

Abstract
Previous studies have shown that dormant licensed replication origins can be exploited to enhance recovery from replication stress. Since tumour cells express high levels of origin licensing proteins, we examined whether depletion of such factors might specifically sensitise tumour versus non-tumour cells. Consistent with previous findings, we observed that three tumour-derived cell lines overexpress ORC1, a licensing component, compared to four non-tumour cell lines and that a greater level of ORC1 was required to maintain viability in the tumour cells. We determined siRNA-mediated knockdown conditions for each line that maximally reduced ORC1 but did not impact upon viability, which we considered would optimally deplete dormant origins. ORC1 depletion hypersensitised the tumour-derived cells to hydroxyurea (HU) and H202 but did not affect the sensitivity of the non-tumour lines. Similar results were observed following depletion of ORC6 or CDC6. Further, co-depletion of p53 and ORC1 modestly impaired viability of 1BR3hTERT non-tumour fibroblasts and more dramatically caused hypersensitivity to HU. Finally, overexpression of the c-Myc oncogene combined with ORC1 depletion in non-tumour BJhTERT cells diminished viability. Collectively, these findings suggest that tumour cells may have a reliance on origin licensing capacity, suggesting that licensing factors could represent a target for drug-based cancer therapy.
Keywords: Origin licensing, ORC1, replication stress, tumour cell lines, c-Myc overexpression
Introduction
To replicate the human genome in a timely manner, replication is initiated bidirectionally from multiple origins. However, this necessitates that replication origins only fire once during each cell cycle to avoid re-replication. Origin licensing occurs from late mitosis to G1-phase and involves assembly of the origin recognition complex (ORC), encompassing ORC1 to ORC6, onto origin sequences (1, 2). Together with CDC6 and CDT1, ORC loads the heterohexameric MCM2-7 complex which provides helicase activity, generating the pre-replication complex (pre-RC) (3-6). Cells exploit several mechanisms to prevent origin refiring, including the inhibition of MCM2-7 loading onto origins during S-G2 and the tight regulation of other pre-RC components via proteasome-mediated degradation (1, 2).
Only a fraction of licensed origins are used for replication, with non-fired origins being considered dormant (7-9). Following replication stress, the activation of checkpoint kinases stabilises stalled replication forks (10). Additionally, dormant origins can be exploited to promote recovery from replication stress (11-15). However, replication fork stalling also activates an intra-S phase checkpoint response that inhibits late-firing origins (16-18). Although apparently conflicting with the notion that replication fork stalling exploits dormant origins, recent studies have shown that origins are organised in clusters, with activation occurring stochastically and inactivating further origins within the cluster (14, 19). Whilst damage response signalling inhibits the firing of origins in new clusters, a distinct process promotes dormant origin firing within a cluster in which double fork stalling has occurred (14). This model is intrinsically appealing since it implies that dormant origins are only activated near a stalled fork where they are needed whilst new replication is diminished elsewhere to preclude further replication in the presence of DNA damage. Support for this model has come from studies involving siRNA-mediated depletion of MCM2-7 in human cells, which suppresses dormant origin usage, inhibits the rate of DNA synthesis, and reduces cell survival in response to replication-inhibiting agents (11-13).
Until recently, studies on dormant origin usage were predominantly undertaken in tumour cells and it was unclear whether the same process occurs in primary cells. Interestingly, an increase in the number of stalled replication forks in unchallenged S-phase cells was observed in mouse embryonic fibroblasts (MEFs) expressing a hypomorphic Mcm4Chaos3 allele, which impairs MCM2-7 complex stability and reduces the number of dormant origins (15). Significantly, Mcm4Chaos3 mice are cancer prone (20). Importantly, Mcm4Chaos3 cells have a normal rate of replication and helicase activity. Thus, reducing the number of dormant origins need not affect replication but can impede recovery from either endogenous or damage-induced replication stress. Although other pathways of replication fork recovery exist, a failure to use dormant origins is proposed to cause genomic instability.
Two recent studies identified mutations in origin licensing components (ORC1, ORC4, ORC6, CDT1 and CDC6) in Meier-Gorlin Syndrome (MGS), a disorder characterised by microcephaly, proportionate dwarfism and bone abnormalities including small or absent patellae (21-23). Cells from MGS patients, despite having substantially reduced origin licensing capacity, grow well in culture consistent with the notion that only a fraction of licensed origins are required to sustain replication (22).
Carcinogenesis necessitates multiple genetic changes to support often rapid and uncontrolled proliferation. Most tumour cells suffer high replication stress, due to uncontrolled proliferation and/or enhanced genomic instability. Interestingly, several studies have reported that origin licensing proteins are overexpressed in tumour-derived cell lines (24-27). Given this, we reasoned that tumour cells might have a greater demand for origin licensing than non-transformed cells, either to sustain rapid replication and/or to enhance recovery from the increased level of replication stalling/collapse. Given the finding that non-transformed cells can grow efficiently with substantially reduced licensing capacity, we considered that ORC proteins might represent targets to specifically sensitise tumour cells. Here, we examine this possibility by investigating the impact of diminished origin licensing capacity in tumour versus non-transformed cells. Strikingly, our results suggest that tumour cells more frequently rely on dormant origin usage following exposure to agents that cause replication stress compared to non-tumour cells.
Materials and Methods
Cell culture
Cell lines were purchased from the American Type Culture Collection (ATCC) or established and authenticated in-house or by scientific collaborators indicated in references. All cell lines were tested for mycoplasma contamination prior to use and assessed for ORC1 expression by immunoblot. Control primary skin fibroblasts (48BR), control hTERT-immortalised fibroblasts 1BR3hTERT or BJhTERT (ATCC), and ORC1-P4hTERT, derived from an ORC1-deficient MGS patient, were cultured in Dulbecco’s Modified Eagle’s medium supplemented with 15% fetal calf serum (FCS) (Invitrogen) (22, 28-30). Medium for BJ-MYC-ER, a derivative of BJhTERT expressing a tamoxifen-inducible c-Myc gene, was supplemented with 2 μg/ml puromycin (Invitrogen). MRC-5 is a primary fetal lung fibroblast cell line. MRC5, U2OS and HeLa cells (ATCC) were cultured in Minimal Essential Medium containing 10% FCS. Cells were transfected with siRNA oligonucleotide pools (Thermo Scientific Dharmacon) (ORC1, p53, ORC6, or CDC6) or Stealth siRNA targeting ORC1 (Invitrogen) (22) using HiPerFect (Qiagen) or DharmaFECT (Thermo Scientific Dharmacon). siControl represents scrambled oligonucleotides (Thermo Scientific Dharmacon).
Viability assay
siRNA-transfected cells were seeded in 96 well dishes, treated as described and viability was assessed using the CellTiter-Blue® assay (Promega). Viability was normalised to the siRNA-transfected but untreated control. The half maximal inhibitory concentration (IC50) values from viability curves were calculated with SigmaPlot (Systat Software, Inc.) using the five-parameter logistic nonlinear regression model. IC50 values represent the mean +/− standard deviation (SD) of three independent experiments.
Immunoblotting
For whole cell extracts, cells were lysed in buffer A (22) containing 500 mM NaCl and supplemented with protease (Sigma-Aldrich) and phosphatase inhibitors (Thermo Fisher Scientific) for one hour on ice and sonicated at 4°C. Fractionation of chromatin-bound and unbound proteins was performed as previously described (22). Lysates were resolved by electrophoresis, transferred onto PVDF (GE Healthcare) and immunoblotted using α-ORC1, ORC6, CDC6, p53 (Santa Cruz Biotechnology, Inc.), or β-actin (Abcam) antibodies.
Drug treatments
siRNA-transfected cells were seeded into dishes and grown for 48 hours. HU (Sigma-Aldrich) or H2O2 (Fisher Scientific/Acros Organics) was added for the indicated times. Cells were washed three times in phosphate buffered saline and incubated in fresh medium as indicated.
Clonogenic survival
Clonogenic survival was assessed as previously described (31). Briefly, cells were transfected with siRNA, treated as described and incubated for 10 days (U2OS) or for 21 days using irradiated feeder cells to enhance plating efficiency (1BR3hTERT). Survival was normalized to the siRNA-transfected but untreated control. Plotted values represent the mean +/− SD of three independent experiments.
DNA fibre assay
Cells were labelled with 25 μM chlorodeoxyuridine (CldU) for 20 min, washed three times with medium, incubated in 2 mM HU for 24 hours, washed three times again, and pulse-labelled with 250 μM iododeoxyuridine (IdU) for 1 hour. Labelled cells were harvested and DNA fibre spreads were prepared as previously described (32). CldU and IdU were detected by incubating acid-treated fibre spreads with rat α-BrdU (1:1000, AbD Serotec) and mouse α-bromodeoxyuridine (BrdUrd) (1:750, Becton Dickinson) monoclonal antibodies for 1 hour. Slides were fixed with 4% paraformaldehyde and incubated with AlexaFluor 555-conjugated goat α-rat IgG (1:500, Molecular Probes) and AlexaFluor 488-conjugated goat α-mouse IgG (1:500, Molecular Probes) for 1.5 hours. Images of DNA fibres were acquired on a Nikon E600 microscope using a 60x (1.3NA) lens, a Hamamatsu digital camera and the Volocity package (Perkin Elmer). For quantification, at least 130 structures were counted per experiment using ImageJ software (National Institutes of Health, http://rsbweb.nih.gov/ij/).
Replication recovery assay
siRNA-transfected cells were treated with HU and labelled with 50 μM BrdU (Becton Dickinson) 30 minutes prior to harvesting. Cells were fixed, BrdU-labelled, propidium iodide (PI)-stained and analysed by FACS as previously described (31).
γH2AX immunofluorescence
Cells were processed for γH2AX analysis as previously described (33) using α-γH2AX and α-CENPF (Abcam) and DAPI labelling of DNA. Cells harbouring S phase-associated DNA damage, referred to as γH2AX+, were detected by bright, pan-nuclear γH2AX and minimal CENPF signal (34, 35). γH2AX+ cells were manually scored in >500 cells per condition. Images of cells were acquired with identical exposure settings on a Zeiss Axioplan2 microscope using a 40x (0.75 NA) lens, a Hamamatsu digital camera, and SimplePCI software (Hamamatsu).
Results
Substantial ORC1 depletion does not impact upon proliferation
We aimed to compare how diminished origin licensing capacity affects recovery from replication stress in tumour-derived versus non-tumour cell lines. We used 1BR3hTERT and U20S osteosarcoma cells as non-tumour and tumour cell line, respectively. 1BR3hTERT, an hTERT immortalized fibroblast line derived from a normal individual, has a stable karyotype, shows genomic stability, and has an intact G1/S checkpoint (unpublished observations). Since ORC1 is essential, we sought knockdown conditions that reduce ORC1 protein levels without impeding proliferation. Viability, monitored using the CellTiter-Blue® assay, was assessed following siRNA-mediated knockdown of ORC1 in 1BR3hTERT and U20S using a range of siRNA oligonucleotide (scrambled or ORC1-specific) concentrations. We observed diminished proliferation with increasing siORC1 concentrations, consistent with the notion that oligonucleotide concentration correlates with knockdown efficiency (Fig. 1A-B). Since tumour and non-tumour cell lines differ in efficiency of siRNA-mediated knockdown and requirement for ORC1, the impact was distinct for each line. The highest siORC1 concentration that did not significantly impede viability was 5 or 0.6 nM for 1BR3hTERT and U20S, respectively (Supplementary Fig. S1A). Immunoblotting revealed that U20S has higher ORC1 protein levels compared to 1BR3hTERT, consistent with previous findings that tumour-derived cells overexpress ORC proteins (Fig. 1C) (24-27). Although α-ORC1 antibodies are inefficient for immunoblotting, a marked reduction in ORC1 protein could be observed in both lines following siORC1 (Fig. 1C). Routinely, low residual ORC1 was detectable in siORC1-treated U20S cells whilst residual ORC1 was not detectable in siORC1-transfected 1BR3hTERT. Since ORC1 is essential, it is likely that 1BR3hTERT retain residual, although undetectable, ORC1. This suggests that U20S cells require a higher level of ORC1 to maintain viability compared to 1BR3hTERT, consistent with the notion that tumour cells have a greater need for origin licensing proteins compared to non-tumour cells. Having identified knockdown conditions that substantially deplete ORC1 without impeding viability, which we anticipated would substantially reduce the level of dormant origins, we proceeded to examine the impact on recovery from damage-induced replication arrest.
Figure 1. siORC1 impairs recovery of U2OS but not 1BR3hTERT cells from HU.

1BR3hTERT (A) or U2OS (B) cells were transfected with siControl or siORC1 oligonucleotides (0.1-20 nM), and viability assessed 7 days later. Results represent mean +/− SD from triplicate samples. Black arrows indicate the oligonucleotide concentration subsequently utilised. (C) 1BR3hTERT and U2OS cells were transfected with 5 or 0.6 nM siORC1, respectively. ORC1 protein levels were assessed by immunoblotting 48 hours later. β-actin was a loading control. (D-E) 1BR3hTERT and U2OS cells were transfected with siRNA for 48 hours and treated with HU (0.03-40 mM) for 24 hours. Viability was assessed 4 days following HU removal. (F-G) 1BR3hTERT and U2OS cells transfected with siRNA as described were treated with 2 mM HU for indicated times and HU-induced S-phase damage was assessed by immunofluorescence labelling of γH2AX. Nuclei containing bright γH2AX pan-nuclear staining were scored as γH2AX+. Black arrows indicate the time of HU treatment required to obtain 100% γH2AX+ cells. (H) Viability was assessed after transfection with siRNA and treatment with HU for 24 or 48 hours as in (D-E). HU IC50 values were estimated from the viability graphs. (I) U2OS cells were treated with siRNA as described above and then with HU (0.05-2 mM) for 24 hours. Clonogenic survival was estimated at 10 days following HU removal. (J) As in (I) except 1BR3hTERT cells were treated with HU for 48 hours and clonogenic survival was estimated 21 days following HU removal. Additional controls are shown in Supplementary Fig. S1. (K) ORC1 protein levels were assessed in chromatin-bound and unbound fractions in 1BR3hTERT and ORC1-P4hTERT by immunoblotting. (L) 1BR3hTERT or ORC1-P4hTERT were treated with HU (0.03-40 mM) for 24 hours. Viability was assessed 4 days post HU removal. (M) 1BR3hTERT or ORC1-P4hTERT cells were treated with 0.5 mM HU for 48 or 72 hours, respectively. Viability was assessed as above.
ORC1 depletion impairs recovery from hydroxyurea in U20S but not 1BR3hTERT
We examined sensitivity to hydroxyurea (HU), which depletes ribonucleotide reductase and enhances fork stalling/collapse, in 1BR3hTERT and U20S following siControl or siORC1. siRNA transfection was conducted as described above and cells were exposed to differing concentrations of HU for 24 hours. HU was removed and viability monitored 4 days later (Fig. 1D-E). To compare the effect of siORC1 between the cell lines, we estimated the HU concentration that reduced viability by 50 % (the IC50 value). The relative IC50 value was calculated by comparison to the IC50 of siControl-transfected cells (Fig. 1H). Strikingly, whereas siORC1 did not affect HU sensitivity in 1BR3hTERT, it significantly enhanced sensitivity in U20S. Similar effects were observed following transfection of cells with a distinct pool of ORC1 siRNA oligonucleotides (Supplementary Fig. S1B-D). To verify that the resistance of 1BR3hTERT cells is not simply a consequence of their slower cell cycle progression, resulting in a lower fraction of cells progressing into S phase, we monitored the population doubling time (Supplementary Fig. S1E-F) and rate of HU-induced γH2AX formation in the two cell lines (Fig. 1F-G). We estimated that by 48 hour all 1BRhTERT cells had undergone replication fork arrest after HU. However, examination of viability following 48 hour HU treatment yielded similar results (Fig. 1H). Thus, the resistance of 1BR3hTERT cells to siORC1-induced hypersensitivity is not explained by their slower cell cycle progression. To verify that the viability assay reflects clonogenicity, we also examined clonogenic survival of U20S following 24 hour HU treatment (Fig. 1I) and of 1BR3hTERT following 48 hour HU treatment (Fig. 1J). Although these two assays monitor different endpoints, a similar impact was observed, validating use of the viability assay.
To substantiate these findings without relying on siRNA-mediated depletion, we also examined an hTERT-immortalised fibroblast line derived from an ORC1-deficient MGS patient (ORC1-P4hTERT) (22). ORC1 is expressed at normal levels in ORC1-P4hTERT but chromatin binding of ORC1 is impaired (22) (Fig. 1K).We observed resistance rather than marked sensitivity of ORC1-P4hTERT cells to HU at higher concentrations potentially due to a lower number of replication origins (Fig. 1L). We also adjusted HU treatment time to achieve complete HU-induced S phase arrest in 1BR3hTERT and ORC-P4hTERT (representing 48 or 72 hour exposures, respectively). Under these conditions, resistance but not sensitivity to 0.5 mM HU was also observed (Fig. 1M).
U20S cells show diminished recovery of DNA synthesis and accumulated DNA damage following siORC1 compared to 1BR3hTERT
To examine whether siORC1 affects replication recovery, 1BR3hTERT or U20S were treated with siRNA as described above and exposed to 2 mM HU for 24 hours. Following HU removal, cells were incubated for 2, 4, or 24 hours, and BrdU added for the final 30 minutes. BrdU incorporation, representing recovery of DNA synthesis, was assessed by FACS (Fig. 2A-B). HU treatment abolished BrdU incorporation in both cell lines, consistent with replication inhibition. Strikingly, in 1BR3hTERT, BrdU incorporation was substantially recovered at 2 hours post HU removal and was similar in siControl or siORC1-treated cells. In marked contrast, although siControl-treated U20S cells also recovered DNA synthesis at 2 hours following HU removal, DNA synthesis was dramatically reduced at this time in siORC1-treated U20S cells. Since this difference is observed at early times (2 hours) post HU removal, this suggests that siORC1 does not impair rapid recovery of replication in HU-treated 1BR3hTERT but does so in U20S.
Figure 2. siORC1 impairs recovery of replication following HU and reduces HU-induced new origin firing in U2OS.
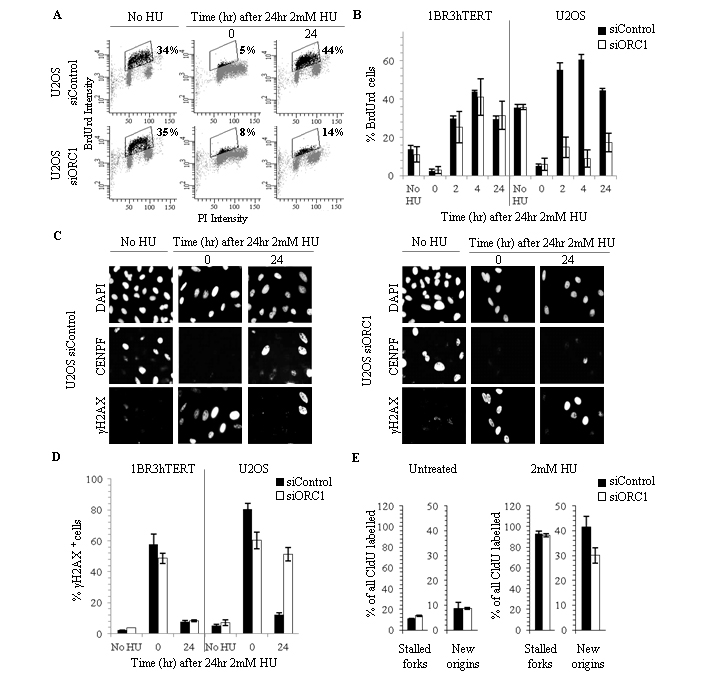
(A-B) 1BR3hTERT and U2OS cells were transfected with siRNA oligonucleotides (5 or 0.6 nM, respectively). 48 hours later, 2 mM HU was added for 24 hours and cells were grown for times shown. BrdU was added 30 minutes prior to processing by FACS. The fraction of replicating (BrdU+) cells was determined. (A) Representative FACS analysis. Boxed regions containing black data points indicate BrdU+ cells; numbers represent BrdU+ cell fraction. Supplementary Fig. S2A shows additional analyses. (B) Quantification from three experiments using 1BR3hTERT or U2OS cells. (C) Representative immunofluorescence images showing DAPI (DNA), CENPF (cell cycle phase) or γH2AX (DNA damage) in U20S cells treated as in (A-B). Representative merged channel images are shown in Supplementary Fig. S2B. (D) Nuclei containing bright γH2AX pan-nuclear staining were scored as γH2AX+. Figure shows the fraction of γH2AX+ cells. (E) 48 hours following transfection of U2OS cells with 0.6 nM siControl or siORC1, cells were pulse labelled with CldU, treated with 2 mM HU for 24 hours and released (or untreated), and pulse labelled with IdU for 1 hour. The number of structures representing fork stalling and new origin firing was normalised to the number of CldU+ replication tracks. The experimental design and representative images are shown in Supplementary Fig. S2C-D. Results represent the mean and SD of > 2 experiments (0mM HU n=2, 2mM HU n=3).
We also examined whether the inability of siORC1-treated U2OS to recover replication causes accumulated DNA damage. Either immediately (0) or 24 hours following treatment with 2 mM HU, cells were examined for γH2AX, a marker of DNA damage, and CENPF, a G2/M-phase marker. Immediately following HU treatment, most cells were CENPF- (consistent with S-phase arrest) and showed pan-nuclear γH2AX staining, demonstrating the presence of collapsed/stalled replication forks; untreated cells had a lower fraction of γH2AX+ cells (Fig. 2C-D). 24 hours post HU removal, few siControl or siORC1-transfected 1BR3hTERT cells were γH2AX+ (Fig. 2D), consistent with the observed recovery of replication. Similarly, the number of γH2AX+ siControl-treated U20S cells was dramatically reduced 24 hours post HU removal (Fig. 2C-D). In stark contrast, approximately 50 % of siORC1-treated U20S cells retained γH2AX staining 24 hours post HU removal. γH2AX+ cells were negative for the G2/M marker, CENPF, consistent with the notion that they represent damaged S-phase cells. This analysis shows that 1BR3hTERT undergo replication fork arrest and activate the DNA damage response after HU treatment but efficiently recover despite substantial depletion of ORC1.
ORC1 depletion reduces new origin firing after HU in U20S
The DNA fibre assay allows replication at new versus pre-existing origins to be monitored (36).We exploited the technique to assess new origin firing in U2OS cells after release from HU exposure. siRNA-transfected cells were exposed to CldU for 20 minutes, then either exposed to IdU for 20 minutes (control) or CldU was washed out and cells were exposed to HU for 24 hours. Following HU removal, IdU was added for 1 hour. CldU+/IdU- replication tracks are considered to represent stalled forks that have not reinitiated replication; CldU-/IdU+ tracks represent ones with newly fired origins (Supplementary Fig. S2C-D). To monitor new origin firing, the fraction of CldU-/IdU+ tracks was assessed. In untreated U20S cells, ORC1 siRNA did not significantly impact upon new origin firing (Fig. 2E). Following HU, although siORC1 did not impact upon the level of stalled forks, new origin firing was substantially diminished. These findings strongly suggest that siORC1 diminishes replication restart by new origin firing after HU whilst not affecting new origin firing in unperturbed cells.
Hypersensitivity of U20S to HU following depletion of additional licensing components
We next examined whether the sensitivity of U2OS cells to HU is impacted following depletion of additional origin licensing factors. We examined ORC6 and CDC6 since they are also causal defects for MGS (22). 0.6 nM siORC6 or CDC6 substantially depleted ORC6 or CDC6 but did not impede cellular proliferation (Fig. 3A; data not shown). Viability assessment revealed a similar level of HU sensitivity following siORC6 or CDC6 to that observed following siORC1 (Fig. 3B-C). However, neither siORC6 nor siCDC6 affected HU sensitivity in 1BR3hTERT (Supplementary Fig. S3).
Figure 3. Depletion of additional origin licensing components in U20S enhances HU sensitivity.

U2OS cells were transfected with 0.6 nM siRNA for 48 hours. (A) Immunoblotting using α-ORC6 and α-CDC6. (B) Viability was assessed as in Fig. 1D-E. (C) Estimated IC50 values.
siORC1 does not affect HU sensitivity in additional non-tumour lines (BJhTERT, 48BR, and MRC-5) but sensitises additional tumour-derived lines (HeLa and MDA-MB-231)
To extend our findings, we examined additional non-tumour fibroblasts (BJhTERT, 48BR, and MRC-5) and tumour-derived lines (HeLa and MDA-MB-231). 48BR and MRC5 represent primary fibroblast lines to complement the analysis of the hTERT-immortalised line. For all lines, we examined the optimum siORC1 oligonucleotide concentration that failed to impact upon viability (Supplementary Fig. S4). BJhTERT cells showed slightly diminished viability above 5 nM siORC1 similar to 1BR3hTERT cells; 5 nM was chosen for analysis (Supplementary Fig. S4A). HeLa cells were resistant to high oligonucleotide concentrations; 0.6 nM was chosen to allow comparison to U20S (Fig. 4A-B, Supplementary Fig. S4D). 48BR, MDA-MB-231, and MRC-5 displayed diminished viability above 1 nM siORC1; 1 nM was chosen for analysis (Supplementary Fig. S4B-C, E). The time of HU treatment required to achieve complete HU-induced S phase damage was also assessed in each cell line (Supplementary Fig. S5). The time indicated was used for subsequent viability experiments. Similar to 1BR3hTERT and U20S cells, although ORC1 protein levels were greater in siORC1-depleted HeLa and MDA-MB-231 cells compared to BJhTERT, 48BR, or MRC-5 cells, siORC1 enhanced HU sensitivity of HeLa and MDA-MB-231 without substantially impacting upon sensitivity of BJhTERT, 48BR or MRC-5 cells (Fig. 4A, C-G). For MRC-5, slightly enhanced sensitivity was observed at high HU doses but there was no impact on the IC50 value.
Figure 4. siORC1 enhances HU sensitivity of additional tumour-derived cell lines (HeLa and MDA-MB-231) but does not affect non-tumour cells (BJhTERT, 48BR, and MRC5).

(A-G) BJhTERT and were transfected with 5nM, 48BR, MRC-5, and MDA-MB-231 with 1 nM, and HeLa with 0.6 nM siRNA oligonucleotides. (A) ORC1 protein was assessed by immunoblotting 48 hours later. (B) Viability was assessed 7 days later. Analyses using different siORC1 oligonucleotide concentrations are shown in Supplementary Fig. S4. (C-G) Viability was assessed as in Fig. 1D-E using indicated HU treatment times to achieve complete HU-induced S phase arrest (Supplementary Fig. S5A-E).
siORC1 diminishes viability of tumour but not primary cells to H202
Having shown that siORC1 hypersensitises U20S and HeLa but not 1BR3hTERT or BJhTERT cells to HU, we used a similar approach to evaluate whether recovery from oxidative damage, which can indirectly induce replication stress, might also involve differential dormant origin usage. Strikingly, whilst siORC1 did not affect sensitivity of 1BR3hTERT or BJhTERT to H202, U20S and HeLa cells showed marked hypersensitivity (Fig. 5A-E). The slightly higher resistance of siORC1-treated 1BR3hTERT cells to H202 compared with siControl cells likely reflects their slightly slower replication. Nonetheless, the distinction between 1BR3hTERT/BJhTERT and U20S/HeLa cells to combined siORC1 and H202 was marked.
Figure 5. siORC1 specifically enhances sensitivity of tumour-derived cell lines to H2O2.

(A-E) 1BR3hTERT, BJhTERT, U20S or HeLa were transfected with siRNA as described in Fig. 1 and 4. 48 hours later, cells were treated with H2O2, with concentrations adjusted to account for substantial differences in sensitivity between cell lines. 24 hours later, H2O2 was removed and viability assessed 4 days later. (A-D) Representative viability plots. (E) Estimated IC50 values.
sip53 mildly sensitises 1BR3hTERT cells to siORC1 without exogenous DNA damage and causes marked sensitivity to HU
p53 loss abrogates the damage-induced G1/S checkpoint, enhancing S-phase progression and replication stalling (37). Additionally, p53 is required for a licensing checkpoint which prevents S-phase entry until sufficient origins have been licensed (38, 39). We examined whether sip53 in 1BR3hTERT affects viability and HU sensitivity following siORC1. 1BR3hTERT cells were transfected with siControl, sip53, siORC1 or combined siORC1+sip53 for 48 hours, then viability assessed in untreated or HU-treated cells as described above. p53 was efficiently depleted in 1BR3hTERT (Fig. 6A). As above, siORC1 did not affect viability of 1BR3hTERT cells (Fig. 6B). sip53 alone slightly inhibited viability but combined sip53+siORC1 diminished viability by approximately1.7 (Fig. 6B). These findings suggest that in undamaged cells, siORC1 more markedly affects viability in the absence of p53. siORC1 did not significantly affect HU sensitivity similar to the findings in Fig. 1D; there was a modest but not statistically significant impact of sip53 on HU sensitivity but a marked decrease following sip53+siORC1 (Fig. 6C-D).
Figure 6. p53 depletion enhances sensitivity of 1BR3hTERT to HU.

(A-D) 1BR3hTERT was transfected with 5 nM siControl, siORC1, sip53 or a combination of siORC1+sip53. (A) 48 hours later, p53 levels were assessed by immunoblotting. (B) Viability was assessed 7 days after siRNA transfection as in Fig. 1A-B. (C-D) Viability was assessed in HU-treated siRNA transfected cells as in Fig. 1D-E. (C) Representative viability curves. (D) Estimated IC50 values.
Depletion of ORC1 confers sensitivity to Myc overexpression
Myc overexpression, which enhances proliferation and replication stress, is frequently observed during carcinogenesis (40-42). We examined whether Myc expression influences the requirement for origin licensing capacity using a BJhTERT derivative that expresses c-Myc fused to a tamoxifen-inducible estrogen receptor (40-42). Firstly, anticipating that tamoxifen concentration affects the level of c-Myc expression, we estimated the tamoxifen concentration promoting endogenous DNA damage (assessed by γH2AX). 24 hours post 2μM tamoxifen, 30 % of siControl-transfected cells were γH2AX+, suggesting that Myc overexpression induces replication stress (Fig. 7A). Following siORC1, 60 % of BJhTERT cells were γH2AX+, raising the possibility that siORC1 causes enhanced or persistent c-Myc-induced replication arrest. Next, assessment of viability 5 days following exposure to different tamoxifen concentrations revealed substantial sensitivity following siORC1 (Fig. 7B-C) suggesting that depletion of origin licensing capacity diminishes the ability to cope with c-Myc-induced replication stress.
Figure 7. siORC1 enhances viability following Myc overexpression.

(A-C) BJhTERT cells stably expressing a tamoxifen-inducible Myc oncogene (BJ-MYC-ER) were transfected with 5 nM siRNA for 48 hours. Tamoxifen was added as indicated. (A) 24 hours following transfection, cells were examined by immunofluorescence for γH2AX (DNA damage), CENPF (cell cycle phase) and DAPI (DNA). Nuclei containing bright γH2AX pan-nuclear staining were scored as γH2AX+. (B) BJ-MYC-ER fibroblasts were transfected with siRNA as in (A) and treated with tamoxifen (0.07-100 μM) to induce Myc expression. Viability was assessed 5 days later. (C) Estimated IC50 values.
Discussion
We previously observed that MGS patient-derived cell lines grow efficiently in culture despite ten-fold lower levels of origin licensing components (21, 22). This ability to sustain substantial proliferation is consistent with findings that only 10 % of licensed origins are utilised during unchallenged replication (7-9). Recent studies have provided evidence that dormant origins can be utilised to promote recovery from replication fork stalling or collapse (11-15). Since tumour cells show elevated oxidative and replicative stress, we predicted that they might have an enhanced reliance on origin licensing capacity compared to normal cells, raising the possibility that targeting origin licensing components could specifically inhibit cancer cell growth. Here, we evaluate this possibility.
Consistent with previous findings, we observed that three tumour-derived cell lines showed high ORC1 expression compared to non-tumour lines (24-27). In general, higher ORC1 levels were required to maintain viability in the tumour lines (although MRC-5 cells also had a requirement for a higher ORC1 level). Nonetheless, the ability to detect higher residual ORC1 in the tumour than non-tumour cell lines demonstrates that our findings cannot simply be explained by more efficient ORC1 knockdown in the tumour versus non-tumour cells.
To enhance the applicability to exploit inhibition of origin licensing for tumour therapy, we examined whether partial ORC1 depletion affected the response to HU, a chemotherapeutic agent. Significantly, we observed marked sensitisation of the tumour-derived lines to HU (or H202) compared to the non-tumour cells. Additionally, we observed that ORC1 depletion enhanced HU sensitivity of p53-depleted non-tumour cells and also conferred sensitivity to Myc overexpression. This important result suggests that enhancing the level of replication stress and/or rate of proliferation, both of which arise following c-Myc expression, increases the demand for origin licensing capacity.
Collectively, using our panel of three tumour and four primary or hTERT-immortalised cell lines, our findings suggest that tumour cells have a greater demand for origin licensing capacity following replication fork arrest compared to non-tumour lines and that loss of p53 or c-Myc expression enhances this demand in non-tumour cells. Our findings could have several explanations. One possibility is that following replication stress, stalled forks more readily collapse in tumour compared with non-tumour cells and that dormant origin firing enhances recovery from replication fork collapse. However, we are not aware of studies supporting this suggestion. Alternatively, it is possible that fork collapse occurs similarly in tumour and non-tumour cells, but that tumour cells more frequently exploit dormant origins for recovery and are hence hypersensitive when this route is unavailable. Although the original studies describing dormant origin usage after replication stress used tumour cells, a recent study involving primary MEFs showed that they also exploit dormant origins for recovery from replicative stress (15). However, it is difficult to evaluate the comparative usage of dormant origins in tumour versus non-tumour cells from these studies. An alternative and appealing possibility is that tumour cells override the origin licensing checkpoint to enhance proliferation and, therefore, enter S-phase with diminished dormant origins. Indeed, the upregulation of origin licensing proteins in tumour cells may reflect their need to effect rapid origin licensing during their short G1 phase. Thus, further reducing licensing capacity may provide a situation where there are insufficient dormant origins to exploit following replication fork arrest. It should be noted that our knockdown conditions were designed to prevent any impact on unperturbed cell growth. This model is consistent with the known function of p53 in enhancing G1-phase progression and/or abolishing the G1/S checkpoint. Importantly in the present context, p53 is also required for the origin licensing checkpoint since co-depletion of p53 and CDC6, another licensing component, in normal fibroblasts permits S-phase entry with insufficient origin capacity (43). c-Myc also enhances G1-phase progression and disrupts p53 activity (44, 45). However, both p53 loss and c-Myc expression have multiple additional impacts including an influence on replication (44-46). Thus, although the latter explanation is appealing, further work is required to define the basis underlying our observations. It is likely, moreover, that there could be multiple impacts. Our findings to date are based on a restricted number of tumour or non-tumour cell lines. Nonetheless, the relationship seems marked and further work will be required to examine the extent to which this represents a phenotype of many tumour cell lines. It should be noted that around 50 % of tumour cell lines show an upregulation of origin licensing components (24-27).
Our goal was to examine whether the origin licensing complex represents a suitable target to specifically sensitise tumour cells. Importantly, we report that three tumour cells require greater origin licensing capacity following exposure to DNA damaging agents than four non-tumour cells. Additionally, we demonstrate that p53 loss (in the presence of HU) or c-Myc expression in non-tumour cells enhances the reliance on ORC1. Interestingly, the BAH domain of ORC1 was recently reported to bind H4K20me2 with high specificity and affinity via an aromatic cage, which could provide a route for drug targeting (47). In summary, we provide evidence that the down-regulation of ORC1 and other origin licensing proteins enhances the sensitivity of tumour but not non-tumour cell lines to replicative stress, providing a potential route for specific sensitisation of tumour cells.
Supplementary Material
Acknowledgements
We thank Dr. M. O’Driscoll for discussions and Dr. O. Fernandez-Capetillo for kindly providing BJ-MYC-ER.
Financial support: The PAJ laboratory is supported by an MRC programme grant, the Association for International Cancer Research, the Wellcome Research Trust and the EMF Biological Research Trust. The E. Petermann laboratory is supported by an MRC project grant, Cancer Research UK and the University of Birmingham.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nature reviews Molecular cell biology. 2005;6:476–86. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes & development. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 3.Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA-replication by a multiprotein complex. Nature. 1992;357:128–34. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 4.Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404:625–8. doi: 10.1038/35007110. [DOI] [PubMed] [Google Scholar]
- 5.Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends in biochemical sciences. 2011;36:405–14. doi: 10.1016/j.tibs.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719–30. doi: 10.1016/j.cell.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anglana M, Apiou F, Bensimon A, Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell. 2003;114:385–94. doi: 10.1016/s0092-8674(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 8.DePamphilis ML, Blow JJ, Ghosh S, Saha T, Noguchi K, Vassilev A. Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol. 2006;18:231–9. doi: 10.1016/j.ceb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert DM. Evaluating genome-scale approaches to eukaryotic DNA replication. Nature reviews Genetics. 2010;11:673–84. doi: 10.1038/nrg2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caspari T, Carr AM. Checkpoints: how to flag up double-strand breaks. Curr Biol. 2002;12:R105–7. doi: 10.1016/s0960-9822(02)00673-5. [DOI] [PubMed] [Google Scholar]
- 11.Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8956–61. doi: 10.1073/pnas.0803978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. The Journal of cell biology. 2006;173:673–83. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–41. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blow JJ, Ge XQ. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009;10:406–12. doi: 10.1038/embor.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, et al. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Molecular cell. 2011;41:543–53. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert S, Carr AM. Checkpoint responses to replication fork barriers. Biochimie. 2005;87:591–602. doi: 10.1016/j.biochi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nature reviews Molecular cell biology. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 18.Branzei D, Foiani M. The DNA damage response during DNA replication. Current opinion in cell biology. 2005;17:568–75. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Lebofsky R, Heilig R, Sonnleitner M, Weissenbach J, Bensimon A. DNA replication origin interference increases the spacing between initiation events in human cells. Molecular biology of the cell. 2006;17:5337–45. doi: 10.1091/mbc.E06-04-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, et al. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nature genetics. 2007;39:93–8. doi: 10.1038/ng1936. [DOI] [PubMed] [Google Scholar]
- 21.Bicknell LS, Bongers EM, Leitch A, Brown S, Schoots J, Harley ME, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43:356–9. doi: 10.1038/ng.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A, Kerzendorfer C, et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet. 2011;43:350–5. doi: 10.1038/ng.776. [DOI] [PubMed] [Google Scholar]
- 23.Gorlin RJ. Microtia, absent patellae, short stature, micrognathia syndrome. J Med Genet. 1992;29:516–7. [PMC free article] [PubMed] [Google Scholar]
- 24.McNairn AJ, Gilbert DM. Overexpression of ORC subunits and increased ORC-chromatin association in transformed mammalian cells. Journal of cellular biochemistry. 2005;96:879–87. doi: 10.1002/jcb.20609. [DOI] [PubMed] [Google Scholar]
- 25.Lau E, Tsuji T, Guo L, Lu SH, Jiang W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2007;21:3786–94. doi: 10.1096/fj.07-8900rev. [DOI] [PubMed] [Google Scholar]
- 26.Di Paola D, Zannis-Hadjopoulos M. Comparative analysis of pre-replication complex proteins in transformed and normal cells. Journal of cellular biochemistry. 2012;113:1333–47. doi: 10.1002/jcb.24006. [DOI] [PubMed] [Google Scholar]
- 27.Karakaidos P, Taraviras S, Vassiliou LV, Zacharatos P, Kastrinakis NG, Kougiou D, et al. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability--evidence of E2F-1 transcriptional control over hCdt1. Am J Pathol. 2004;165:1351–65. doi: 10.1016/S0002-9440(10)63393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata A, Barton O, Noon AT, Dahm K, Deckbar D, Goodarzi AA, et al. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol Cell Biol. 2010;30:3371–83. doi: 10.1128/MCB.01644-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, et al. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nature genetics. 1999;21:115–8. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- 30.Keyse SM, McAleer MA, Davies DJ, Moss SH. The response of normal and ataxia-telangiectasia human fibroblasts to the lethal effects of far, mid and near ultraviolet radiations. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1985;48:975–85. doi: 10.1080/09553008514552101. [DOI] [PubMed] [Google Scholar]
- 31.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–87. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, et al. XRCC3 and Rad51 Modulate Replication Fork Progression on Damaged Vertebrate Chromosomes. Mol Cell. 2003;11:1109–17. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 33.Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011;39:6986–97. doi: 10.1093/nar/gkr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nature structural & molecular biology. 2011;18:1331–5. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, et al. gamma H2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle. 2010;9:662–9. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 36.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Molecular cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wahl GM, Linke SP, Paulson TG, Huang LC. Maintaining genetic stability through TP53 mediated checkpoint control. Cancer Surv. 1997;29:183–219. [PubMed] [Google Scholar]
- 38.Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002;21:6624–32. doi: 10.1038/sj.onc.1205910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ge XQ, Blow JJ. The licensing checkpoint opens up. Cell Cycle. 2009;8:2320–2. [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson K, Asawachaicharn N, Galloway DA, Grandori C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PloS one. 2009;4:e5951. doi: 10.1371/journal.pone.0005951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herold S, Herkert B, Eilers M. Facilitating replication under stress: an oncogenic function of MYC? Nature reviews Cancer. 2009;9:441–4. doi: 10.1038/nrc2640. [DOI] [PubMed] [Google Scholar]
- 42.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 43.Nevis KR, Cordeiro-Stone M, Cook JG. Origin licensing and p53 status regulate Cdk2 activity during G(1) Cell cycle. 2009;8:1952–63. doi: 10.4161/cc.8.12.8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Molecular cell. 2002;9:1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 45.Ceballos E, Munoz-Alonso MJ, Berwanger B, Acosta JC, Hernandez R, Krause M, et al. Inhibitory effect of c-Myc on p53-induced apoptosis in leukemia cells. Microarray analysis reveals defective induction of p53 target genes and upregulation of chaperone genes. Oncogene. 2005;24:4559–71. doi: 10.1038/sj.onc.1208652. [DOI] [PubMed] [Google Scholar]
- 46.Kumari A, Schultz N, Helleday T. p53 protects from replication-associated DNA double-strand breaks in mammalian cells. Oncogene. 2004;23:2324–9. doi: 10.1038/sj.onc.1207379. [DOI] [PubMed] [Google Scholar]
- 47.Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–9. doi: 10.1038/nature10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.