

Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder caused by inactivating mutations in either the TSC1 or TSC2 genes. It is characterized by the development of multiple, benign tumors in several organs throughout the body. Lesions occur in the brain, kidneys, heart, liver, lungs, and skin and result in seizures and epilepsy, mental retardation, autism, and renal and pulmonary organ system dysfunction, as well as other complications. Elucidation of the molecular pathways and etiological factors responsible for causing TSC has led to a paradigm shift in the management and treatment of the disease. TSC1 or TSC2 mutations lead to constitutive upregulation of the mammalian target of rapamycin pathway, which affects many cellular processes involved in tumor growth. By targeting mammalian target of rapamycin with everolimus, an orally active rapamycin derivative, clinically meaningful and statistically significant reductions in tumor burden have been achieved for the main brain (subependymal giant cell astrocytoma) and renal manifestations (angiomyolipoma) associated with TSC. This review provides an overview of TSC, everolimus, and the clinical trials that led to its approval for the treatment of TSC-associated subependymal giant cell astrocytoma and renal angiomyolipoma.
Keywords: everolimus, subependymal giant cell astrocytoma, angiomyolipomas, lymphangioleiomyomatosis, facial angiofibromas, tuberous sclerosis complex
Epidemiology and etiology of subependymal giant cell astrocytomas, angiomyolipomas, skin lesions, and lymphangioleiomyomatosis associated with tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder with an approximate birth incidence of 1:6,000 newborns.1–3 Up to 1 million people worldwide are affected.4 Although TSC can be inherited, approximately two-thirds of all cases are sporadic.5 Patients with TSC have a severely debilitating disorder that is associated with multiple benign tumors (hamartomas), most commonly in the brain, kidney, lungs, and skin.4,6 Common symptoms include seizures (90%); renal tumors, also known as angiomyolipomas (up to 80%); skin lesions, ie, ash leaf macules and cutaneous fibromas (∼70%); pulmonary involvement, specifically lymphangioleiomyomatosis (LAM) (≤40%); autism and autism spectrum disorders (30%–40%); and neurocognitive impairments (50%–60%), which often occur in individuals with a normal intelligence quotient.5,7,8
In the brain, up to 20% of TSC patients develop subependymal giant cell astrocytomas (SEGAs)—slow-growing glioneuronal tumors that typically arise near the foramen of Monro.2,8,9 SEGAs may grow and obstruct cerebrospinal fluid and increase cerebral pressure, causing headaches, blurred vision, or sudden death from acute hydrocephalus (Figure 1).8–10 The extent of elevated intracranial pressure and hydrocephalus is proportional to tumor diameter.
Figure 1.
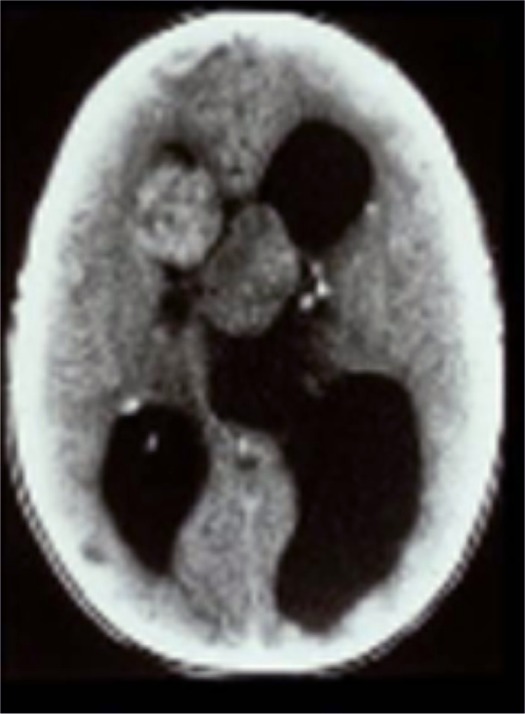
Computed tomography scan demonstrating acute presentation of bilateral subependymal giant cell astrocytomas with marked obstructive hydrocephalus. After emergency surgical resection, the patient with tuberous sclerosis complex was left blind and mentally retarded.
In the kidney, angiomyolipoma is one of the leading causes of death in TSC patients.11 Angiomyolipomas occur in up to 80% of patients and typically are multiple and located bilaterally in the kidneys. They tend to increase in size over time, with some becoming >25–30 cm in their longest dimension (Figure 2). Patients are at risk of serious complications such as life-threatening hemorrhage (25%–50%) and often present in the emergency room with shock (~20%–30%).4,7,12 As angiomyolipomas enlarge, they often develop micro- and macroaneurysms. Angiomyolipomas ≥4 cm in diameter are at increased risk for spontaneous bleeding as a result of ruptured vessels. The presence of aneurysms >5 mm is a strong predictor of spontaneous hemorrhage.13 Renal failure resulting in end-stage renal disease is largely due to encroachment of angiomyolipomas on normal renal parenchyma. Many patients require dialytic replacement therapy.7
Figure 2.
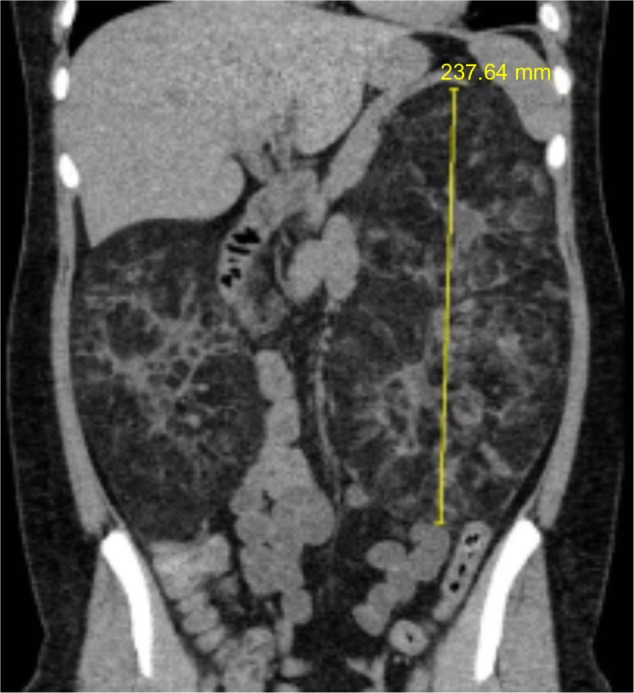
Renal computed tomography scan demonstrating significant angiomyolipomata burden in both kidneys of a patient with tuberous sclerosis complex.
Skin lesions are detected in >70% of patients with TSC and include hypomelanotic macules (ash leaf spots), shagreen patches, confetti-like lesions, forehead fibrous plaque, facial angiofibromas, and periungual and ungual fibromas (Figure 3A and B).5,14,15 Facial angiofibromas can result in decreased quality of life for patients because they affect appearance, may cause disfigurement, and are prone to bleeding, which increases the possibility of infection.14,16
Figure 3.
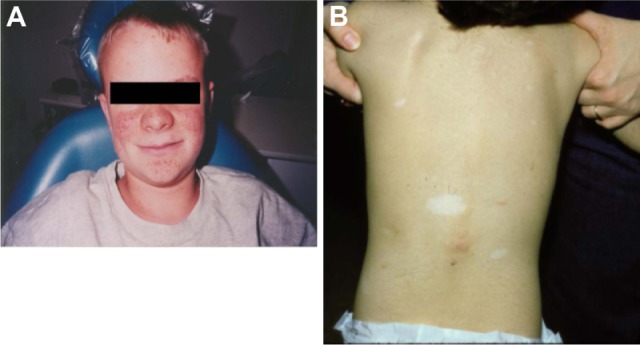
Examples of tuberous sclerosis complex-associated skin lesions: (A) angiofibromas and (B) shagreen patches and ash leaf macules.
Pulmonary LAM occurs in up to 40% of TSC patients and predominantly affects women.1,7,17 LAM occurs more rarely in patients without TSC (sporadic LAM). It is characterized by cystic destruction of the lung caused by infiltration of smooth muscle cells (Figure 4).7
Figure 4.
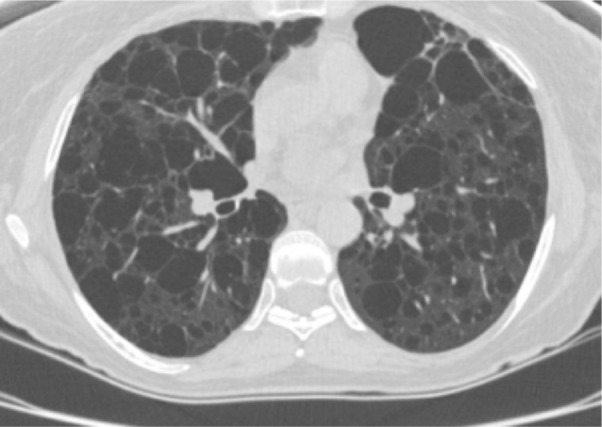
Chest computed tomography scan showing parenchymal destruction associated with lymphangioleiomyomatosis in a patient with tuberous sclerosis complex.
The molecular pathogenesis of TSC is thought to be caused by mutations in either the TSC1 (encoding hamartin) or TSC2 (encoding tuberin) genes, both of which are integral to the normal functioning of the mammalian target of rapamycin (mTOR) pathway.18–20 The mTOR pathway integrates numerous cellular inputs to affect a multitude of downstream signaling cascades that are involved in cellular processes such as cellular metabolism, growth, proliferation, angiogenesis, and survival.1,6,8,21 Hamartin and tuberin interact to form a dimer that activates a GTPase, preventing phosphorylation of the GAP protein—Ras homolog enriched in brain (Rheb)—and inhibiting activation of mTOR complex 1 (mTORC1), a serine threonine kinase.22,23 In patients with TSC, the hamartin/tuberin dimer is unable to form because of mutations in TSC1 and TSC2. As a result, mTORC1 is constitutively activated, which leads to hyperactive mTOR signaling of the downstream kinase signaling cascade and consequent abnormalities in many cellular processes, including cell cycle progression, transcription, translation, and metabolic control (Figure 5).22,24
Figure 5.

The mammalian target of rapamycin signaling pathway.
Note: Franz DN et al. Journal of Child Neurology. 28(5):602–607, Copyright © 2013 by Sage. Reprinted by Permission of SAGE Publications.22
Abbreviations: 4E-BP1, 4E binding protein 1; Abl, Abelson kinase; Akt, protein kinase B; eIF-4E, eukaryotic initiation factor 4E; IGF-1R, insulin-like growth factor-1 receptor; PI3K, phosphoinositide 3-kinase; Pten, phosphatase and tensin homolog; RAS, rat sarcoma; S6K1, 40 S ribosomal S6 kinase; TSC1, tuberous sclerosis complex 1; TSC2, tuberous sclerosis complex 2.
TSC is thought to develop from complete loss of functional TSC1 or TSC2 and follows Knudson’s two-hit hypothesis whereby the first “hit,” causing a mutation in one allele of the TSC1 or TSC2 gene, is followed by a second “hit,” somatic mutation, in the corresponding TSC wildtype allele.8 Disease severity can be variable, even within families, and may reflect differential expression of normal and mutant TSC alleles.25
TSC2 mutations, which occur in 70%–80% of cases, are more common than TSC1 mutations, which occur in 20%–30% of cases.26 Studies of TSC patients have found that the clinical manifestations of TSC are more frequent and often more severe in patients with TSC2 mutations.18,19 Data from genotype/phenotype studies suggest that different types and locations of TSC germline mutations may be associated with distinct neurocognitive phenotypes.19,27,28
Management and current therapeutic strategies
Recently, a subcommittee at the Tuberous Sclerosis Complex Clinical Consensus Conference (sponsored by the Tuberous Sclerosis Alliance) reviewed and updated the recommendations for surveillance and management of patients with TSC.29 Specific guidance on screening and follow-up evaluations, as well as clearly defined time intervals for clinical evaluations, is expected in the TSC guidelines and will help to standardize and improve the clinical care of TSC patients. These guidelines will soon be published and available to clinicians. Historically, treatment of TSC-associated hamartomas has focused on a combination of active and watchful waiting, palliative treatments with drug therapy, and surgical procedures to reduce tumor burden (Table 1).
Table 1.
Common treatment modalities for SEGA, angiomyolipoma, skin lesions, and pulmonary LAM
| Clinical issue | Treatment | Additional comment |
|---|---|---|
| SEGA30–32,36,61,65 | • Active surveillance | • Periodic neuroimaging is recommended to monitor lesion size |
| • Surgery | • Timing of surgery is debatable | |
| • Gamma Knife stereotactic radiosurgery | • Incomplete excision of SEGAs results in tumor recurrence | |
| • Pharmacotherapy with mTOR inhibitors | • Postoperative complications include limit to the efficacy of surgery | |
| Angiomyolipoma7,43,60 | • Arterial embolization | • Repeat embolization required |
| • Partial/total nephrectomy | • Postembolization syndrome | |
| • Pharmacotherapy with mTOR inhibitors | ||
| Skin lesions5,14 | • Cryosurgery | • Variable recurrence rate |
| • Curettage | • Repeat procedures needed | |
| • Dermabrasion | ||
| • Chemical peeling | ||
| • Excision | ||
| • Laser therapy | ||
| Pulmonary LAM1,45 | • Bronchodilators | • Treatments are palliative only |
| • Oxygen support | ||
| • Estrogen antagonists |
Abbreviations: LAM, lymphangioleiomyomatosis; mTOR, mammalian target of rapamycin; SEGA, subependymal giant cell astrocytoma.
SEGA
Active surveillance with serial neuroimaging is recommended even in asymptomatic patients, because SEGAs have the potential to grow over time.30,31 Surgical resection has been the standard treatment option for symptomatic SEGAs; however, scientific evidence for the effectiveness of SEGA surgery in reducing SEGA-related conditions is limited.30,32–35 Postoperative morbidity is variable and may occur in ~50% of patients.36 Complications include increased prevalence of seizures, hydrocephalus, vision disorders, headaches, stroke, hemiparesis, and autism.32,33,35,37
Tumor recurrence invariably is seen in the absence of gross total resection. This may occur with very large, locally invasive, or bilateral lesions.32,33 The timing of surgical intervention for SEGA is controversial, with some surgeons advocating early surgery, and others waiting for symptomatic lesions or development of hydrocephalus, which can facilitate a transcortical approach to the lesion.30,32,38,39 If surgery is chosen, gradual formation of an operative corridor using balloon dilation is an example of a minimally invasive technique.40 This approach has resulted in gross total resection with resolution of ventricular dilation and minimal cortical disruption.40 Clinical recommendations made by a panel of European experts who met in Rome, Italy, in March 2012, endorse surgery in symptomatic patients and asymptomatic patients with documented tumor growth.41 The panel also recommends everolimus (Afinitor®, Novartis, East Hanover, NJ, USA) for the treatment of adults and children ≥3 years of age with SEGA associated with TSC who require therapeutic intervention but are not amenable to surgery.41 Contraindications to surgery include an inability to tolerate anesthesia and cases in which the risks of surgery outweigh the benefits (total resection is unachievable or poses significant risk to the patient).41 Endoscopic procedures are associated with lower morbidity, but they are limited to lesions with a diameter ≤2 cm.32 Gamma Knife stereotactic radiosurgery has been used, but its role in treating SEGA has not been clearly defined because efficacy and safety data are sufficient to support its use.31,37 Standard chemotherapy is not advised owing to the fact that therapeutic efficacy data are lacking and risk of remote, second malignancies is increased.42
Angiomyolipomas
Appropriate management of angiomyolipomas is determined by lesion size and symptoms. An association between angiomyolipoma size (>4 cm), aneurysm size (≥5 mm), and risk of hemorrhage has been noted.13 Embolization is preferred in the setting of recent or active hemorrhage, or when large or multiple aneurysms are present. Embolization may require multiple procedures over time and is associated with modest reductions in tumor bulk.43 Complications of embolization include a condition called postembolization syndrome, which is characterized by pain, fever, and malaise resulting from the presence of necrotic tissue in the retroperitoneum.41 This can be prevented by the use of prophylactic steroids.44
Skin lesions
Currently, treatments for skin lesions such as facial angiofibromas include dermabrasion, cryosurgery, curettage, chemical peeling, electrodesiccation, excision, and laser therapy; however, repeated treatment is often needed because lesions may recur.5,14
Pulmonary LAM
Bronchodilators are used to help control symptoms in patients with reversible air flow obstruction. Other treatment approaches include oxygen support and treatment with estrogen antagonists; however, effective treatment options for this condition are lacking.1,45
Pharmacology and mode of action of everolimus
Pharmacodynamics
The pivotal role of the mTOR pathway in the etiology of TSC and its associated conditions provides a strong rationale for the use of mTOR inhibition as a targeted therapy. Everolimus is a rapamycin derivative that inhibits the mTOR pathway by acting on mTORC1 (Figure 5).46,47 It binds to FKBP-12, forming an inhibitory complex with mTORC1, thereby inhibiting mTOR kinase activity and downstream pathways.46,48,49 Everolimus reduces the phosphorylation of downstream effectors of mTOR, such as the translational repressor eukaryotic elongation factor 4E binding protein and the S6 ribosomal protein kinase 1, which are involved in protein translation.46,50,51
Pharmacokinetics
In healthy volunteers, everolimus was rapidly absorbed within 30 minutes and reached a maximum blood concentration of 44.2 ± 13.3 μg/L after a single 4 mg oral dose (time to reach maximal concentration [Tmx] = 0.5 hour [range 0.5–1 hour]) (Table 2).52 In adult patients with solid tumors, once-daily dosing with 5 mg or 10 mg of everolimus resulted in rapid absorption within 1 hour.53 The mean (standard deviation) maximum steady-state blood concentration was 32 ng/L (9 ng/L) and 61 ng/L (17 ng/L) after the 5 mg and 10 mg dose, respectively. The median (range) time to maximum serum concentration at the 5 mg and 10 mg dose was 1 hour (1 hour) and 1 hour (1–6 hours), respectively53 For these same doses, the mean (standard deviation) area under the curve at steady state was 238 ng·h/mL (77 ng·h/mL) and 514 ng·h/mL (231 ng·h/mL), respectively53 In healthy human subjects, a high-fat meal delayed the median time to maximum concentration by 1.25 hours (median) and reduced the maximum serum concentration by 60% and the area under the concentration–time curve by 16%.54 Therefore, everolimus should be consistently administered with or without food to avoid fluctuations in therapeutic drug.
Table 2.
Pharmacokinetic properties of everolimus in various patient populations
| Patient population | Tmax (h) | Cmax (ng/mL) | AUC (ng/mL · h) 0–∞ | Half-life (h) |
|---|---|---|---|---|
| Healthy subjectsa (n = 24) 2.0 mg dose54 |
0.5 (0.5–2.0) | 17.9 ± 5.9 | 122 ± 52 | 31.5 ± 6.4 |
| Healthy subjectsa (n = 12) 4.0 mg dose66 |
0.5 (0.5–1.0) | 44.2 ± 13.3 | 219 ± 69 | 32.2 ± 6.1 |
| Refractory advanced solid tumor patients aged ≥18 years (n = 4)a 5.0 mg dose53 |
1 (1) | 32 ± 9 | 238 ± 77 | — |
| Refractory or recurrent solid tumor patients aged ≥3 years to <22 years (n = 6)b 2.1 mg/m2 67 |
1 (0.5–1.2) | 21.5 (16.1–78.5) | 139.8 (94.6–197.7) | 18.0 (15.2–19.9) |
| Refractory or recurrent solid tumor patients aged ≥3 years to <22 years (n = 8)b 3.0 mg/m2 67 |
1.1 (0.6–2.3) | 43.5 (18–58) | 197.6 (59.6–249.4) | 15.9 (13.9–22.5) |
Notes:
Values are mean ± standard deviation, except for Tmax, which is median (range)
all values are median (range).
Abbreviations: AUC, area under the concentration–time curve from zero to infinity; Cmax, maximum concentration; Tmax, time to reach maximum concentration.
Everolimus is a substrate of CYP3A4 and P-glycoprotein. Therefore, administration of CYP3A4 inhibitors or inducers affects its metabolism.46 A comparison of the relative amount of parent compound in blood after 1.5 mg/kg oral and 2-hour 1.0 mg/kg intravenous administration of everolimus suggests that intestinal metabolism (first-pass effect) accounts for a large percentage of metabolism.55
Everolimus is eliminated mainly in bile and, to a lesser extent, in urine.52 The mean elimination half-life is approximately 30 hours. In healthy subjects, the elimination half-life after a 4 mg dose of everolimus was 32.2 ± 6 hours.46,52 Hepatic impairment increases exposure of everolimus.46,52 The extent of exposure depends on whether hepatic impairment is mild, moderate, or severe. Dose reductions should be implemented in TSC patients with SEGA and/or renal angiomyolipoma and mild to moderate hepatic impairment.46 If the benefit outweighs the risk, reductions in doses of everolimus are warranted in TSC patients with severe hepatic impairment and renal angiomyolipoma; however, in TSC patients with SEGA and severe hepatic impairment, everolimus should be avoided.46
Clinical efficacy
The efficacy of everolimus in treating TSC patients was first demonstrated in an open-label Phase I/II clinical trial in 28 patients with SEGA associated with TSC (NCT00411619).56 The core treatment phase lasted 6 months, after which patients could transition into an extension phase. This trial led to US Food and Drug Administration approval of everolimus for the treatment of SEGA in TSC patients; it was the first trial to show marked reductions in SEGA volume within the first 3 months of treatment that were sustained as long as therapy continued.56 Patients (median age 11 years [range 3–34 years]) received everolimus initiated at a dose of 3 mg/m2 and subsequently adjusted to whole blood trough concentrations of 5–15 ng/mL.56 The median duration of treatment was 21.5 months (range 4.7–34.4 months).56 Clinically meaningful reductions in the volume of primary SEGA, as assessed by independent central review, were achieved, and the median reduction was −0.80 cm3 (95% confidence interval [CI]: 0.4–1.2; P < 0.001).56 No new lesions, evidence of increased intracranial pressure, worsened hydrocephalus, or need for surgical resection or any other therapy was reported. Everolimus was associated with a significant reduction in the overall frequency of clinical and subclinical seizures (median change −1 seizure; P = 0.02).56 Quality of Life in Childhood Epilepsy scores improved over time (58.74 ± 14 at baseline; 63.4 ± 12.4 at 3 months; 62.1 ± 14.2 at 6 months).53 This trial continues in the extension phase, and results for patients treated up to 3 years were recently reported by Krueger et al.57 Of the original 28 patients enrolled 25 were continuing treatment at the data cutoff for the 3-year extension study57 After a median exposure of 34.2 months, the primary SEGA volume was reduced from 1.74 cm3 at baseline to 0.97 cm3 at 36 months.57 Reductions ≥30% from baseline at 24 months, 30 months, and 36 months were seen in 79.2%, 64.7%, and 77.8% of patients, respectively, and reductions ≥50% were seen in 50.0%, 41.2%, and 55.6% of patients, respectively57 At 24 months, improvements in facial angiofibromas compared with the previous visit were observed in eight of nine patients (88.9%).57 A subgroup analysis of patients enrolled in this trial reported a significant change in white matter diffusion in patients treated with everolimus, suggesting that treatment alters the genetic defects in normal-appearing white matter in the brain of TSC patients.58 Significant changes in fractional anisotropy and radial diffusivity were observed in the corpus callosum, internal capsule, and geniculocalcarine regions of patients treated with everolimus, and no changes were observed in the age- and gender-matched control group of patients with TSC.58 Mean (95% CI) fractional anisotropy significantly increased by 0.04 (0.019–0.062) in all three brain regions after 12–18 months of everolimus treatment (P < 0.05), and this was driven by a decrease in radial diffusivity in the corpus callosum and the geniculocalcarine tract (P < 0.01 for each).58
A Phase III, randomized double-blind placebo-controlled trial, A Randomised Double-blind Placebo-controlled Study of Everolimus in the Treatment of Patients With Subependymal Gaint Cell Astrocytomas (SEGA) Associated With Tuberous Sclerosis Complex (TSC) (EXIST-1)(NCT 0078982859), investigated the efficacy of everolimus versus placebo in 117 TSC patients with SEGA (Table 3).36 Patients were randomized to everolimus 4.5 mg/m2 per day (n = 78) or placebo (n = 39), and baseline demographics and disease characteristics were generally well balanced with the exception of a higher proportion of males and the presence of hydrocephalus in the everolimus group.36 The median age of patients was 9.5 years (range 0.8–26.6 years), and 51% and 64% of patients in the everolimus and placebo arms, respectively, had one target SEGA lesion; 44% and 36% of patients in the everolimus and placebo arms, respectively, had two SEGA lesions.36 One or more skin lesions were present in 110 patients (92% [n = 72] of patients in the everolimus arm and 97% of patients in the placebo arm [n = 38]), and 38% (n = 30) of patients in the everolimus arm and 36% (n = 14) of patients in the placebo arm had one or more angiomyolipomas.36 After a median follow-up of 9.7 months, the SEGA response rate—the proportion of patients with a reduction in sum of volumes of target SEGA lesions ≥50% relative to baseline in the absence of nontarget lesion worsening, new lesions ≥1 cm in diameter, or new/worsening hydrocephalus—was 35% (95% CI: 24%–46%) in the everolimus group and 0% (95% CI: 0%–9%) in the placebo group (difference in response rate 35%, 95% CI: 15%–52%; P < 0.0001).36
Table 3.
Efficacy and safety analyses from EXIST-1 and EXIST-2
| Trial | Efficacy | Safety |
|---|---|---|
| EXIST-136 | Primary end point | • AEs were mostly grade 1/2 in severity |
| • TSC patients (n = 117) with ≥ 1 SEGA ≥ 1 cm and either serial growth of SEGA, a new lesion of ≥ 1 cm, or new or worsening hydrocephalus | • SEGA response rate: 35% vs 0% (EVE vs PBO; P < 0.0001) | |
| Key secondary end points | • Most common AEs were mouth ulceration (32% vs 5% EVE vs PBO), stomatitis (31% vs 21% EVE vs PBO), convulsions (23% vs 26% EVE vs PBO), and pyrexia (22% vs 15% EVE vs PBO) | |
| • Change from baseline to week 24 in the total number of seizures per 24 hours | ||
| • Oral 4.5 mg/m2/day everolimus titrated to target whole blood trough level of 5–15 ng/mL | – No significant difference in seizure frequency was found in the everolimus arm relative to placebo (P = 0.20) | |
| • Time to progression of SEGA | ||
| – No EVE patients progressed vs six PBO patients (15.4%) (P = 0.0002) | ||
| • Skin lesion response rate (among patients with ≥ 1 skin lesion at baseline; n = 110) | ||
| – 42% vs 11% (EVE vs PBO; P = 0.0004) | ||
| Exploratory end points | ||
| • Angiomyolipoma response rate (among patients with ≥ 1 angiomyolipoma ≥ 1 cm in longest diameter at baseline; n = 44) 53% vs 0% (EVE vs PBO) | ||
| EXIST-260 | Primary end point | • AEs were mostly grade 1/2 in severity |
| • TSC or sporadic lymphangioleiomyomatosis patients (n = 118) with ≥ 1 angiomyolipoma ≥3 cm in longest diameter, no requirement for angiomyolipoma-related surgery, or no angiomyolipoma-related bleeding or embolization in past 6 months | • Angiomyolipoma response rate | |
| – 42% vs 0% (EVE vs PBO; P < 0.0001) Key secondary end points | • Most common AEs were stomatitis (48% vs 8% EVE vs PBO), nasopharyngitis (24% vs 31% EVE vs PBO), and acne-like skin lesions (22% vs 5% EVE vs PBO) | |
| Key secondary end points | ||
| • Time to progression of angiomyolipoma | ||
| – EVE was superior to PBO (hazard ratio: 0.08; 95% CI: 0.02–0.37; P < 0.0001) | ||
| • Oral 10 mg/day EVE | • Skin lesion response rate (among patients with ≥ 1 skin lesion at baseline; n = 114): 26% vs 0% (EVE vs PBO; P = 0.0002) |
Abbreviations: AE, adverse event; CI, confidence interval; EVE, everolimus; EXIST, EXamining everolimus In a Study of TSC; PBO, placebo; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis complex; vs, versus.
The median time to SEGA progression was not reached in either treatment arm, and estimated progression-free rates at 6 months were 100% for everolimus and 86% for placebo.36 Analysis of change in seizure frequency was inconclusive—a fact that was attributed to the method of assessment, a single 24-hour video electroencephalography, which likely limited the number of evaluable patients. The median change from baseline to week 24 in seizure frequency was 0 in both treatment arms (P = 0.2004), yielding an inconclusive analysis, as most patients had no seizures at baseline or follow-up.36 Thirty patients (42%) in the everolimus arm and four patients (11%) in the placebo arm had a skin lesion response (P = 0.0004), and 16 patients (53%) had an angiomyolipoma response, defined as reduction in the sum of volumes of all target lesions ≥50% relative to baseline; no patient in the placebo arm had a response.36
In another large, Phase III, randomized, double-blind, placebo-controlled—A Randomized, Double-blind, Placebo-controlled Study of RAD0001 in the Treatment of Angiomyolipoma in Patients With Either Inberous Sclerusis Complex (TSC) or Sporadic Lymphangioleiomysmatosis (LAM) (EXIST 2)—the efficacy of everolimus in treating angiomyolipoma associated with TSC or sporadic LAM was assessed in 118 patients.60 Patients (median age 31 years [range 18.0–61.0 years]) were randomized to receive everolimus 10 mg per day (n = 79) or placebo (n = 39).60 The percentages of patients with angiomyolipoma lesions with longest diameter ≥4 cm but not larger than 8 cm in the everolimus and placebo arms were 57% and 49%, respectively.60 Of note, lesions ≥4 cm are at risk for hemorrhagic rupture. SEGAs were noted as a major feature in a higher percentage of everolimus patients (54%) compared with placebo patients (36%) at baseline, but patient demographics were well balanced between the treatment arms and were generally representative of disease epidemiology.60 Skin lesion response was determined for the 114 patients with one or more skin lesions at baseline.
The angiomyolipoma response rate, defined as the proportion of patients with confirmed ≥50% reductions in the sum of volumes of all target angiomyolipomas relative to baseline, was 42% (95% CI: 31%–53%) for everolimus compared with 0% (95% CI: 0%–9%) for placebo, translating to a significant difference of 42% (95% CI: 23.5%–58.4%; P < 0.0001).60 A higher proportion of patients in the everolimus arm had a ≥50% reduction from baseline in the sum of volumes of target angiomyolipoma lesions at week 24 (55% versus 0%).60 Similarly, a higher proportion of patients in the everolimus arm had a reduction in the sum of volumes of target angiomyolipoma lesions ≥30% at this same time point (80% versus 3%).60 The median time to angiomyolipoma response with everolimus was 2.9 months.60
The everolimus arm had a significantly higher skin lesion response rate: response rates were 26% (95% CI: 17%–37%) in the everolimus arm and 0% (95% CI: 0%–10%) in the placebo arm (P = 0.0002).60 Time to angiomyolipoma progression was significantly longer with everolimus versus placebo (hazard ratio: 0.08 [95% CI: 0.02–0.37]), and progression-free rates for everolimus versus placebo, respectively, were 98% (95% CI: 89%–100%) versus 83% (95% CI: 65%–93%) at 6 months, and 92% (95% CI: 65%–98%) and 25% (95% CI: 1%–64%) at 12 months.60 The median time to angiomyolipoma progression was 11.4 months for placebo and was not reached for everolimus.60
Safety and tolerability
In all three clinical trials, everolimus yielded a comparable safety profile: adverse events (AEs) were generally mild and grade 1 or 2 in severity, as assessed by the National Cancer Institute Common Terminology Criteria for Adverse Events.36,57,60,61
Stomatitis (79%) and upper respiratory tract infection (79%) accounted for the majority of AEs in the Phase I/II trial; grade 3 AEs were reported in ten patients, and one grade 4 event (convulsion) occurred in one patient.61 No consistent laboratory abnormalities were noted, with the exception of increases in the concentrations of total cholesterol, low-density lipoprotein cholesterol, and triglycerides.61 The safety profile in the extension phase continues to be consistent with that reported in the original analysis, and no patient discontinued treatment because of an AE.57 No patients discontinued because of AEs in EXIST-1, and few patients discontinued because of AEs in EXIST-2.36,60 Reasons for discontinuation in the everolimus arm included one case of grade 2 blood phosphorus decrease; one patient with concurrent grade 3 hypersensitivity, grade 3 angioedema, and grade 3 bronchospasm; and one patient with convulsion due to intractable seizures deemed not related to study drug.60 In the EXIST-1 study, the most common AEs in the everolimus versus placebo groups, respectively, were mouth ulceration (32% versus 5%), stomatitis (31% versus 21%), convulsion (23% versus 26%), and pyrexia (22% versus 15%).36 The most common grade 3 AEs in the everolimus versus placebo group were stomatitis (8% versus 3%), pyrexia (6% versus 0%), and convulsion (5% versus 5%).36 In the EXIST-2 study, the most common AEs in the everolimus versus placebo group, respectively, were stomatitis (48% versus 8%), nasopharyngitis (24% versus 31%), acne-like skin lesions (22% versus 5%), and headache (22% versus 18%).60 The most common grade 3 AEs in the everolimus versus placebo group were aphthous stomatitis (3% versus 0%) and mouth ulceration (3% versus 0%).60 Grade 4 events were rare in both studies. Secondary amenorrhea was observed in EXIST-1 and EXIST-2. In EXIST-1, three of eight everolimus-treated and no placebo-treated female patients experienced amenorrhea.36 In EXIST-2, seven of 52 everolimus-treated females and one of 26 placebo-treated females experienced amenorrhea.60 As a result, amenorrhea is being further investigated as a potential AE associated with everolimus.
Biomarkers
Loss of the TSC1 or TSC2 gene has been associated with increased production of vascular endothelial growth factor (VEGF), and inhibitors of mTOR have been shown to have an inhibitory effect on production of VEGF, tumor growth, and angiogenesis, both in vitro and in vivo.62,63
The effect of everolimus on several angiogenic biomarkers is being assessed in the EXIST-1 and EXIST-2 trials. Compared with placebo, a sustained ~30% and ~60% increase in VEGF-A was observed in the everolimus arm of EXIST-1 and EXIST-2, respectively.64 A concomitant decrease in collagen type IV (~25% EXIST-1; ~45% EXIST-2) and soluble VEGF receptor (sVEGFR2) (~25% both trials) was observed in the everolimus arm.64 A sustained decrease (~60%) in VEGF-D was observed in the everolimus arm of EXIST-2, but not in EXIST-1.64 In both studies, no change was observed in placental growth factor, sVEGFR1, or c-Kit plasma concentrations in the everolimus arm or in any biomarkers evaluated in the placebo arm.64 Baseline sVEGFR2 and VEGF-D were ~40% and ~four-fold higher, respectively, while VEGF-A was ~50% lower in EXIST-2 compared with in EXIST-1.64 A similar baseline plasma concentration for the other biomarkers was noted in both studies.64 The inclusion of biomarkers in future clinical trials may provide new predictive or prognostic biomarkers when evaluating disease burden and everolimus efficacy and safety.
Conclusion
Everolimus is approved for the treatment of adults with renal angiomyolipoma and TSC not requiring immediate surgery, and of adults and children ≥3 years of age with SEGA associated with TSC who require therapeutic intervention but are not candidates for curative surgical resection. The therapeutic efficacy and safety of everolimus have been demonstrated in multicenter, international, randomized, double-blind, placebo-controlled trials. These studies continue in their extension phases with the goal of establishing the long-term safety and efficacy profile of everolimus in patients with TSC. Everolimus, an orally administered mTOR inhibitor, is the first pharmacological therapy approved for the treatment of TSC. Targeted systemic therapies such as everolimus are particularly useful in that they treat the multiple manifestations of TSC and offer patients both a means of symptom relief and a treatment alternative to invasive surgical procedures.
Acknowledgments
Dr Franz acknowledges ApotheCom for medical editorial assistance, and Novartis for supporting this trial and for funding medical editorial assistance.
Footnotes
Disclosure
Dr Franz has received honoraria and travel support from Novartis and Lundbeck. His institution, Cincinnati Children’s Hospital, has received research support and consulting fees from Novartis.
References
- 1.Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics. 2010;41(5):199–208. doi: 10.1055/s-0030-1269906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner SG, Peters KB, Vredenburgh JJ, Desjardins A, Friedman HS, Reardon DA. Everolimus tablets for patients with subependymal giant cell astrocytoma. Expert Opin Pharmacother. 2011;12(14):2265–2269. doi: 10.1517/14656566.2011.601742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hallett L, Foster T, Liu Z, Blieden M, Valentim J. Burden of disease and unmet needs in tuberous sclerosis complex with neurological manifestations: systematic review. Curr Med Res Opin. 2011;27(8):1571–1583. doi: 10.1185/03007995.2011.586687. [DOI] [PubMed] [Google Scholar]
- 4.Budde K, Gaedeke J. Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR inhibition. Am J Kidney Dis. 2012;59(2):276–283. doi: 10.1053/j.ajkd.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz RA, Fernandez G, Kotulska K, Jozwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57(2):189–202. doi: 10.1016/j.jaad.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 7.Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924–934. doi: 10.1111/j.1523-1755.2004.00838.x. [DOI] [PubMed] [Google Scholar]
- 8.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 9.Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol. 2009;16(6):691–696. doi: 10.1111/j.1468-1331.2009.02567.x. [DOI] [PubMed] [Google Scholar]
- 10.Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004;63(8):1457–1461. doi: 10.1212/01.wnl.0000142039.14522.1a. [DOI] [PubMed] [Google Scholar]
- 11.Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66(8):792–796. doi: 10.1016/s0025-6196(12)61196-3. [DOI] [PubMed] [Google Scholar]
- 12.Dixon BP, Hulbert JC, Bissler JJ. Tuberous sclerosis complex renal disease. Nephron Exp Nephrol. 2011;118(1):e15–e20. doi: 10.1159/000320891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamakado K, Tanaka N, Nakagawa T, Kobayashi S, Yanagawa M, Takeda K. Renal angiomyolipoma: relationships between tumor size, aneurysm formation, and rupture. Radiology. 2002;225(1):78–82. doi: 10.1148/radiol.2251011477. [DOI] [PubMed] [Google Scholar]
- 14.Hofbauer GF, Marcollo-Pini A, Corsenca A, et al. The mTOR inhibitor rapamycin significantly improves facial angiofibroma lesions in a patient with tuberous sclerosis. Br J Dermatol. 2008;159(2):473–475. doi: 10.1111/j.1365-2133.2008.08677.x. [DOI] [PubMed] [Google Scholar]
- 15.Seibert D, Hong CH, Takeuchi F, et al. Recognition of tuberous sclerosis in adult women: delayed presentation with life-threatening consequences. Ann Intern Med. 2011;154(12):806–813. doi: 10.1059/0003-4819-154-12-201106210-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yates JR. Tuberous sclerosis. Eur J Hum Genet. 2006;14(10):1065–1073. doi: 10.1038/sj.ejhg.5201625. [DOI] [PubMed] [Google Scholar]
- 17.Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591–594. doi: 10.4065/75.6.591. [DOI] [PubMed] [Google Scholar]
- 18.Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype:phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13(6):731–741. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- 20.Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9(2):88–100. doi: 10.1097/gim.0b013e31803068c7. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franz DN, Agricola KD, Tudor CA, Krueger DA. Everolimus for tumor recurrence after surgical resection for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. J Child Neurol. 2013;28(5):602–607. doi: 10.1177/0883073812449904. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Inoki K, Guan KL. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol. 2004;24(18):7965–7975. doi: 10.1128/MCB.24.18.7965-7975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804(3):433–439. doi: 10.1016/j.bbapap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Jentarra GM, Rice SG, Olfers S, Saffen D, Narayanan V. Evidence for population variation in TSC1 and TSC2 gene expression. BMC Med Genet. 2011;12:29. doi: 10.1186/1471-2350-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franz DN. Everolimus: an mTOR inhibitor for the treatment of tuberous sclerosis. Exp Rev Anticancer Ther. 2011;11(8):1181–1192. doi: 10.1586/era.11.93. [DOI] [PubMed] [Google Scholar]
- 27.van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet. 2012;20(5):510–515. doi: 10.1038/ejhg.2011.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansen AC, Sancak O, D’Agostino MD, et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann Neurol. 2006;60(5):528–539. doi: 10.1002/ana.21037. [DOI] [PubMed] [Google Scholar]
- 29.Krueger D, Northrup H. TSC Clinical Consensus Conference: summary of updates diagnosis, treatment and surveillance guidelines. Silver Spring, MD; Tuberous Sclerosis Alliance; 2012. 2012. [Google Scholar]
- 30.Berhouma M. Management of subependymal giant cell tumors in tuberous sclerosis complex: the neurosurgeon’s perspective. World J Pediatr. 2010;6(2):103–110. doi: 10.1007/s12519-010-0025-2. [DOI] [PubMed] [Google Scholar]
- 31.Roach ES, DiMario FJ, Kandt RS, Northrup H. Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol. 1999;14(6):401–407. doi: 10.1177/088307389901400610. [DOI] [PubMed] [Google Scholar]
- 32.Beaumont TL, Limbrick DD, Smyth MD. Advances in the management of subependymal giant cell astrocytoma. Childs Nerv Syst. 2012;28(7):963–968. doi: 10.1007/s00381-012-1785-x. [DOI] [PubMed] [Google Scholar]
- 33.Sun P, Kohrman M, Liu J, Guo A, Rogerio J, Krueger D. Outcomes of resecting subependymal giant cell astrocytoma (SEGA) among patients with SEGA-related tuberous sclerosis complex: a national claims database analysis. Curr Med Res Opin. 2012;28(4):657–663. doi: 10.1185/03007995.2012.658907. [DOI] [PubMed] [Google Scholar]
- 34.Cuccia V, Zuccaro G, Sosa F, Monges J, Lubienieky F, Taratuto AL. Subependymal giant cell astrocytoma in children with tuberous sclerosis. Childs Nerv Syst. 2003;19(4):232–243. doi: 10.1007/s00381-002-0700-2. [DOI] [PubMed] [Google Scholar]
- 35.Sun P, Krueger D, Liu J, Guo A, Rogerio J, Kohrman M. Surgical resection of subependymal giant cell astrocytomas (SEGAs) and changes in SEGA-related conditions: a US national claims database study. Curr Med Res Opin. 2012;28(4):651–656. doi: 10.1185/03007995.2012.658908. [DOI] [PubMed] [Google Scholar]
- 36.Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 37.Campen CJ, Porter BE. Subependymal giant cell astrocytoma (SEGA) treatment update. Curr Treat Options Neurol. 2011;13(4):380–385. doi: 10.1007/s11940-011-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Ribaupierre S, Dorfmüller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60(1):83–89. doi: 10.1227/01.NEU.0000249216.19591.5D. [DOI] [PubMed] [Google Scholar]
- 39.Amin S, Carter M, Edwards RJ, et al. The outcome of surgical management of subependymal giant cell astrocytoma in tuberous sclerosis complex. Eur J Paediatr Neurol. 2013;17(1):36–44. doi: 10.1016/j.ejpn.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 40.Levine NB, Collins J, Franz DN, Crone KR. Gradual formation of an operative corridor by balloon dilation for resection of subependymal giant cell astrocytomas in children with tuberous sclerosis: specialized minimal access technique of balloon dilation. Minim Invasive Neurosurg. 2006;49(5):317–320. doi: 10.1055/s-2006-950391. [DOI] [PubMed] [Google Scholar]
- 41.Jozwiak S, Nabbout R, Curatolo P. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2013;17(4):348–352. doi: 10.1016/j.ejpn.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 42.Matsumura H, Takimoto H, Shimada N, Hirata M, Ohnishi T, Hayakawa T. Glioblastoma following radiotherapy in a patient with tuberous sclerosis. Neurol Med Chir (Tokyo) 1998;38(5):287–291. doi: 10.2176/nmc.38.287. [DOI] [PubMed] [Google Scholar]
- 43.Sooriakumaran P, Gibbs P, Coughlin G, et al. Angiomyolipomata: challenges, solutions, and future prospects based on over 100 cases treated. BJU Int. 2010;105(1):101–106. doi: 10.1111/j.1464-410X.2009.08649.x. [DOI] [PubMed] [Google Scholar]
- 44.Bissler JJ, Racadio J, Donnelly LF, Johnson ND. Reduction of postembolization syndrome after ablation of renal angiomyolipoma. Am J Kidney Dis. 2002;39(5):966–971. doi: 10.1053/ajkd.2002.32770. [DOI] [PubMed] [Google Scholar]
- 45.Kohrman MH. Emerging treatments in the management of tuberous sclerosis complex. Pediatr Neurol. 2012;46(5):267–275. doi: 10.1016/j.pediatrneurol.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 46.Afinitor (everolimus) tablets for oral administration [summary of product characteristics] Stein, Switzerland: Novartis Pharma AG; 2012. [Google Scholar]
- 47.O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3(2):65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebwohl D, Thomas G, Lane HA, et al. Research and innovation in the development of everolimus for oncology. Expert Opin Drug Discov. 2011;6(3):323–338. doi: 10.1517/17460441.2011.558079. [DOI] [PubMed] [Google Scholar]
- 49.Houghton PJ. Everolimus. Clin Cancer Res. 2010;16(5):1368–1372. doi: 10.1158/1078-0432.CCR-09-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lane HA, Wood JM, McSheehy PM, et al. mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin Cancer Res. 2009;15(5):1612–1622. doi: 10.1158/1078-0432.CCR-08-2057. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka C, O’Reilly T, Kovarik JM, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008;26(10):1596–1602. doi: 10.1200/JCO.2007.14.1127. [DOI] [PubMed] [Google Scholar]
- 52.Kirchner GI, Meier-Wiedenbach I, Manns MP. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet. 2004;43(2):83–95. doi: 10.2165/00003088-200443020-00002. [DOI] [PubMed] [Google Scholar]
- 53.O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1588–1595. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 54.Kovarik JM, Hartmann S, Figueiredo J, et al. Effect of food on everolimus absorption: quantification in healthy subjects and a confirmatory screening in patients with renal transplants. Pharmacotherapy. 2002;22(2):154–159. doi: 10.1592/phco.22.3.154.33542. [DOI] [PubMed] [Google Scholar]
- 55.Crowe A, Bruelisauer A, Duerr L, Guntz P, Lemaire M. Absorption and intestinal metabolism of SDZ-RAD and rapamycin in rats. Drug Metab Dispos. 1999;27(5):627–632. [PubMed] [Google Scholar]
- 56.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 57.Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long-term safety and efficacy in subependymal giant-cell astrocytoma. Neurology. 2013;80(6):574–580. doi: 10.1212/WNL.0b013e3182815428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tillema JM, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology. 2012;78(8):526–531. doi: 10.1212/WNL.0b013e318247ca8d. [DOI] [PubMed] [Google Scholar]
- 59.Novartis Pharmaceuticals Efficacy and Safety of Everolimus (RAD001) in Patients of All Ages With Subependymal Giant Cell Astrocytoma Associated With Tuberous Sclerosis Complex (TSC)(EXIST-1) Available from http://www.clinicaltrials.gov/ct2/show/NCT00789828. NLM identifier: NCT00789828Accessed September 11, 2013
- 60.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 61.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26(13):1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 62.El-Hashemite N, Walker V, Zhang H, Kwiatkowski DJ. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003;63(17):5173–5177. [PubMed] [Google Scholar]
- 63.Dabora SL, Franz DN, Ashwal S, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and. PLoS One. 2011;6(9):e23379. doi: 10.1371/journal.pone.0023379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franz DN, Kingswood C, Jozwiak S, et al. Effect of everolimus on angiogenic biomarkers in patients with tuberous sclerosis complex (TSC): results From EXIST-1 and EXIST-2; Poster presented at: American Society of Clinical Oncology; June 1–5, 2012; Chicago, IL. [Google Scholar]
- 65.Ekici MA, Kumandas S, Per H, et al. Surgical timing of the subependymal giant cell astrocytoma (SEGA) with the patients of tuberous sclerosis complex. Turk Neurosurg. 2011;21(3):315–324. doi: 10.5137/1019-5149.JTN.4169-11.0. [DOI] [PubMed] [Google Scholar]
- 66.Kovarik JM, Hartmann S, Figueiredo J, Rouilly M, Port A, Rordorf C. Effect of rifampin on apparent clearance of everolimus. Ann Pharmacother. 2002;36(6):981–985. doi: 10.1345/aph.1A384. [DOI] [PubMed] [Google Scholar]
- 67.Fouladi M, Laningham F, Wu J, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol. 2007;25(30):1806–1812. doi: 10.1200/JCO.2007.11.4017. [DOI] [PubMed] [Google Scholar]