

Abstract
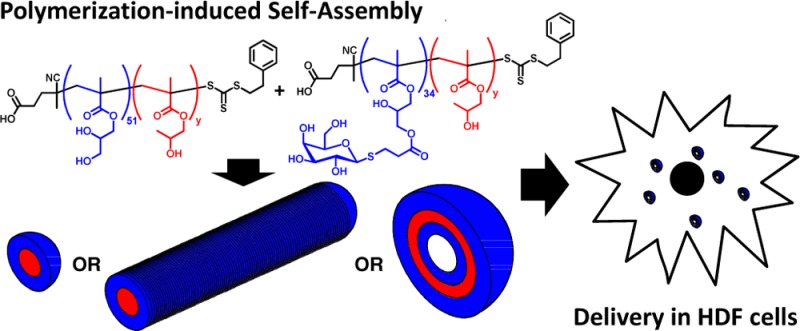
Recent advances in polymer science are enabling substantial progress in nanobiotechnology, particularly in the design of new tools for enhanced understanding of cell biology and for smart drug delivery formulations. Herein, a range of novel galactosylated diblock copolymer nano-objects is prepared directly in concentrated aqueous solution via reversible addition–fragmentation chain transfer polymerization using polymerization-induced self-assembly. The resulting nanospheres, worm-like micelles, or vesicles interact in vitro with galectins as judged by a turbidity assay. In addition, galactosylated vesicles are highly biocompatible and allow intracellular delivery of an encapsulated molecular cargo.
Introduction
Controlled radical polymerization (CRP) techniques have enabled synthetic chemists to prepare a remarkably wide range of functional copolymers.1 Glycopolymers2 have particularly benefited from the versatility offered by CRP syntheses, with many well-defined architectures3 (e.g., block,4 surface grafting,5 hyperbranched,6 or dendritic7) being reported. Indeed, glycosylated macromolecules and nano-objects are very promising tools to study biological processes and/or design novel therapeutics in the field of nanobiotechnology.8−11 Glycotargeting exploits interactions of specific glycan receptors with carbohydrate ligands. Given the vast density of information that sugars can encode, these interactions offer enhanced specificity and affinity compared to many other ligand-binding systems.12 Glycotargeting was first demonstrated more than four decades ago.13 However, despite its manifest advantages, the therapeutic potential of glycotargeting strategies has yet to be properly exploited. Of particular relevance to the present study is the design of galectin-binding nanoparticles, for which there are a growing number of biological applications. Mammalian galectins are a family of lectins that exhibit strong affinity for β-galactose-containing glycoconjugates.14 All galectins share a core sequence consisting of about 130 amino acids, many of which are highly conserved. This core sequence is known as the carbohydrate recognition domain (CRD) and is responsible for the binding of specific sugars.15 The biological significance of specific carbohydrate-ligand recognition by various galectins is not fully understood, but this may in part explain why individual galectins preferentially bind to different glycoprotein counter-receptors, which implies specific targeting. Galectins are ubiquitous within the cell and are highly mobile; they can be found in the cytosol and nuclear region and, although secretion signal peptides have not been found in the sequence of galectins, they are also present in the extracellular space.16 It appears that galectins may be targeted for secretion by nonclassical mechanisms, possibly by direct translocation across the plasma membrane.15 Galectins bind to the cell-surface and extracellular matrix glycans and are known to play key roles in numerous cellular processes, such as apoptosis, cell adhesion17 and receptor turnover and endocytosis.18 Galectins also have important functions in many physiological and pathological processes, including immune and inflammatory responses,18 tumorigenesis,19 neural degeneration, atherosclerosis, and wound repair.15 Furthermore, galectin-mediated cellular receptor internalization and recycling processes are very rapid. Moreover, although the precise internalization mechanism remains unknown, it appears to avoid the degradative environment of the endosomes.20 Thus, galectin targeting has the potential to offer new therapeutic avenues for nanomedicine.
The self-assembly of amphiphilic diblock copolymers21 allows access to a wide range of nano-objects, such as spherical micelles,22 worm-like micelles,23,24 and vesicles,22,25−27 which have applications in nanomedicine, cell biology, electronics, and energy.28−30 For example, self-assembled glycopolymer-based nano-objects are very attractive for the development of novel gene delivery vectors and vaccines.31 Block copolymer nano-objects are typically obtained via postpolymerization processing of soluble copolymer chains using traditional solvent switch,22 pH switch32 or thin film rehydration techniques.33 These techniques have been successfully employed to prepare a range of self-assembled nanostructures based on glycopolymers.34 Li et al. reported one of the first examples of glycosylated self-assembled polymeric morphologies using polystyrene-b-poly[(2-β-d-glucopyranosyloxy)ethyl acrylate] block copolymers obtained by atom transfer radical polymerization (ATRP).4 Similarly, Alexander and Pasparakis prepared temperature-sensitive glucose-decorated vesicles from a double-hydrophilic block copolymer obtained by ATRP and reversible addition–fragmentation chain transfer (RAFT) polymerization and demonstrated that these glycosylated vesicles strongly interact with bacteria that express glucose-binding proteins on their surface.31b Hedrick, Dubois, Yang and co-workers reported the formation of new biodegradable polycarbonate-based micelles displaying either glucose or galactose surface moieties via self-assembly in aqueous media.35 Schlaad reported the formation of well-defined glycosylated morphologies by aqueous self-assembly of partially glycosylated 1,2-polybutadiene-b-polystyrene36 and 1,2-polybutadiene-b-poly(ethylene oxide)37 prepared by a combination of thiol–ene chemistry and anionic polymerization. Interestingly, the latter vesicles possessed asymmetric membranes, with the poly(ethylene oxide) chains being confined to the inner surface, while the sugar moieties were expressed at the outer surface. Lecommandoux and Heise utilized the ring-opening polymerization of N-carboxyanhydrides to synthesize glycosylated peptide-based block copolymers that formed well-defined spherical, worm-like micelles or vesicles in aqueous solution.38 Finally, rod–coil glycosylated block copolymer self-assembly in water has also been demonstrated for biosensor applications.39
However, such traditional self-assembly strategies invariably only allow the formation of block copolymer nano-objects in relatively dilute solution (<1%). In contrast, the recent introduction of polymerization-induced self-assembly (PISA) formulations based on RAFT polymerization40 enables well-defined block copolymer nano-objects to be prepared directly at up to 25% solids without recourse to any postpolymerization processing.41,42
Herein we exploit this PISA approach to prepare a range of new galactose-functionalized diblock copolymer nano-objects that interact in vitro with galactose-specific lectins. The effect of copolymer morphology on the sensitivity of a simple turbidimetric binding assay is explored. Furthermore, encapsulation of a molecular cargo and effective intracellular delivery while escaping the endolysosome environment are also demonstrated.
Materials and Methods
All reagents were purchased from Sigma-Aldrich (U.K.) and were used as received, unless otherwise noted. 4,4′-Azobis-4-cyanopentanoic acid (ACVA, >98%) was used as an initiator. 2-Hydroxypropyl methacrylate (HPMA, 97%) was kindly donated by GEO Specialty Chemicals (Hythe, U.K.) and comprises ∼75% 2-hydroxypropyl methacrylate and 25 mol % 2-hydroxyisopropyl methacrylate. According to HPLC analysis, this monomer also contained about 0.10 mol % dimethacrylate impurity. All solvents were purchased from Fisher Scientific (U.K.) as HPLC grade and were used as received. Deionized water was used in all experiments. Silica gel 60 (0.0632–0.2 mm) was obtained from Merck (Darmstadt, Germany). All NMR solvents (D2O and CD3OD) were purchased from Goss Scientific Instruments Ltd. (U.K.). Dialysis membrane (molecular weight cutoff, MWCO = 1000) was purchased from Fisher Scientific (U.K.). The PETTC RAFT agent was prepared as described previously.41c
Synthesis of Galactose Methacrylate (GalSMA)
1-Thio-β-d-galactose (GalSH) was first prepared according to the method described by Floyd et al.43 in an overall yield of 70%. GalSH (5.00 g, 25.48 mmol) was placed in a round-bottomed flask and dissolved in DMF (15 mL). A solution of 3-(acryloyloxy)-2-hydroxypropyl methacrylate (6.00 g, 28.03 mmol) in DMF (5.0 mL) was then added to this GalSH solution. Dimethylphenylphosphine (10 μL, 7.0 × 10–2 mmol) was then added to the reaction solution. After 30 min, the DMF solution was precipitated into excess diethyl ether. The viscous residue was dissolved in DMF and precipitated again into diethyl ether. The resulting viscous oil was then purified by flash chromatography using 9:1 methanol/dichloromethane, and this solvent mixture was then evaporated under vacuum. The monomer was not isolated but instead was stored at −20 °C as a 77 wt % concentrated solution in methanol. After purification, the yield was estimated by 1H NMR to be around 90%. The overall yield based on β-d-galactose pentaacetate was 63%. 1H NMR (400.13 MHz, D2O, 298 K) δ (ppm): 1.93 (s, 3H, -CH3); 2.83 (t, 2H, -CH2-COO); 2.92–3.06 (m, 2H, -CH2-S); 3.54 (t, 1H, H2); 3.61–3.64 (dd, 1H, H3); 3.67–3.82 (m, 4H, H5, H6, -CH2-CHOH-CH2-); 3.96 (d, 1H, H4); 4.20–4.30 (m, 4H, -CH2-CHOH-CH2-); 4.48 (d, 1H, H1); 5.73 (s, 1H, vinyl), 6.16 (s, 1H, vinyl). 13C NMR (400.13 MHz, D2O, 298 K) δ (ppm): 18.0 (CH3-); 25.8 (-S-CH2); 35.5 (-S-CH2-CH2-); 61.6 (C6); 65.9 (2C, -CH2-CHOH-CH2-); 67.5, 69.4, 70.2, 74.5, 79.5, 86.7 (6C, C1, C2, C3, C4, C5, -CH2-CHOH-CH2-); 127.8, 136.2 (2C, vinyl), 169.9, 174.7 (2C, carbonyls). (M + H+): calcd mass = 411.1307, actual mass found = 411.1325.
RAFT Homopolymerization of GalSMA
GalSMA (10.65 g of a 77 wt.% methanolic solution; 8.20 g, 19.97 mmol) was placed in a round-bottomed flask containing a magnetic bar, PETTC (0.226 g, 666 μmol), and ACVA (18.60 mg, 66.4 μmol; PETTC/ACVA molar ratio = 10). Phosphate buffer solution (22.15 g, 150 mM, pH 7.2) was added, and the final solution was degassed by nitrogen bubbling. After 30 min, the round-bottomed flask was placed in a preheated oil bath at 70 °C. The reaction was quenched after 150 min (97% conversion), and the polymer was purified by dialysis (MWCO = 1000) against deionized water, followed by freeze-drying overnight. DMF GPC analysis gave Mn = 16 300 g mol–1, Mw/Mn = 1.13. End-group analysis via 1H NMR spectroscopy indicated a mean degree of polymerization of 34 (Mn = 14 300 g mol–1), which corresponds to a RAFT CTA efficiency of 85% for the PETTC.
RAFT Homopolymerization of Glycerol Monomethacrylate (GMA)
GMA (7.00 g, 43.70 mmol) was added to a round-bottomed flask containing a magnetic bar, PETTC (269.78 mg, 795.00 μmol), and ACVA (22.27 mg, 79.50 μmol). Ethanol (7.00 g) was added to this solution, which was then degassed by nitrogen bubbling. After 30 min, the round-bottomed flask was placed in a preheated oil bath at 70 °C. The polymerization was quenched after 5 h (conversion = 88%), the polymer was purified by dialysis (MWCO = 1000) against deionized water, and freeze-dried overnight. DMF GPC analysis indicated Mn = 16 200 g mol–1 and Mw/Mn = 1.15. End-group analysis using 1H NMR gave a mean degree of polymerization of 51 (Mn = 8500 g mol–1). This indicated a RAFT CTA efficiency of 94% for the PETTC.
Polymerization-Induced Self-Assembly (PISA)
A typical RAFT aqueous dispersion polymerization was performed as follows: HPMA (377.0 mg, 2.61 mmol, target DP = 201) and deionized water (1.78 mL) were added to a sample vial containing a magnetic stir bar, PGMA51 macro-CTA (100 mg, 11.7 μmol), PGalSMA34 macro-CTA (18.7 mg; 1.31 μmol), and ACVA initiator (200 μL of a 13.0 mM aqueous solution; macro-CTA/initiator molar ratio = 5.0). The reaction solution was degassed by nitrogen bubbling for 15 min and then placed in a preheated oil bath at 70 °C. The polymerization was quenched after 6 h (>99% conversion, as judged by 1H NMR spectroscopy). Similar polymerizations were conducted targeting alternative PHPMA block lengths, which allowed access to either spherical, worm-like, or vesicular copolymer morphologies. See note in the Supporting Information.
Gel Permeation Chromatography (GPC)
Homopolymer and diblock copolymer molecular weight distributions were determined by DMF GPC. The GPC setup comprised two Polymer Laboratories PL gel 5 μm Mixed-C columns maintained at 60 °C in series with a Varian 390 LC refractive index detector. The flow rate was 1.0 mL min–1, and the mobile phase contained 10 mM LiBr. Ten near-monodisperse PMMA standards (Mp = 625–618 000 g mol–1) were used for calibration.
1H NMR Spectroscopy
All 1H NMR and 13C NMR spectra were recorded in either CD3OD, d6-DMSO, or D2O using either a 250 MHz Bruker Avance 250 or a 400 MHz Bruker Avance 400 spectrometer.
Transmission Electron Microscopy
TEM images were acquired using a Philips CM100 instrument operating under UHV at 100 kV. To prepare TEM samples, 5.0 μL of a dilute aqueous copolymer solution was placed onto a carbon-coated copper grid, stained using uranyl formate, and then dried under ambient conditions.
Dynamic Light Scattering
DLS measurements were conducted at 25 °C using a scattering angle of 173° with a Malvern Instruments Zetasizer Nanoseries instrument equipped with a 4 mW He–Ne laser operating at 633 nm, an avalanche photodiode detector with high quantum efficiency, and an ALV/LSE-5003 multiple t digital correlator electronics system. The intensity-average diameter and polydispersity of the diblock copolymer particles were calculated by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20.
Rheology Studies
The storage modulus (G′) and loss modulus (G′′) curves for the (1:9 PGalSMA34 + PGMA51)-PHPMA150 diblock copolymer worm gel were determined using a TA Instruments AR-G2 rheometer equipped with a Peltier heating/cooling plate. A cone-and-plate geometry (40 mm, 2° aluminum cone) was used for the measurements. Temperature sweeps were conducted at a fixed strain of 1.0% using an angular frequency of 1 rad s–1. Stepwise sweeps were conducted at increments of 1 °C, using an equilibration time of 3 min for each step and an equilibration time of 5 min at 25 and 1 °C.
UV–vis Spectroscopy
Turbidimetry studies were conducted at 20 °C using a Cary 50 UV–vis spectrophotometer at a wavelength of 420 nm. All lectin interaction studies were performed in HEPES buffer (HEPES 10 mM, NaCl 150 mM, MnCl2 1 mM, CaCl2 1 mM) at pH 7.4. For the lectin assay, a cuvette containing 0.50 mL of a 2 µM solution of RCA120 in HEPES buffer was placed in the spectrometer. 0.50 mL of a 1.0 wt% aqueous diblock copolymer dispersion was added to the cuvette, and the absorbance at 420 nm was monitored over time. For the negative control, a cuvette containing 0.50 mL of a 2 µM solution of RCA120 in HEPES buffer was placed in the spectrometer. 0.50 mL aliquots of 50 µM homopolymer solutions (either PGMA51 or PGalSMA34, see Figure S5) were added to the cuvette, and the absorbance at 420 nm was monitored over time.
Preparation of Sterile (1:9 PGalSMA34 + PGMA51)-PHPMA270 Vesicles via Film Rehydration
Copolymers were dissolved in a 2:1% v/v methanol/chloroform mixture to form a 1 mM solution. A 0.05 mM Rhodamine B octadecyl ester perchlorate (Sigma-Aldrich) solution in 2:1% v/v methanol/chloroform mixture was prepared. Equal volumes of these two solutions were mixed, the resulting solution was then filter-sterilized using a 0.20 μm Nylon filter (Millipore), and the solvent mixture evaporated under sterile conditions to form a thin copolymer film. This film was subsequently rehydrated under sterile conditions using phosphate buffer saline (100 mM PBS) at pH 7.4 with constant stirring for 5 days to form a 1.0% w/w copolymer suspension. The vesicle dispersion was sonicated daily for up to 30 min under controlled temperature (20 °C). Vesicles were purified via preparative GPC, using a size exclusion column containing Sepharose 4B and using PBS at pH 7.4 as an eluent.
Cell Culture
Primary human dermal fibroblasts (HDFs) were obtained from LGC standards (Teddington, U.K.). Cells were maintained in DMEM (Biosera, U.K.) supplemented with 10% v/v fetal calf serum, 2 mM l-glutamine, 100 IU/mL penicillin, 100 mg/mL streptomycin, and 0.625 μg/mL amphotericin B (all from Sigma-Aldrich, U.K.). Cells were subcultured routinely using 0.02% (w/v) trypsin-EDTA (Sigma-Aldrich, U.K.) and used for experimentation between passages 4 and 8.
Cell Viability via MTT-ESTA Assay
The well-known 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was used to measure cellular metabolic potential of treated cells after exposure to nanovesicles.44 Briefly, 3–4 × 104 HDF cells per well were cultured in 24-well plates until 70% confluence (typically 48 h). Cells were incubated for 24 h with varying concentrations of vesicles. After treatment, cell cultures were thoroughly washed in PBS and then incubated with MTT solution (0.50 mg/mL MTT in PBS, 1.0 mL per well for 24-well plates) for 45 min at 37 °C and in a 95% air/5% CO2 environment. Intracellular dehydrogenase activity reduces MTT to form a purple formazan salt. After 45 min, the solution was aspirated, and the insoluble intracellular formazan product was solubilized and released from cells by adding acidified isopropanol (0.30 mL per well of 24-well plate or 1 mL per cm–2 cultured tissue) and incubated for 10 min. The optical density at 570 nm was then recorded using a plate reading spectrophotometer (with a reference filter at 630 nm). For statistical analysis (student’s t test), experiments were performed in triplicate wells with a total of N = 3 independent experiments.
Live Fluorescence Microscopy Imaging of the Cellular Uptake of Sterilized (1:9 PGalSMA34 + PGMA51)-PHPMA270 Vesicles Loaded with Rhodamine B Octadecyl Ester
Cells were seeded at a density of 5 × 103 cells/well in BD Falcon 96-well imaging plates and grown until 50% confluence. Cells were treated overnight (typically 16 h) with a 1.0 mg/mL aqueous vesicle dispersion encapsulating ∼10 μmol of rhodamine B octadecyl ester perchlorate (Sigma-Aldrich, U.K.) per mmol of copolymer solution. The cells were washed three times with PBS and nuclei stained using 1.0 μg/mL of Hoechst 33342 solution (Thermo Scientific, U.K.) for 10 min. Stained cells were washed once more, and imaging medium (culture medium without phenol red) was added to each well for subsequent live imaging experiments, which were performed using a Zeiss LSM510 Meta instrument (40× magnification).
Results and Discussion
The synthesis, isolation, and purification of glycomonomers is notoriously difficult. For the present study, a novel route to a methacrylic glycomonomer was devised. This method (see Figure 1a) relies on the very high efficiency and regioselectivity offered by thia-Michael addition to acrylates.45 1-Thio-β-d-galactose was readily prepared on a multigram scale according to literature protocols43 and subsequently reacted with 3-(acryloyloxy)-2-hydroxypropyl methacrylate to produce the desired galactose methacrylate (GalSMA) in 63% yield (Figure S1). GalSMA tends to autopolymerize during long-term storage, hence this monomer was kept under nitrogen at −20 °C as a concentrated methanolic solution and utilized shortly after its synthesis. PGalSMA was synthesized by RAFT polymerization in a 9:1 mixture of PBS buffer (150 mM, pH 7.2) and methanol at 70 °C using a trithiocarbonate-based PETTC (4-cyano-4-(2-phenylethane sulfanylthiocarbonyl) sulfanylpentanoic acid)41c RAFT agent and ACVA (4,4′-azobis(4-cyanopentanoic acid)) initiator. This afforded a well-defined PGalSMA34 macro-CTA (Figure S2), which was then chain-extended using HPMA under RAFT aqueous dispersion polymerization conditions.41
Figure 1.
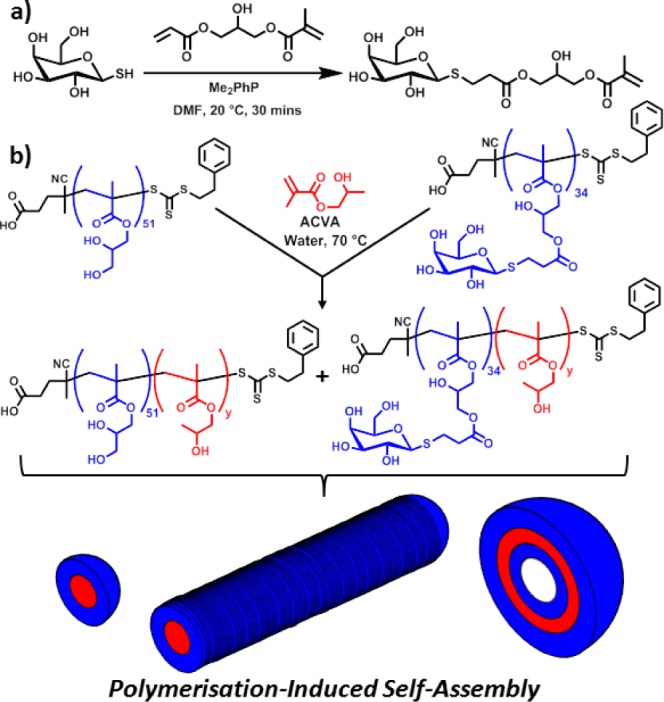
(a) Synthesis of a new GalSMA monomer via thia-Michael addition. (b) Preparation of self-assembled block copolymer nano-objects (spheres, worms or vesicles) via PISA of HPMA using a binary mixture of RAFT macro-CTAs based on PGMA and PGalSMA.
When PGalSMA34 was used as the sole macro-CTA, self-assembled spherical nanoparticles, worm-like micelles, or vesicles were obtained, depending on the target diblock copolymer composition and the copolymer concentration (Figure S3). Well-defined spherical nanoparticles were readily formed over a wide range of reaction conditions, but the other two morphologies were generally only obtained as components of mixed phases. In particular, when targeting DPs of around 700 for the core-forming PHPMA block, large vesicles and tubular structures featuring relatively thick walls were often observed as major components of these mixtures (Figure S3). However, according to the literature46 biological activity is unlikely to require surface expression of a high density of galactose residues. Thus a binary mixture of PGalSMA34 and poly(glycerol methacrylate) (PGMA51) macro-CTAs was utilized for the RAFT aqueous dispersion polymerization of HPMA (Figure 1b). We have previously reported that using binary mixtures of a nonionic and a polyelectrolytic macro-CTA leads to hybrid nano-objects with mixed coronal stabilizer layers.41c,41d As the HPMA polymerization progressed, the initially homogeneous aqueous milieu gradually became first translucent and then increasingly turbid, depending on the mean DP targeted for the core-forming PHPMA block. This turbidity corresponds to the onset of copolymer aggregation; such nucleation is usually accompanied by an enhanced rate of polymerization, which is believed to be due to the preferential partitioning of unreacted HPMA monomer within the growing micellar aggregates.41,42a These RAFT syntheses proved to be both efficient and well-controlled: >99% monomer conversion and low copolymer polydispersities were routinely achieved as judged by 1H NMR and DMF GPC studies (Figure S4 and Table S1). The mean DP of the PHPMA block and the copolymer solids content were systematically varied to construct a phase diagram that enables the reproducible preparation of morphologically pure nano-objects (Figure 2a). Representative TEM images for the various galactose-functionalized nano-objects are shown in Figure 2b. Using a 1:9 binary mixture of PGalSMA34 and PGMA51 macro-CTAs produces well-defined spherical, worm-like, or vesicular phases as well as various mixed phases. The (1:9 PGalSMA34 + PGMA51)-PHPMA150 worm-like micelles47 form translucent free-standing gels at 20 °C similar to those previously reported for PGMA-PHPMA diblock copolymers.48 However, cooling to 3 °C leads to degelation, with the gel phase reforming on returning to ambient temperature. Rheology and DLS studies were undertaken to further characterize this thermo-reversible transition (Figure 3). More specifically, the temperature dependence of the storage (G′) and loss (G′′) moduli was monitored (Figure 3b) for a 20% w/w aqueous dispersion of (1:9 PGalSMA34 + PGMA51)-PHPMA150 worm-like micelles.
Figure 2.

(a) Phase diagram constructed for (1:9 PGalSMA34 + PGMA51)-PHPMAx diblock copolymer nano-objects prepared by RAFT aqueous dispersion polymerization at 70 °C. The target PHPMA DP and the total solids content were systematically varied and the post mortem copolymer morphologies obtained at >98% HPMA conversion were determined by TEM. N.B. S, SW, W, V, and FS denote spheres, short worm-like micelles, worm-like micelles, vesicles, and frustrated (i.e., kinetically trapped) spheres, respectively. (b) Representative TEM images obtained for (1:9 PGalSMA34 + PGMA51)-PHPMAx copolymer nano-objects prepared by RAFT aqueous dispersion polymerization of HPMA at 70 °C. The targeted DP (x) for the PHPMA block (herein denoted by ‘H’ for brevity) and the copolymer solids content % is indicated on each image.
Figure 3.

(a) Digital photographs recorded for a 20% w/w aqueous dispersion of (1:9 PGalSMA34 + PGMA51)-PHPMA150 worm-like micelles recorded at (i) 20 °C (gel) and (ii) 3 °C (fluid). (b) Variation of storage modulus (G′, black symbols) and loss modulus (G′′, red symbols) for the same 20% w/w diblock copolymer worm gel during temperature cycling in 1 °C increments: (i) cooling from 25 to 1 °C (G′ = inverted black triangles, G′′ = inverted red triangles) and (ii) subsequent warming from 1 to 25 °C in 1 °C increments (G′ = black triangles, G′′ = red triangles). (c) and (d) Temperature-dependent DLS studies of the intensity-average diameter and scattered light intensity respectively, for the same aqueous dispersion of diblock copolymer worms diluted to 1.0% w/w.
For this particular dispersion, G′ exceeds G′′ between 25 and 7.5 °C, which indicates the formation of a soft viscoelastic gel. Below 7.5 °C, G′ is reduced by ∼2 orders of magnitude and these curves cross over; this is consistent with the formation of a free-flowing viscous liquid at 3 °C (Figure 3a). During the heating cycle, crossover occurs at the same critical temperature, which suggests minimal hysteresis. However, a somewhat weaker final gel is obtained, indicating some irreversibility. This sol–gel transition is ascribed to the transformation of worm-like micelles into spherical micelles upon cooling; such an order–order transition has also been observed for PGMA-PHPMA diblock copolymers.48 The molecular origin for this sol–gel transition is the well-known thermoresponsive nature of the PHPMA block.48 This leads to a subtle variation in the degree of hydration of these hydrophobic chains, which in turn causes a change in the molecular packing parameter that dictates the overall copolymer morphology.48,49 DLS studies of a 1.0% aqueous dispersion of (1:9 PGalSMA34 + PGMA51)-PHPMA150 worm-like micelles are shown in Figure 3c,d.
The intensity-average diameter and scattered light intensity are both significantly reduced at lower temperature, suggesting a worm-to-sphere morphological transition. This is consistent with the observation of degelation, since, unlike anisotropic worms, the isotropic spheres can no longer form multiple interparticle entanglements/contacts. However, on allowing the cold copolymer solution to warm up to ambient temperature, the sphere-to-worm transition is not observed on the time scale of the experiment. This is simply because of the reduced probability of efficient sphere–sphere 1D fusion at this relatively low copolymer concentration.41b
The interaction of these new galactose-functionalized diblock copolymer nano-objects with RCA120, a galactose-specific (and N-acetylgalactosamine-specific) lectin, was then examined using turbidimetry and DLS. Such lectin assays are well-established and can be used to assess the bioavailability of the galactose moieties on the surface of the self-assembled nano-objects. The spheres, worm-like micelles and vesicles all interact strongly with RCA120, leading to the rapid formation of much larger aggregates (Figure 4). Moreover, control experiments confirm that PGalSMA34 homopolymer interacts strongly with RCA120, whereas PGMA51 homopolymer produces no detectable interaction (Figure S5). Taken together, these data indicate that galactose residues are expressed at the surface of the diblock copolymer nano-objects, as expected. It is also striking that the galactose-functionalized worm-like micelles and vesicles are characterized by a much stronger (and faster) optical response on exposure to RCA120 than the galactose-functionalized spheres (Figure 4). This is important, because it suggests that the copolymer morphology profoundly influences the lectin assay sensitivity.
Figure 4.
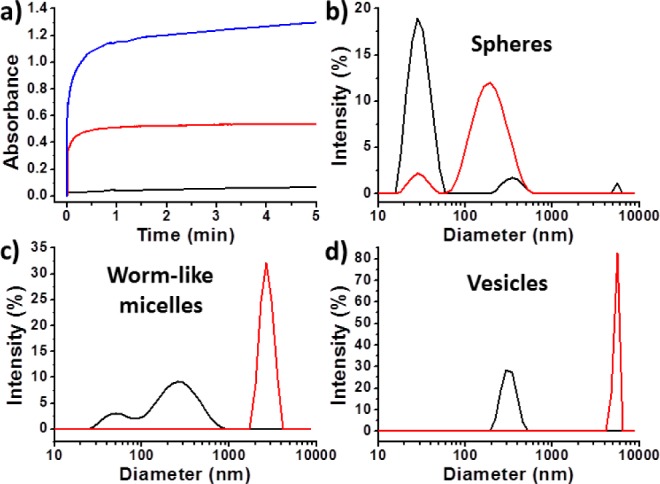
Galactose-specific lectin interactions with three types of galactose-functionalized diblock copolymer nano-objects (each originally prepared at 20% solids). (a) Turbidimetric assays for (1:9 PGalSMA34 + PGMA51)-PHPMA75 spheres (black curve), (1:9 PGalSMA34 + PGMA51)-PHPMA150 worm-like micelles (red curve), and (1:9 PGalSMA34 + PGMA51)-PHPMA270 vesicles (blue curve). DLS size distributions recorded for the same nano-objects: (b) spheres, (c) worm-like micelles, and (d) vesicles, recorded both in the absence (black curves) and presence (red curves) of RCA120. Assay conditions: [RCA120] = 1 μM and [copolymer] = 0.50 wt % in 10 mM HEPES buffer at pH 7.2.
Finally, the interaction of galactose-functionalized vesicular nano-objects with living cells was examined. (1:9 PGalSMA34 + PGMA51)-PHPMA270 vesicles (synthesized by RAFT aqueous dispersion polymerization at 15% solids) were dialyzed against deionized water prior to freeze drying. Vesicles were then generated via thin-film rehydration. DLS measurements show that the dimensions of such reconstituted vesicles are not significantly different from that of the vesicles initially obtained by PISA (see Figure S6). This purification protocol ensured the elimination of any potentially toxic volatile chemicals remaining in the reaction solution at the end of the polymerization. PISA syntheses conducted under sterile conditions are also now under investigation and will be reported elsewhere in due course. Vesicle biocompatibility, cellular uptake, and cargo delivery studies were then performed on HDF cells. These relatively delicate primary cells are a sensitive model for testing nanoparticle-induced toxicity50 and are known to express galectins.51 Cytotoxicity studies were performed using an MTT-ESTA assay and confirmed cell viabilities of >95% when exposed to varying concentrations of vesicles for 24 h, with no significant differences compared to the untreated control cells (Figure S7). These viability data confirm that thin-film rehydration and the associated purification steps efficiently eliminate toxic solvents (e.g., chloroform and methanol) and produce sterile, highly biocompatible vesicles that are suitable for subsequent cell uptake studies.
Rhodamine B octadecyl ester-loaded (1:9 PGalSMA34 + PGMA51)-PHPMA270 vesicles were avidly internalized by HDF cells. Subsequent release of their fluorescent dye cargo led to extensive staining of the cell membranes (Figure 5). In the absence of a suitable vesicle carrier, it is known that this particular amphiphilic rhodamine dye cannot enter HDF cells.52 Hence the observation of positive intracellular staining confirms both cellular uptake of the vesicles and intracellular delivery of their dye cargo. Many membrane-rich organelles are stained, such as the mitochondria and endocytic compartments. This was not unexpected, as the mechanism of uptake of these vesicles is likely to be endocytosis and rhodamine dyes have a well-known affinity for the mitochondria.53 However, staining of the endomembrane system, including the nuclear membrane (Figure 5c) and even co-staining within the nuclear region, was also observed. Since the nuclear region lacks endolysosomal compartments, this observation suggests the intracellular release of the dye cargo and its escape from the normal endocytic pathway.
Figure 5.
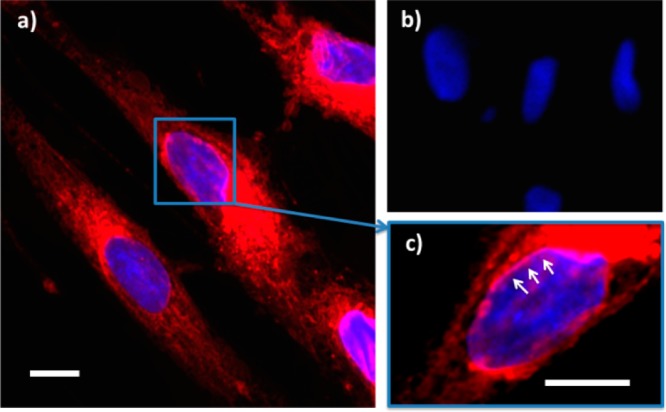
Effective intracellular delivery of rhodamine B octadecyl ester in HDF cells mediated by (1:9 PGalSMA34 + PGMA51)-PHPMA270 vesicles (prepared via thin film rehydration to ensure sterility and enable loading with a fluorescent dye). Cells were incubated for 16 h with 1.0 mg/mL rhodamine B octadecyl ester-loaded vesicles. (a) Confocal microscopy image of live HDF cells: note the intracellular staining of membranes (red) after exposure to the rhodamine-loaded vesicles, cell nuclei are counter-stained blue using Hoechst 33342. (b) HDF cells treated with the same vesicles containing no rhodamine dye (negative control). (c) Higher magnification image obtained for (a): effective intracellular delivery of rhodamine dye allows selective staining of the nuclear membrane (white arrows). Scale bar: 50 μm.
Galectin-mediated receptor turnover is known to be a rapid process that can potentially avoid degradation in the lysosomal environment.20 Nevertheless, our observation of release of the rhodamine dye from the vesicles and its subsequent location outside the endocytic compartments (within the cell nuclei) is rather surprising.
Previous work using other diblock copolymers containing weakly hydrophobic PHPMA chains54 suggests that the stability of such colloidal aggregates is concentration-dependent. Thus reducing the aggregate concentration below the critical micelle concentration of the copolymer leads to their spontaneous dissociation to form molecularly dissolved copolymer chains. Hence we suggest that dilution-triggered vesicle dissociation is the most likely mechanism for the in situ release of the rhodamine dye label within the cells. It is noteworthy that this release mechanism is not necessarily exhibited by other biocompatible methacrylic diblock copolymers comprising significantly more hydrophobic membrane-forming chains than that utilized in the present study.55
Conclusion
In summary, PISA has been exploited for the convenient synthesis of a range of new glycopolymer-decorated block copolymer nano-objects (spheres, worm-like micelles, or vesicles) directly in concentrated aqueous solution. As expected, these nano-objects interact strongly with galactose-specific lectins in vitro, demonstrating high specificity. The sensitivity of a simple turbidimetric lectin binding assay is strongly dependent on the copolymer morphology: For a given copolymer concentration, the use of vesicles and worms leads to much greater turbidity than spherical nanoparticles. Furthermore, galactosylated vesicles were rapidly taken up by primary HDF cells with little or no cytotoxicity. Such vesicles can encapsulate and subsequently release their payloads intracellularly. These preliminary results may lead to new opportunities for targeted drug delivery.
Acknowledgments
We thank EPSRC for postdoctoral support of V.L. and I.C. (EP/G007950/1, EP/I012060/1 and EP/E012949/1). Prof. Armes also acknowledges an ERC Advanced Investigator grant (PISA 320372).
Supporting Information Available
1H NMR spectrum, additional TEM and DMF GPC data, lectin interaction control experiments, and MTT-ESTA assay details.
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Institut Charles Gerhardt de Montpellier (UMR 5253, ENSCM-CNRS-UM2) ENSCM, 8, rue de l′école normale, 34296 Montpellier, France.
Author Present Address
‡ Institut Européen des Membranes (UMR 5635, ENSCM-CNRS-UM2), Université Montpellier 2,C.C. 047, Place E. Bataillon, 34095 Montpellier Cedex 05, France.
The authors declare no competing financial interest.
Supplementary Material
References
- Braunecker W. A.; Matyjaszewski K. Prog. Polym. Sci. 2007, 32, 93. [Google Scholar]
- Ladmiral V.; Melia E.; Haddleton D. M. Eur. Polym. J. 2004, 40, 431. [Google Scholar]
- Voit B.; Appelhans D. Macromol. Chem. Phys. 2010, 211, 727. [Google Scholar]
- Li Z.-C.; Liang Y.-Z.; Li F.-M. Chem. Commun. 1999, 16, 1557. [Google Scholar]
- Ejaz M.; Ohno K.; Tsujii Y.; Fukuda T. Macromolecules 2000, 33, 2870. [Google Scholar]
- Semsarilar M.; Ladmiral V.; Perrier S. Macromolecules 2010, 43, 1438. [Google Scholar]
- Xu J.; Boyer C.; Bulmus V.; Davis T. P. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 4302. [Google Scholar]
- a Brown C. D.; Rusek M. S.; Kiessling L. L. J. Am. Chem. Soc. 2012, 134, 6552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Becer C. R.; Gibson M. I.; Geng, Ilyas R.; Wallis R.; Mitchell D. A.; Haddleton D. M. J. Am. Chem. Soc. 2010, 132, 15130. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ting S. R. S.; Chen G.; Stenzel M. H. Polym. Chem. 2010, 1, 1392. [Google Scholar]
- Belardi B.; O’Donoghue G. P.; Smith A. W.; Groves J. T; Bertozzi C. R. J. Am. Chem. Soc. 2012, 134, 9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godula K.; Bertozzi C. R. J. Am. Chem. Soc. 2012, 134, 15732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ahmed M.; Deng Z.; Liu S.; Lafrenie R.; Kumar A.; Narain R. Bioconjugate Chem. 2009, 20, 2169. [DOI] [PubMed] [Google Scholar]; b Pfaff A.; Schallon A.; Ruhland T. M.; Majewski A. P.; Schmalz H.; Freitag R.; Müller A. H. E. Biomacromolecules 2011, 12, 3805. [DOI] [PubMed] [Google Scholar]
- Bertozzi C. R.; Kiessling L. L. Science 2001, 291, 2357. [DOI] [PubMed] [Google Scholar]
- Rogers J. C.; Kornfeld S. Biochem. Biophys. Res. Commun. 1971, 45, 622. [DOI] [PubMed] [Google Scholar]
- Ambrosi M.; Cameron N. R.; Davis B. G. Org Biomol Chem. 2005, 9, 1593. [DOI] [PubMed] [Google Scholar]
- Yang R. Y.; Rabinovich G. A.; Liu F. T. Expert Rev. Mol. Med. 2008, 10, 1. [DOI] [PubMed] [Google Scholar]
- Cooper D. N. Biochim. Biophys. Acta 2002, 1572, 231. [DOI] [PubMed] [Google Scholar]
- Hughes R. C. Biochimie 2001, 83, 667. [DOI] [PubMed] [Google Scholar]
- Rabinovich G. A.; Toscano M. A.; Jackson S. S.; Vasta G. R. Curr. Opin. Struct. Biol. 2007, 17, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.-T.; Rabinovich G. A. Nature Reviews Cancer 2005, 5, 29. [DOI] [PubMed] [Google Scholar]
- a Delacour D.; Koch A.; Jacob R. Traffic 2009, 10, 1405. [DOI] [PubMed] [Google Scholar]; b Stechly L.; Morelle W.; Dessein A.-F.; André S.; Grard G.; Trinel D.; Dejonghe M.-J.; Leteurtre E.; Drobecq H.; Trugnan G.; Gabius H. J.; Huet G. Traffic 2009, 10, 438. [DOI] [PubMed] [Google Scholar]
- Mai Y.; Eisenberg A. Chem. Soc. Rev. 2012, 41, 5969. [DOI] [PubMed] [Google Scholar]
- Zhang L. F.; Eisenberg A. Science 1995, 268, 1728. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S. S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Nat. Nanotechnol. 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Y. Y.; Davis H. T.; Bates F. S. Science 1999, 283, 960. [DOI] [PubMed] [Google Scholar]
- Discher B. M.; Won Y. Y.; Ege D. S.; Lee J. C. M.; Bates F. S.; Discher D. E.; Hammer D. A. Science 1999, 284, 1143. [DOI] [PubMed] [Google Scholar]
- Discher D. E.; Eisenberg A. Science 2002, 297, 967. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Hernandez J.; Lecommandoux S. J. Am. Chem. Soc. 2005, 127, 2026. [DOI] [PubMed] [Google Scholar]
- Riess G. Prog. Polym. Sci. 2003, 28, 1107. [Google Scholar]
- Kim H-C; Park S-M; Hinsberg W. D. Chem. Rev. 2010, 110, 146. [DOI] [PubMed] [Google Scholar]
- Orilall M. C.; Wiesner U. Chem. Soc. Rev. 2011, 40, 520. [DOI] [PubMed] [Google Scholar]
- a Schatz C.; Lecommandoux S. Macromol. Rapid Commun. 2010, 31, 1664. [DOI] [PubMed] [Google Scholar]; b Pasparakis G.; Alexander C. Angew. Chem., Int. Ed. 2008, 47, 4847. [DOI] [PubMed] [Google Scholar]; c Parry A. L.; Clemson N. A.; Ellis J.; Bernhard S. S. R.; Davis B. G.; Cameron N. R. J. Am. Chem. Soc. 2013, 135, 9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J. Z.; Tang Y. P.; Lewis A. L.; Armes S. P. J. Am. Chem. Soc. 2005, 127, 17982. [DOI] [PubMed] [Google Scholar]
- Kita-Tokarczyk K.; Grumelard J.; Haefele T.; Meier W. Polymer 2005, 46, 3540. [Google Scholar]
- a Narain R.; Armes S. P. Chem. Commun. 2002, 23, 2776. [DOI] [PubMed] [Google Scholar]; b Narain R.; Armes S. P. Macromolecules 2003, 36, 4675. [Google Scholar]; c Narain R.; Armes S. P. Biomacromolecules 2003, 4, 1746. [DOI] [PubMed] [Google Scholar]
- Suriano F.; Pratt R.; Tan J. P.K.; Wiradharma N.; Nelson A.; Yang Y.-Y.; Dubois P.; Hedrick J. L. Biomaterials 2010, 31, 2637. [DOI] [PubMed] [Google Scholar]
- You L.; Schlaad H. J. Am. Chem. Soc. 2006, 128, 13336. [DOI] [PubMed] [Google Scholar]
- Schlaad H.; You L.; Sigel R.; Smarsly B.; Heydenreich M.; Mantione A.; Mašic A. Chem. Commun. 2009, 12, 1478. [DOI] [PubMed] [Google Scholar]
- Huang J.; Bonduelle C.; Thévenot J.; Lecommandoux S.; Heise A. J. Am. Chem. Soc. 2012, 134, 119. [DOI] [PubMed] [Google Scholar]
- a Kim B.-S.; Hong D.-J.; Bae J.; Lee M. J. Am. Chem. Soc. 2005, 127, 16333. [DOI] [PubMed] [Google Scholar]; b Dal Bó A. G.; Soldi V.; Giacomelli F. C.; Travelet C.; Jean B.; Pignot-Paintrand I.; Borsali R.; Fort S. Langmuir 2012, 28, 1418. [DOI] [PubMed] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; Rizzardo E.; Thang S. H. Macromolecules 1998, 31, 5559. [Google Scholar]
- a Li Y.; Armes S. P. Angew. Chem., Int. Ed. 2010, 49, 4042. [DOI] [PubMed] [Google Scholar]; b Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. J. Am. Chem. Soc. 2011, 133, 16581. [DOI] [PubMed] [Google Scholar]; c Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Langmuir 2012, 28, 914. [DOI] [PubMed] [Google Scholar]; d Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Langmuir 2013, 29, 7416. [DOI] [PubMed] [Google Scholar]
- a Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Macromolecules 2012, 45, 5091. [Google Scholar]; b Boissé S.; Rieger J.; Belal K.; Di-Cicco A.; Beaunier P.; Li M.-H.; Charleux B. Chem. Commun. 2010, 46, 1950. [DOI] [PubMed] [Google Scholar]; c Zhang X.; Boissé S.; Zhang W.; Beaunier P.; D’Agosto F.; Rieger J.; Charleux B. Macromolecules 2011, 44, 4149. [Google Scholar]; d Cai W.; Wan W.; Hong C.; Huang C.; Pan C.-Y. Soft Matter 2010, 6, 5554. [Google Scholar]; e He W.-D.; Sun X.-L.; Wan W.-M.; Pan C.-Y. Macromolecules 2011, 44, 3358. [Google Scholar]
- Floyd N.; Vijayakrishnan B.; Koeppe J. R.; Davis B. G. Angew. Chem., Int. Ed. 2009, 48, 7798. [DOI] [PubMed] [Google Scholar]
- Mossman T. J. Immunol. Methods 1983, 65, 55.6606682 [Google Scholar]
- a McKee J. R.; Ladmiral V.; Niskanen J.; Tenhu H.; Armes S. P. Macromolecules 2011, 44, 7692. [Google Scholar]; b Mazzolini J.; Boyron O.; Monteil V.; D’Agosto F.; Boisson C.; Sanders G. C.; Heuts J. P. A.; Duchateau R.; Gigmes D.; Bertin D. Polym. Chem. 2012, 3, 2383. [Google Scholar]
- a Davis B. G.; Robinson M. A. Curr. Opin. Drug Discovery Dev. 2002, 5, 279. [PubMed] [Google Scholar]; b Gabor F.; Bogner E.; Weissenboeck A.; Wirth M. Adv. Drug Delivery Rev. 2004, 56, 459. [DOI] [PubMed] [Google Scholar]
- Dreiss C. A. Soft Matter 2007, 3, 956. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Verber R.; Mykhaylyk O. O.; Ryan A. J.; Heath J. Z.; Douglas C. W. I.; Armes S. P. J. Am. Chem. Soc. 2012, 134, 9741. [DOI] [PubMed] [Google Scholar]
- Madsen J.; Armes S. P.; Lewis A. L. Macromolecules 2006, 39, 7455. [Google Scholar]
- Chang J. S.; Chang K. L.; Hwang D. F.; Kong Z. L. Environ. Sci. Technol. 2007, 41, 2064. [DOI] [PubMed] [Google Scholar]
- Akimoto Y.; Hirabayashi J.; Kasai K.-I.; Hirano H. Cell Tissue Res. 1995, 280, 1. [DOI] [PubMed] [Google Scholar]
- Massignani M.; Canton I.; Sun T.; Hearnden V.; MacNeil S.; Blanazs A.; Armes S. P.; Lewis A.; Battaglia G. PLoS One 2010, 5, e10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonenko Y. N.; Avetisyan A. V.; Cherepanov D. A.; Knorre D. A.; Korshunova G. A.; Markova O. V.; Ojovan S. M.; Perevoshchikova I. V.; Pustovidko A. V.; Rokitskaya T. I.; Severina I. I.; Simonyan R. A.; Smirnova E. A.; Sobko A. A.; Sumbatyan N. V.; Severin F. F.; Skulachev V. P. J. Biol. Chem. 2011, 286, 17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J.; Armes S. P.; Bertal K.; MacNeil S.; Lewis A. L. Biomacromolecules 2009, 10, 1875. [DOI] [PubMed] [Google Scholar]
- a Canton I.; Massignani M.; Patikarnmonthon N.; Chierico L.; Robertson J.; Renshaw S . A.; Warren N. J.; Madsen J. P.; Armes S P.; Lewis A. L.; Battaglia G. FASEB J. 2013, 1, 98. [DOI] [PubMed] [Google Scholar]; b Lomas H.; Canton I.; MacNeil S.; Du J.; Armes S. P.; Ryan A. J.; Lewis A. L.; Battaglia G. Adv. Mater. 2007, 19, 4238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.