly-L-lysine solution for 10–15 minutes and then rinsed 3 times with milliQ water and dried by ultra-pure nitrogen flow.
To prepare serum-treated coverslips 25 μL of serum-containing media were placed in between two clean coverslips, incubated for 30 minutes, rinsed with water and dried by ultra-pure nitrogen flow. Fifteen μL of virus sample was placed in between two coverslips with same surface treatment and incubated for 15 minutes at room temperature before the cover slips were imaged and the number of bound particles determined. A minimum of two different virus preparations of each virus sample were used. At least 20 images at three different amounts, 2.4×108, 1.2×107, and 2.4×106 of virus particles were used to calculate the surface density of attached virus particles for each sample. The concentration of virus samples was chosen so that the density of the virus particles on the surface was between 0.15–0.30 viruses/μm2.
2.9 Atomic Force Microscopy
Liquid cell atomic force microscopy imaging was performed on a temperature-stabilized Cypher system (Asylum Research, Inc.). To mount the sample, 50 μl virus solution at a concentration 1.6×109 virus particles/ml were deposited on a freshly cleaved, highly ordered pyrolytic graphite disk, mounted on the piezo holder, and incubated for 10 min before probing. A droplet holder was used to minimize evaporation and maintain a stable concentration buffer system. Silicon cantilevers (0.1 N/m, 30 kHz, BioLeverMini; Olympus) with a tip radius of ~9 nm were used. Topographic images (80 nm×80 nm) were obtained in the alternative contact mode using the lowest possible set point amplitudes to minimize damage to the virus features by the tip. Two different biological preparations of virus particles were imaged and a total of at least 11 images per sample were analyzed.
2.10 Bioinformatic and structural comparison between SINV-Apple and SINV-Venus
Clustal W (Larkin et al., 2007) was used to align different FP and determine the percent identity. Three-dimensional model of mApple was built by the program 3D JIGSAW (Bates et al., 2001). Molecular graphics and analyses were performed with the UCSF Chimera package (Pettersen et al., 2004) and The PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC.
3. RESULTS AND DISCUSSION
3.1 SINV-Apple and SINV-Venus have similar infectivity and morphology as SINV-WT
mApple and Venus FP genes were inserted between the coding sequences of the E3 and E2 proteins in SINV (Figure 1A) (Lustig et al., 1988) to produce SINV-Apple and SINV-Venus respectively. This region was chosen so it would not obstruct the receptor binding site at the distal end of the E2 protein (Davis et al., 1987; Meyer and Johnston, 1993; Strauss et al., 1991). Furthermore, the spike polyprotein would still be translocated to the ER via the E3 protein, and cleavage of E3 by furin can still occur in the trans-Golgi, both necessary steps for infectious virus propagation reviewed in (Strauss and Strauss, 1994). The FP mApple and Venus were selected because they are both monomeric proteins and the combination of relatively high brightness and long photostability (compared to other FPs), makes them ideal for single particle tracking experiments (Nagai et al., 2002; Rizzo et al., 2004; Shaner et al., 2004; Shaner et al., 2008; Tsien, 1998).
Figure 1. SINV-Apple and SINV-Venus are viable.

mApple and Venus were inserted in the SINV genome between the E3 and E2 proteins. A) Schematic showing the placement of the FP and adjacent linkers in SINV. Amino acids for the end of E3, the linker, and the beginning of E2 are shown. The furin cleavage site, RSKR, is in bold and underlined. B) Multi-step growth curves of SINV-WT, SINV-Apple, and SINV-Venus. BHK cells were infected with each virus at an MOI=5. Every three hours, a sample of media was removed and titered to determine the number of infectious particles. C) Negative-stained images of SINV-WT and SINV-FP viewed by TEM, scale bar is 100 nm. Particles were purified by pelleting followed by sucrose gradient as described in the text. (D) Histograms of fluorescence intensity of purified SINV-Apple and (E) SINV-Venus particles respectively and images of single virus particles on a cover slip (insets).
At 24 hours post-infection, SINV-Apple and SINV-Venus mutants showed a 1.5-log decrease in titer compared to SINV-WT (Figure 1B). The total particle-to-infectious particle ratio (particle-to-PFU ratio) for SINV-Apple and SINV-Venus were around 100, close to that of SINV-WT which is ~80 (Sokoloski et al., 2012). Together, this data demonstrates that addition of FP to the glycoprotein spike somewhat reduced the total amount of particles assembled, but the fraction of infectious particles produced was not altered in the presence of FP. Repeated passaging of SINV-Apple and SINV-Venus, resulted in no change in plaque size or titer after 3 rounds of infection indicating FP-labeled viruses were stable over several rounds of infection (data not shown).
Purified SINV-Apple and SINV-Venus virus particles showed no signs of aggregation by TEM and had spherical morphologies with a diameter of approximately 70 nm, similar to SINV-WT viruses (Figure 1C). Purified SINV-Apple and SINV-Venus show sharp, single-peak fluorescence intensity histograms (Figure 1D, E) indicating a negligible amount of particle dimers or aggregates.
3.2 Photobleaching kinetics suggests SINV-Apple and SINV-Venus have different surface topologies
We determined rates of photobleaching decay to further characterize the FP in SINV-Apple and SINV-Venus. Time constants of single exponential bleaching decay were 24.2 ± 5.5 s for SINV-Apple and 22.7 ± 3.3 s for SINV-Venus (Figure 2), using the laser intensities reported in 2.6 Single-particle data collection. When normalized to the absorbed power, the bleaching decay rate provides an indication of chromophore accessibility by photobleaching reactive species in solution, and thus, indirectly, about the local chromophore environment. The power-normalized bleaching rate for SINV-Apple was lower than for SINV-Venus, SINV-Apple was at 0.078 s−1 W−1and for SINV-Venus was at 0.186 s−1 W−1. This suggests that the chromophore in SINV-Apple is approximately 2.5 times more photostable than in SINV-Venus. These results are in contrast to previous studies which demonstrated that Venus by itself is approximately 3 times more photostable than mApple (Nagai et al., 2002; Shaner et al., 2008). Therefore, since chromophore photobleaching rate depends on the environment via exposure to reactive species, bleaching decay rates suggest that SINV-Apple chromophores may be less exposed on average than SINV-Venus. Therefore, despite apparent morphological and biological similarities, SINV-Apple and SINV-Venus have interfacial properties that may be unlike and thus differently influence the efficiency of certain viral processes. Hence the question: what viral processes are affected by these differences, if any?
Figure 2. Photobleaching properties of SINV-Apple and SINV-Venus.
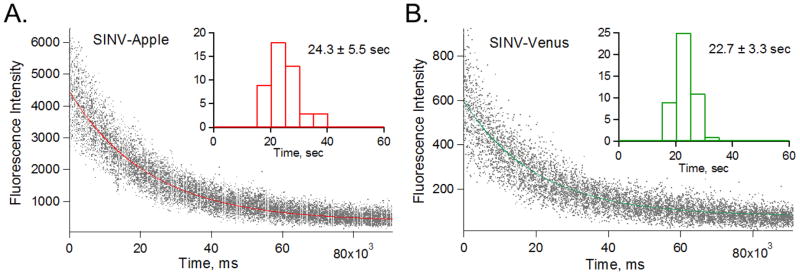
Purified SINV-Apple (A) and SINV-Venus (B) were adhered to a coverslip and fluorescence over time was measured to determine photobleaching. Curves were fit with single-exponential using IgorPro software. Histograms of the rate of photobleaching are shown in the insets. SINV-Apple showed an average decay of 24.3±5.5 sec and SINV-Venus was 22.7±3.3 sec.
3.3 SINV-Venus has higher binding affinity to cells than SINV-Apple
The first step in the virus-host interaction is the attachment of the virion to receptors on the cell surface. SINV has been reported to enter the host cell by penetration at the plasma membrane (Paredes et al., 2004) and receptor-mediated clathrin-coated endocytosis (Marsh, Kielian, and Helenius, 1984; White, Kielian, and Helenius, 1983). Along with several identified receptors (Smith and Tignor, 1980) including laminin (Wang et al., 1992), DC-SIGN (Klimstra et al., 2003), and NRAMP (Rose et al., 2011), lab strains of SINV can bind heparan sulfate possibly as an attachment factor (Klimstra, Ryman, and Johnston, 1998). The attachment step is then followed by entry of the particle into the cell or detachment and release of the particle back into the media. To compare the relative frequency of these outcomes for SINV-Apple and SINV-Venus, particles were added to a monolayer of cover glass-adhered BHK cells at a ratio of ~ 1 infectious particle per cell and the trajectories of initially membrane-bound particles were recorded for 10 minutes. With SINV-WT, approximately 50% of the virions are thought to enter the cell within this time frame (Kielian and Jungerwirth, 1990; White, Kielian, and Helenius, 1983).
Single particle trajectories of SINV-Apple and SINV-Venus were separated into three groups (1) Attachment only defined as the virus particles binding to the cells without showing additional movement during a 10 minute observation period; (2) Virus entry represented by virus particles attaching, then entering the cell; (3) Detachment defined by virus particles attaching, remaining fixed on the membrane, then detaching from the cell and returning into the medium within the 10 minute period (Figure 3A). To measure the differences in the surface interactions between the SINV-Apple and SINV-Venus with the cell, 15 biological replicates were performed each using at least 3 different SINV-Apple and SINV-Venus virus preps.
Figure 3. SINV-Apple binds and enters cells less frequently than SINV-Venus.
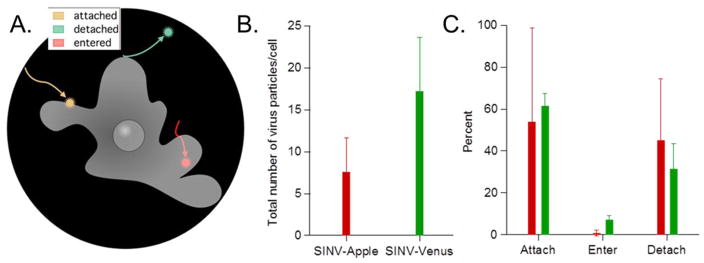
SINV-Apple and SINV-Venus were added to BHK cells and over 10 minutes the number of particles per cell that attached, entered, and detached were monitored. Three different SINV-Apple and SINV-Venus preparations were used with at least 15 different biological replicates and at least 100 particles were observed. A) Schematic illustrating the three stages attachment, entry, and detachment. B) Total number of virus particles per cell that attached, entered, or detached. On average, SINV-Apple had 7/cell and SINV-Venus had 17/cell. C) Out of the total number of particles, the percentage of which attached, entered, or detached for SINV-Apple and SINV-Venus are shown. SINV-Venus had more particles enter the cell compared to SINV-Apple. Average number and standard deviation are shown.
Our first observation was that overall fewer SINV-Apple particles bound to cells than SINV-Venus particles, SINV-Apple averaged 7 particles bound per cell compared to SINV-Venus which averaged 17 particles bound per cell. This finding supports the hypothesis that there are different surface conformations on the two mutant particles, and while the insertion of the FP was designed not to interfere with the receptor-binding site of the virus particle, virus-cell binding is reduced differentially. Our second observation was that, of the total number of virus particles that bound to the cells, the fraction of SINV-Venus particles that entered the cells was significantly higher than SINV-Apple (Figure 3B). However, once the virus particles entered cells, both SINV-Venus and SINV-Apple exhibited similar intracellular kinetics (Figure S2). Thus the difference between SINV-Apple and SINV-Venus particles was only evident at the attachment step and did not propagate to the internal transport of the particle within the cell. It is not known at this point whether the mechanism of endosomal fusion was differentially affected as well.
A possible explanation for the difference in the entry efficiency, but similarity of intracellular transport could be that both SINV-Venus and SINV-Apple enter the cell by receptor-mediated endocytosis, which requires multivalent interactions between virion and the membrane (Fries and Helenius, 1979; Wickham et al., 1990). Receptor interactions are sensitive to proper folding and interfacial chemistry at the virus surface which is different between SINV-Apple and SINV-Venus. Once internalized, the virus motion is endosomal, and virus surface properties become less important (Kielian, Marsh, and Helenius, 1986). Thus, if the membrane binding valency is reduced by altered ligand presentation or misfolding, for instance, one would expect the rate of detachment to increase. Furthermore, the rate of invagination required for endosomal formation could decrease. Another possibility is an altered surface property like charge, which could raise the kinetic barrier against internalization. Obviously, such surface charge, steric, and misfolding effects could arise in combination.
Whatever their origin, differences in attachment do not seem to impact overall viral growth (Figure 1B) or plaque size morphology. This is probably because the titers of SINV-Apple and SINV-Venus (Figure 1B) were determined after cells were incubated with virus for 1 hour at an MOI=5 when most cells were saturated with virus.
3.4 Surface potential is different on SINV-Apple and SINV-Venus particles
Although Apple and Venus are structurally similar, amino acid comparison of the loop regions show the free loops of Venus protein containing more negatively charged residues compared to Apple. This suggests net surface charges may be different (Figure S3A, S3B), the estimated pI values for the two proteins being 5.58 for Venus and 6.02 for Apple (Artimo et al., 2012).
To determine whether surface/virus interaction differences have a predominantly electrostatic origin, we utilized native gel electrophoresis. Initial tests with the whole virus particles showed that samples did not migrate from the well, likely because of the large size of the virus particle. The spikes were then isolated from SINV-Apple and SINV-Venus while maintaining their trimeric structure and fluorescent proteins, and ran on a 0.5% agarose gel (Figure S3C). Migration of proteins in agarose was in presence of non-denaturing buffer and thus, in the assumption of similar morphology, the separation should be based on the net native protein charge. Gel mobility shift assay indicated that Venus-containing viral spikes had a greater mobility compared to Apple-containing viral spikes, which is consistent with Apple having a higher pI.
3.5 Surface support binding properties of SINV-Apple and SINV-Venus
To determine whether the surface potential differences and non-specific binding are the leading cause for different rates of entry we tested non-specific virus binding to well-defined substrates. If the charge differences in the spike proteins of SINV-Apple and SINV-Venus contribute significantly to their cell binding properties, then we reasoned that when we incubate the virus particles with coverslips that had different surface properties, we would observe differences in binding to coverslip. If charge was not a predominant factor in cell binding, there would be no difference between SINV-Apple and SINV-Venus binding to different surface chemistries.
Three types of substrates were prepared to observe particle-surface binding, (1) clean cover slips providing a polar surface with an overall negative charge, (2) APTES or poly-L-lysine coated, providing a positive charge on the surface, and (3) cover slips treated with serum-containing media to test for non-specific binding of SINV-Apple and SINV-Venus to a putative adsorbate layer originating from the cell imaging buffer. Regardless of the chemical nature and, in particular, surface charge of the coverslip surface, more SINV-Apple particles than SINV-Venus particles were observed to bind to the cover slips after 15 minutes incubation time (Figure 4). These results suggest SINV-Apple is more prone to bind non-specifically, perhaps a consequence of a partially misfolded or multiple Apple-E2 conformations and electrostatics is of secondary importance. However, as data in Figure 3 shows, despite SINV-Apple having a higher affinity for all surfaces, its entry efficiency is lower. One possible explanation is that entry depends on specific binding to cell receptors which in SINV-Apple is decreased. In contrast, SINV-Venus could have a fold more consistent with cellular receptor binding, including co-receptors like heparin, and thus enter the host cell at a higher rate. Note that neither SINV-Apple, nor SINV-Venus bound significantly to cover slips treated with serum-containing media. This last result correlates with the observation that, during cell binding experiments (Figures 3 and S3), only few particles bound to the coverslip surface. Therefore, differences in cell binding are due to cell/virus interactions and not to competing binding events with the glass support.
Figure 4. SINV-Apple binds to multiple surfaces whereas SINV-Venus is more selective.

Purified SINV-Apple and SINV-Venus were added to glass cover slips treated with APTES, poly-L-lysine, serum and untreated and the number of virus particles that were bound was determined. SINV-Apple bound to all surfaces suggesting multiple conformations of the spikes on the virus surface or partially unfolded/denatured regions of the spike. SINV-Venus, in contrast, was discriminatory in its binding. Average number of particles bound and standard deviation are shown.
3.6 Spike folding in SINV-Apple and SINV-Venus particles
To further investigate the biochemical origin of the observed surface-interaction differences, we hypothesized that spike assembly and spike conformations may be different between the two FP-tagged viruses. In order to determine if Apple-E2 and Venus-E2 were forming similar spike proteins, purified particles were run on 10% SDS-PAGE gel and probed using an anti-E2 polyclonal antibody. E2 migrates at a molecular weight of 50 kDa, FP at 27 kDa, and FP-E2 at 77 kDa. In both wild-type and SINV-Venus, single bands corresponding to E2 and Venus+E2 respectively were detected (Figure 5). In contrast, for SINV-Apple multiple E2 bands were observed. This suggests that although Apple and Venus have 29% identity/48% amino acid similarity (Larkin et al., 2007), the differences are large enough to induce a different folding of the E2 protein between the two viruses. The large molecular weight bands could represent E3+FP+E2 where the furin cleavage site is inaccessible. The smaller molecular weight bands could be a consequence of misfolded E2 protein and proteolytic cleavage. The change in folding could account for the observed differences in host cell attachment as well.
Figure 5. SINV-Apple forms multiple E2 conformations when produced from high MOI infections.

SINV-WT, SINV-Apple, and SINV-Venus were purified and run on SDS-PAGE. A. Virus samples were produced from MOI=5 infections. Gels were probed with anti-E2 antibodies. SINV-Apple had multiple E2 bands. B. When SINV-Apple was produced from MOI=0.2 infections, only one E2 band appeared. This blot was also probed with anti-E1 antibody to verify its presence in the sample. E2 migrates at a molecular weight of ~50 kDa and FP-E2 at ~77 kDa.
One explanation of the multiple Apple-E2 bands in SINV-Apple particles could be different glycosylation modifications occur in the Apple-E2 protein than in the Venus-E2 protein. Through sequence analysis, we found no additional N-X-S/T motifs or potential N-linked glycosylation motifs present in Apple-E2 but we cannot eliminate potential O-linked gylcosylation. A second explanation for the multiple Apple-E2 bands could be that additional furin cleavage sites are present in E3-Apple-E2, but not in E3-Venus-E2. During viral infection, the E3 protein is cleaved from E2 in the trans-Golgi by the celluar protease furin (Jain, DeCandido, and Kielian, 1991). Furin cleaves at the recognition sequence BBXXBB where B is a basic residue. However, we found no additional furin-like recognition motifs in Apple-E2 thus negating the possibility of alternative furin cleavage of the E3-Apple-E2 protein.
Previous results have demonstrated that regions of unfolded proteins are deleted in the presence of low concentrations of trypsin, trimming the protein to a core domain (Choi et al., 1991; Kar et al., 2011). We treated SINV-Apple with low concentrations of trypsin protease to determine if the Apple-E2 bands would converge on such a common size species. Treatment of SINV-Apple with trypsin did not show the reduction to a core domain, which suggests SINV-Apple spikes may be folded in several, heterogeneous conformations (data not shown).
We did observe that a more homogeneous population of E2-Apple was formed when an MOI of 0.2 or lower was used to propagate virus (Figure 5). While the number of total particles and infectious particles produced was reduced, the homogeneity of the E2 protein was increased. SINV-Venus could be propogated at an MOI of 5 or 10 and still produced a single E2-Venus species.
3.7 Surface topology of SINV-Apple and SINV-Venus particles by atomic force microscopy
Our results so far suggest that Apple-E2 folds in multiple conformations but Venus-E2 folds predominantly in one conformation. Furthermore, the FP in SINV-Apple may be in a different chemical environment, either buried within the viral spike complex or partially denatured as evident by photobleaching studies and the ability of the particles to bind to cover slips treated with different agents.
Because of the possibility of multiple conformational states, a morphological, single-particle comparison between SINV-Apple and SINV-Venus was performed. To this end we utilized fluid cell atomic force microscopy (AFM) (Kuznetsov and McPherson, 2011). Figure 6 shows the physical arrangement of the spikes obtained by AFM from single SINV-WT, SINV-Apple, and SINV-Venus in a physiological buffer. The tip radius limits the spatial resolution of the measurement, but broad structural features which can be described as dimples placed in an icosahedral array can be observed. These provided the ground for quantitative statistical measurements and comparison. Histograms of dimple height showed quantitative and qualitative differences between SINV-Apple and SINV-Venus and SINV-WT. For SINV-Venus, the average dimple depth is larger than for SINV-WT (3.1 nm versus 1.6 nm). This could correspond to the Venus protein being attached in a way that extends the spike (Figure 5). The dimples of SINV-Apple particles have smaller average depth and a broader, possibly bimodal distribution. Together with the results discussed up to here, the AFM data indicates in the case of SINV-Apple a more heterogeneous interaction between the FP and the spike than for to SINV-Venus.
Figure 6. Surface spike arrangement of SINV viruses.

Atomic force microscopy (AFM) was used to measure the surface topology of SINV-WT, SINV-Apple, and SINV-Venus. The depth between the spikes were measured (called dimple depth) to determine if the spikes in each virus sample were arranged in a similar manner. A) Representative AFM images of SINV-WT, SINV-Apple, and SINV-Venus are shown. B) Histograms showing the dimple depth of the different viruses. Two different biological preparations of virus particles were imaged and a total of at least 11 images per sample were analyzed. Numbers represent average depth and standard deviation.
4. CONCLUDING REMARKS
Our results demonstrate that fusion of two different FP to the E2 proteins in SINV, SINV-Apple and SINV-Venus, show no gross morphological differences when compared to SINV-WT. However, upon closer examination of the virus particles in interaction with the cell surface, there was a difference in the number of SINV-Apple particle that bound to cells and the fraction of SINV-Apple particles entering the cell compared to SINV-Venus. Once internalized, SINV-Apple and SINV-Venus behave similar. The differences in host binding could be attributed to differences in surface charge of the FP and/or multiple conformations of the spike protein. The effects of the different FP were only observed at one stage of the virus lifecycle, cell binding. It is worth to note that such effects could have been overlooked when solely relying on routine infectivity test or when comparing intracellular trafficking and cellular localization.
Atomic force microscopy and electrophoretic mobility analysis suggested that the folding of the spike proteins is different between SINV-Apple and SINV-Venus, with SINV-Apple exhibiting increased departure from SINV-WT spike proteins. The difference(s) in binding could be due to the FPs fused to the E2 protein altered the conformation of the spike proteins as well as modifying the net surface charge of individual virus particles. SINV, like many other viruses, is believed to bind to one receptor and then other free receptors on the cell surface diffuse to the virus-cell binding site before the particle is endocytosed. Structural and chemical changes on the spikes may alter the affinity between the virus and receptor and ultimately cell binding and membrane translocation kinetics (Fries and Helenius, 1979; Wickham et al., 1990).
Our findings are consistent with recent work examining the effects of FPs on cellular localization of ClpX and ClpP and MreB in E. coli (Landgraf et al., 2012), (Swulius and Jensen, 2012) and how different FP behaved differently. In addition, GFP is secreted from B. subtilis via a different secretion mechanism than mCherry or superfolder GFP (Dinh and Bernhardt, 2011) suggesting minute changes in the FP may be significant enough to warrant different secretion mechanisms by the cell. These results along with the data presented here emphasize that even subtle differences in structurally similar FPs may lead to discrete differences in the microbe.
Supplementary Material
Highlights.
mApple and Venus fluorescent proteins were fused to Alphavirus E2 glycoprotein
General infectivity between two mutants was the same
We observed differences in host cell binding between the mutants
Fluorescent proteins cannot always be interchanged despite very similar structure
Acknowledgments
We thank the members of the IU virology group for stimulating discussions and comments on the manuscript, and John Murray for help in constructing our Sindbis virus mutants. This work was supported by NIH award 5R01GM081029 to B.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(Web Server issue):W597–603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates PA, Kelley LA, MacCallum RM, Sternberg MJ. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins Suppl. 2001;5:39–46. doi: 10.1002/prot.1168. [DOI] [PubMed] [Google Scholar]
- Byrnes AP, Griffin DE. Binding of Sindbis Virus to Cell Surface Heparan Sulfate. Journal of Virology. 1998;72(9):7349–7356. doi: 10.1128/jvi.72.9.7349-7356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng RH, Kuhn RJ, Olson NH, Rossmann MG, Choi HK, Smith TJ, Baker TS. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell. 1995;80(4):621–30. doi: 10.1016/0092-8674(95)90516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HK, Tong L, Minor W, Dumas P, Boege U, Rossmann MG, Wengler G. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature. 1991;354(6348):37–43. doi: 10.1038/354037a0. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90(3):1103–63. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Davis NL, Pence DF, Meyer WJ, Schmaljohn AL, Johnston RE. Alternative forms of a strain-specific neutralizing antigenic site on the Sindbis virus E2 glycoprotein. Virology. 1987;161(1):101–8. doi: 10.1016/0042-6822(87)90175-9. [DOI] [PubMed] [Google Scholar]
- de Curtis I, Simons K. Dissection of Semliki Forest virus glycoprotein delivery from the trans-Golgi network to the cell surface in permeabilized BHK cells. Proc Natl Acad Sci U S A. 1988;85(21):8052–6. doi: 10.1073/pnas.85.21.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh T, Bernhardt TG. Using superfolder green fluorescent protein for periplasmic protein localization studies. J Bacteriol. 2011;193(18):4984–7. doi: 10.1128/JB.00315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries E, Helenius A. Binding of Semliki Forest virus and its spike glycoproteins to cells. Eur J Biochem. 1979;97(1):213–20. doi: 10.1111/j.1432-1033.1979.tb13105.x. [DOI] [PubMed] [Google Scholar]
- Gibbons DL, Erk I, Reilly B, Navaza J, Kielian M, Rey FA, Lepault J. Visualization of the target-membrane-inserted fusion protein of Semliki Forest virus by combined electron microscopy and crystallography. Cell. 2003;114(5):573–83. doi: 10.1016/s0092-8674(03)00683-4. [DOI] [PubMed] [Google Scholar]
- Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427(6972):320–5. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- Jain SK, DeCandido S, Kielian M. Processing of the p62 envelope precursor protein of Semliki Forest virus. J Biol Chem. 1991;266(9):5756–61. [PubMed] [Google Scholar]
- Jose J, Snyder JE, Kuhn RJ. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009;4:837–56. doi: 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar AK, Mao Y, Bird G, Walensky L, Sodroski J. Characterization of a core fragment of the rhesus monkey TRIM5alpha protein. BMC Biochem. 2011;12:1. doi: 10.1186/1471-2091-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian M, Jungerwirth S. Mechanisms of enveloped virus entry into cells. Mol Biol Med. 1990;7(1):17–31. [PubMed] [Google Scholar]
- Kielian MC, Marsh M, Helenius A. Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. Embo J. 1986;5(12):3103–9. doi: 10.1002/j.1460-2075.1986.tb04616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra WB, Nangle EM, Smith MS, Yurochko AD, Ryman KD. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J Virol. 2003;77(22):12022–32. doi: 10.1128/JVI.77.22.12022-12032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra WB, Ryman KD, Johnston RE. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J Virol. 1998;72(9):7357–66. doi: 10.1128/jvi.72.9.7357-7366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers GJ, Gilbert SG, Cranfill PJ, Davidson MW, Piston DW. Fluorescent proteins at a glance. J Cell Sci. 2011;124(Pt 2):157–60. doi: 10.1242/jcs.072744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov YG, McPherson A. Atomic force microscopy in imaging of viruses and virus-infected cells. Microbiol Mol Biol Rev. 2011;75(2):268–85. doi: 10.1128/MMBR.00041-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nat Meth. 2012;9(5):480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. Molecular basis of Sindbis virus neurovirulence in mice. J Virol. 1988;62(7):2329–36. doi: 10.1128/jvi.62.7.2329-2336.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Kielian MC, Helenius A. Semliki forest virus entry and the endocytic pathway. Biochem Soc Trans. 1984;12(6):981–3. doi: 10.1042/bst0120981. [DOI] [PubMed] [Google Scholar]
- Meyer WJ, Johnston RE. Structural rearrangement of infecting Sindbis virions at the cell surface: mapping of newly accessible epitopes. J Virol. 1993;67(9):5117–25. doi: 10.1128/jvi.67.9.5117-5125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Mapping the Structure and Function of the E1 and E2 Glycoproteins in Alphaviruses. Structure. 2006;14(1):63–73. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20(1):87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Navaratnarajah CK, Kuhn RJ. Functional characterization of the Sindbis virus E2 glycoprotein by transposon linker-insertion mutagenesis. Virology. 2007;363(1):134–47. doi: 10.1016/j.virol.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar A, Koblet H. Semliki Forest virus particles containing only the E1 envelope glycoprotein are infectious and can induce cell-cell fusion. Virology. 1988;166(1):17–23. doi: 10.1016/0042-6822(88)90141-9. [DOI] [PubMed] [Google Scholar]
- Owen KE, Kuhn RJ. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J Virol. 1996;70(5):2757–63. doi: 10.1128/jvi.70.5.2757-2763.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes AM, Ferreira D, Horton M, Saad A, Tsuruta H, Johnston R, Klimstra W, Ryman K, Hernandez R, Chiu W, Brown DT. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virology. 2004;324(2):373–86. doi: 10.1016/j.virol.2004.03.046. [DOI] [PubMed] [Google Scholar]
- Parrott MM, Sitarski SA, Arnold RJ, Picton LK, Hill RB, Mukhopadhyay S. Role of conserved cysteines in the alphavirus E3 protein. J Virol. 2009;83(6):2584–91. doi: 10.1128/JVI.02158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Davidson MW, Piston DW. Fluorescent protein tracking and detection: applications using fluorescent proteins in living cells. Cold Spring Harb Protoc. 2009a;(12):pdb top64. doi: 10.1101/pdb.top64. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Davidson MW, Piston DW. Fluorescent protein tracking and detection: fluorescent protein structure and color variants. Cold Spring Harb Protoc. 2009b;(12):pdb.top63. doi: 10.1101/pdb.top63. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotech. 2004;22(4):445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, Ross SR, Hardy RW, Bambina SA, Heise MT, Cherry S. Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe. 2011;10(2):97–104. doi: 10.1016/j.chom.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotech. 2004;22(12):1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5(6):545–51. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Tignor GH. Host cell receptors for two strains of Sindbis virus. Arch Virol. 1980;66(1):11–26. doi: 10.1007/BF01315041. [DOI] [PubMed] [Google Scholar]
- Sokoloski KJ, Hayes CA, Dunn MP, Balke JL, Hardy RW, Mukhopadhyay S. Sindbis virus infectivity improves during the course of infection in both mammalian and mosquito cells. Virus Res. 2012;167(1):26–33. doi: 10.1016/j.virusres.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss EG, Stec DS, Schmaljohn AL, Strauss JH. Identification of antigenically important domains in the glycoproteins of Sindbis virus by analysis of antibody escape variants. J Virol. 1991;65(9):4654–64. doi: 10.1128/jvi.65.9.4654-4664.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58(3):491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swulius MT, Jensen GJ. The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-Terminal yellow fluorescent protein tag. J Bacteriol. 2012;194(23):6382–6. doi: 10.1128/JB.00505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annual Review of Biochemistry. 1998;67(1):509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Wahlberg JM, Bron R, Wilschut J, Garoff H. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J Virol. 1992;66(12):7309–18. doi: 10.1128/jvi.66.12.7309-7318.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg JM, Garoff H. Membrane fusion process of Semliki Forest virus. I: Low pH-induced rearrangement in spike protein quaternary structure precedes virus penetration into cells. J Cell Biol. 1992;116(2):339–48. doi: 10.1083/jcb.116.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17(7):691–5. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- Wang KS, Kuhn RJ, Strauss EG, Ou S, Strauss JH. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J Virol. 1992;66(8):4992–5001. doi: 10.1128/jvi.66.8.4992-5001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengler G, Rey FA. The isolation of the ectodomain of the alphavirus E1 protein as a soluble hemagglutinin and its crystallization. Virology. 1999;257(2):472–82. doi: 10.1006/viro.1999.9661. [DOI] [PubMed] [Google Scholar]
- White J, Kielian M, Helenius A. Membrane fusion proteins of enveloped animal viruses. Q Rev Biophys. 1983;16(2):151–95. doi: 10.1017/s0033583500005072. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Granados RR, Wood HA, Hammer DA, Shuler ML. General analysis of receptor-mediated viral attachment to cell surfaces. Biophys J. 1990;58(6):1501–16. doi: 10.1016/S0006-3495(90)82495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 4.4 A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. Embo J. 2011;30(18):3854–63. doi: 10.1038/emboj.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. Placement of the structural proteins in Sindbis virus. J Virol. 2002;76(22):11645–58. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.