

Background: NO reductase (NOR) takes up protons from the opposite side of the membrane compared with other heme-copper oxidases.
Results: NOR is sensitive to mutations along the suggested proton pathway 1 but not the others.
Conclusion: Only pathway 1 is used for proton transfer.
Significance: Although no energy is conserved, proton transfer still occurs through a specific pathway.
Keywords: Bioenergetics/Electron Transfer Complex, Electron Transfer, Enzyme Kinetics, Membrane Biophysics, Nitric Oxide, Proton Transport, Flow-Flash, Heme-Copper Oxidase, Kinetic Isotope Effect
Abstract
The NO reductase from Paracoccus denitrificans reduces NO to N2O (2NO + 2H+ + 2e− → N2O + H2O) with electrons donated by periplasmic cytochrome c (cytochrome c-dependent NO reductase; cNOR). cNORs are members of the heme-copper oxidase superfamily of integral membrane proteins, comprising the O2-reducing, proton-pumping respiratory enzymes. In contrast, although NO reduction is as exergonic as O2 reduction, there are no protons pumped in cNOR, and in addition, protons needed for NO reduction are derived from the periplasmic solution (no contribution to the electrochemical gradient is made). cNOR thus only needs to transport protons from the periplasm into the active site without the requirement to control the timing of opening and closing (gating) of proton pathways as is needed in a proton pump. Based on the crystal structure of a closely related cNOR and molecular dynamics simulations, several proton transfer pathways were suggested, and in principle, these could all be functional. In this work, we show that residues in one of the suggested pathways (denoted pathway 1) are sensitive to site-directed mutation, whereas residues in the other proposed pathways (pathways 2 and 3) could be exchanged without severe effects on turnover activity with either NO or O2. We further show that electron transfer during single-turnover reduction of O2 is limited by proton transfer and can thus be used to study alterations in proton transfer rates. The exchange of residues along pathway 1 showed specific slowing of this proton-coupled electron transfer as well as changes in its pH dependence. Our results indicate that only pathway 1 is used to transfer protons in cNOR.
Introduction
Cytochrome c-dependent NO reductase (cNOR)5 from Paracoccus denitrificans is an integral membrane protein complex that reduces nitric oxide to nitrous oxide (2NO + 2H+ + 2e− → N2O + H2O). Nitric oxide reductases (NORs) are mostly found in denitrifying bacteria that stepwise reduce NO3− to N2 gas (via NO2−, NO and N2O). NORs are also expressed in various pathogenic non-denitrifying bacteria in order to inactivate the toxic NO that is produced by the immune system of the host. NORs are members of the heme-copper oxidase (HCuO) superfamily. Most members of this superfamily reduce O2 to H2O and conserve the liberated free energy by pumping protons across the membrane, thus maintaining an electrochemical gradient. In addition, electrons are donated from the (positive, lower pH) outside, and protons needed for water formation are taken up strictly from the (negative, higher pH) inside. For this purpose, the O2-reducing HCuOs use defined proton transfer pathways (one or two) from the cytoplasm into the active site and a route for pumped protons to the outside that is less well defined (for recent reviews on cNOR and proton transfer pathways in the HCuO superfamily, see Refs. 1 and 2). cNOR, on the other hand, has been shown not to contribute to the electrochemical proton gradient, although the amount of free energy available from NO reduction is similar to that for O2 reduction (3–5). Electrons are donated by soluble carriers in the periplasm, so for the overall reaction to be non-electrogenic, protons used for NO reduction must also originate in the periplasmic (outside) solution.
cNOR is isolated as a complex of two subunits, NorB and NorC (see Fig. 1A). NO reduction takes place in the NorB subunit that contains three redox centers: two b-type hemes (hemes b and b3) and a non-heme iron (FeB). Heme b3 and the non-heme iron form the binuclear center, where nitric oxide is bound and reduced. NorC is a membrane-anchored cytochrome c with one c-type heme, which presumably forms the site of electron entry. In addition to NO reduction, cNOR can also catalyze O2 reduction (6–8). Site-directed mutations that affect NO reduction affect O2 reduction in a similar manner (7, 9, 10); thus, presumably the same catalytic components are used for both reactions. Furthermore, proton transfer occurs from the same side with similar rates and amplitudes during single-turnover reactions between the fully reduced cNOR with NO (11, 12) and O2 (8).
FIGURE 1.

Structure of cNOR from P. aeruginosa (Protein Data Bank code 3O0R) (13) with the predicted proton transfer pathways. The NorB (light gray, transparent) and NorC (black, transparent in B–D) subunits are shown in a helical representation. A, the residues that are predicted to be involved in proton transfer are indicated with sticks, in cyan (pathway 1), magenta (pathway 2), or wheat (pathway 3). The location of the K-pathway (not present in cNOR but important in other HCuOs) is indicated in dark red. B–D, close-up of the proton transfer pathways. The blue arrows indicate the point of entrance for waters from the bulk. The predicted proton transfer pathways are indicated with stick representations in cyan (pathway 1; A), in magenta (pathway 2; B), or wheat (pathway 3; C). The corresponding residues in P. denitrificans cNOR are indicated in parenthesis in case they are different or if they were exchanged in this study. Residues in NorC are indicated with a C in superscript. This figure was prepared with PyMOL (Schrödinger, LLC, New York).
Recently, the crystal structure of a cNOR from Pseudomonas aeruginosa (13), highly homologous (52% sequence identity) to cNOR from P. denitrificans, was determined, and based on this structure and molecular dynamics (MD) simulations, three different proton transfer pathways were proposed, all leading from the periplasmic side of the membrane (see Fig. 1) (13–15).
Proton transfer pathway (PW) 1 was predicted (13, 15) to involve the following residues (P. aeruginosa numbering with P. denitrificans numbers in parenthesis): Glu-135 (Glu-122), Asp-198 (Asp-185), Lys-53C (Lys-54C; the superscript “C” indicates the NorC subunit), and Glu-57C (Glu-58C) (see Fig. 1, A and B, and Table 1). MD simulations supported the formation of a well hydrated, hydrogen-bonded network and indicated that additional residues were involved: Arg-134 (Arg-121), Lys-199 (Lys-186), and Glu-70C (Glu-71C) (15). The entrance of the pathway was kept open by rigid hydrogen bonding networks between Glu-57C, Lys-53C, Arg-134, and Asp-198 (15).
TABLE 1.
Overview of the conservation of residues in predicted proton transfer pathways
Ps. aer., Pseudomonas aeruginosa; Pa. den., Paracoccus denitrificans.
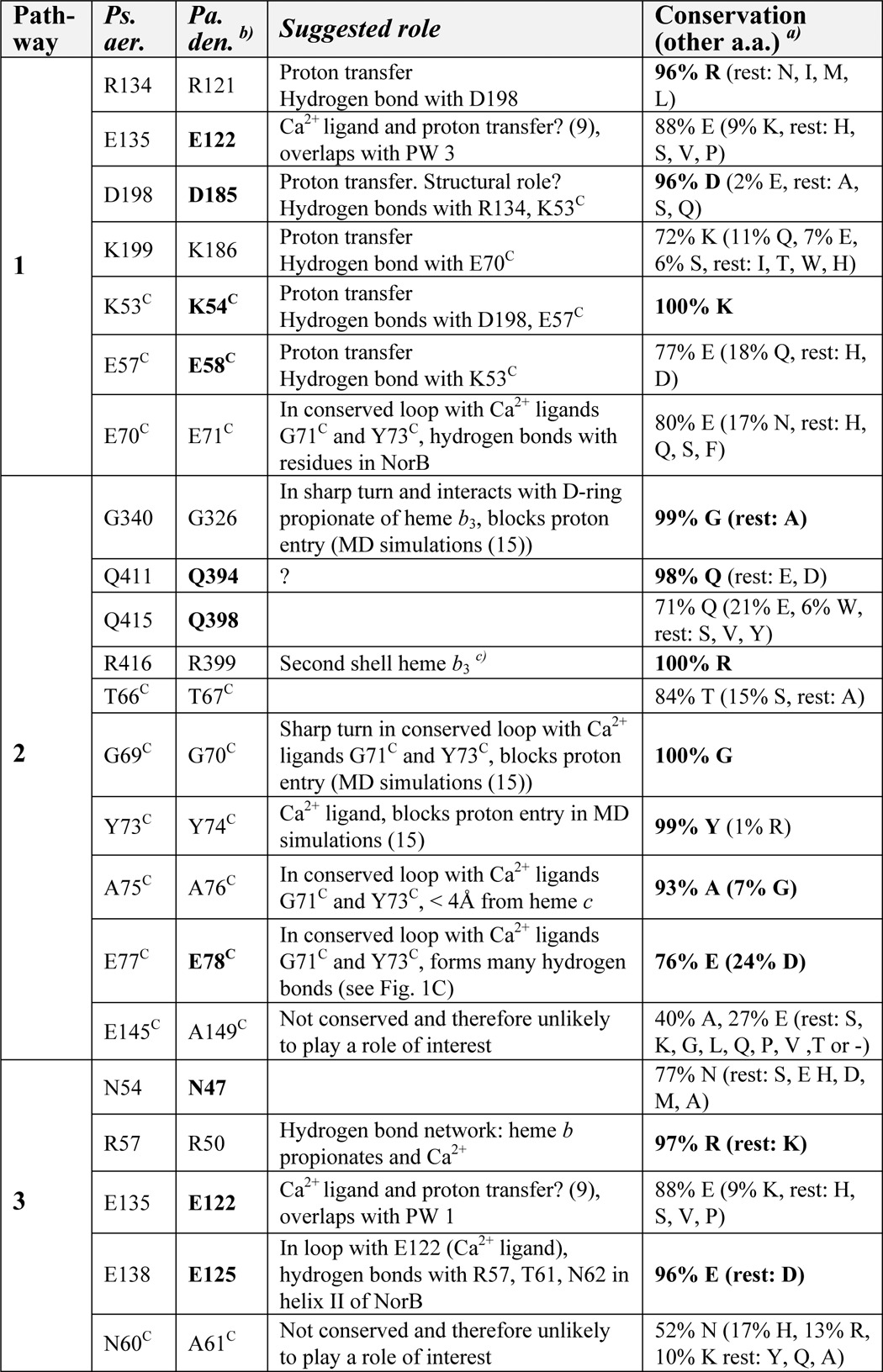
a Percentage conservation was calculated from the alignment in Fig. S5 of Ref. 18, 141 sequences. The value for Glu-77C (E77C) deviates slightly from the reported conservation in Ref. 18.
b The roles of the residues in boldface type have been investigated in this study or in Ref. 9.
c Hydrogen bonds with backbone His-339, which hydrogen-bonds with D propionate of heme b3.
PW 2 was predicted (13) to go from the heme c propionate via Gln-415 (Gln-398), Gln-411 (Gln-394), Gln-340 (Gln-326), Ala-75C (Ala-76C), Thr-66C (Thr-67C), and Gly-69C (Gly-70C) (see Fig. 1, A and C, and Table 1). Glu-145C in the P. aeruginosa cNOR was also predicted to be involved, but the P. denitrificans cNOR has an Ala at the equivalent position (Ala-149C). In MD simulations (15), a large hydrophilic region around Gln-415, Glu-77C, Arg-416, and Thr-66C was formed that is connected to the bulk solvent. However, the two loops with Gly-340 and Gly-69C together with the Tyr-73C (which ligates a Ca2+; see Fig. 1) prevented water from this cavity reaching the water cluster around the heme b3 propionates further on in the path. Pathway 2 was therefore concluded to be unlikely (15), but it could not be excluded without experimental data.
PW 3, not observed in the crystal structure but suggested from MD simulations (15), is lined by Glu-135 (Glu-122, shared with PW 1), Glu-138 (Glu-125), Arg-57 (Arg-50), Asn-54 (Asn-47), and Asn-60C. The Asn-60C is not conserved and is an alanine in P. denitrificans (Ala-61C) (Fig. 1, A and D, and Table 1). Asn-54 and Asn-60C were predicted to form a gate that could open and provide connectivity between the bulk water and an internal hydrated cavity.
When studying the role of individual amino acids for proton transfer, the flow-flash technique has provided extensive information about the HCuOs. Here a single turnover of fully reduced enzyme with O2 is studied time-resolved so that individual transitions can be resolved. We have previously studied the oxidation of the fully reduced cNOR by O2 using this technique and found a transition that involves proton-coupled electron transfer from the hemes b and c to the active site, modeled as rate-limited by proton transfer from an internal group (e.g. amino acid, H2O molecule, or part of cofactor), crucial for proton transfer to the catalytic site, with a pKa of 6.6 (8). This transition should thus be specifically sensitive to changes in the rate of proton transfer, and in this work we further demonstrate that it shows a kinetic isotope effect (when H2O was exchanged for D2O as the solvent) of ∼4 for the maximum rate constant, indicating that the transition is limited by the rate of proton transfer.
Before the crystal structure was known, we had constructed a model of the NorB from P. denitrificans based on the homology to structurally defined HCuOs, and a proton pathway was predicted based on this model, sequence conservation, and biochemical studies (5). This pathway involved residues Glu-135 (Glu-122) and Glu-138 (Glu-125) and was supported by data on P. denitrificans cNOR Glu-122 and Glu-125 variants, which showed effects on catalytic turnover (7, 16) as well as proton-coupled electron transfer (9). The crystal structure of the cNOR from P. aeruginosa (13) later showed that the Glu-135 is in fact coordinating a Ca2+ (see Fig. 1), and the Glu-138 stabilizes the loop in which the Glu-135 is located. These residues, although predicted to line the recently proposed proton transfer pathway 1 (Glu-135) and 3 (Glu-135 and Glu-138), thus have structural roles that make interpretations about their roles in proton transfer difficult.
In this work, based on the pathways proposed from the crystal structure and MD simulations, we exchanged individual residues along each of the three proposed pathways for non-protonatable residues. We chose the residues furthest away from the Ca2+, FeB, or hemes, because they are less likely to have structural roles. These variants were characterized in terms of their catalytic turnover with both NO and O2, their ligand binding properties with CO and O2, and finally their proton-coupled electron transfer rates and amplitudes. Our results strongly favor pathway 1 as the only functional proton pathway in cNOR.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
The pNOREX plasmid with the norB and norC genes was used for cNOR expression (7). Mutations were introduced directly in pNOREX with the use of the QuikChange XL site-directed mutagenesis kit (Stratagene). Correctly mutated pNOREX was transformed to the E. coli JM109 strain together with the pEC86 vector as described (7). E. coli was grown, and cNOR was expressed and purified essentially as described (9) with a few alterations for the cNOR variants and the wild type used for the comparison with the cNOR variants (but not for the experiments with wild type in D2O). The altered purification protocol was as follows. Membrane vesicles from a 6-liter cell culture were solubilized in 100 ml of buffer containing 100 mm Tris, pH 7.6, 50 mm NaCl, and 1% (w/v) n-dodecyl-β-d-maltoside (DDM). The solution was incubated at 4 °C with constant stirring for 1 h. Unsolubilized material was removed by centrifugation (30 min, 185,000 × g, 4 °C), and the supernatant was filtered over a 0.2-μm filter. The filtrate was diluted twice (with 100 mm Tris, pH 7.6, and 50 mm NaCl) to lower the DDM concentration and loaded at 2 ml/min on a 110-ml Q-Sepharose high performance (GE Healthcare) column pre-equilibrated with the same buffer and 0.05% (w/v) DDM. The column was washed with ∼300 ml of 100 mm Tris, 50 mm NaCl, and 0.05% (w/v) DDM at 5 ml/min. cNOR was eluted from the column in a 475-ml gradient of 200–500 mm NaCl with 20 mm Tris, pH 7.6, and 0.05% (w/v) DDM at 4 ml/min. 5-ml fractions were collected and diluted 3× in 20 mm Tris, pH 7.6, and 0.05% (w/v) DDM. The absorbance spectra of the collected fractions were analyzed via a dip probe connected to a Cary 50 Bio spectrophotometer (Varian). Fractions with an A280 nm/A410 nm of <2 were collected and concentrated over a 100 kDa cut-off filter (Millipore). 20 mm Tris, pH 7.6, and 0.05% (w/v) DDM were added so that the NaCl concentration was below 50 mm. 25–100-μl fractions of 20–150 μm purified protein (A280 nm/A410 nm ∼1) were flash frozen in liquid nitrogen and stored at −80 °C.
The presence of correctly inserted b and c hemes in all the cNOR variants was verified via UV-visible spectra from 260 to 700 nm on a Cary 50 or 400 spectrophotometer (Varian). The cNOR concentration was calculated from ϵ550 nm red-ox = 70 mm−1 cm−1.
Multiple Turnover
The reduction rates of the cNOR variants with O2 and NO were essentially determined as described in (10), but all measurements were conducted at 30 °C, and NO measurements were done with a specific NO electrode (World Precision Instruments) and recorded via the LabScribe2 software (World Precision Instruments). There were also small differences in the reaction set-up. For the multiple-turnover measurements with NO, NO-saturated water (2 mm NO) was added in five steps of 5 μl (10 μm/addition) to a deoxygenated solution of 50 mm HEPES, pH 7.5, 50 mm KCl, 0.05% (w/v) DDM, 30 mm glucose, 20 units/ml catalase, 1 unit/ml glucose oxidase, 500 μm N,N,N′,N′-tetramethyl-p-phenylenediamine, and 20 μm cytochrome c (horse heart; Sigma-Aldrich). This resulted in five equal steps in the NO signal. Then 3 mm ascorbate was added (this results in some background signal), followed by 25–100 nm cNOR. Because of substrate inhibition at high [NO], the obtained maximum rate was calculated from the slope at low [NO] (∼5 μm). At this part of the curve, the ascorbate background is negligible (as verified by a measurement without the addition of cNOR). O2 turnover was measured in a solution of 50 mm HEPES, pH 7.5, 50 mm KCl, 0.05% (w/v) DDM, 500 μm N,N,N′,N′-tetramethyl-p-phenylenediamine, 20 μm cytochrome c, and 3 mm ascorbate with a Clark-type electrode (Hansatech). The maximal rate was obtained directly after cNOR (at ∼250 nm) addition, and the background rate, recorded for ∼1 min just before the cNOR addition, was subtracted.
Flash Photolysis and Flow-Flash
Samples of ∼5 μm cNOR were prepared, and measurements were made as described in Ref. 8 on a set-up described in Ref. 17. In short, samples of ∼5 μm cNOR, 10 mm HEPES, pH 7.5, 50 mm KCl, 0.05% (w/v) DDM, 30 mm glucose, 20 units/ml catalase, 0.2 μm N-methylphenazinium methosulfate were prepared in a modified Thunberg cuvette. The sample was made anaerobic with N2(g), 0.5 unit/ml glucose oxidase was added to remove the remaining oxygen, and 2 mm ascorbate was added to reduce cNOR. The reduced sample was put under 100% (v/v) CO(g) and incubated overnight at 4 °C. CO recombination was studied by flash photolysis; the sample was illuminated with a short laser flash (10 ns, 200 mJ, 523 nm, Nd-YAG laser, Quantel), and the kinetic traces were recorded at the indicated wavelength on a digital oscilloscope. The CO concentration was then lowered to ∼30% (v/v, 70% N2(g)) until the average rebinding time constant was ∼100 μs. ∼50 μm dithionite was added to the cNOR sample, and the protein sample was connected to the stopped-flow syringe that was preincubated with 100 mm dithionite and washed with anaerobic water. The other syringe contained an oxygenated buffer with 50 mm HEPES, pH 7.5, 50 mm KCl, and 0.05% (w/v) DDM. To look at pH dependence, the HEPES was exchanged for different buffers at various pH values: MES (pH 6.0–7.0, HEPES (pH 7–8.5), Tris (pH 8.5), and citric acid (pH <6). The protein and the buffer samples were mixed in a 1:5 ratio (protein/oxygenated buffer) in a modified stopped-flow apparatus (Applied Photophysics), and after a 200-ms delay, the laser flash was applied to dissociate CO and allow O2 to bind and initiate the reaction.
Data Handling and Analysis
The time course of the reaction was studied from microseconds to seconds (via two channels, one recording 10 ms and one channel, prefiltered at 30 kHz, recording 2 s) at different wavelengths in the Soret and α regions. At each wavelength, 100,000 data points were collected, and the data set was then reduced to ∼1000 points by averaging over a progressively increasing number of points. The time-resolved absorbance changes were fitted individually or globally to a model of consecutive irreversible reactions with the software package Pro-K (Applied Photophysics).
The pH dependence of the proton-coupled electron transfer phase was fitted with the following equations (8),
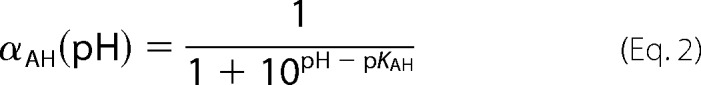 |
where kobs represents the obtained rate constant at a certain pH, and kH is the maximum rate at low pH. kH is the rate-limiting internal proton transfer (in Ref. 8 presumed to be from a group, AH (assumed to be in rapid equilibrium with bulk pH), to the active site). αAH is the fraction of protonated AH, determined by its pKa and the pH. For wild type, a small background rate, k0, was added to Equation 1 (8).
Kinetic Isotope Effect
For the flow-flash experiments in D2O, H2O in the solution was exchanged for D2O (99% (v/v); Cambridge Isotope Laboratories Inc.) by concentration and dilution in 10 mm Tris, 50 mm KCl, 0.05% (w/v) DDM, pH 7, on a 100 kDa cut-off filter (Millipore). The oxygenated buffers were also made with D2O. The reported pH* values in D2O are the pH meter readings, not the pD value (which is ∼0.4 pH units higher).
RESULTS
Conservation of Residues in the Predicted Proton Transfer Pathways
In Table 1, we have listed the conservation pattern of the residues involved in the three predicted proton transfer pathways (based on the alignment of 141 cNOR sequences in Ref. 18). Some, but not all, residues of pathway 1 are highly conserved. There are also some highly conserved residues in pathways 2 and 3, but for most of them, we suggest a role that is not related to proton transfer (Table 1), based on the crystal structure (13).
cNOR Variants Made
In pathway 1, we constructed variants for the two amino acids at the entrance of the pathway: K54AC (Lys-53C in P. aeruginosa) and E58QC (Glu-57C). We also substituted the following aspartate, Asp-185 (Asp-198), for a glutamate (to maintain the negative charge but change the side chain length), asparagine (to maintain the side chain length but remove the charge), or alanine (for both a drastic change in side chain length and the removal of charge).
In pathway 2, the initial glutamate, Glu-145C (P. aeruginosa; Fig. 1C), is an alanine in the P. denitrificans cNOR (Table 1). We therefore exchanged the next polar residue in the path, the equivalent of Gln-415, for a hydrophobic leucine (Q398L). We also changed the following glutamine (the equivalent of Gln-411; Fig. 1C and Table 1) for a hydrophobic methionine (Q394M).
In pathway 3, Asn-54 and Asn-60C were predicted to form a gate that could open and provide connectivity between the bulk water and an internal hydrated cavity (15). The Asn-60C is not conserved and is an alanine in P. denitrificans (Ala-61C) (Fig. 1, A and D, and Table 1). We constructed cNOR variants for the other asparagine: N47F and N47L (Asn-54 in P. aeruginosa cNOR; Fig. 1D). All other PW 3 residues have structural roles (Table 1), and their importance was therefore not analyzed in this study.
Expression, Optical Spectra, and Multiple Turnover
Most cNOR variants along the three proposed pathways could be expressed and resulted in stable protein complexes, except for E78FC (in pathway 2; Table 2). The stable cNOR variants were characterized with respect to their optical spectra (oxidized and reduced) and their catalytic turnover rates with NO and O2. All variants showed wild type-like optical spectra (except for N47L, which was therefore not studied further), showing that hemes b and c were integrated normally into the cNOR. All cNOR variants with mutations in pathway 1 were significantly affected in the ability to reduce NO and O2 (Table 2), although the mutations were >7 Å away from both the active site and the Ca2+. The expressed and stable cNOR variants with mutations in pathway 2 (Q394M and Q398L) or pathway 3 (N47F) reduced both NO and O2 with rates that were more similar to wild type (Table 2). N47F showed wild type rates for both NO and O2 reduction. Q398L reduced NO as wild type but had a slightly slower rate with O2 (∼61%). Q394M reduced NO at ∼67% and O2 at ∼33% of the wild type rate.
TABLE 2.
Overview of cNOR variants constructed
Ps. aer., Pseudomonas aeruginosa; Pa. den., Paracoccus denitrificans.
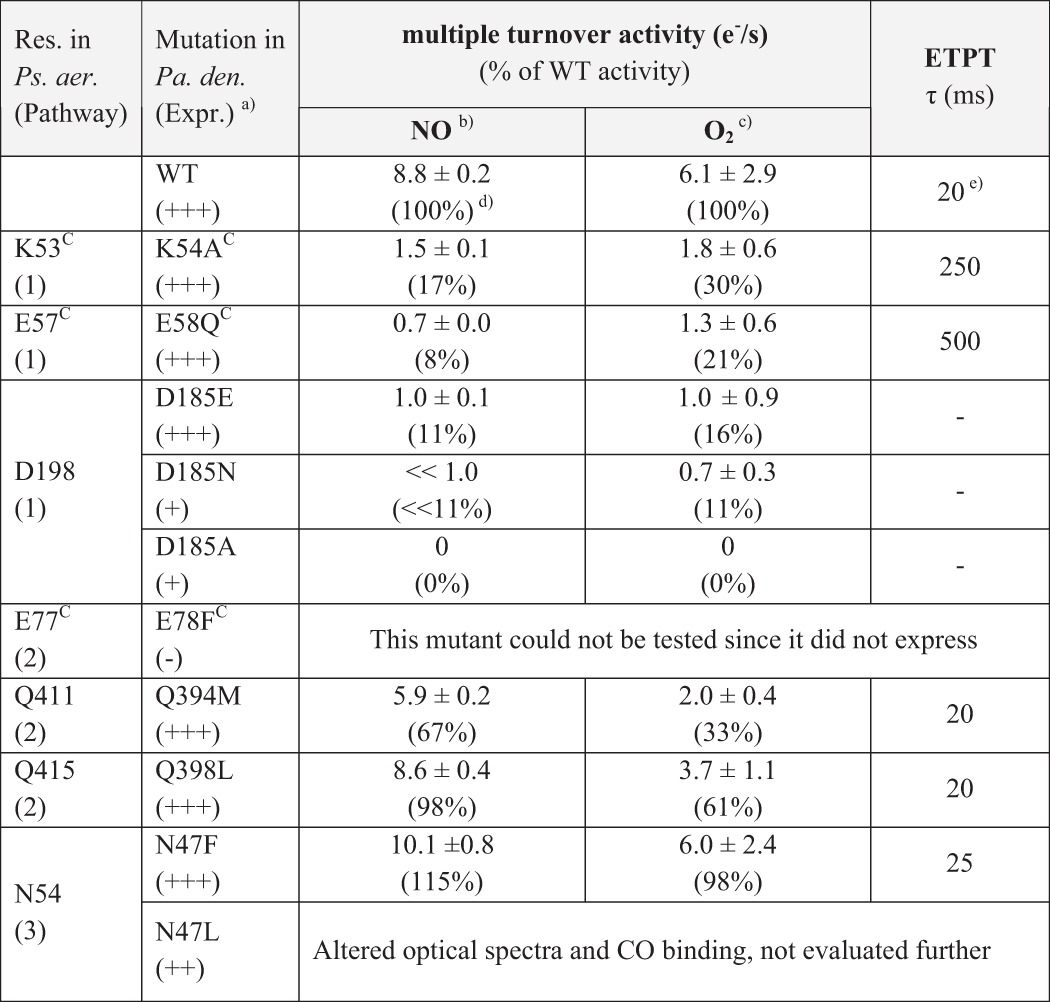
a Expression (Expr.) indicated as +++ for WT-like levels of cNOR.
b The error indicates the range for two measurements for NO multiple turnover.
c S.D. value is indicated for O2 multiple turnover for 5–7 measurements.
d The NO multiple turnover activity of the wild type was lower than reported previously (7, 10), possibly because of slight alterations in the purification protocol. The cNOR variants were prepared in the same way as the wild type, such that the percentage activity reports a valid comparison.
e ETPT stands for the proton-coupled electron transfer during single-turnover O2 reduction. The ETPT in wild type was slightly faster than before (25 ms in Ref. 8).
Single-turnover O2 Reduction
Multiple-turnover rates cannot be used to discriminate between changes in proton transfer rates and any other change in the reaction cycle. Therefore, we studied the transitions as the fully reduced cNOR is oxidized by O2. In this reaction for wild type (WT) cNOR, O2 binds to heme b3 with a time constant of ∼50 μs at 1 mm O2 (kobs ∼2 × 104 s−1), followed by proton-coupled electron transfer from the hemes b and c to the active site with a time constant of ∼20 ms at pH 7.5 (kobs ∼60 s−1; Figs. 2 and 3 and Table 2). These time constants deviate slightly from the ∼40 μs for O2 binding and ∼25 ms for the second phase as reported previously (8), probably because of the slightly altered purification conditions. Based on its pH dependence (Fig. 2B), the 20–25-ms phase was modeled to be rate-limited by proton transfer from an internal group with a pKa of 6.6 (see Ref. 8 and Equations 1 and 2). In this paper, we will refer to this phase as the proton-coupled electron transfer (ETPT), but we would like to stress that this term is not intended to imply the order of the reactions.
FIGURE 2.
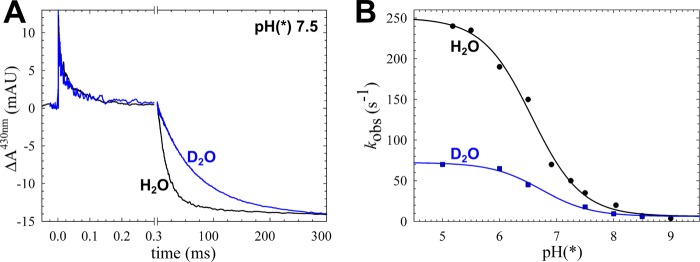
The kinetic (deuterium) isotope effect on ETPT during the reaction between fully reduced cNOR and O2. A, trace obtained at 430 nm of wild type cNOR in H2O or D2O at pH(*) 7.5, showing the change in absorbance (ΔA) over time (with the laser flash set at t = 0). The data were normalized to the COoff step at t = 0 for the rapid time scale and to the amplitude of the ETPT (which varies slightly between experiments) for the longer time scale. The laser artifact at t = 0 has been removed for clarity. The COoff reaction results in a rapid increase of absorbance, and O2 binding (with τ of ∼40–50 μs) then results in a decrease. On the longer time scale, the ETPT is seen as a further negative ΔA (τ of ∼20–25 ms in H2O). B, the rate of the ETPT in D2O (blue) and H2O (black) as a function of pH(*). The D2O data were fitted to a pKa* of 6.7 ± 0.1 and a kH (maximal rate at low pH) of 66 ± 3 s−1, and the H2O data (which are from Ref. 8) were fitted to a pKa of 6.6 ± 0.1 and a kH of 244 ± 7 s−1. mAU, milliabsorbance units.
FIGURE 3.

The reaction between fully reduced cNOR variants and O2. The traces show the change in absorbance (ΔA) over time (with the laser flash set at t = 0) for cNOR wild type and the constructed variants. A–C, variants in proton transfer PW 2 and 3; D–F, variants in PW 1. Data were recorded at 430 nm (A and D), 420 nm (B and E), and 550 nm (C and F). The laser artifact at t = 0 has been removed for clarity. At 420 nm, the COoff results in a rapid decrease in absorbance on the fast time scale, and the subsequent oxidation of the hemes (ETPT) results in a further slower negative ΔA. The amplitude of the ETPT varied slightly between experiments for both WT and variants and was normalized at 550 nm (reporting on the heme c) and 430 nm to the same ΔA for easier comparison of the rates. For the 420 nm trace and the shorter time scale at 430 nm, the COoff step was used for normalization. The ΔA in the Asp-185 variants are only normalized to the COoff step, because their ETPT ΔA values deviate from WT much more than the variation between measurements. A–C, WT (black trace), Q394M (blue trace), Q398L (green trace), and N47F (red trace). Gln-384 and Gln-398 are located in PW 2, and Asn-47 is in PW 3. D–F, wild type (black trace), E58QC (blue trace), K54AC (red trace), D185E (green trace), D185N (gray trace), and D185A (yellow trace). mAU, milliabsorbance units.
Kinetic Isotope Effect
To further investigate if the ETPT is indeed limited by proton transfer, we studied its kinetic (deuterium) isotope effect at various pH/pH* values (i.e. reporting the pH meter reading for both H2O and D2O solutions). The obtained rate constants are plotted in Fig. 2B. The observed rate constants in H2O were previously fitted to a pKa of 6.6 ± 0.1 and a kH of 244 ± 7 s−1 (8). In D2O, the corresponding values were pKa* = 6.7 ± 0.1 and kH* = 66 ± 3 s−1. The maximum rate constant kH thus has a kinetic isotope effect (ratio of rate constants in H2O and D2O) of 3.7, indicating that this phase is indeed limited by proton transfer (e.g. see Ref. 19). The small change in pKa when exchanging H2O for D2O follows the empirical formula (pKH = 0.929 × pKH* + 0.42) determined in Ref. 20.
CO and O2 Binding Rates
In order to further probe the integrity of the active site in our cNOR variants, we determined the time constant for CO and O2 binding. O2 binding was found to be the same as in wild type, with τ ranging from ∼40 to ∼50 μs (Fig. 3). Only the D185N and D185A had slightly slower O2 binding with a τ of ∼60 or ∼65 μs, respectively (Fig. 3). CO binding in the D185A and the D185E variants was slower than in wild type, whereas it was essentially unchanged for any of the other variants with normal optical spectra (data not shown). The changes in O2 and/or CO binding in the Asp-185 variants could indicate an altered environment of the b3 heme. D185N and D185A were also less well expressed (Table 2), indicating that the Asp-185 is needed to produce/maintain a stable and functional cNOR.
Proton-coupled Electron Transfer
Because we established that the ETPT is indeed limited by proton transfer, we used it to indicate whether proton transfer was affected in the cNOR variants.
Pathways 2 and 3
The pathway 2 substitutions Q398L and Q394M (Gln-415 and Gln-411 in P. aeruginosa cNOR) had no effect on the ETPT rate (Fig. 3 and Table 2). Also in the N47F cNOR variant (Asn-54 in P. aeruginosa cNOR, PW 3), the ETPT rate was similar to WT (Fig. 3 and Table 2).
Pathway 1
For the Asp-185 (Asp-198 in P. aeruginosa cNOR) variants, the amplitude for the ETPT is small (430 and 550 nm) or even absent (420 nm), indicating that the reaction is completely inhibited. In the E58QC (Glu-57C in P. aeruginosa cNOR) variant, the ETPT rate constant is ∼30 times slower than in WT at pH 7.5 (with a kobs of ∼2 s−1, τ of ∼500 ms). The K54AC (Lys-53C in P. aeruginosa cNOR) has an ETPT rate constant that is ∼10 times slower (with a kobs of ∼4 s−1 and τ of ∼250 ms) than in WT at pH 7.5.
pH Dependence of the Proton-coupled Electron Transfer in Affected cNOR Variants
For the cNOR variants that had slower ETPT rate constants at pH 7.5, we further investigated this reaction at various pH values. However, this could not be done for the Asp-185 variants because their ETPT is small or even absent (and the rates can therefore not be fitted).
In the cNOR variant E58QC, the reaction displayed severely slower rates in the entire pH range that we tested, with a kH (maximum rate constant) of ∼50 s−1 (i.e. ∼5 times slower than for WT) (Fig. 4). The pKa in E58QC shifted from 6.6 to ∼5.8.
FIGURE 4.
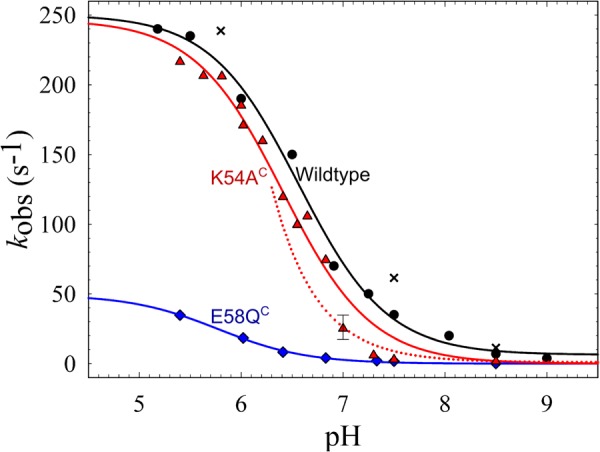
The pH dependence of the ETPT in cNOR wild type and variants. The rates of the ETPT for cNOR wild type and the constructed variants that were affected at pH 7.5 are plotted as a function of pH: wild type (black; circles for the data from Ref. 8 and crosses for the data with the slightly altered purification protocol), K54AC (red triangles), E58QC (blue diamonds). The WT data (from Ref. 8) were fitted to a pKa of 6.61 ± 0.05 and a kH of 244 ± 7 s−1 (black line); K54AC data were fitted to a pKa = 6.4 ± 0.1 and a kH = 247 ± 11 s−1 (red line); and E58QC data were fitted to a pKa = 5.8 ± 0.1 and a kH = 49 ± 2 s−1 (blue line). However, data points for K54AC did not follow the fit around pH 7–7.5, possibly because the one-exponential fit is an oversimplification for this mutant. As a comparison, also plotted (as a dotted red line) is the diffusion rate, assuming kdiff of ∼2 × 108 m−1 s−1 (see “Results” for details). The error bar at pH 7.0 for K54AC indicates that at this pH the range of possible fits is rather large.
The K54AC cNOR variant, however, has a similar kH as wild type (∼250 s−1), but the pKa is shifted from 6.6 to 6.4. The ETPT rate constant in the K54AC variant was fitted to a single exponential at all pH values for comparison with wild type, although the fit was better with two exponentials at pH values from 6.5 to 7.5. This could explain why the obtained rate constants at pH 7–7.5 were not fitted well to a single pKa transition (Fig. 4). This behavior can be explained by a model in which at high pH the assumption that the internal protonatable group (AH, Equations 1 and 2) is in rapid equilibrium with the bulk pH is not valid for this cNOR variant (because the entry point to the pathway is altered). Thus, at higher pH, proton diffusion to AH becomes rate-limiting, and this proton diffusion rate (tentatively fitted to ∼2 × 108 m−1 s−1) is plotted as a red dotted line in Fig. 4. A small background rate k0 of ∼1 s−1 was added to this fit. This altered model could also explain the biphasic nature of the ETPT in this cNOR variant because in the enzyme population with AH protonated (and not in rapid equilibrium with bulk pH), ETPT can occur with the maximum rate kH. With a forced one-exponential fit, we would see an average of the two rate constants (kH and the diffusion-limited rate constant) in the pH range where both populations (protonated/deprotonated AH) contribute significantly. However, we did not pursue a full biphasic fit of the data because of the small number of points that could not be fitted reasonably well with one exponential.
DISCUSSION
The multiple-turnover data with NO and O2 show severe effects for variants with pathway 1, but not pathway 2 (Q398L, Q394M) or 3 (N47F), residues modified (Table 2). Furthermore, all cNOR variants that are affected in their NO turnover are also affected in their O2 turnover (Table 2). The correlation between O2 and NO turnover has been observed before (although the extent of the change can differ (7, 10)) and shows the validity of using O2 as an alternative substrate.
The ET reaction (in this paper called ETPT) during single-turnover reduction of O2 by fully reduced cNOR was previously suggested to be rate-limited by proton transfer based on the uptake of protons from solution with the same rate constant and the pH dependence of the reaction (8). In this work, we show that the kmax (kH) is decreased, from ∼250 s−1 to ∼70 s−1 (i.e. by a factor of ∼4) in D2O (Fig. 2), which indicates that the rate of proton transfer indeed limits the overall rate constant for this reaction. We thus have strengthened the basis for using this ETPT as a “reporter” for effects on proton transfer rates in cNOR. The kinetic isotope effect is higher in A-type HCuOs (the F → O transition has a kinetic isotope effect of 7 (21)), where there are conformational changes needed in the proton pumping process. In cNOR we do not expect large conformational changes, because cNOR does not pump protons, and no “gating” is required.
The effects on the ETPT in the cNOR variants studied here were qualitatively in agreement with the multiple-turnover data, except for Q394M (PW 2), where the turnover rate with O2 is slower than wild type, whereas the ETPT is unaffected. Presumably, rereduction is slowed in this cNOR variant but for reasons unknown at this point. We note, however, that Gln-394 is a highly conserved residue (Table 1). For the other variants in PWs 2 and 3 (Fig. 1), there are no effects on the ETPT (Fig. 3 and Table 2) and no (large) effects on multiple turnover. It could be argued that very few variants were made in PWs 2 and 3, but because we aimed at residues away from the Ca2+ and the hemes without other roles, as suggested by the structure (Table 1), few residues were good candidates (especially for PW 3, which shares parts with PW 1). Even so, the reason why the cNOR variant E78FC (in PW 2) was not expressed and N47L (in PW 3) had altered spectra and CO binding properties is possibly because of their close proximity to heme c (4.9 and 3.5 Å for Glu-78C and Asn-47, respectively). Both suggested PWs 2 and 3 in the P. aeruginosa cNOR contain one residue that is not conserved in P. denitrificans cNOR (as in many other cNORs; see Table 1 and Ref. 18), also indicating that PWs 2 and 3 are not used for proton transfer.
The variants in PW 1 all affect catalytic turnover (Table 2) and the rate of the ETPT (Fig. 3). None of the modified residues have obvious structural roles as, for example, Ca2+ ligands (Table 1), and all are >7.5 Å away from the redox centers and the Ca2+ site. Therefore, it is unlikely that all of these residues have structural roles and/or control the heme potentials. The variants where the Asn-185 (Asn-198 in P. aeruginosa cNOR) was exchanged for a non-protonatable residue (D185A and D185N) retain some residual activity in multiple turnover. In the single-turnover reaction with O2, however, only O2 binding is observed, and there is no ETPT (Fig. 3). This might be due to a limited “window” in which the ETPT reaction can be observed, because on the longer time scale at 420 nm (Fig. 3E), a phase that we attribute to CO rebinding is observed, presumably due to a slow re-equilibration between CO and the O2 at the active site (9, 10). Although D185E had higher multiple-turnover rates than D185A and D185N, its rates were still much lower than those of wild type. The effects on the ETPT seemed too severe for the residue to have a “simple” role in proton transfer because an Asp is only a methyl group shorter than a Glu. The chain length might, however, influence the pathway because the Asp-198 could form hydrogen bonds with Lys-53C (Lys-54C), Arg-134 (Arg-121), and various waters (15) (Fig. 5). An alternative explanation is that the aspartate has a more structural role, because the D185N and D185A variants were only expressed at low levels, and ligand binding was also slowed in these variants.
FIGURE 5.
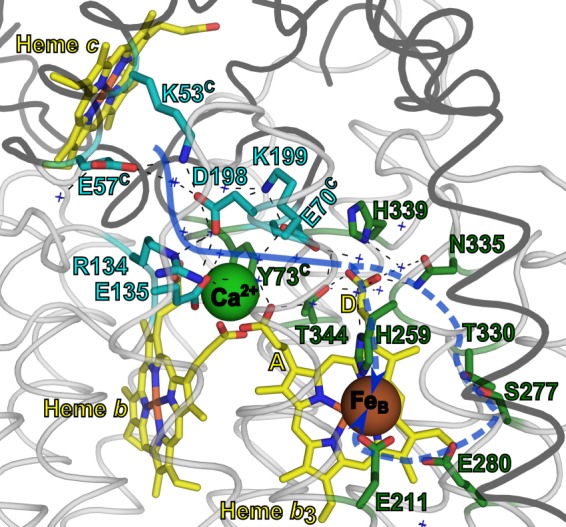
The proton transfer pathway in cNOR as indicated in this work. The structure is the same as in Fig. 1 (cNOR from P. aeruginosa (Protein Data Bank code 3O0R) (13)) and rendered in the same way (except that for clarity a loop instead of helical representation is used) with the residues in the start of pathway 1 indicated with sticks in cyan (pathway 1) and the ones predicted to be involved in the continuation of the proton pathway to the active site in green. The suggested pathway is indicated with a blue (dotted) arrow. Crystallographic waters in or around the pathway are indicated with blue crosses. The black dotted lines indicate hydrogen bonds. The yellow A and D indicate the heme b3 propionate A and D, respectively.
The K54A cNOR variant reaches the same maximum rate at low pH, but it has a shifted pKa and is ∼10 times slower than wild type at pH 7.5. Thus, the Lys-54C (Lys-53C in P. aeruginosa) is important to support rapid proton transfer at the growth pH used for denitrifying conditions (pH 7.5 (22)), where the full denitrification chain is active (23).
In the E58QC variant (Glu-57C in P. aeruginosa), even at low pH, the rate constant of the ETPT is still ∼5 times slower than in WT, and the pH dependence was fitted with a tentative pKa of ∼5.8. There is thus a severe slowing of the proton transfer rate constants at all pH values, making the effect much more drastic than in K54AC. The Glu-58C seems thus to be a very important part of the pathway, presumably forming its entry point (Fig. 5). We note that the Lys-54C is actually more conserved than the Glu-58C (100% compared with 77%; Table 1 and Ref. 18). Although we do not have an explanation for this at the moment, we do note that the sequences that do not have Glu-58C cluster together in the alignment and belong to different subgroups of the cNOR family than the subgroup of P. aeruginosa and P. denitrificans (18). The replacing residues are hydrophilic (Gln, His, or Asp), and there are charged residues in the flanking sequence (3 or less residues away) that might take over the role of the Glu-58C in these cNORs.
The identity of the internal proton donor (AH in Equations 1 and 2, pKa = 6.6 in wild type) remains unknown. There was a large pKa shift (>3 pH units upshifted) in the ETPT observed in variants with an exchanged Ca2+ ligand (Glu-122 in P. denitrificans cNOR, Glu-135 in P. aeruginosa, also in PW 1) (9). This shift was much larger than the pKa shifts observed with the variants studied in this work and indicates that the donor is located in the vicinity of the Glu-135. However, the coordination of the Ca2+ with multiple groups (one propionate each from the b and b3 hemes, Tyr-73C, Gly-71C, and an H2O) gives many possibilities. Because these residues all have structural roles as Ca2+ ligands, it is difficult to study any additional roles. We rule out the Arg-134 (Arg-121 in P. denitrificans) as AH, although it is very close and highly conserved (Fig. 1B and Table 1) because arginines have been shown to keep their high pKa values (∼12.5) even when embedded in protein interiors (24). The heme b propionate and the A propionate of heme b3 are possible candidates for AH, because one of them could, in principle, transiently dissociate from the Ca2+ and be protonated (Fig. 5). The Asp-198 (Asp-185 in P. denitrificans) is also a possible candidate, being ∼10 Å from the Glu-135, hydrogen-bonded to the Lys-53C, and giving such severe effects of mutation. However the pKa shift in the variant of the Lys-53C equivalent (K54AC) seems too modest (only 0.2 units) for a residue directly hydrogen-bonded to the proton donor. Although not in direct contact with the Glu-135 or the Ca2+, the D propionate of heme b3 (Fig. 5) seems like a better candidate for the proton donor for several reasons. First, precisely because it is not coordinated to Ca2+, it could adopt a higher pKa, possibly around 6.6, and it could still be affected by a change in ligation of the other propionate. Second, the D propionate is better positioned in the path to form a connection “onward” (Fig. 5), toward the reaction intermediate bound at (or between) the irons of heme b3 and FeB. MD simulations (15) postulated that protons could move 1) directly from the water cluster around the A and D propionates of heme b3 to a water cluster around the active site, 2) from the D propionate of heme b3 via the His-259 to the active site, or 3) from the D propionate of heme b3 via Thr-330, Ser-277, Glu-280, and Glu-211 to the active site (Fig. 5). It is tempting to suggest that the proton donor in cNOR in the path from the periplasm to the active site became (or originated from, depending on the rooting of the evolutionary tree; see, for example, Ref. 25) the site for extrusion of pumped protons in the O2-reducing HCuOs, the so-called proton loading site. The A propionate has recently been suggested by several research groups (e.g. see Refs. 26 and 27) to be this proton-loading site in O2-reducing HCuOs (A-, B-, and C-type, where it should be noted that the location of the A propionate of the active site heme corresponds to the location of the D propionate of the b3 heme in cNOR). Pathway 1 in cNOR is not conserved to the closest O2-reducing HCuO, the C-type, where instead parts of the PW 3 are conserved (15). However, the PW 3 residues conserved to C-type HCuOs (Glu-135 (also involved in PW 1), Glu-138, and Arg-57 in P. aeruginosa cNOR) all are involved in defining the Ca2+ site (Table 1). This could explain why they are conserved to C-type HCuOs, which have a Ca2+ bound in a similar manner (28).
Taken together, our data presented here strongly suggest that cNORs, at least the one from P. denitrificans, do use a specific pathway for proton transfer from the periplasm into the active site and that this is the suggested pathway 1 (Figs. 1 and 5) and that the other suggested pathways cannot take over the role of pathway 1. We note that a different study, where several residues were mutated in the cNOR from Thermus thermophilus (18), reached the conclusion that there is no preferred pathway in (this) cNOR. They did not investigate PW 3, but they observed effects on turnover rates when changing the residue at the entrance of PW 2, D209E/N/LC, which is an alanine in P. denitrificans (Ala-149C) and a glutamate in P. aeruginosa (Glu-145C). Their results also differed for residues along pathway 1. Mutating the equivalent of Asp-198 (P. aeruginosa numbering) of PW 1 in the T. thermophilus cNOR to either Glu or Asn had no effect on turnover (although, when changed into a leucine, the cNOR was not assembled). The Glu-57C is a Gln in T. thermophilus cNOR. Lys-53C is present in T. thermophilus cNOR, but its role was not investigated. The reason for the differences between the results of our study and the study with T. thermophilus cNOR (18) is not known. It is possible that PW 1 is used in the P. denitrificans cNOR (and P. aeruginosa cNOR) but not in T. thermophilus cNOR. The P. aeruginosa cNOR and the P. denitrificans cNORs are much more closely related to each other (51% identity between the NorB subunits, both belonging to the Proteobacteria phylum) than to the only distantly related T. thermophilus cNOR (belonging to the Thermus phylum and with 38% identity in NorB to both P. aeruginosa and P. denitrificans). There might be larger differences in the proton transfer pathways between subfamilies in cNOR compared with the O2-reducing HCuOs because no gating is necessary. It is also possible that effects on the proton transfer rates that we see in the P. denitrificans cNOR for D185N/D185E would not have been observed in the multiple-turnover experiments with NO in T. thermophilus cNOR, given that the overall turnover rate is orders of magnitude lower in the T. thermophilus enzyme (∼0.1 s−1 compared with ∼10 s−1 for the P. denitrificans cNOR used in this study; Table 2).
In principle, cNOR would not need to provide a specific pathway for protons, it could have protons “leak” into the active site from multiple routes. However, the use of a single, specific route can have different reasons. One is the evolutionary relationship to the other heme-copper oxidases. If cNOR evolved from an O2-reducing HCuO that had a specific path for the extrusion of the pumped protons, then this pathway could have been “reversed” in cNOR. Also, there is an energetic cost in terms of protein stability involved in creating a polar, water-filled pathway capable of proton transfer in an otherwise hydrophobic protein interior. This might minimize the number of proton transfer pathways to those that are absolutely necessary for function.
Acknowledgments
Dr. Nicholas Watmough (University of East Anglia) is acknowledged for sending the plasmids pNOREX and the pNORXX16 (used originally for mutagenesis, not described here). We thank Liesa Westner for making the first mutants (not described here) in the larger pNOREX plasmid. Anne Roehrig is acknowledged for making the first Asp-185 mutant in the pNORXX16 (not described here). We thank Lina Salomonsson for help with supervising N. K.
This work was supported by grants from the Faculty of Science at Stockholm University and the Swedish Research Council (to P. Ä.).
- cNOR
- cytochrome c-dependent NO reductase
- NOR
- NO reductase
- HCuO
- heme-copper oxidase
- DDM
- n-dodecyl-β-d-maltoside
- MD
- molecular dynamics
- PW
- pathway
- AH
- internal proton donor
- ETPT
- proton-coupled electron transfer reaction (not intended to indicate the order of the reactions) during the reduction of O2 by fully reduced cNOR.
REFERENCES
- 1. Shiro Y. (2012) Structure and function of bacterial nitric oxide reductases. Nitric oxide reductase, anaerobic enzymes. Biochim. Biophys. Acta 1817, 1907–1913 [DOI] [PubMed] [Google Scholar]
- 2. Lee H. J., Reimann J., Huang Y., Ädelroth P. (2012) Functional proton transfer pathways in the heme-copper oxidase superfamily. Biochim. Biophys. Acta 1817, 537–544 [DOI] [PubMed] [Google Scholar]
- 3. Bell L. C., Richardson D. J., Ferguson S. J. (1992) Identification of nitric oxide reductase activity in Rhodobacter capsulatus. The electron transport pathway can either use or bypass both cytochrome c2 and the cytochrome bc1 complex. J. Gen. Microbiol. 138, 437–443 [DOI] [PubMed] [Google Scholar]
- 4. Shapleigh J. P., Payne W. J. (1985) Nitric oxide-dependent proton translocation in various denitrifiers. J. Bacteriol. 163, 837–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reimann J., Flock U., Lepp H., Honigmann A., Ädelroth P. (2007) A pathway for protons in nitric oxide reductase from Paracoccus denitrificans. Biochim. Biophys. Acta 1767, 362–373 [DOI] [PubMed] [Google Scholar]
- 6. Fujiwara T., Fukumori Y. (1996) Cytochrome cb-type nitric oxide reductase with cytochrome c oxidase activity from Paracoccus denitrificans ATCC 35512. J. Bacteriol. 178, 1866–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butland G., Spiro S., Watmough N. J., Richardson D. J. (2001) Two conserved glutamates in the bacterial nitric oxide reductase are essential for activity but not assembly of the enzyme. J. Bacteriol. 183, 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flock U., Watmough N. J., Ädelroth P. (2005) Electron/proton coupling in bacterial nitric oxide reductase during reduction of oxygen. Biochemistry 44, 10711–10719 [DOI] [PubMed] [Google Scholar]
- 9. Flock U., Thorndycroft F. H., Matorin A. D., Richardson D. J., Watmough N. J., Ädelroth P. (2008) Defining the proton entry point in the bacterial respiratory nitric-oxide reductase. J. Biol. Chem. 283, 3839–3845 [DOI] [PubMed] [Google Scholar]
- 10. Flock U., Lachmann P., Reimann J., Watmough N. J., Ädelroth P. (2009) Exploring the terminal region of the proton pathway in the bacterial nitric oxide reductase. J. Inorg. Biochem. 103, 845–850 [DOI] [PubMed] [Google Scholar]
- 11. Hendriks J. H., Jasaitis A., Saraste M., Verkhovsky M. I. (2002) Proton and electron pathways in the bacterial nitric oxide reductase. Biochemistry 41, 2331–2340 [DOI] [PubMed] [Google Scholar]
- 12. Lachmann P., Huang Y., Reimann J., Flock U., Ädelroth P. (2010) Substrate control of internal electron transfer in bacterial nitric-oxide reductase. J. Biol. Chem. 285, 25531–25537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hino T., Matsumoto Y., Nagano S., Sugimoto H., Fukumori Y., Murata T., Iwata S., Shiro Y. (2010) Structural basis of biological N2O generation by bacterial nitric oxide reductase. Science 330, 1666–1670 [DOI] [PubMed] [Google Scholar]
- 14. Shiro Y., Sugimoto H., Tosha T., Nagano S., Hino T. (2012) Structural basis for nitrous oxide generation by bacterial nitric oxide reductases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1195–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pisliakov A. V., Hino T., Shiro Y., Sugita Y. (2012) Molecular dynamics simulations reveal proton transfer pathways in cytochrome c-dependent nitric oxide reductase. PLoS Comput. Biol. 8, e1002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thorndycroft F. H., Butland G., Richardson D. J., Watmough N. J. (2007) A new assay for nitric oxide reductase reveals two conserved glutamate residues form the entrance to a proton-conducting channel in the bacterial enzyme. Biochem. J. 401, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brändén M., Sigurdson H., Namslauer A., Gennis R. B., Ädelroth P., Brzezinski P. (2001) On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 98, 5013–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schurig-Briccio L. A., Venkatakrishnan P., Hemp J., Bricio C., Berenguer J., Gennis R. B. (2013) Characterization of the nitric oxide reductase from Thermus thermophilus. Proc. Natl. Acad. Sci. U.S.A. 110, 12613–12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krishtalik L. I. (2000) The mechanism of the proton transfer. An outline. Biochim. Biophys. Acta 1458, 6–27 [DOI] [PubMed] [Google Scholar]
- 20. Krezel A., Bal W. (2004) A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 98, 161–166 [DOI] [PubMed] [Google Scholar]
- 21. Karpefors M., Ädelroth P., Brzezinski P. (2000) Localized control of proton transfer through the D-pathway in cytochrome c oxidase. Application of the proton-inventory technique. Biochemistry 39, 6850–6856 [DOI] [PubMed] [Google Scholar]
- 22. Harms N., de Vries G. E., Maurer K., Veltkamp E., Stouthamer A. H. (1985) Isolation and characterization of Paracoccus denitrificans mutants with defects in the metabolism of one-carbon compounds. J. Bacteriol. 164, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bergaust L., Mao Y., Bakken L. R., Frostegård A. (2010) Denitrification response patterns during the transition to anoxic respiration and posttranscriptional effects of suboptimal pH on nitrous [corrected] oxide reductase in Paracoccus denitrificans. Appl. Environ. Microbiol. 76, 6387–6396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harms M. J., Schlessman J. L., Sue G. R., García-Moreno B. (2011) Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. U.S.A. 108, 18954–18959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gribaldo S., Talla E., Brochier-Armanet C. (2009) Evolution of the haem copper oxidases superfamily. A rooting tale. Trends Biochem. Sci. 34, 375–381 [DOI] [PubMed] [Google Scholar]
- 26. Kaila V. R., Sharma V., Wikström M. (2011) The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochim. Biophys. Acta 1807, 80–84 [DOI] [PubMed] [Google Scholar]
- 27. Chang H. Y., Choi S. K., Vakkasoglu A. S., Chen Y., Hemp J., Fee J. A., Gennis R. B. (2012) Exploring the proton pump and exit pathway for pumped protons in cytochrome ba3 from Thermus thermophilus. Proc. Natl. Acad. Sci. U.S.A. 109, 5259–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buschmann S., Warkentin E., Xie H., Langer J. D., Ermler U., Michel H. (2010) The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 329, 327–330 [DOI] [PubMed] [Google Scholar]