

Background: B-domain fragments of factor V (FV) were used to assess the mechanism by which it is maintained as a procofactor.
Results: A basic region fragment binds to FV containing an intact acidic region and inhibits FXa binding.
Conclusion: B-domain sequences function as cis- and trans-acting elements to suppress FV(a) procoagulant function.
Significance: The results provide mechanistic insight into FV autoinhibition and activation.
Keywords: Coagulation Factors, Enzyme Mechanisms, Hemostasis, Protein Engineering, Recombinant Protein Expression, Autoinhibition, Factor V, Factor Va, Prothrombinase
Abstract
Coagulation factor V (FV) circulates as an inactive procofactor and is activated to FVa by proteolytic removal of a large inhibitory B-domain. Conserved basic and acidic sequences within the B-domain appear to play an important role in keeping FV as an inactive procofactor. Here, we utilized recombinant B-domain fragments to elucidate the mechanism of this FV autoinhibition. We show that a fragment encoding the basic region (BR) of the B-domain binds with high affinity to cofactor-like FV(a) variants that harbor an intact acidic region. Furthermore, the BR inhibits procoagulant function of the variants, thereby restoring the procofactor state. The BR competes with FXa for binding to FV(a), and limited proteolysis of the B-domain, specifically at Arg1545, ablates BR binding to promote high affinity association between FVa and FXa. These results provide new insight into the mechanism by which the B-domain stabilizes FV as an inactive procofactor and reveal how limited proteolysis of FV progressively destabilizes key regulatory regions of the B-domain to produce an active form of the molecule.
Introduction
Hemostasis is achieved through spatially and temporally regulated thrombin generation following vascular injury. The final protease of the coagulation cascade, thrombin, is generated by the enzyme complex prothrombinase, composed of the serine protease factor Xa (FXa)3 associated with FVa on activated cellular surfaces. FVa is essential for thrombin generation, as it increases the catalytic activity of FXa toward prothrombin by several orders of magnitude (1). Like most clotting factors, FVa is generated from a functionally inert precursor following vascular injury. Its precursor, FV, is a single-chain protein in which the heavy and light chains of FVa flank a large, heavily glycosylated B-domain (domain organization A1-A2-B-A3-C1-C2) (2, 3). Unlike FVa, FV does not bind to FXa at physiological concentrations and thus cannot enhance thrombin generation (4–7). FV is activated by limited proteolysis at three discrete sites (Arg709, Arg1018, and Arg1545), releasing the B-domain as two large fragments to produce the procoagulant heterodimer FVa (7–11).
Given the profound effect FVa has on thrombin generation, FV activation represents an important step in hemostasis. Unlike serine proteases such as FXa, in which discrete proteolysis induces a well defined series of conformational changes that produce an active enzyme (12), the specific contributions of B-domain proteolysis to FV activation have remained largely uncharacterized. A prevailing hypothesis is that the B-domain inhibits procoagulant function through nonspecific steric constraints that are relieved by proteolysis. Surprisingly however, recent studies have shown that FV is regulated by an autoinhibitory region contained within the B-domain. Initial evidence of this came from studies using the recombinant variant FV-810 (FVΔ811–1491), which contains a truncated B-domain and exhibits constitutive FVa-like activity even in the absence of proteolysis (13–15). Subsequent generation of a panel of B-domain truncation variants led to the identification of an evolutionarily conserved basic region (BR) that is necessary to stabilize FV as a procofactor (16). More recently, it was discovered that the BR is in fact part of a bipartite autoinhibitory domain that also contains an evolutionarily conserved acidic region (AR) (17). Together, the BR and AR, which we have termed the procofactor regulatory region (PRR), constitute the minimal inhibitory B-domain sequence that is necessary to stabilize FV as a procofactor.
Although identification of the PRR provides new insights into FV regulation, important mechanistic questions remain. For example, how does the PRR inhibit FV procoagulant function, and how does B-domain proteolysis produce an active cofactor? Although systematic mutagenesis of the B-domain has been useful to identify critical regulatory B-domain elements, this approach is unlikely to bear fruit in defining the mechanism of FV autoinhibition and activation. In this study, we report important new insights into the mechanism of suppression of FV procoagulant activity and provide new ideas regarding how proteolysis of the B-domain produces an active cofactor.
EXPERIMENTAL PROCEDURES
Materials
The peptidyl substrate H-d-phenylalanyl-l-pipecolyl-l-arginyl-p-nitroanilide (S2238) was from Diapharma (West Chester, OH). Benzamidine, 4-amidinophenylmethanesulfonyl fluoride hydrochloride, isopropyl β-d-1-thiogalactopyranoside, BSA, and poly-l-lysine (average Mr = 1000–5000) were from Sigma. Dansylarginine N-(3-ethyl-1,5-pentanediyl)amide (DAPA) was from Hematologic Technologies (Essex Junction, VT). All tissue culture reagents were from Invitrogen, except insulin/transferrin/sodium selenite was from Roche Applied Science. Small unilamellar phospholipid vesicles composed of 75% (w/w) hen egg l-α-phosphatidylcholine and 25% (w/w) porcine brain l-α-phosphatidylserine (PCPS; Avanti Polar Lipids, Alabaster, AL) were prepared and characterized as described (18).
Proteins
Human prothrombin, thrombin, FX, and FV were isolated from plasma and prepared as described (19–22). Recombinant FX (rFX), rFXa, rFVa, plasma-derived FVa, FV-810, and the FV-810 variants FV-810R709Q, FV-810R1545Q, and FV-810QQ were prepared, purified, and characterized as described (15, 23, 24). Prethrombin-2 was prepared by proteolysis of human prothrombin and purified as described (25). Molecular weights and extinction coefficients (E0.1% = 280 nm) of the various proteins have been reported previously (16, 17). All functional assays were performed at 25 °C in assay buffer (20 mm HEPES, 150 mm NaCl, 5 mm CaCl2, and 0.1% polyethylene glycol 8000, pH 7.5).
Construction of FV B-domain Peptides
BR cDNA-encoding residues Ser951–Lys1008 of FV was amplified from FV cDNA using primers A (5′-GCAAGGTCTCAAGGTTCACGTGCTTGGGGAGAAAGCACC-3′, forward) and B (5′-GCTTGTCGACTTACTTCTCTTTTTTCTTTTTTCGTGTCTTAATGAGAAACTGG-3′, reverse). The BR+AR peptide, encoding the juxtaposed BR and AR sequences from the FV variant FV-B104 (17), was amplified from FV-B104 cDNA using primers C (5′-GCAAGGTCTCAAGGTAGTGGCCACCCAAAGTTTCCTAGAG-3′, forward) and D (5′-GCTTGTCGACTTAGTTGTCAGGATCTCTGGAGGAGTTGATGTTTGTCC-3′, reverse). Bovine BR (Bos taurus Ser938–Lys996), green anole BR (Anolis carolinensis Ser1331–Lys1393), and zebrafish BR (Danio rerio Ser1260–Lys1318) cDNAs were synthesized by GenScript (Piscataway, NJ). Amplified cDNAs were digested with the restriction enzymes BsaI (5′) and SalI (3′) and ligated into the pE-SUMO bacterial expression vector (LifeSensors, Malvern, PA), which had been digested with the same restriction enzymes. All constructs were verified by DNA sequencing.
Expression and Purification of B-domain Peptides
Sequence-verified bacterial expression constructs were transformed into chemically competent BL21(DE3) cells (EMD Millipore, Billerica, MA), and single colonies were used to inoculate liquid LB cultures containing 50 μg/ml kanamycin (LB-Kan50). Starter cultures were subcultured into 1 liter of LB-Kan50 and incubated at 37 °C until A600 reached 0.5, at which point, isopropyl β-d-1-thiogalactopyranoside was added at a final concentration of 1 mm. After 2 h, cells were pelleted, resuspended in lysis buffer (20 mm Tris, 150 mm NaCl, and 1% Triton X-100, pH 8), and lysed with a Misonix Sonicator 3000 system (Qsonica, Newtown, CT). Cell debris was pelleted, and the small ubiquitin-like modifier (SUMO) fusion proteins were purified on HisTrap FF columns (GE Healthcare) following the manufacturer's instructions. Purified SUMO fusion proteins were incubated with SUMO protease (LifeSensors) for 2 h at 30 °C to remove the SUMO fusion, and cleaved peptides were purified by cation exchange chromatography. Protein purity was assessed by SDS-PAGE using 4–12% gradient gels (Invitrogen) in MES buffer, followed by staining with Coomassie Brilliant Blue R-250.
Peptide Acetylation and Fluorescent Labeling
The BR peptide was acetylated by incubation with a 20-fold molar excess (relative to amine groups) of N-hydroxysulfosuccinimide acetate (Thermo Scientific) for 1 h at 25 °C in 20 mm HEPES, 150 mm NaCl, 2 mm CaCl2, and pH 7.4. Mass spectroscopy data of the acetylated BR peptide were consistent with quantitative modification of all Lys residues and the N terminus (data not shown). Fluorescent labeling of the BR peptide was performed by incubating the BR peptide containing an N-terminal Cys (Cys-BR) for 10 min at 25 °C with a 10-fold molar excess of tris(2-carboxyethyl)phosphine HCl (Thermo Scientific) to reduce disulfide bonds, followed by 2 h at 25 °C with a 20-fold molar excess of either Oregon Green 488 maleimide or QSY7 C5-maleimide (Invitrogen). The reactions were quenched by the addition of excess DTT, and labeled BR peptides were purified by gel filtration through Bio-Gel P-6DG resin (Bio-Rad) to remove excess labeling reagents.
Prothrombin and Prethrombin-2 Activation Assays
Steady-state initial velocities of prothrombin cleavage were determined discontinuously at 25 °C as described (23). Reaction mixtures containing PCPS (50 μm), DAPA (3 μm), prothrombin (1.4 μm), and either rFVa or the indicated FV-810 derivatives were incubated with B-domain peptides (0–10 μm) in assay buffer. Reactions were initiated with FXa (2 nm), and aliquots were quenched in buffer containing 50 mm EDTA at multiple time points. Prothrombin activation was determined using the chromogenic thrombin substrate S2238 as described (23). Prethrombin-2 activation was measured similarly to prothrombin using the following reaction conditions: 50 μm PCPS, 1.4 μm prethrombin-2, 3 μm DAPA, 5 nm rFVa or FV-810 derivatives, 1–50 nm FXa, and 0–1000 nm BR peptide.
Clotting Assays
FV derivatives (500 nm) were prepared in assay buffer. Where noted, FV derivatives were pretreated with 10 nm thrombin for 15 min at 37 °C, followed by the addition of 15 nm hirudin. Samples were diluted to 0.25 nm in assay buffer with 0.1% BSA, and the specific clotting activity was measured in FV-deficient plasma (George King Bio-Medical, Overland Park, KS) with TriniCLOT PT Excel (Tcoag, Wicklow, Ireland) as described (26). The data are presented as the means ± S.D.
Fluorescence Anisotropy Measurements
Steady-state fluorescence anisotropy was measured at 25 °C in a PTI QuantaMaster fluorescence spectrophotometer (Photon Technology International, Birmingham, NJ) using excitation and emission wavelengths of 480 and 520 nm, respectively, with long-pass filters (KV500, CVI Melles Griot) in the emission beam. Reaction mixtures (2.5 ml) containing fixed concentrations (20–40 nm as indicated) of Oregon Green 488 maleimide-modified BR (OG488-BR) and 50 μm PCPS in assay buffer were prepared in 1-cm2 quartz cuvettes to which increasing concentrations of FVa or FV-810 were added. Fluorescence anisotropy measurements, including controls, were performed as described (20, 27).
Analytical Ultracentrifugation
Analytical ultracentrifugation of QSY7 C5-maleimide-modified BR (QSY7-BR) was performed in a Beckman Optima XL-I analytical ultracentrifuge using absorbance optics. Sedimentation velocity was measured at 25,000 or 45,000 rpm with two-sector cells in an An-60 Ti rotor at 20 °C. Sedimentation of QSY7-BR was followed measuring absorbance at 560 nm in cells containing 5 μm QSY7-BR alone or with 7 μm FV-810 or rFVa in 20 mm HEPES, 150 mm NaCl, and 2 mm CaCl2, pH 7.4. Sedimentation coefficients and molecular weights were determined by g(s*) analysis performed using DCDT+ (28).
Data Analysis
Data were analyzed by nonlinear least squares regression analysis using the Marquardt algorithm (29), and the quality of each fit was assessed as described (30). Equilibrium dissociation constants (Kd) and stoichiometries (n) for the interaction between OG488-BR and FV-810 in saturation binding measurements were obtained from the change in OG488-BR anisotropy over increasing concentrations of FV-810, which was corrected for the overall change in fluorescence intensity (31, 32). Displacement binding experiments, in which unlabeled BR or FXaS195A were titrated into preformed complexes of OG488-BR·FV-810, were analyzed to determine Kd and n as described (27). In prothrombin-2 activation reactions, Kd values for the interactions of BR and FXa with FV-810 were obtained by global analysis of prethrombin-2 activation rates at varying concentrations of FXa and BR fit to a model of tight binding using DYNAFIT (33).
RESULTS
Inhibition of Cofactor-like FV Variants by B-domain Fragments
We previously identified a minimal inhibitory motif termed the PRR within the FV B-domain that consists of evolutionarily conserved basic and acidic elements (17). To define how these elements inhibit FV function, B-domain fragments were expressed as SUMO fusions in Escherichia coli and purified by ion exchange chromatography following removal of the SUMO tag (Fig. 1). The inhibitory effects of the purified fragments were determined in assays containing either FVa or the cofactor-like FV variant FV-810 (Fig. 2A). In reconstituted prothrombin activation reactions, the BR peptide effectively inhibited prothrombinase containing FV-810 (Fig. 2B). The clotting activity of FV-deficient plasma supplemented with FV-810 was similarly inhibited in the presence of the BR peptide (Fig. 2C), indicating that the BR peptide reconstituted a functional inhibitory PRR within FV-810. Importantly, these results recapitulate our previous observation that the cofactor-like activity of FV-810 can be inhibited by expanding its B-domain to include the BR (16). In contrast to FV-810, the BR peptide had no inhibitory effect on rFVa in prothrombin activation assays or clotting assays (Fig. 2, B and D). Furthermore, whereas a minimal B-domain almost exclusively composed of tandem BR and AR elements is sufficient to stabilize FV as a procofactor (17), rFVa was not inhibited by the BR+AR peptide (Fig. 2D). Thus, whereas the BR can act in trans to reconstitute the PRR, it appears that the AR must be covalently attached to mediate inhibition of procoagulant activity.
FIGURE 1.
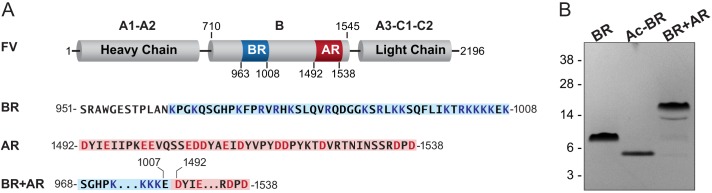
Design and expression of FV B-domain fragments. A, structural organization of FV. The BR (Lys963–Lys1008) is indicated in blue, and the AR (Thr1492–Asn1538) is indicated in red. Amino acid sequences of the BR and AR used to design B-domain fragments are shown below. B, SDS-PAGE analysis of purified B-domain fragments. Purified proteins (2 μg/lane) were resolved on 4–12% gels under reducing conditions and stained with Coomassie Brilliant Blue. First lane, the BR peptide; second lane, the acetylated BR peptide (Ac-BR); third lane, the BR+AR peptide. The apparent molecular weights of the protein standards are indicated.
FIGURE 2.

Inhibition of FV variants by B-domain fragments. A, structures of FV, FV-810 and FVa, indicating the presence of the BR (blue) and AR (red). B, the BR peptide (0–8 μm) was titrated into reaction mixtures containing 1.4 μm prothrombin, 3 μm DAPA, 50 μm PCPS, and 0.1 nm rFVa (○) or FV-810 (●) in assay buffer at 25 °C. Reactions were initiated with 2 nm FXa, and prothrombin activation was measured as described under “Experimental Procedures.” C and D, specific clotting activity was measured in FV-deficient plasma supplemented with 0.25 nm FV-810 (C) or rFVa (D) and the indicated peptides at 5 μm. Ac-BR, the acetylated BR peptide.
The complementary charge states of the BR and AR elements suggest the electrostatic forces likely contribute to PRR function. Accordingly, the BR peptide that had been acetylated to neutralize the positive charge no longer inhibited FV-810 (Fig. 2C). However, positive charge alone was insufficient to reconstitute a functional PRR, as neither low molecular weight poly-l-lysine (average Mr = 1000–5000) (Fig. 2C) nor the cationic platelet factor 4 (data not shown) affected FV-810 activity. Thus, inhibition of FV by the PRR is both charge- and sequence-dependent.
Direct Binding of the BR Peptide to FV Variants
The ability of the BR peptide to inhibit FV-810 suggests that the peptide binds to FV-810 to reconstitute a functional PRR. To test this, we measured direct binding of the BR peptide to FV-810 or rFVa using multiple approaches. First, the BR peptide was fluorescently labeled with Oregon Green 488 maleimide, and changes in fluorescence anisotropy were monitored. Titration of FV-810 into reactions containing fixed concentrations of OG488-BR produced saturable binding curves (Fig. 3) with calculated equilibrium binding values of Kd = 2.07 ± 0.2 nm and n = 1.27 ± 0.02 mol of FV-810/mol of OG488-BR. The binding of OG488-BR to FV-810 was calcium-dependent, as no binding was observed when 10 mm EDTA was added to the buffer. Titrating the unlabeled BR peptide into reactions containing a preformed complex of OG488-BR and FV-810 reduced the anisotropy signal toward the base line (Fig. 3, inset). From these displacement curves, the equilibrium binding values for the unlabeled BR peptide were calculated to be Kd = 2.1 ± 0.2 nm and n = 1.0 ± 0.06 mol of BR/mol of FV-810, essentially identical to the labeled peptide. In contrast to FV-810, rFVa showed no detectable binding to OG488-BR (Fig. 3).
FIGURE 3.
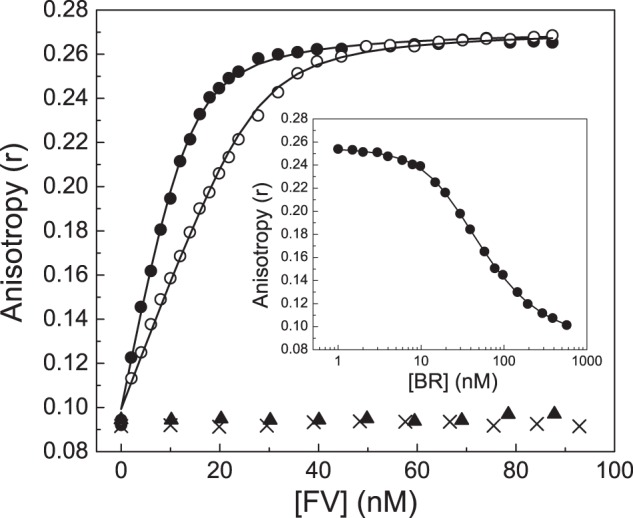
Direct binding of the BR peptide to FV-810. FV-810 was titrated into reaction mixtures containing 20 nm (●) or 40 nm (○) OG488-BR peptide and 50 μm PCPS in assay buffer at 25 °C. Changes in fluorescence anisotropy of OG488-BR were measured as described under “Experimental Procedures.” Lines were drawn after analysis to independent, non-interacting sites with the fitted constants Kd = 2.07 ± 0.2 nm and n = 1.27 ± 0.02 mol of FV-810/mol of OG488-BR at saturation. Control experiments were performed by titrating FV-810 into buffer containing 10 mm EDTA (×) or by titrating rFVa (▴). Inset, the unlabeled BR peptide was titrated into reaction mixtures containing 30 nm OG488-BR, 20 nm FV-810, and 50 μm PCPS. The fitted constants for the unlabeled BR peptide were determined as Kd = 2.1 ± 0.2 nm and n = 1.0 ± 0.06 mol of BR/mol of FV-810 assuming the constants determined above for OG488-BR.
As a complementary approach, we also monitored binding between the BR peptide and FV-810 by analytical ultracentrifugation. Sedimentation velocity experiments were performed with 5 μm QSY7-BR either alone (Fig. 4A) or with 7 μm FV-810 (Fig. 4B). In the absence of FV-810, the sedimentation coefficient (s20,w0) of QSY7-BR was 0.98 s, identical to the previously determined value for recombinant hirudin, which is similar in size (34). In the presence of FV-810, the QSY7-BR sedimentation coefficient shifted to 9.8 s, somewhat larger than our previously determined value for FV-810 of 8.43 s (15). This increase in the sedimentation coefficient is consistent with a 1:1 stoichiometry between QSY7-BR and FV-810, agreeing with the calculated stoichiometry from fluorescence measurements. Molecular weight determination yielded Mr = 7210 ± 40 for the QSY7-labeled peptide and Mr = 199,000 ± 3000 for QSY7-BR·FV-810 complex, which are in reasonable agreement with expected values. QSY7-BR sedimentation velocity was also performed with rFVa or with FV-810 in buffer containing 10 mm EDTA as controls. The data from these control experiments indicated a weak interaction (Kd ≥ 1 μm) (data not shown), consistent with our anisotropy data showing that no detectable binding was observed using ≤100 nm FVa (Fig. 3).
FIGURE 4.
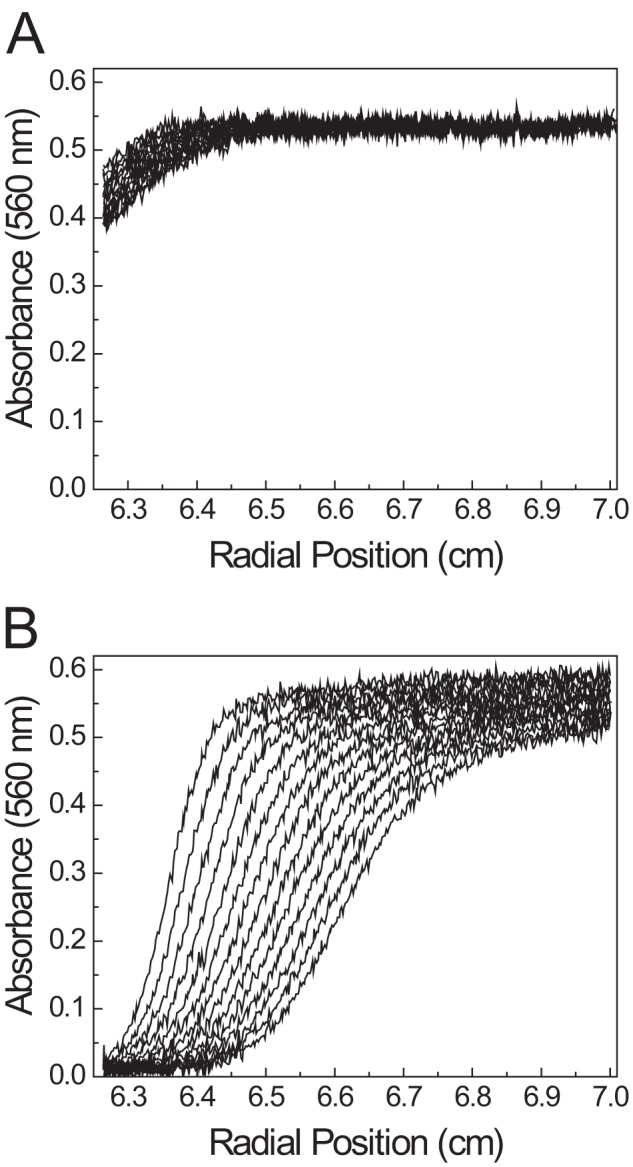
Sedimentation velocity of the BR peptide. The sedimentation velocity of 5 μm QSY7-BR was measured as described under “Experimental Procedures” either alone (A) or in the presence of 7 μm FV-810 (B). The panels show 14 scans taken at 8-min intervals.
PRR Sequence Specificity
To assess the sequence specificity of the PRR, we generated BR fragments from several vertebrate species and compared their effect on FV-810 with that of the human BR. The bovine BR, which is highly conserved with the human BR (Fig. 5A), inhibited FV-810 weakly in both prothrombin activation reactions and clotting assays compared with the human BR, whereas the more divergent BR fragments from lizard (A. carolinensis) and zebrafish (D. rerio) had no effect on FV-810 activity (Fig. 5C and data not shown). In displacement binding experiments, the bovine BR exhibited ∼10-fold weaker binding to FV-810 than did the human BR (Kd = 28.3 ± 0.6 versus 2.2 ± 0.2 nm, respectively) (Fig. 5D). These data show that even minor changes within the BR sequence are sufficient to dramatically reduce PRR assembly and function, suggesting a high degree of sequence specificity.
FIGURE 5.

Effect of BR peptides from several species on FV-810 activity. A, multiple sequence alignment of the BR elements from human (Homo sapiens), bovine (B. taurus), lizard (A. carolinensis), and zebrafish (D. rerio) FV proteins. Lysine and arginine residues are shaded in blue. B, purified BR fragments (2 μg each) were resolved by reducing SDS-PAGE and stained with Coomassie Brilliant Blue. C, human (●), bovine (♦), lizard (▴), and zebrafish (■) BR peptides were titrated into reactions containing 1.4 μm prothrombin, 3 μm DAPA, 50 μm PCPS, and 0.1 nm FV-810 in assay buffer at 25 °C. Prothrombin activation was measured as described in the legend to Fig. 1A. D, human (●) and bovine (♦) BR peptides were titrated into reactions containing 30 nm OG488-BR, 20 nm FV-810, and 50 μm PCPS at 25 °C. Fluorescence anisotropy was measured, and equilibrium binding constants were determined assuming a stoichiometry of 1 mol of FV-810/mol of BR peptide: the human BR, Kd = 2.2 ± 0.2 nm; and the bovine BR, Kd = 28.3 ± 0.6 nm.
Competition between the BR and FXa
A fundamental difference between procofactor-like and cofactor-like FV proteins is the ability of the latter to bind to FXa with high affinity. We previously demonstrated that a minimal B-domain consisting almost exclusively of the PRR is sufficient to maintain the procofactor state (17). We hypothesized that the PRR occludes a high affinity FXa-binding site on FV; thus, inhibition of FV-810 procoagulant function by the BR peptide could reflect competitive binding between the BR and FXa to FV(a). Consistent with this, when catalytically inactive FXaS195A was titrated into fluorescence binding assays, it displaced OG488-BR from FV-810 (Fig. 6A). Using the previously determined binding constants for OG488-BR, we calculated that FXaS195A bound to FV-810 with Kd = 1.8 nm and n = 1.1 mol FXa/mol of FV-810, consistent with previously reported equilibrium binding values for FXa and FV-810 (15). As a control, titration of zymogen FXS195A at concentrations up to 1 μm had little effect on OG488-BR anisotropy (Fig. 6A).
FIGURE 6.
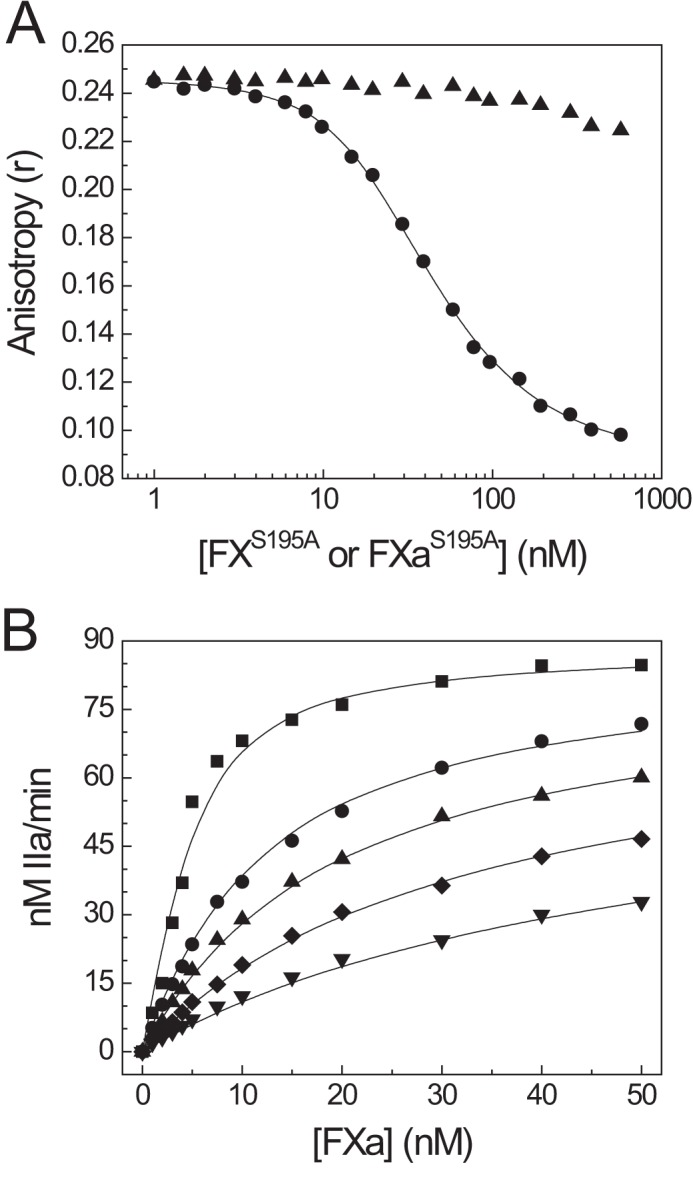
Competitive binding of the BR peptide and FXa to FV-810. A, FXaS195A (●) or zymogen FXS195A (▴) was titrated into reaction mixtures containing 30 nm OG488-BR, 20 nm FV-810, and 50 μm PCPS in assay buffer at 25 °C. Changes in OG488-BR anisotropy were measured, and lines were drawn as described with the fitted constants Kd = 1.8 ± 0.2 nm for FXa and n = 1.1 ± 0.07 mol of FXa/mol of FV-810 assuming the binding constants for OG488-BR determined in Fig. 2A. B, reactions containing 1.4 μm prethrombin-2, 3 μm DAPA, 50 μm PCPS, 5 nm FV-810, and 0 nm (■), 125 nm (●), 250 nm (▴), 500 nm (♦), or 1 μm (▾) BR peptide were prepared in assay buffer at 25 °C. Reactions were initiated by the addition of 1–50 nm FXa, and thrombin generation was monitored as described under “Experimental Procedures.” Experimental data were fitted to a model for tight binding with calculated values of Kd = 2.0 ± 0.2 nm for FXa and Kd = 34.2 ± 3.6 nm for the BR.
We also assessed competition between the BR peptide and FXa by monitoring prethrombin-2 activation in reactions containing variable concentrations of FXa and the BR peptide at a fixed concentration of FV-810. Under these reaction conditions, the BR and FXa followed a model of competitive binding (Fig. 6B), with fitted equilibrium binding values Kd = 2.0 ± 0.2 nm for FXa and Kd = 34.2 ± 3.6 nm for the BR. The calculated Kd for the BR from these reactions is somewhat higher than that observed in direct binding measurements (Fig. 3). One likely explanation for this discrepancy may be generation of FVa in the reactions from feedback proteolysis of FV-810 by thrombin (see below). Together with the fluorescence binding data, these results support a model of competitive binding of the BR and FXa to FV-810.
Role of Site-specific Proteolysis in PRR Stability and Inhibition
The observation that rFVa activity is not inhibited by any of the B-domain peptides suggests that proteolysis of the FV B-domain by thrombin irreversibly disrupts the PRR, thereby producing the active cofactor FVa. We assessed the effect of proteolysis at individual thrombin cleavage sites on PRR function by comparing the ability of the BR peptide to inhibit FV-810 cleaved by thrombin at either Arg709 or Arg1545. FV-810 variants containing the R709Q or R1545Q mutation were preincubated with thrombin to generate cleavage products in which the B-domain was still tethered to either the heavy and/or light chains (Fig. 7, A and B). Prior to thrombin cleavage, all FV-810 variants were potently inhibited by the basic peptide (Fig. 7C). However, following incubation with thrombin, only FV-810R1545Q and FV-810QQ were still inhibited by the BR peptide (Fig. 7D). Furthermore, when binding of the thrombin-cleaved FV-810 variants to OG488-BR was measured by fluorescence anisotropy, only FV-810R1545Q and FV-810QQ still bound OG488-BR, whereas FV-810 and FV-810R709Q did not (Fig. 8). Thus, cleavage of FV at Arg1545 specifically disrupts the ability of the BR to bind to FV-810 and establish a functional PRR. Interestingly, cleavage at Arg709 appears to partially destabilize the PRR, as FV-810R1545Q displayed a weakened binding affinity for OG488-BR after thrombin cleavage (Kd = 30 versus 2 nm). This observation suggests that proteolytic activation of FV progressively disrupts the structural integrity of the PRR, thereby reducing its inhibitory effect.
FIGURE 7.

Effect of the BR peptide on FV-810 thrombin cleavage site variants. A, schematic of the partial cleavage products generated by incubating FV-810 containing the R709Q and/or R1545Q mutation with thrombin. B, reactions containing 600 nm FV-810, FV-810R709Q, FV-810R1545Q, FV-810QQ, or rFVa were incubated for 15 min at 37 °C with buffer or 10 nm thrombin and then quenched with 20 nm hirudin. Samples were resolved by 4–12% gradient SDS-PAGE under reducing conditions and stained with Coomassie Brilliant Blue. C and D, specific clotting activities of FV-810 variants after incubation with buffer (C) or thrombin (D). FV-deficient plasma was supplemented with the quenched reaction products at 0.25 nm, and specific activity was measured in the absence (white bars) or presence (gray bars) of 5 μm BR peptide.
FIGURE 8.
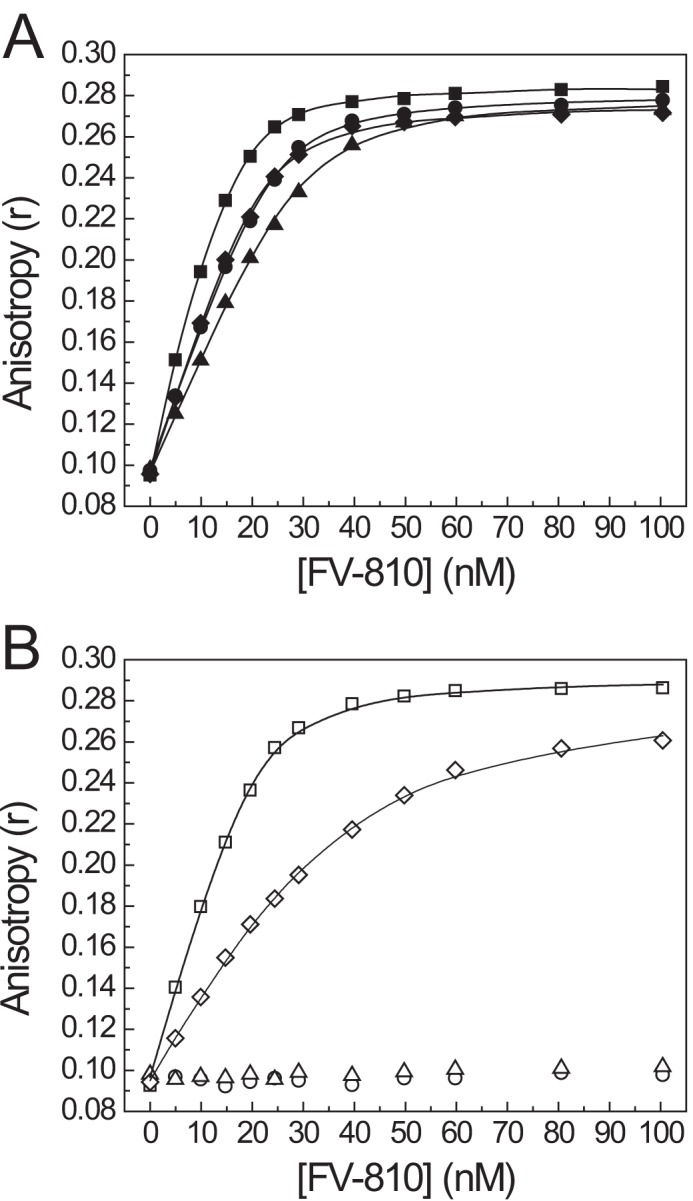
Direct binding of OG488-BR to thrombin-cleaved FV-810 variants. FV-810 (●), FV-810R709Q (▴), FV-810R1545Q (♦), and FV-810QQ (■) were pretreated with buffer (A) or thrombin (B) as described in the legend to Fig. 7. Quenched FV-810 species were titrated into reaction mixtures containing 30 nm OG488-BR and 50 μm PCPS in assay buffer, and changes in the fluorescence anisotropy signal were measured. Binding constants were calculated assuming a stoichiometry of n = 1 of mol FV-810/mol of OG488-BR: FV-810, Kd = 2.1 ± 0.3 nm; FV-810R709Q, Kd = 7.1 ± 1.6 nm; FV-810R1545Q, Kd = 2.0 ± 0.4 nm; and FV-810QQ, Kd = 0.31 ± 0.29 nm. After incubation with thrombin, neither FV-810 nor FV-810R709Q had detectable binding to OG488-BR, whereas FV-810QQ bound with a calculated Kd of 1.3 ± 0.1 nm, and FV-810R1545Q bound with a Kd of 30.3 ± 4.1 nm.
DISCUSSION
Despite decades of work attempting to draw correlations between FV B-domain cleavage and generation of procoagulant activity, the mechanism responsible for keeping FV inactive has largely remained a mystery. In this study, we undertook a completely different approach using recombinant B-domain fragments to reassemble functionally important regions of the FV B-domain. Our results emphasize the modular nature of specific regions of the B-domain. We found that the BR functions as a trans-acting regulator that binds with high affinity to an obligate cis-acting AR element located immediately N-terminal to the A3-domain. Importantly, these B-domain fragments have proved invaluable in deciphering how B-domain proteolysis destabilizes the PRR to produce a high affinity cofactor for FXa. Previous studies correlating selective cleavage of the Arg709, Arg1018, or Arg1545 peptide bond with FV activation demonstrated that maximal activation occurs upon Arg1545 cleavage (7, 14, 26, 35–41). Our data now provide a mechanistic explanation for these observations. Cleavage of the Arg1545 peptide bond disrupts the PRR by releasing the obligate cis-acting AR element from the A3-domain. We speculate that cleavage of the Arg709 and Arg1018 peptide bonds facilitates FV activation by progressively destabilizing the PRR, thereby increasing the accessibility of the molecule for FXa to form prothrombinase. Accordingly, cleavage of FV-810 at Arg709 noticeably reduced its binding affinity for OG488-BR (Fig. 8). In endogenous FV, we would expect subsequent cleavage at Arg1018 to dramatically weaken PRR stability by releasing the BR as part of the Ser710–Arg1018 cleavage product.
The affinity (low nanomolar Kd) with which the BR fragment binds to FV-810 is unexpectedly high. Perhaps the most relevant comparison is the interaction between thrombin and hirugen, the anionic dodecapeptide derived from the C terminus of hirudin. Whereas full-length hirudin binds thrombin with femtomolar affinity, hirugen binds with micromolar affinity mediated by electrostatic interactions throughout exosite I (42, 43). Given the complementary charges of the BR and AR, it would be reasonable to assume that PRR assembly is predominantly driven by electrostatic interactions. Accordingly, acetylating the BR peptide ablated its ability to inhibit FV-810. We also observed that elevated NaCl concentrations (≥250 mm) impaired OG488-BR binding to FV-810 (data not shown). However, despite the highly conserved amino acid sequence and net charge between the human and bovine BRs, the bovine fragment demonstrated considerably weaker binding and inhibition of FV-810. This striking effect from relatively minor changes in the BR strongly suggests that the PRR has a defined structure that depends on both electrostatic forces and additional sequence-dependent contributions. Unfortunately, the only structural information about the FV B-domain is from electron microscopy studies, in which the B-domain appears as an appendage extending from a globular core of heavy and light chains (44–49). Although an x-ray crystal structure has been resolved for activated protein C-inactivated bovine FVa (50), it provides no structural information about the B-domain. Preliminary NMR studies from our group indicate that the free BR appears largely unstructured in solution (data not shown); however, this does not preclude the possibility that a fully assembled PRR is structured. In fact, this scenario has been demonstrated by x-ray crystallography studies of hirugen, where free hirugen is disordered but adopts a well defined structure when bound to thrombin (51, 52).
Although we have generally discussed the PRR as an interaction between the BR and AR, it is important to note that some of our data actually suggest that the AR, although necessary, is not sufficient to bind the BR. For example, thrombin-cleaved FV-810R709Q did not show any detectable binding to OG488-BR, despite the AR being present on the partially cleaved B-domain (Fig. 8B). This could potentially be explained by additional sequences proximal to the AR contributing to a docking surface for the BR. In fact, a scenario in which the BR docks to an extended surface consisting of, for example, additional heavy or light chain sequences could also explain the competitive nature by which the BR and FXa bind to FV-810. Although we cannot eliminate the possibility that the PRR inhibits FXa binding allosterically, our working hypothesis is that the PRR directly occludes a FXa-binding site. Studies using site-directed mutagenesis and glycosylation have implicated the region surrounding the activated protein C cleavage site at Arg506 within the A2-domain in FXa binding, with additional contributions from residues in the A3-domain (53–56). Although the AR is distant from Arg506 in the primary structure of FV, sequences proximal to Arg506 in the tertiary structure could conceivably allow the BR to occlude critical FXa-binding sites when docked.
The ability of the BR to function as a trans-acting inhibitor of FV(a) activity offers a novel target for modulating FV(a) activity in vivo. Specifically, ligands that mimic the BR could function as anticoagulants by opposing or delaying full FV activation in the early stages of coagulation. A physiological ligand that may function this way is the Kunitz-type tissue factor pathway inhibitor (TFPI). TFPI is best known for its ability to oppose the initial stages of coagulation by forming a ternary complex with FXa and tissue factor/FVIIa, thereby attenuating the extrinsic pathway (57). Recent studies have also highlighted an intriguing link between TFPI and FV. Mast and Broze (58) previously observed that TFPI can inhibit thrombin generation in reactions initiated with FV but not FVa, suggesting a role for the B-domain in TFPI-mediated inhibition. More recently, Duckers et al. (59, 60) demonstrated a physical association between TFPI and FV in plasma that correlates with decreased TFPI levels in FV-deficient patients. Interestingly, the C terminus of the splice variant TFPIα shares substantial sequence homology with the FV BR (57, 61) and was recently shown to mediate the interaction of TFPIα with FV (62). Thus, by mimicking the FV BR, TFPIα may be able to delay FVa generation by prolonging the existence of a functional PRR. This could be particularly effective in modulating thrombin generation at nascent thrombi, where a distinct pool of partially activated FV is released from the α-granules of activated platelets (63). Platelets exclusively express TFPIα but not other TFPI isoforms (64, 65), and although platelet-derived TFPIα does not co-localize with FV in α-granules, their concomitant release from activated platelets could allow TFPIα to modulate platelet FV-mediated thrombin generation. Conversely, molecules that disrupt the PRR could potentially function as procoagulants by increasing FV cofactor activity and/or accelerating FV activation. Experimental evidence from Morrissey and co-workers (66–68) has implicated platelet-derived polyphosphate as an accelerator of FV activation by thrombin; although the mechanism is not currently defined, one possible explanation is that polyphosphate destabilizes the PRR by weakening the interaction between the BR and AR elements, thereby accelerating cleavage at Arg1545 to activate FV.
In summary, the data presented in this study provide new mechanistic insight to explain how the B-domain stabilizes FV as an inactive procofactor and how discrete proteolysis of FV generates an active cofactor for FXa. The bipartite PRR, composed of cis- and trans-acting acidic and basic elements, respectively, suppresses cofactor activity by competitively inhibiting FXa binding to FV. Discrete proteolysis of the B-domain by thrombin destabilizes the PRR, with cleavage of the Arg1545 peptide bond causing irreversible dissolution of PRR integrity and fully exposing a high affinity FXa-binding site. These observations offer a mechanistic explanation for why maximal activation requires cleavage at Arg1545 and suggest intriguing new strategies to regulate thrombin generation by modulating FV activation.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL88010 and P01 HL74124 (Project 2; to R. M. C.) and Grant T32 HL-07439 from NHLBI (to M. W. B.).
- FX
- factor Xa
- rFX
- recombinant FX
- BR
- basic region
- AR
- acidic region
- PRR
- procofactor regulatory region
- DAPA
- dansylarginine N-(3-ethyl-1,5-pentanediyl)amide
- PCPS
- l-α-phosphatidylcholine/l-α-phosphatidylserine
- SUMO
- small ubiquitin-like modifier
- OG488-BR
- Oregon Green 488 maleimide-modified BR
- QSY7-BR
- QSY7 C5-maleimide-modified BR
- TFPI
- tissue factor pathway inhibitor.
REFERENCES
- 1. Mann K. G., Nesheim M. E., Church W. R., Haley P., Krishnaswamy S. (1990) Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76, 1–16 [PubMed] [Google Scholar]
- 2. Mann K. G., Kalafatis M. (2003) Factor V: a combination of Dr. Jekyll and Mr. Hyde. Blood 101, 20–30 [DOI] [PubMed] [Google Scholar]
- 3. Camire R. M., Bos M. H. (2009) The molecular basis of factor V and VIII procofactor activation. J. Thromb. Haemost. 7, 1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Esmon C. T., Owen W. G., Duiguid D. L., Jackson C. M. (1973) The action of thrombin on blood clotting factor V: conversion of factor V to a prothrombin-binding protein. Biochim. Biophys. Acta 310, 289–294 [DOI] [PubMed] [Google Scholar]
- 5. Foster W. B., Nesheim M. E., Mann K. G. (1983) The Factor Xa-catalyzed activation of Factor V. J. Biol. Chem. 258, 13970–13977 [PubMed] [Google Scholar]
- 6. Nesheim M. E., Taswell J. B., Mann K. G. (1979) The contribution of bovine Factor V and Factor Va to the activity of prothrombinase. J. Biol. Chem. 254, 10952–10962 [PubMed] [Google Scholar]
- 7. Suzuki K., Dahlbäck B., Stenflo J. (1982) Thrombin-catalyzed activation of human coagulation factor V. J. Biol. Chem. 257, 6556–6564 [PubMed] [Google Scholar]
- 8. Esmon C. T. (1979) The subunit structure of thrombin-activated factor V. Isolation of activated factor V, separation of subunits, and reconstitution of biological activity. J. Biol. Chem. 254, 964–973 [PubMed] [Google Scholar]
- 9. Jenny R. J., Pittman D. D., Toole J. J., Kriz R. W., Aldape R. A., Hewick R. M., Kaufman R. J., Mann K. G. (1987) Complete cDNA and derived amino acid sequence of human factor V. Proc. Natl. Acad. Sci. U.S.A. 84, 4846–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nesheim M. E., Mann K. G. (1979) Thrombin-catalyzed activation of single chain bovine factor V. J. Biol. Chem. 254, 1326–1334 [PubMed] [Google Scholar]
- 11. Nesheim M. E., Foster W. B., Hewick R., Mann K. G. (1984) Characterization of factor V activation intermediates. J. Biol. Chem. 259, 3187–3196 [PubMed] [Google Scholar]
- 12. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kane W. H., Devore-Carter D., Ortel T. L. (1990) Expression and characterization of recombinant human factor V and a mutant lacking a major portion of the connecting region. Biochemistry 29, 6762–6768 [DOI] [PubMed] [Google Scholar]
- 14. Keller F. G., Ortel T. L., Quinn-Allen M. A., Kane W. H. (1995) Thrombin-catalyzed activation of recombinant human factor V. Biochemistry 34, 4118–4124 [DOI] [PubMed] [Google Scholar]
- 15. Toso R., Camire R. M. (2004) Removal of B-domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J. Biol. Chem. 279, 21643–21650 [DOI] [PubMed] [Google Scholar]
- 16. Zhu H., Toso R., Camire R. M. (2007) Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J. Biol. Chem. 282, 15033–15039 [DOI] [PubMed] [Google Scholar]
- 17. Bos M. H., Camire R. M. (2012) A bipartite autoinhibitory region within the B-domain suppresses function in factor V. J. Biol. Chem. 287, 26342–26351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Higgins D. L., Mann K. G. (1983) The interaction of bovine factor V and factor V-derived peptides with phospholipid vesicles. J. Biol. Chem. 258, 6503–6508 [PubMed] [Google Scholar]
- 19. Baugh R. J., Krishnaswamy S. (1996) Role of the activation peptide domain in human factor X activation by the extrinsic Xase complex. J. Biol. Chem. 271, 16126–16134 [DOI] [PubMed] [Google Scholar]
- 20. Buddai S. K., Toulokhonova L., Bergum P. W., Vlasuk G. P., Krishnaswamy S. (2002) Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J. Biol. Chem. 277, 26689–26698 [DOI] [PubMed] [Google Scholar]
- 21. Katzmann J. A., Nesheim M. E., Hibbard L. S., Mann K. G. (1981) Isolation of functional human coagulation Factor V by using a hybridoma antibody. Proc. Natl. Acad. Sci. U.S.A. 78, 162–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mann K. G. (1976) Prothrombin. Methods Enzymol. 45, 123–156 [DOI] [PubMed] [Google Scholar]
- 23. Camire R. M. (2002) Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J. Biol. Chem. 277, 37863–37870 [DOI] [PubMed] [Google Scholar]
- 24. Toso R., Zhu H., Camire R. M. (2008) The conformational switch from the factor X zymogen to protease state mediates exosite expression and prothrombinase assembly. J. Biol. Chem. 283, 18627–18635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mann K. G., Elion J., Butkowski R. J., Downing M., Nesheim M. E. (1981) Prothrombin. Methods Enzymol. 80, 286–302 [DOI] [PubMed] [Google Scholar]
- 26. Camire R. M., Kalafatis M., Tracy P. B. (1998) Proteolysis of factor V by cathepsin G and elastase indicates that cleavage at Arg1545 optimizes cofactor function by facilitating factor Xa binding. Biochemistry 37, 11896–11906 [DOI] [PubMed] [Google Scholar]
- 27. Betz A., Krishnaswamy S. (1998) Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J. Biol. Chem. 273, 10709–10718 [DOI] [PubMed] [Google Scholar]
- 28. Philo J. S. (2000) A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 279, 151–163 [DOI] [PubMed] [Google Scholar]
- 29. Bevington P. R., Robinson K. D. (1992) Data Reduction and Error Analysis for the Physical Sciences, pp. 141–167, McGraw-Hill, New York [Google Scholar]
- 30. Straume M., Johnson M. L. (1992) Analysis of residuals: criteria for determining goodness-of-fit. Methods Enzymol. 210, 87–105 [DOI] [PubMed] [Google Scholar]
- 31. Buddai S. K., Layzer J. M., Lu G., Rusconi C. P., Sullenger B. A., Monroe D. M., Krishnaswamy S. (2010) An anticoagulant RNA aptamer that inhibits proteinase-cofactor interactions within prothrombinase. J. Biol. Chem. 285, 5212–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krishnaswamy S. (1990) Prothrombinase complex assembly: contributions of protein-protein and protein-membrane interactions toward complex formation. J. Biol. Chem. 265, 3708–3718 [PubMed] [Google Scholar]
- 33. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 34. Otto A., Seckler R. (1991) Characterization, stability and refolding of recombinant hirudin. Eur. J. Biochem. 202, 67–73 [DOI] [PubMed] [Google Scholar]
- 35. Thorelli E., Kaufman R. J., Dahlbäck B. (1998) Cleavage requirements of factor V in tissue-factor induced thrombin generation. Thromb. Haemost. 80, 92–98 [PubMed] [Google Scholar]
- 36. Steen M., Dahlbäck B. (2002) Thrombin-mediated proteolysis of factor V resulting in gradual B-domain release and exposure of the factor Xa-binding site. J. Biol. Chem. 277, 38424–38430 [DOI] [PubMed] [Google Scholar]
- 37. Siigur J., Aaspõllu A., Tõnismägi K., Trummal K., Samel M., Vija H., Subbi J., Siigur E. (2001) Proteases from Vipera lebetina venom affecting coagulation and fibrinolysis. Haemostasis 31, 123–132 [DOI] [PubMed] [Google Scholar]
- 38. Segers K., Dahlbäck B., Bock P. E., Tans G., Rosing J., Nicolaes G. A. (2007) The role of thrombin exosites I and II in the activation of human coagulation factor V. J. Biol. Chem. 282, 33915–33924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kane W. H., Majerus P. W. (1981) Purification and characterization of human coagulation factor V. J. Biol. Chem. 256, 1002–1007 [PubMed] [Google Scholar]
- 40. Kalafatis M., Beck D. O., Mann K. G. (2003) Structural requirements for expression of factor Va activity. J. Biol. Chem. 278, 33550–33561 [DOI] [PubMed] [Google Scholar]
- 41. Segers K., Rosing J., Nicolaes G. A. (2006) Structural models of the snake venom factor V activators from Daboia russelli and Daboia lebetina. Proteins 64, 968–984 [DOI] [PubMed] [Google Scholar]
- 42. Dennis S., Wallace A., Hofsteenge J., Stone S. R. (1990) Use of fragments of hirudin to investigate thrombin-hirudin interaction. Eur. J. Biochem. 188, 61–66 [DOI] [PubMed] [Google Scholar]
- 43. Stone S. R., Hofsteenge J. (1986) Kinetics of the inhibition of thrombin by hirudin. Biochemistry 25, 4622–4628 [DOI] [PubMed] [Google Scholar]
- 44. Fowler W. E., Fay P. J., Arvan D. S., Marder V. J. (1990) Electron microscopy of human factor V and factor VIII: correlation of morphology with domain structure and localization of factor V activation fragments. Proc. Natl. Acad. Sci. U.S.A. 87, 7648–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lampe P. D., Pusey M. L., Wei G. J., Nelsestuen G. L. (1984) Electron microscopy and hydrodynamic properties of blood clotting factor V and activation fragments of factor V with phospholipid vesicles. J. Biol. Chem. 259, 9959–9964 [PubMed] [Google Scholar]
- 46. Laue T. M., Johnson A. E., Esmon C. T., Yphantis D. A. (1984) Structure of bovine blood coagulation factor Va. Determination of the subunit associations, molecular weights, and asymmetries by analytical ultracentrifugation. Biochemistry 23, 1339–1348 [DOI] [PubMed] [Google Scholar]
- 47. Mosesson M. W., Nesheim M. E., DiOrio J., Hainfeld J. F., Wall J. S., Mann K. G. (1985) Studies on the structure of bovine factor V by scanning transmission electron microscopy. Blood 65, 1158–1162 [PubMed] [Google Scholar]
- 48. Mosesson M. W., Church W. R., DiOrio J. P., Krishnaswamy S., Mann K. G., Hainfeld J. F., Wall J. S. (1990) Structural model of factors V and Va based on scanning transmission electron microscope images and mass analysis. J. Biol. Chem. 265, 8863–8868 [PubMed] [Google Scholar]
- 49. Nesheim M. E., Myrmel K. H., Hibbard L., Mann K. G. (1979) Isolation and characterization of single chain bovine factor V. J. Biol. Chem. 254, 508–517 [PubMed] [Google Scholar]
- 50. Adams T. E., Hockin M. F., Mann K. G., Everse S. J. (2004) The crystal structure of activated protein C-inactivated bovine factor Va: implications for cofactor function. Proc. Natl. Acad. Sci. U.S.A. 101, 8918–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rydel T. J., Tulinsky A., Bode W., Huber R. (1991) Refined structure of the hirudin-thrombin complex. J. Mol. Biol. 221, 583–601 [DOI] [PubMed] [Google Scholar]
- 52. Skrzypczak-Jankun E., Carperos V. E., Ravichandran K. G., Tulinsky A., Westbrook M., Maraganore J. M. (1991) Structure of the hirugen and hirulog 1 complexes of α-thrombin. J. Mol. Biol. 221, 1379–1393 [PubMed] [Google Scholar]
- 53. Steen M., Tran S., Autin L., Villoutreix B. O., Tholander A. L., Dahlbäck B. (2008) Mapping of the factor Xa binding site on factor Va by site-directed mutagenesis. J. Biol. Chem. 283, 20805–20812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gale A. J., Yegneswaran S., Xu X., Pellequer J. L., Griffin J. H. (2007) Characterization of a factor Xa binding site on factor Va near the Arg-506 activated protein C cleavage site. J. Biol. Chem. 282, 21848–21855 [DOI] [PubMed] [Google Scholar]
- 55. Heeb M. J., Kojima Y., Hackeng T. M., Griffin J. H. (1996) Binding sites for blood coagulation factor Xa and protein S involving residues 493–506 in factor Va. Protein Sci. 5, 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steen M., Villoutreix B. O., Norstrøm E. A., Yamazaki T., Dahlbäck B. (2002) Defining the factor Xa-binding site on factor Va by site-directed glycosylation. J. Biol. Chem. 277, 50022–50029 [DOI] [PubMed] [Google Scholar]
- 57. Broze G. J., Jr., Girard T. J. (2012) Tissue factor pathway inhibitor: structure-function. Front. Biosci. 17, 262–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mast A. E., Broze G. J., Jr. (1996) Physiological concentrations of tissue factor pathway inhibitor do not inhibit prothrombinase. Blood 87, 1845–1850 [PubMed] [Google Scholar]
- 59. Duckers C., Simioni P., Spiezia L., Radu C., Gavasso S., Rosing J., Castoldi E. (2008) Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood 112, 3615–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duckers C., Simioni P., Spiezia L., Radu C., Dabrilli P., Gavasso S., Rosing J., Castoldi E. (2010) Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood 115, 879–886 [DOI] [PubMed] [Google Scholar]
- 61. Mast A. E. (2011) Alternatively spliced tissue factor pathway inhibitor: functional implications. Front. Biosci. 3, 1457–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ndonwi M., Girard T. J., Broze G. J., Jr. (2012) The C-terminus of tissue factor pathway inhibitor α is required for its interaction with factors V and Va. J. Thromb. Haemost. 10, 1944–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gould W. R., Silveira J. R., Tracy P. B. (2004) Unique in vivo modifications of coagulation factor V produce a physically and functionally distinct platelet-derived cofactor: characterization of purified platelet-derived factor V/Va. J. Biol. Chem. 279, 2383–2393 [DOI] [PubMed] [Google Scholar]
- 64. Maroney S. A., Haberichter S. L., Friese P., Collins M. L., Ferrel J. P., Dale G. L., Mast A. E. (2007) Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood 109, 1931–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Novotny W. F., Girard T. J., Miletich J. P., Broze G. J., Jr. (1988) Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood 72, 2020–2025 [PubMed] [Google Scholar]
- 66. Smith S. A., Mutch N. J., Baskar D., Rohloff P., Docampo R., Morrissey J. H. (2006) Polyphosphate modulates blood coagulation and fibrinolysis. Proc. Natl. Acad. Sci. U.S.A. 103, 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smith S. A., Choi S. H., Davis-Harrison R., Huyck J., Boettcher J., Rienstra C. M., Morrissey J. H. (2010) Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood 116, 4353–4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yun T. H., Morrissey J. H. (2009) Polyphosphate and omptins: novel bacterial procoagulant agents. J. Cell. Mol. Med. 13, 4146–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]