

Abstract
Crohn's disease (CD) is a chronic, immune-mediated, inflammatory disorder of the intestine that has been linked to numerous susceptibility genes, including the immunity-related GTPase (IRG) M (IRGM). IRGs comprise a family of proteins known to confer resistance to intracellular infections through various mechanisms, including regulation of phagosome processing, cell motility, and autophagy. However, despite its association with CD, the role of IRGM and other IRGs in regulating intestinal inflammation is unclear. We investigated the involvement of Irgm1, an ortholog of IRGM, in the genesis of murine intestinal inflammation. After dextran sodium sulfate exposure, Irgm1-deficient [Irgm1 knockout (KO)] mice showed increased acute inflammation in the colon and ileum, with worsened clinical responses. Marked alterations of Paneth cell location and granule morphology were present in Irgm1 KO mice, even without dextran sodium sulfate exposure, and were associated with impaired mitophagy and autophagy in Irgm1 KO intestinal cells (including Paneth cells). This was manifested by frequent tubular and swollen mitochondria and increased LC3-positive autophagic structures. Interestingly, these LC3-positive structures often contained Paneth cell granules. These results suggest that Irgm1 modulates acute inflammatory responses in the mouse intestine, putatively through the regulation of gut autophagic processes, that may be pivotal for proper Paneth cell functioning.
Keywords: experimental colitis, immunity-related GTPases, autophagy
crohn's disease (CD) consists of a heterogeneous disease complex characterized by chronic, relapsing inflammation that can occur throughout the gastrointestinal tract (1, 27). Considerable evidence suggests that this disease results from dysregulated immune responses to commensal enteric bacteria in genetically susceptible hosts. Genome-wide association studies have identified multiple CD risk alleles that encode components of the innate immune system. These components include cytokines (IL-23) (28), bacterial recognition receptors (NOD2) (28), and recently identified autophagy-associated genes such as ATG16L1 (21, 58a) and immunity-related GTPase (IRG) M (IRGM) (38, 58a). While the function of many of these molecules in regulating intestinal inflammation has been explored with mouse models (9, 10, 41), the role of IRGM in this context remains uncertain.
The IRGs comprise a family of vertebrate proteins that coordinate immunity to intracellular pathogens (33, 46, 49). Much knowledge of IRG proteins comes from studies of the mouse proteins, which are divided into Irga, Irgb, Irgc, Irgd, and Irgm subfamilies (4). These 47- to 48-kDa proteins are expressed in a variety of immune and nonimmune cells, and their absence results in enhanced susceptibility to multiple bacterial and protozoan infections (14, 49). Although it has not been formally demonstrated, the biochemical function of IRG proteins is thought to be similar to that of classical dynamins in regulating membrane fusion, vesiculation, and/or trafficking events. This putative role underscores several immune functions that have been attributed to mouse IRG proteins, such as regulating the processing of pathogen-containing phagosomes (29, 31, 34, 52), inflammatory cytokine production (3), cell motility (22), and T cell homeostasis (16, 17). For example, current models suggest that mouse Irg proteins act coordinately to drive breakdown of the Toxoplasma gondii vacuole, leading to eradication of this pathogen (29, 34).
Additional data suggest that Irgm1 performs other functions that are important for bacterial resistance, most notably the regulation of autophagy (20). This is also true for the human ortholog of Irgm1, IRGM (47), which has been found to regulate autophagy, as well as a specific autophagic process known as mitophagy (48). Several genome-wide association studies have identified the IRGM gene as a CD susceptibility allele (38, 58a); however, the direct impact of IRGM or other IRGs on regulation of intestinal inflammation in vivo has not been explored. In the present study, we used mice lacking Irgm1 [Irgm1 knockout (KO)] to examine the role of IRG proteins in suppressing experimental colitis. We found that Irgm1 is indeed a key regulator of small intestinal and colonic inflammatory responses. Moreover, intestinal autophagy and Paneth cell function were also disrupted in Irgm1 KO mice, suggesting a role for Irgm1 in modulating these processes within the context of acute intestinal inflammation.
MATERIALS AND METHODS
Mice.
Irgm1 KO mice used in these experiments have been described previously (14, 49) and were backcrossed to C57BL/6NCr1 mice for nine generations. All protocols were approved by the Institutional Animal Care and Use Committee of the Durham Veterans Affairs and Duke University Medical Centers. The mice were maintained in the Durham Veterans Affairs Medical Center Animal Facility under conventional housing conditions.
Dextran sodium sulfate-induced colitis.
Acute colitis was established according to standard protocols (36, 60) by addition of 3% (wt/vol) dextran sodium sulfate (DSS; ICN Biomedicals, Aurora, OH) to the drinking water of mice for 7 days. Control mice received water without DSS. Each day, the mice were weighed, and fecal blood and stool consistency were assessed. Fecal blood was assayed with a Hemoccult test (catalog no. 60151, Beckman Coulter, Fullerton, CA) according to the following scale: 0, no color; 1, faintly blue; 2, light blue; 2.5, medium blue; 3, dark blue; and 4, gross bleeding. Stool consistency was quantified as follows: 0, normal; 1, pasty; 2, soft, but formed; 3, soft, no form; and 4, diarrhea. At necropsy, the colons of the mice were dissected and lengths were measured. Tissue was also isolated from the ileum and the distal, proximal, and transverse colon for histological analysis following formalin fixation, paraffin embedding, and hematoxylin-and-eosin staining. Blinded histological scores were assigned using validated scales (36, 60). In the colon, the scores were as follows: 0, no inflammation; 1, low inflammation with scattered infiltrating cells (1–2 foci); 2, moderate inflammation with multiple foci (with epithelial hyperplasia and mild loss of goblet cells); 3, high inflammation with increased vascular density and marked wall thickening (with obvious epithelial hyperplasia and goblet cell depletion); and 4, maximal inflammation (with transmural leukocyte infiltration and loss of goblet cells). In the ileum, the scores were as follows: 0, no inflammation and normal villus architecture; 1, mild focal cellular infiltration and normal villus architecture; 2, mild lamina propria cellular infiltration and early crypt epithelial hyperplasia with normal villus architecture; 3, more pronounced cellular filtration, thickened mucosa, marked epithelial hyperplasia, and moderate distortion of villus architecture; and 4, extensive cellular infiltration throughout the section and severe architectural distortion.
Immunohistochemistry and immunofluorescence.
Paraffin-embedded sections were deparaffinized by two 10-min incubations in xylene (Fisher Scientific, Pittsburgh, PA), rehydrated by passage through graded ethanol concentrations, and washed in PBS. The sections were processed by boiling in antigen retrieval solution (0.01 M citrate buffer solution, pH 6.0) in a microwave oven for 10 min. Endogenous peroxidase activity was inhibited by incubation of sections in 3% (vol/vol) hydrogen peroxide for 10 min. Sections were then blocked with 1% BSA-0.3% Triton X-100-PBS (blocking solution) overnight at 4°C and incubated for 1 h at room temperature in a 1:800 dilution of rabbit anti-Muc2 (Santa Cruz Biotechnology), a 1:100 dilution of goat anti-lysozyme (Lyz; Santa Cruz Biotechnology), or a 1:300 dilution of anti-LC3 [catalog no. L7543 (Sigma) or M152-3B (MBL), used interchangeably] antibody in blocking solution. Subsequently, the sections were washed and incubated for 1 h at room temperature with a 1:300 dilution of donkey fluorescein-conjugated secondary antibody raised against rabbit, mouse, or goat, as appropriate. The sections were mounted using Prolong Gold Anti-Fade reagent with 4′,6-diaminido-2-phenylindole (Invitrogen).
For LC3 immunohistochemical studies, sections were processed as described above and blocked overnight with blocking solution. Anti-LC3 antibody was applied at a 1:300 dilution in the blocking solution, and LC3 was detected using the Vectastain Elite ABC kit (Vector Laboratories) and a 10-s incubation in the diaminobenzidine tetrahydrochloride solution. All sections were counterstained with hematoxylin. In experiments involving quantification of LC3-positive puncta in Paneth cells, ≥15 crypts per section were counted.
Mouse embryonic fibroblasts.
Wild-type (WT) and Irgm1 KO mouse embryonic fibroblasts were derived from day 14–17 mouse embryos, cultured in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% FBS (Hyclone, Logan, UT), and immortalized by the standard 3T3 procedure (53). They were exposed to 100 U/ml IFN-γ (catalog no. 407320, Calbiochem, EMD Biosciences, San Diego, CA) in the culture medium for 24 h. Where appropriate, cells were pulsed with Mito Tracker Red (Molecular Probes/Invitrogen) for 30 min. Transfections of pEGFP-Mito (Clontech, Mountain View, CA) were carried out using FuGENE (Roche). Cells were fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.2% saponin in PBS for 10 min, and blocked with 10% FBS in permeabilization buffer. Some cells were stained with anti-Irgm1 monoclonal antiserum (8) followed by Alexa Fluor-conjugated secondary antibodies (Molecular Probes) in blocking buffer. Images were collected on an Olympus IX70 microscope equipped with a Hamamatsu digital camera (model C8484-03G01) using MetaMorph version 6.2.3.5 software. Cells were magnified at ×1,000.
Paneth cell enumeration.
Sections were stained with anti-Lyz, as described above. They were examined in a blinded fashion, with properly oriented, longitudinal crypt-villus units identified and divided into four regions (crypt base, midcrypt, upper crypt, and villus). The number of Lyz-positive cells in each location was recorded for ≥10 crypt/villus units per section. Any cell with Paneth cell-like granules outside the crypt-base position were considered ectopic cells.
Electron microscopy.
Tissue was fixed in 2% paraformaldehyde-2.5% glutaraldehyde in 0.15 M sodium phosphate, pH 7.4. The University of North Carolina-Chapel Hill Microscopy Services Laboratory processed tissues for transmission electron microscopy (TEM) according to standard techniques. Ultrathin (70-nm) sections were cut with a diamond knife and collected on 200-mesh copper grids. TEM grids were observed and photographed using a transmission electron microscope (Zeiss EM-910, LEO Electron Microscopy, Thornwood, NY) and photographed using a digital camera (Bioscan, Gatan, Pleasanton, CA).
Quantitative RT-PCR.
Total RNA was extracted from ileal tissue using an RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was generated using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). Quantitative RT-PCR was performed for each sample in duplicate using TaqMan Gene Expression Master Mix (Applied Biosystems, Pleasanton, CA). The following primer/probe sets were obtained from Applied Biosystems: Defa3 (Mm04205962_gH), Defa5 (Mm00651548_g1), Defa20 (Mm00842045_g1), Lyz (Mm00727183_s1), Reg3γ (Mm00441127_m1), Ang4 (Mm03647554_g1), Vil1 (Mm00494156_m1), Scd1 (Mm00772290_m1), Adipoq (Mm00456425_m1), Saa1 (Mm00656927_g1), Lep (Mm00434759_m1), Actb (Mm00607939_s1), and Gapdh (Mm99999915_g1). Relative gene expression was determined using the comparative threshold cycle (CT) method (44). Briefly, fold change was calculated as 2−ΔΔCT, where ΔΔCT = [(CT gene of interest − CT β-actin)Irgm1 KO − mean(CT gene of interest − CT β-actin)Control WT]. β-Actin was used as an internal control. The control WT (non-DSS-treated) group was set at a baseline of “1” by dividing all final values by the average of this group. TNF-α mRNA expression was assessed using the following primers: 5′-ACCCTCACACTCAGATCATCTTCTC-3′ (forward) and 5′-TGAGATCCATGCCGTTGG-3′ (reverse). These were amplified using SYBR Green PCR Master Mix (Applied Biosystems). For these assays, Gapdh was used as the internal control, and relative gene expression was determined as described above.
Statistical analyses.
Statistical comparisons were completed using Excel (Microsoft) or Prism 5 software (GraphPad, San Diego, CA). Values are means ± SE. Means were compared using a two-tailed Student's t-test.
RESULTS
Irgm1 deficiency leads to exaggerated intestinal inflammation.
Irgm1 KO mice did not consistently exhibit spontaneous intestinal inflammation. To assess the role of Irgm1 in the development of acutely induced intestinal inflammation, we administered low-dose (3%) DSS in the drinking water of Irgm1 KO and WT mice for 7 days. Clinically, male Irgm1 KO mice exhibited greater degrees of weight loss, altered stool consistency, and rectal bleeding than did WT mice (Fig. 1, A–C). Furthermore, at necropsy, the Irgm1 KO mice demonstrated increased colonic inflammation and epithelial injury, including shortening of the colon and elevated histological scores (Fig. 1, D–F). Female Irgm1 KO mice exhibited the same trends as male Irgm1 KO mice, although exaggerated weight loss was the only statistically significant finding (data not shown). This disparity was due to the highly variable responses of female WT mice to DSS and is consistent with a male gender bias to DSS susceptibility that has previously been reported (32).
Fig. 1.

Enhanced colonic inflammation in immunity-related GTPase (Irg) M (Irgm)-deficient (Irgm1 KO) mice receiving dextran sodium sulfate (DSS). Male wild-type (WT) and Irgm1 KO mice were given 3% DSS in drinking water (DSS) or maintained on standard drinking water (control) for 7 days. A–C: clinical indicators of inflammation [average body weight, average stool consistency score, and average bleeding (Hemoccult) index] at days 0–7. D: average colon length at necropsy. E: representative histological tissue sections from transverse colon (hematoxylin-eosin stain; ×100 magnification). F: average histological scores from proximal (Prox), transverse (Trans), and distal (Dist) colon and cecum. Values are means ± SE; data were combined from 3 separate studies. Combined cohort sizes were as follows: n = 9 WT control and WT DSS and 7 KO control and KO DSS. *P < 0.05, **P < 0.005 (KO DSS vs. WT DSS).
Contrary to expectation, DSS-treated Irgm1 KO mice also displayed significant ileal injury not classically reported in DSS-treated animals (Fig. 2). This was manifested as increased infiltration by lamina propria inflammatory cells, focal epithelial denudation, mild goblet cell depletion, and mild crypt epithelial cell hyperplasia (Fig. 2, A and B). The inflammation was underscored by a sharp increase in ileal mRNA expression of TNF-α (Fig. 2C), a proinflammatory cytokine that is pivotal in establishing intestinal inflammation (35). These small intestinal findings are particularly relevant, given that the majority of patients with CD have some degree of ileal involvement (30); moreover, an association of IRGM risk polymorphisms with ileal location of CD has been reported (40). Hence, Irgm1 appears to play a prominent role in regulating ileal and colonic inflammation in response to acute epithelial injury.
Fig. 2.

Enhanced ileal inflammation in Irgm1 KO mice receiving DSS. Male WT and Irgm1 KO mice were given 3% DSS in drinking water (DSS) or maintained on standard drinking water (control) for 7 days. A: representative histological tissue sections from ileum of WT control, WT DSS, KO control, and KO DSS mice (hematoxylin-eosin stain, ×100 magnification). B: average histological scores from ileum. C: results of quantitative RT-PCR measurement of ileal tissue TNF-α transcript levels in DSS-treated and control mice, normalized to Gapdh and expressed as fold change relative to WT control group. Values are means ± SE; data were combined from ≥3 experiments. For A and B, combined cohort sizes were as follows: n = 9 WT control and WT DSS and 7 KO control and KO DSS; for C, combined cohort sizes were n = 13 WT control, 11 KO control, 10 WT DSS, and 9 KO DSS. *P < 0.02, **P < 0.006.
Irgm1 deficiency leads to changes in Paneth cell morphology and function.
Because of the ileal phenotype observed in DSS-treated Irgm1 KO mice, we speculated that the function of Paneth cells, a small intestine epithelial cell type that is thought to be critical for controlling the microbiota through secretion of antimicrobial peptides (AMPs), may have been altered in these animals (39). By histological examination, the overall architecture of ileal tissues obtained from untreated Irgm1 KO mice was normal, with no change in the frequency or height of the villi; however, numerous alterations of the Paneth cell compartment were apparent (Fig. 3). First, the morphology of Paneth cell granules in Irgm1 KO vs. WT mice was strikingly different, with the granules of Irgm1 KO mice being finer, irregular in size, and less dense in appearance (Fig. 3A). This was particularly evident by TEM, which showed that the granules in the Irgm1 KO mice were smaller, with a halo appearance, potentially representing partial degranulation of their electron-dense contents (Fig. 3B). Second, increased numbers of granulated Lyz-positive cells were observed in Irgm1 KO mice, as determined by enumeration of anti-Lyz-positive staining cells. This was observed with and without DSS treatment (Fig. 3, C and D). Interestingly, the Irgm1 KO mice also had high numbers of ectopically positioned Lyz-positive cells that were outside their normal location in the base of the crypts. These ectopic, granulated, Lyz-positive cells were present in untreated Irgm1 KO mice, as well as DSS-treated animals (Fig. 3, C and D). Importantly, untreated Irgm1 KO mice did not have significant histological inflammation, suggesting that altered Paneth cell function may be a primary susceptibility factor for intestinal injury in this model, rather than an acquired response to the inflammatory process.
Fig. 3.

Paneth cell abnormalities in Irgm1 KO mice. A and B: histological staining with hematoxylin and eosin (A; ×400 magnification) and transmission electron microscopy (B; ×5,000 magnification) of ileal tissues from untreated WT and Irgm1 KO mice. C: lysozyme (Lyz) immunohistochemical staining of ileal tissue from control and DSS-treated WT and KO mice (×200 magnification). D: total number of lysozyme (Lyz)-positive cells (left) and Lyz-positive cells above the base of the crypt (ectopic, right). Values are means ± SE; data were collected from 3 individual mice for each group, with ≥10 crypt-villus units per mouse. *P < 0.05.
To further characterize the ectopically positioned Lyz-positive cells in Irgm1 KO mice, additional histological and TEM studies were performed. By light microscopy, the ectopic granulated cells were globular in shape and appeared to contain mucin, as well as Paneth cell granules (Fig. 4A). This was confirmed by immunofluorescent costaining with anti-Muc2 and anti-Lyz antibodies (Fig. 4B). Further examination by TEM also demonstrated subcellular mucin deposition and dense Paneth cell-like components within these ectopic cells (Fig. 4C). These findings are consistent with an intermediate cell phenotype. Intermediate cells have been described to occur rarely in normal mice, and it is unclear whether they represent a common Paneth/goblet cell progenitor or an independent cell type with distinct biological functions (24).
Fig. 4.

Overproduction and ectopic placement of ileal intermediate cells in Irgm1 KO mice. Ileal tissue from Irgm1 KO mice was processed for hematoxylin-and-eosin staining (A; ×600 magnification), immunofluorescence staining with anti-Muc2 (green) and anti-Lyz (red) antibodies (B; ×600 magnification), and transmission electron microscopy [C; ×2,500 (left) and ×7,500 (right) magnification]. All modalities show numerous intermediate cells with properties of both goblet and Paneth cells (white arrows).
To assess Paneth cell AMP production in Irgm1 KO mice, transcript levels of representative mouse AMPs were measured. Major subtypes of Paneth cell AMPs in C57BL/6 mice include Lyz, Reg3γ, angiogenin 4, and the α-defensins (5, 19). A selective reduction of Lyz expression was observed in Irgm1 KO mice, independent of DSS treatment, while Reg3γ and Ang4 levels were not significantly different across the experimental groups (Fig. 5A). Because the total number of Lyz-staining cells was increased in Irgm1 KO mice (Fig. 3, C and D), the decrease in Lyz expression in Irgm1 KO tissue was even lower on a per cell basis. Similar to Lyz expression, quantitative RT-PCR for selected α-defensin isoforms (Fig. 5B) showed reduced expression of Defa20 in Irgm1 KO mice relative to WT animals with and without DSS treatment. Conversely, mRNA levels of Defa3 and Defa5 were similar among all groups, indicating a selective reduction of specific α-defensin isoforms in Irgm1 KO mice. This is likely biologically relevant, as murine α-defensins (cryptdins) account for the majority of antimicrobial activity in Paneth cell secretions (2). Expression of villin, a marker of epithelial cell mass, was equivalent among experimental groups, indicating that the differences in AMP expression were not due to differential epithelial cell loss (Fig. 5C). Thus, in summary, Irgm1 KO mice displayed marked Paneth cell alterations, including cellular hyperplasia, ectopic positioning of the cells along the villi, abnormal development of secretory granules, and decreased expression of selected AMPs.
Fig. 5.

Altered antimicrobial peptide production in Irgm1 KO mice. Male WT and Irgm1 KO mice were given 3% DSS in drinking water (DSS) or maintained on standard drinking water (cont) for 7 days. Quantitative RT-PCR was used to measure transcript levels of selected molecules from several mouse antimicrobial peptide classes, including Lyz, Reg3γ, and Ang4 (A), representative α-defensin isoforms, including Defa3, Defa5, and Defa20 (B), and villin (Vil1, C), as an indicator of total epithelial cell mass. Values (means ± SE) are normalized to β-actin and expressed as fold change relative to the WT control group. Cohort sizes were as follows: n = 13 WT control, 11 KO control, 10 WT DSS, and 9 KO DSS. *P ≤ 0.05.
Autophagy and mitophagy are altered in the absence of Irgm1.
We next addressed alterations in cellular processes that might lead to the Paneth cell phenotype in the Irgm1 KO mice. As mentioned above, Irgm1 has been assigned several diverse functions related to innate immunity. However, among these, we speculated that the modulation of autophagy may be important in Paneth cells for at least two reasons: 1) the primary function assigned to the CD-associated gene and Irgm1 ortholog IRGM has been the regulation of autophagy (47) and mitophagy (48), and 2) mice hypomorphic for Atg16L1 (an essential regulator of autophagy) also demonstrate increased ileal inflammation in response to DSS, as well as similar abnormalities in Paneth cell function (9, 10). To examine these processes more rigorously, we performed immunostaining for LC3, an autophagic marker, in WT and Irgm1 KO mouse intestine. In WT and KO mice, we found substantial numbers of Paneth cells that were positive for LC3 (Fig. 6, A–D). However, while the proportion of LC3-positive Paneth cells was not significantly different between the two groups, we found more LC3-positive granules on a per cell basis in Irgm1 KO Paneth cells. Findings were similar in mice exposed to DSS (Fig. 6, E and F). These data suggest that formation or processing of Paneth cell granules may involve an autophagic pathway and, furthermore, that this process is altered in the absence of Irgm1.
Fig. 6.
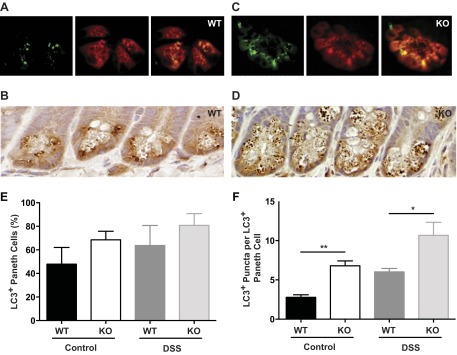
Altered autophagy in Irgm1 KO intestinal cells. Ileal tissue from untreated WT and Irgm1 KO mice was processed for immunofluorescence staining with anti-LC3 (green) and anti-Lyz (red) antibodies (A and C) or immunohistochemical staining with anti-LC3 (brown) followed by hematoxylin counterstain (purple; B and D). Images are representative of 8 mice examined per genotype. Magnification ×400. Quantitative analyses from control and DSS-treated WT and Irgm1 KO mice include average percentage of Paneth cells that were LC3-positive (E; 5 mice analyzed per genotype, with 150 cells examined per mouse) and total LC3-positive punctae per LC3-positive Paneth cell (F; 4 mice analyzed per genotype, with 40 LC3-positive cells examined per mouse, counted on ≥15 crypts per section). Values are means ± SE. *P < 0.05, **P < 0.001.
In addition to alterations in autophagy, abnormalities were also found in the mitochondria of Irgm1 KO Paneth and other intestinal cells. In Irgm1 KO intestinal tissue, we found numerous swollen mitochondria that likely represented defective organelles that had not been removed from the cell (Fig. 7B). In addition, elongated/tubular mitochondria were commonly observed in Irgm1 KO intestinal cells (Fig. 7C), but not in WT tissue. Mitochondria normally cycle between punctate and tubular forms, with this cycle being highly regulated by a network of mitochondrial fission and fusion proteins, as well as being linked to the process of mitophagy (for reviews see Refs. 12 and 57). IRGM has previously been shown to regulate mitochondrial fission, thereby influencing mitophagy (48). We explored a similar function for Irgm1 in cultured fibroblasts. In WT fibroblasts, Irgm1 localized strongly to the surface of mitochondria in punctate concentrations (Fig. 7D), as reported previously by others (13). Similar to Irgm1 KO enterocytes, we found a much higher percentage of fused/tubular mitochondrial morphologies in Irgm1 KO fibroblasts (Fig. 7, E and F). Collectively, these data suggest that Irgm1 may mediate mitochondrial fission and that this process is required for efficient removal of damaged mitochondria.
Fig. 7.

Altered mitophagy in Irgm1 KO cells. A–C: representative transmission electron micrographs from WT mouse ileal enterocytes (A), Irgm1 KO ileal enterocytes (B), and Irgm1 KO goblet cells (C). Magnification ×20,000. Note swollen mitochondria (arrowheads) and tubular mitochondria (outlined with dashed line) in Irgm1 KO cells in B and C, respectively. These phenotypes were seen in multiple Irgm1 KO enterocytes and goblet and Paneth cells and were not cell-specific. D: representative WT mouse embryonic fibroblasts transfected with pMito to label mitochondria, exposed to IFN-γ for 24 h, and then processed for staining with anti-Irgm1 antibodies. Magnification ×1,000. E: representative WT and Irgm1 KO mouse embryonic fibroblasts exposed to IFN-γ for 24 h, stained with Mito Tracker Red to label mitochondria, and then imaged. Magnification ×1,000. F: quantified punctate, tubular, or mixed mitochondrial morphologies assessed blindly from >50 cells per treatment group per experiment, with the averages of 4 experiments shown. Values are means ± SE. *P < 0.05.
In a final set of studies, because of the similar phenotypes of Irgm1 KO and Atg16L1HM mice, we sought to determine if additional cellular processes were regulated by both of these molecules. Specifically, previous work demonstrated increased expression of transcripts involved in peroxisome proliferator-activated receptor pathways, adipocytokine signaling, lipid metabolism, and the acute-phase reactant response in Atg16L1-deficient Paneth cells (9). To determine if Irgm1 regulates similar processes, we assessed ileal expression of four transcripts that were enriched in Atg16L1-deficient Paneth cells and are relevant to these pathways. As shown in Fig. 8, there were no significant differences in mRNA expression of these molecules between Irgm1 KO and WT mice, although there were trends toward decreased expression in the Irgm1 KO samples, particularly in adiponectin and leptin (Fig. 8, B and D).
Fig. 8.

Transcript expression of selected genes altered in Atg16L1HM mice. Atg16L1-deficient Paneth cells have been shown to exhibit increased expression of genes involved in peroxisome proliferator-activated receptor (PPAR) pathways, adipocytokine signaling, lipid metabolism, and acute-phase reactant responses. Ileal mRNA expression of specific transcripts enriched in Atg16L1-deficient Paneth cells, relevant to these pathways, was evaluated in untreated WT and Irgm1 KO mice by quantitative RT-PCR. A: stearoyl-coenzyme A desaturase 1 (Scd1), involved in lipid metabolism. B: adiponectin (Adipoq), involved in adipocytokine signaling. C: serum amyloid A1 (Saa1), involved in acute-phase reactant response. D: leptin (Lep), involved in adipocytokine signaling and the PPAR pathway. Values are means ± SE; n = 9 WT and KO. No significant differences were observed for any of the transcripts tested (P > 0.2).
DISCUSSION
Although several studies have linked IRGM polymorphisms to CD (38, 58a), the precise mechanisms by which IRGM dysfunction leads to chronic intestinal inflammation remain unclear. Understanding such mechanisms will be imperative for development of focused, patient-specific therapies for this debilitating disease (56). To accomplish this, it is essential to establish mouse models in which these mechanisms can be probed. This is particularly important, given the inherent limitations to investigating human cells and tissue systems. In this study we use a gene deletion mouse model to demonstrate that Irgm1, a mouse ortholog of IRGM, is a key regulator of inflammation in the small intestine and colon. Furthermore, we offer preliminary insight into the mechanisms by which altered Irgm1 function drives intestinal inflammatory processes.
The key abnormality we identified in the ilea of Irgm1 KO mice was substantially altered Paneth cell morphology and function, most notably a marked alteration in the appearance of their secretory granules. Paneth cells are intestinal epithelial cells that defend against enteric pathogens (7, 59) and modulate the intestinal microbiota by forming and releasing secretory granules that are rich in AMPs (42). The marked decrease in size of the secretory granules in Irgm1 KO cells, along with the increased incidence of secretory granules in an LC3-positive autophagic compartment in those cells, suggests that Irgm1 may be an important regulator of autophagic processes that are required for proper intracellular processing of secretory granules (see further discussion of Irgm1 and autophagy below). An additional finding in the Paneth cell compartment of Irgm1 KO mice was a deficiency in the expression of selective AMPs, despite overall increased numbers of Lyz-positive cells. Although transcriptional regulation of Paneth cell α-defensins is poorly understood (37), it is possible that this effect contributes to the increased inflammation in the mice (6). Nevertheless, it is not clear whether these transcriptional changes are a primary result of Irgm1 deficiency or a compensatory effect, perhaps secondary to the impaired secretory granule processing and, ultimately, decreased microbial control in the intestinal lumen and mucosa. We point out that autophagic processes are known to have direct impact on signaling pathways that affect gene expression [e.g., the effects on cytokine production (41)]; thus, impaired autophagy due to Irgm1 deficiency could have primary impact on Paneth cell function at multiple levels. Finally, many of the Lyz-positive cells in Irgm1 KO mice were ectopically positioned and had characteristics of both Paneth and goblet secretory cells, the hallmarks of intermediate cells. Although their significance is not fully understood, increased numbers of intermediate cells have been observed in response to intestinal helminth infection (23), as well as chronic ileitis in the SAMP1/YitFc mouse strain (55). Nevertheless, we found no evidence of parasitic infection or spontaneous inflammation in our untreated Irgm1 KO mice.
These deficiencies in the Paneth cell compartment are clinically significant, in that numerous lines of evidence implicate Paneth cell dysfunction in the pathogenesis of CD. First, CD occurs most commonly in the distal ileum, where Paneth cell abundance is greatest (26). Second, several CD susceptibility loci contain genes such as NOD2, ATG16L1, and XBP1 that regulate Paneth cell function and AMP expression (9, 18, 25, 58). Therefore, it is possible that the Paneth cell abnormalities documented in Irgm1 KO mice mediate the increased susceptibility to small intestinal and colonic inflammation in these animals, particularly since Paneth cell abnormalities are present in Irgm1 KO mice prior to the onset of intestinal inflammation induced by DSS. Finally, CD is thought to result from dysregulated immune responses to the intestinal microbiota (43, 56). By modulating the gut commensal bacteria, Paneth cells are in a key position to mediate pathogenic and homeostatic inflammatory responses. While the present study does not examine the downstream effects of decreased AMP production in Irgm1 KO mice on the composition of the gut microbiota, such work will be the focus of future investigations.
In addition to altered Paneth cell function and the presence of intermediate cells, we also provide evidence that autophagy and mitophagy are altered in Irgm1 KO mice. While the association between dysfunctional mitochondrial processing and intestinal inflammation is emerging, defects in autophagy have been linked to CD, primarily through genome-wide association studies that have most notably identified the autophagy gene ATG16L1 as a CD risk allele (21, 58a). Our data have several parallels to data published for mice hypomorphic for Atg16L1, which also exhibit increased DSS-induced ileal inflammation, abnormalities in Paneth cell granule appearance and distribution, and altered mitochondria (9, 41). However, these parallels are not absolute: the increased frequency of intermediate cells noted in the Irgm1 KO mice was not reported in the Atg16L1HM mice, and the impaired resistance to Listeria monocytogenes previously found in Irgm1 KO mice (14) was not found in Atg16L1HM mice (9). In addition, somewhat different spectra of Paneth cell gene transcripts were altered in the two mouse models (9, 10), although this might be partially explained by methodological differences: laser capture analysis of Paneth cells in the Atg16L1HM mice in the previous studies vs. quantitative PCR analysis of total ileal tissue in Irgm1 KO mice in the studies reported here. Nevertheless, our data suggest at least a partial overlap in the autophagic pathway that is affected by the two proteins. Subsequent studies in Atg16L1HM mice have indicated that the inflammatory and Paneth cell phenotypes require reduced Atg16L1 function and a microbial component, specifically a murine norovirus strain (10). We have not determined whether the intestinal inflammatory syndrome in Irgm1 KO mice requires a microbial component as well, although initial testing indicated that norovirus was present in our Irgm1 KO mouse colony. Future studies are planned to examine alterations in the gut microbial communities of these animals.
The present study supports previously published data (20, 47, 54) demonstrating that Irgm1 modulates autophagy. The accumulation of LC3-positive exocytic granules in Paneth cells of Irgm1 KO mice allows speculation that Irgm1 is necessary for autophagic progression or flux, which in turn is necessary for proper processing of the granules. A central role for autophagy in exocytic granule processing is consistent with data from the Atg16L1HM mice and with a recent report showing increased LC3 puncta in Paneth cells and altered processing of Paneth cell granules in pediatric CD patients (51). While this model is attractive, the precise role of Irgm1 in intestinal autophagy remains speculative, as definitive experiments are needed to demonstrate whether Irgm1 is involved in autophagic initiation or flux or, indeed, whether this relates directly to the regulation of intestinal inflammatory responses.
Our data contribute to growing evidence that Irgm1 and IRGM modulate autophagy and mitophagy, a point we emphasize because of suggestions that various IRG protein functions have been lost during evolution in humans (4). Indeed, there are key differences between the IRG protein family in mice and humans, including the highly truncated nature of the human IRGM protein sequence compared with that of mouse Irgm1 (4). Furthermore, from a functional standpoint, mouse IRG-driven resistance to T. gondii (14, 29, 50, 61) appears to be lacking in humans [although the closely related guanylate-binding proteins may drive resistance to T. gondii through a similar mechanism in humans (15, 45)]. Nevertheless, there appears to be significant functional overlap between Irgm1 and IRGM. Published studies have shown that decreased expression of IRGM or Irgm1 leads to decreased entry of Mycobacteria into an LC3-positive autophagosomal compartment, while overexpression of Irgm1 has the expected opposite effect (20, 47, 48). In the present study, we draw another key parallel between human IRGM and mouse Irgm1 function: we show that the absence of Irgm1 in intestinal tissue and cultured cells leads to an increased tubular mitochondrial population and accumulation of damaged/swollen mitochondria. This is analogous to reports that human IRGM localizes to mitochondria and drives mitochondrial fission and depolarization, which correlate with autophagy of that organelle (48). Thus, while certain aspects of mouse IRG protein function do not translate to human IRG proteins, our findings support similar roles of Irgm1 and IRGM in the regulation of autophagy and mitophagy. This further validates the use of Irgm1 KO mice to examine the role of IRGM function in controlling intestinal inflammation.
In conclusion, our data demonstrate the importance of IRG proteins in modulating host responses to injury and inflammation. Furthermore, our findings provide preliminary insight into the mechanisms by which IRGM dysfunction may predispose to intestinal inflammation, supporting the developing notion that regulation of autophagy is critical for Paneth cell homeostasis, particularly during recovery from stress and injury.
GRANTS
This work was supported by National Institutes of Health Grants AI-57831 (G. A. Taylor), DK-053347 (R. B. Sartor), DK-034987 (Robert Sandler), and K08 DK-09517 (A. S. Gulati), a North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Foundation/Crohn's and Colitis Foundation of America Young Investigator Award (A. S. Gulati), a Crohn's and Colitis Foundation of America Microbiome Consortium Grant (R. B. Sartor), and a Department of Veterans Affairs Merit Review Grant (G. A. Taylor).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.L., A.S.G., R.B.S., and G.A.T. are responsible for conception and design of the research; B.L., A.S.G., V.C., S.C.H., E.A.S., X.D., E.G., and A.A.S. performed the experiments; B.L., A.S.G., V.C., S.C.H., E.A.S., X.D., E.G., A.A.S., R.B.S., and G.A.T. analyzed the data; B.L., A.S.G., R.B.S., and G.A.T. edited and revised the manuscript; B.L., A.S.G., V.C., S.C.H., E.A.S., X.D., E.G., A.A.S., R.B.S., and G.A.T. approved the final version of the manuscript; A.S.G., R.B.S., and G.A.T. interpreted the results of the experiments; A.S.G. and G.A.T. prepared the figures; A.S.G., R.B.S., and G.A.T. drafted the manuscript.
ACKNOWLEDGMENTS
We thank Thaddeus Stappenbeck for valuable conversations concerning these studies.
REFERENCES
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med 361: 2066–2078, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 1: 113–118, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Bafica A, Feng CG, Santiago HC, Aliberti J, Cheever A, Thomas KE, Taylor GA, Vogel SN, Sher A. The IFN-inducible GTPase LRG47 (Irgm1) negatively regulates TLR4-triggered proinflammatory cytokine production and prevents endotoxemia. J Immunol 179: 5514–5522, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, Howard JC. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 6: R92, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9: 356–368, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, Kobayashi KS. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci USA 107: 14739–14744, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. J Exp Med 204: 1891–1900, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butcher BA, Greene RI, Henry SC, Annecharico KL, Weinberg JB, Denkers EY, Sher A, Taylor GA. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect Immun 73: 3278–3286, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456: 259–263, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 141: 1135–1145, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Chang CP, Yang MC, Lei HY. Concanavalin A/IFN-γ triggers autophagy-related necrotic hepatocyte death through IRGM1-mediated lysosomal membrane disruption. PLos One 6: e28323, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Woude GF, Sher A, Taylor GA. Inactivation of LRG-47 and IRG-47 reveals a family of interferon-γ-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194: 181–188, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Degrandi D, Kravets E, Konermann C, Beuter-Gunia C, Klumpers V, Lahme S, Wischmann E, Mausberg AK, Beer-Hammer S, Pfeffer K. Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc Natl Acad Sci USA 110: 294–299, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell 2: 83–89, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng CG, Zheng L, Jankovic D, Bafica A, Cannons JL, Watford WT, Chaussabel D, Hieny S, Caspar P, Schwartzberg PL, Lenardo MJ, Sher A. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-γ-induced cell death. Nat Immunol 9: 1279–1287, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn's disease: NOD2, autophagy and ER stress converge. Gut 60: 1580–1588, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gulati AS, Shanahan MT, Arthur JC, Grossniklaus E, von Furstenberg RJ, Kreuk L, Henning SJ, Jobin C, Sartor RB. Mouse background strain profoundly influences Paneth cell function and intestinal microbial composition. PLos One 7: e32403, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39: 207–211, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Henry SC, Traver M, Daniell X, Indaram M, Oliver T, Taylor GA. Regulation of macrophage motility by Irgm1. J Leukoc Biol 87: 333–343, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamal M, Dehlawi MS, Brunet LR, Wakelin D. Paneth and intermediate cell hyperplasia induced in mice by helminth infections. Parasitology 125: 275–281, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Kamal M, Wakelin D, Ouellette AJ, Smith A, Podolsky DK, Mahida YR. Mucosal T cells regulate Paneth and intermediate cell numbers in the small intestine of T. spiralis-infected mice. Clin Exp Immunol 126: 117–125, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134: 743–756, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keshav S. Paneth cells: leukocyte-like mediators of innate immunity in the intestine. J Leukoc Biol 80: 500–508, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 474: 307–317, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JC, Parkes M. Genome-wide association studies and Crohn's disease. Brief Funct Genomics 10: 71–76, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med 203: 2063–2071, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn's disease according to the Vienna classification: changing pattern over the course of the disease. Gut 49: 777–782, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302: 654–659, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol Gastrointest Liver Physiol 274: G544–G551, 1998 [DOI] [PubMed] [Google Scholar]
- 33.Martens S, Howard J. The interferon-inducible GTPases. Annu Rev Cell Dev Biol 22: 559–589, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 1: e24, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neurath MF, Fuss I, Pasparakis M, Alexopoulou L, Haralambous S, Meyer zum, Buschenfelde KH, Strober W, Kollias G. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur J Immunol 27: 1743–1750, 1997 [DOI] [PubMed] [Google Scholar]
- 36.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702, 1990 [DOI] [PubMed] [Google Scholar]
- 37.Ouellette AJ. Paneth cell α-defensins in enteric innate immunity. Cell Mol Life Sci 68: 2215–2229, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Cardon L, Mathew CG. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 39: 830–832, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted Paneth cell. Cell Mol Life Sci 59: 156–170, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun 9: 561–565, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11: 76–83, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 134: 577–594, 2008 [DOI] [PubMed] [Google Scholar]
- 44.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Selleck EM, Fentress SJ, Beatty WL, Degrandi D, Pfeffer K, Virgin HW, Macmicking JD, Sibley LD. Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii. PLoS Pathog 9: e1003320, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD. Emerging themes in IFN-γ-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212: 771–784, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313: 1438–1441, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, Komatsu M, Deretic V. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12: 1154–1165, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor GA. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol 9: 1099–1107, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, Taylor LS, Eagleson B, Secrest L, Southon EA, Reid SW, Tessarollo L, Bray M, McVicar DW, Komschlies KL, Young HA, Biron CA, Sher A, Vande Woude GF. Pathogen-specific loss of host resistance in mice lacking the IFN-γ-inducible gene IGTP. Proc Natl Acad Sci USA 97: 751–755, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, Codogno P, Barreau F, Ogier-Denis E, Berrebi D, Viala J. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn's disease. Gastroenterology 142: 1097–1099, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat Immunol 10: 907–917, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17: 299–313, 1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Traver MK, Henry SC, Cantillana V, Oliver T, Hunn JP, Howard JC, Beer S, Pfeffer K, Coers J, Taylor GA. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J Biol Chem 286: 30471–30480, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vidrich A, Buzan JM, Barnes S, Reuter BK, Skaar K, Ilo C, Cominelli F, Pizarro T, Cohn SM. Altered epithelial cell lineage allocation and global expansion of the crypt epithelial stem cell population are associated with ileitis in SAMP1/YitFc mice. Am J Pathol 166: 1055–1067, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Virgin HW, Todd JA. Metagenomics and personalized medicine. Cell 147: 44–56, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy 7: 297–300, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, Schroder JM, Bevins CL, Fellermann K, Stange EF. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal α-defensin expression. Gut 53: 1658–1664, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58a.Wellcome Trust Case Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 286: 113–117, 1999 [DOI] [PubMed] [Google Scholar]
- 60.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNγ-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 5: e1000288, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]