

Abstract
Tuberculosis caused 20% of all human deaths in the Western world between the 17th and 19th centuries, and remains a cause of high mortality in developing countries. In analogy to other crowd diseases, the origin of human tuberculosis has been associated with the Neolithic Demographic Transition, but recent studies point to a much earlier origin. Here we used 259 whole-genome sequences to reconstruct the evolutionary history of the Mycobacterium tuberculosis complex (MTBC). Coalescent analyses indicate that MTBC emerged about 70 thousand years ago, accompanied migrations of anatomically modern humans out of Africa, and expanded as a consequence of increases in human population density during the Neolithic. This long co-evolutionary history is consistent with MTBC displaying characteristics indicative of adaptation to both low- and high host densities.
Tuberculosis killed one in five adults in Europe and North-America between the 17th and 19th centuries1, and remains today a cause of high morbidity and mortality in much of the developing world2. Infectious diseases of humans can be divided into two broad categories3. Crowd diseases are generally highly virulent and depend on high host population densities to maximize pathogen transmission and reduce the risk of pathogen extinction through exhaustion of susceptible hosts4. Many crowd diseases emerged during the Neolithic Demographic Transition (NDT) starting around ten thousand years ago (kya), as the development of animal domestication increased the likelihood of zoonotic transfer of novel pathogens to humans, and agricultural innovations supported increased population densities that helped sustain the infectious cycle3. In contrast, older human infections are often characterized by slow progression to disease, sometimes involving reactivation after many years of latent or asymptomatic infection; these characteristics have been proposed to reflect adaptation to low host population densities by allowing repletion of the reservoir of susceptible individuals5. Tuberculosis is reminiscent of a typical crowd disease in killing up to 50% of individuals when left untreated3,6, and having evolved a mode of aerosol transmission that is promoted by high host densities. However, tuberculosis also displays a pattern of chronic progression, latency and reactivation that is characteristic of a pre-NDT disease7. Human tuberculosis was traditionally believed to have originated from animals4, but more recent phylogenetic analyses of MTBC have suggested that strains adapted to cause tuberculosis in animals diverged from the major human strains before NDT8–13. Moreover, human-associated MTBC is an obligate human pathogen with no known animal or environmental reservoir, suggesting that changes in human demography are likely to affect the evolution of MTBC. Here we used a population genomics approach to explore the evolutionary history of human MTBC, with a particular focus on the impact of changing host population sizes over time. Our results suggest a model that allows reconciliation of the apparent discrepancy between MTBC features characteristic of crowd diseases versus those indicative of adaptation to low host densities.
The global diversity of human-adapted MTBC
We generated the genome sequences of 220 strains representative of the global diversity of MTBC (Supplementary Table 1) and 39 additional strains corresponding to the lineage 2 "Beijing" family. In the global dataset, after excluding repetitive and mobile elements, we identified 34,167 polymorphic sites (SNPs) (Supplementary Table 2), which we used to reconstruct the phylogenetic relationships between these strains (Fig. 1A). This genome-based phylogeny was congruent with previous phylogenies based on other markers and resolved seven major lineages, with animal-adapted strains clustering together with the strains from Lineage 68. The phylogeny includes the recently described Lineage 7, which to date has only been observed in Ethiopia or recent Ethiopian emigrants14. Principal component analysis confirmed all main MTBC lineages, and highlighted the close phylogenetic relationship between the Eurasian Lineages 2, 3 and 4. These three lineages have collectively been referred to as evolutionarily “modern” (Fig. 1B) in the past, because of their comparably more derived position on the MTBC phylogeny and because they are thought to have spread more recently8,11. The maximum genetic distance between any two strains was 2,188 SNPs and involved a human and an animal strain, and 1,856 SNPs when only human clinical isolates were considered. Only 387 (1.1%) of the SNPs were homoplastic. Homoplasy can arise as a consequence of false-positive SNP calls, because of positive selection, recurrent mutations or because of recombination as recently suggested15. However, the fact that only 1.1% of the sites are homoplastic supports the view that the population structure of MTBC is largely clonal with little ongoing recombination occurring between strains16,17.
Figure 1. Genome-based phylogeny of MTBC mirrors that of human mitochondrial genomes.

A, Whole-genome phylogeny of 220 strains of Mycobacterium tuberculosis complex (MTBC). Support values for the main branches after inference with Neighbour-joining (left) and Maximum-likelihood (right) are shown. B, Principal Component Analysis (PCA) of the 34,167 SNPs. The first three PCA axes are shown; these discriminate between evolutionarily “modern” (highlighted in grey) and “ancient” (all other) strains. Individual lineages are shown following the colour coding of Fig. 1A. C and D, Comparison of MTBC phylogeny (C) and a phylogeny derived from 4,955 mitochondrial genomes representative of the main human haplogroups (D). The colour coding highlights the similarities in tree topology and geographic distribution of MTBC strains and main human mitochondrial macro-haplogroups (black - African clades: MTBC Lineage 5 and 6, human mitochondrial macro-haplogroups L0-L3; pink – South-East Asian and Oceanian clades: MTBC Lineage 1, human mitochondrial macro-haplogroup M; blue – Eurasian clades: MTBC Lineage 2, 3, and 4, human mitochondrial macro-haplogroup N). The MTBC Lineage 7 has only been found in Ethiopia and its correlation with any of the three main human haplogroups remains unclear.
African origin and co-divergence of MTBC with modern humans
Several studies have proposed an African origin for MTBC8,10,12. We decided to formally test this hypothesis using our new whole-genome data. We used three independent phylogeographical analyses to determine the likely geographic origin of the most recent common ancestor of MTBC. Two different Bayesian analyses identified Africa as the most likely origin of MTBC, with East- and West Africa showing a combined posterior probability of 90% and 67%, respectively (Supplementary Figs. 1, 2, and 3). Similarly, a Maximum Parsimony approach predicted 100% probability of an African origin. Taken together, these data support an African origin for MTBC.
Next we sought to determine the putative age of the association between MTBC and its human host. Given that human-adapted MTBC is limited to humans, and both anatomically modern humans and MTBC originated in Africa, we tested whether MTBC and humans might have diverged in parallel; this would be particularly likely if the association between the two predates the NDT, as previously postulated8,10,12. To explore this possibility, we first compared our new MTBC phylogeny to a corresponding tree constructed from 4,955 mitochondrial genomes representative of the main human haplogroups (Supplementary Table 3)18. We observed striking similarities (Figs. 1C and 1D). In both cases, the early branching clades are found exclusively in Africa. Moreover, the trichotomy formed by branching of the Out-of-Africa M and N mitochondrial macro-haplogroups from the L3 African source population is mirrored in the MTBC phylogeny by a similar relationship between Lineage 1, Eurasian Lineages 2/3/4, and the African Lineages 5 and 6. In addition to this qualitative similarity, comparison of the most common mitochondrial haplogroups with the most frequent MTBC lineages in the same country revealed a strong quantitative association (Parsimony Score and Association Index tests; P < 0.01 in all cases) (Supplementary Fig. 4, Supplementary Table 5 and Supplementary Table 6). Taken together, these data are consistent with MTBC evolving in parallel with its human host.
Age of the association of MTBC and humans
The similarities in tree topology and phylogeographic distribution suggest that MTBC already infected the early human populations of Africa. To further explore the association between MTBC and its human host, we tested for possible imprints of ancient human divergence times on the main phylogenetic lineages of MTBC using a Bayesian approach19. Several approaches have been used to date bacterial phylogenies (see refs. 20–22 for some examples). Unfortunately, none of these were applicable here because of the following reasons. First, although ancient DNA has been used to study the evolutionary history of other bacteria20, and similar studies have been performed in tuberculosis in the past23, no relevant whole-genome data are currently available from ancient DNA of MTBC strains. Second, although a mutation rate for MTBC has recently been estimated based on a macaque infection model and molecular epidemiological data24,25, it is well known that such short-term mutation rates cannot easily be extrapolated to long-term substitution rates relevant for the time-scale discussed here26,27. Third, and related to the previous point, although the isolation dates of some of the strains included in our analysis were known, at best they would allow calculation only of a short-term mutation rate. Moreover, when performing a tip-to-date analysis of those strains (N = 49), we found that in contrast to several other bacterial species21,28–30, no significant correlation between isolation time and phylogenetic divergence exists in MTBC (correlation coefficient = 0.047).
Because of these limitations, we used an alternative approach to date our MTBC phylogeny. Specifically, we used as initial calibration points several key dates in human evolution. We tested three alternative models in which the coalescent time of the most basal MTBC Lineages 5 and 6 was calibrated against: i) the emergence of anatomically modern humans 185 +/−20 kya (MTBC-185)31, ii) the coalescent time of the L3 mitochondrial haplogroup 70 +/−10 kya (MTBC-70)32, and iii) the beginning of the NDT 10 +/−2 kya (MTBC-10)3 (Table 1). We compared the timing of the branching points predicted by each of the models with estimated dates of known events in human history. A recent model based on human whole-genome analyses suggests that the global dispersal of modern humans occurred through two major waves; an initial eastern dispersal around the Indian Ocean starting 62–75 kya, and a later dispersal into Eurasia 25–38 kya33. Our MTBC-70 model showed a striking correlation with these human migration events by dating a first split of Lineage 1 at 67 kya (95% highest probability density (HPD): 48–88 kya) coinciding with the first wave of human migration31, and a second split at 46 kya (95% HPD: 31–61 kya) matching the later dispersal throughout Eurasia (Fig. 2a, Supplementary Fig 5)34,35. Coalescent dates for the branch leading to Lineages 4 and 2 in the MTBC-70 model (30–46 kya and 32–42 kya, respectively) show a good correlation with archaeological evidence of presence of modern humans in Europe35 and East Asia36. In contrast, our alternate model MTBC-185 postulates initial branching of Out-of-Africa lineages as early as 126–174 kya when focusing on the branch leading to 'modern' strains (Supplementary Fig. 6), which would suggest the global dispersal of MTBC preceded that of anatomically modern humans. MTBC-10, by definition, implies global dispersal within the last 10 ky (Supplementary Fig. 7). While MTBC has been spread by trade and conquest in recent centuries8, the pattern of this dispersal does not match the phylogeographic distribution discussed above. Finally, a fourth model (MTBC-65) using the coalescent time of mitochondrial haplogroup M as a calibration time-point for MTBC Lineage 1 generated very similar results to MTBC-70 (Table 1). In summary, our phylogenetic analysis based on a 70 ky timeframe shows that MTBC has been infecting humans at least for the last 70 ky.
Table 1.
Comparison of different dating scenarios of MTBC evolution.
| Dating scenario | MTBC-70 | MTBC-185 | MTBC-10 | MTBC-65 | |
|---|---|---|---|---|---|
| Rationale | Emergence of MTBC with human mtDNA haplogroup L3 | Emergence of MTBC with anatomically modern humans | Emergence of MTBC during Neolithic Demographic Transition | Emergence of Out-of-Africa MTBC with human mtDNA haplogroup M | |
| Dates inferred from models (in kya) * | |||||
| most recent common ancestor of MTBC | 73 (50–96) | 198 (170–229) | 11 (9–14) | 67 (44–91) | |
| coalescent time for Lineage 5/6 | 70 (48–88)** | 184 (164–203)** | 10 (8–12)** | 61 (40–81) | |
| coalescent time for Lineage 1 | 67 (46–88) | 183 (160–207) | 10 (8–12) | 62 (42–82)** | |
| coalescent time for Lineage 2/3/4 | 46 (31–61) | 126 (104–148) | 7 (6–10) | 41 (26–55) | |
| period of maximum logistic growth | 4–7 | 31–34 | 1 | 4–7 | |
| Substitution rate (SNPs/site/ky) *** | 3.37E-4 (2.38E-4-4.65E-4) | 1.23E-4 (1.04E-4-1.46E-4) | 2.17E-3 (1.71E-3-2.68E-3) | 3.78E-4 (2.62E-4-5.36E-4) | |
dates are shown as the median value and 95% highest posterior density interval predicted in the corresponding Bayesian analysis
value provided as prior input in Bayesian analysis
BEAST predicted rate of SNP accumulation (per polymorphic position and thousand year). In the main text we use the estimated genomic substitution rate (per position and year) for comparative purposes with published estimations from other bacterial species.
Figure 2. Out-of-Africa and Neolithic expansion of MTBC.
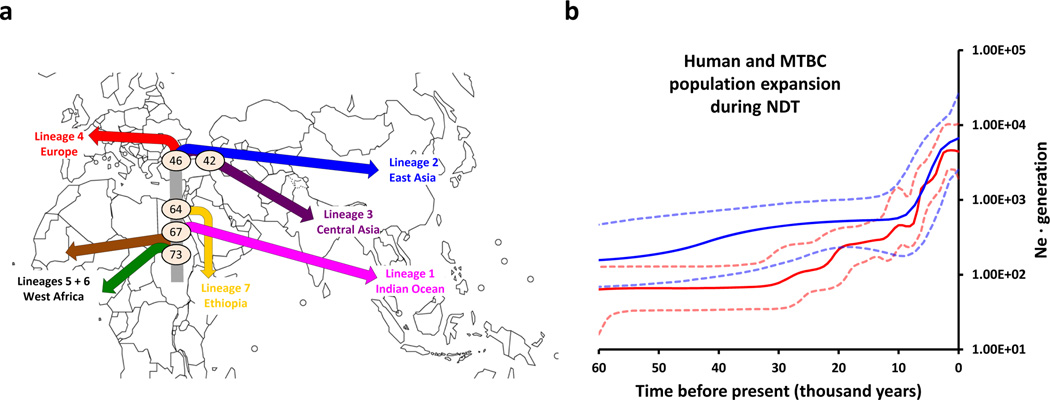
A, Map summarizing the results of the phylogeographic and dating analyses of MTBC. The colour codes used for lineages are according to Fig. 1A. Major splits are annotated with the median value (in kya) of the dating of the relevant node. Lineage 7 (yellow) has so far been isolated exclusively from patients with known country of origin in the Horn of Africa14. Lineage 7 diverged subsequent to the proposed Out-of-Africa migration of MTBC; it may have arisen amongst a human population that remained in Africa, or a population that returned to Africa. B, Bayesian skyline plots illustrating changes in population diversity of MTBC (red line) and humans based on mitochondrial DNA (blue lines) during the last 60 ky. Dashed lines represent the 95% highest probability density (HPD) intervals for the estimated population sizes.
Neolithic co-expansion of MTBC and humans
All the data presented so far strongly support the notion that human tuberculosis indeed predates NDT. How then could the features of tuberculosis typical of crowd diseases have arisen? To address this question, we used Bayesian skyline plots to estimate the changes in effective population size over time of the pathogen and human populations19. Our MTBC dataset revealed a main signal of population size increase starting 10 kya to 2.5 kya (Fig. 2B), suggesting that the expansion of MTBC occurred as a consequence of the increase in population densities that followed the establishment of first human settlements during the NDT37, and not just because of a general increase in the total number of humans peopling the planet at the time. To test if the human population dynamics around that period coincide with that of the MTBC we used a dataset previously described to maximize the information on human demographics during the Neolithic (Supplementary Table 6)38. The resulting skyline plot shows a Neolithic expansion of humans around 4–8 kya (Supplementary Fig. 8) coinciding with that of MTBC (Spearman R = 0.99, p <0.00001; Fig. 2B, Supplementary Fig. 8). Taken together, these findings indicate that the Neolithic contributed to the success of MTBC, not by enhancing the likelihood of zoonotic transfer to humans as previously proposed, but because of a combined increase in host population size and density.
The evolutionary history of MTBC at a regional scale
To analyze MTBC evolution at a regional level, we focused on Lineage 2, which includes the “Beijing” family of strains. These strains have received particular attention because of their hyper-virulence in laboratory models, their recent dissemination in human populations, and their association with drug resistance39. Supplementing our global diversity set with an additional 39 Lineage 2 genomes from China, we observed a strong correlation between skyline plots derived from the Lineage 2 genomes and a set of human mitochondrial genomes enriched with haplogroups from East Asia of likely origin just before, during or after the Neolithic (Spearman R = 0.97, p <0.001; Fig. 3A, Supplementary Fig. 9). MTBC-70 dating for Lineage 2 is consistent with an initial arrival coincident with archeological evidence of anatomically modern humans in East Asia36 (32–42 kya, Supplementary Fig. 5), a first expansion (6–11 kya, Figs. 3B and 3C) alongside the emergence of agriculture in China 8 kya40, and a subsequent main expansion of the "Beijing" strains (3–5 kya, Supplementary Fig. 9) coinciding with the spread of agriculture to neighbouring regions (Figs. 3B and 3C)37.
Figure 3. Neolithic expansion and spread of MTBC Lineage 2 “Beijing” in East Asia.

A, Bayesian skyline indicating changes in Lineage 2 diversity over time (red line) as compared to human mtDNA haplogroups from East Asia (blue line). 95% HPD intervals for the population size estimations are shown in dashed lines. B, Dated Bayesian phylogeny of the MTBC Lineage 2 based on coalescent analysis. C, Map of the parallel origin and migration of MTBC and humans in East Asia indicating the first archaeological evidence of modern human in the region 32–42 kya, coinciding with the migration of MTBC from Central to East Asia, the start of the Neolithic in the region indicated by the first evidence of domesticated crop in China coinciding with the origin of the MTBC “Beijing family” 8 kya (6–11 kya), and the co-expansion of agriculture and MTBC “Beijing family” into neighbouring countries 3–5 kya.
In summary, our data on the global and regional expansion of MTBC during the NDT supports the view that while NDT was not the only period leading to strong increases in human population sizes, it was the period where in addition to human population growth, the densities of human populations increased following the first establishment of permanent human settlements. Hence, in addition to providing a springboard for global domination by modern humans, NDT was also central to the success of MTBC by generating growing numbers of susceptible hosts living in increasingly crowded conditions.
Concluding remarks
The common origin in Africa, the congruence in phylogeography, and the dating of major branching events, lead us to conclude that MTBC has been co-evolving with anatomically modern humans for tens of thousands of years. The marked expansion of MTBC during the NTD, but not during earlier human expansion events41,42, suggests that the success of this pathogen was primarily driven by increases in human host density, which is typical of crowd diseases. However, the striking match between the MTBC and human mitochondrial phylogenies supports a much older association between MTBC and its host, and suggest that carriage of MTBC was ubiquitous in hunter-gatherer populations migrating out of Africa well before NDT. The fidelity of this match is surprising. Considering their vulnerability and small numbers (some of today’s hunter-gatherers live in groups of 20 or less43), it might have been anticipated that tuberculosis disease would have had a significant detrimental impact on these groups, and might therefore have precipitated its own extinction. In fact, the correspondence between MTBC phylogeny with early human migration is strikingly similar to that observed with low virulence Helicobacter pylori44. Perhaps latent infection with MTBC imparted some degree of immunity against more lethal pathogens encountered in the new environment or in contact with archaic human populations? The ongoing analyses of the human microbiota highlight the fuzzy boundaries between commensalism and pathogenecity during health and disease45. A recent study has suggested that co-infection with H. pylori might protect against active tuberculosis disease46. Conversely, whether latent tuberculosis infection protects against gastric ulcers or stomach cancer caused by H. pylori in individuals infected with both bacteria is unknown but an intriguing possibility. In such a case, a positive feedback between both infections would result in an asymptomatic individual benefiting from being infected by both bacterial species.
Alternatively, one could think of a model in which early populations carried the infection in a less virulent form, with transmission sustained by reactivation disease in elderly individuals after reproductive age. The possibility that disease characteristics might have changed over time as different MTBC populations were selected in different human societies may help to explain current epidemiological trends associated with increased dissemination of the "Beijing" family of MTBC39, and decreased rates of disease caused by evolutionarily “ancient” lineages of MTBC47. In addition to changes in population density, it can be anticipated that the pathology of tuberculosis during NDT would have been influenced by co-infections with novel crowd diseases and by variations in key nutrients such as vitamin D48. Similarly, it is important to consider the possibility of reciprocal adaptive changes to the human genome as a result of prolonged co-evolution with MTBC49.
In this study, we have compared MTBC phylogenic diversity to human diversity inferred from mitochondrial genome data. One advantage of using mitochondrial data is that it has been used extensively to study recent human evolution in the past. Furthermore, such data is available from almost any region of the world, and there is a large body of work studying human migrations based on the distribution of mitochondrial haplogroups. However, mitochondrial DNA is also limited in that it contains little phylogenetic information, and the existing data sets suffer from potential sampling bias. Increasingly, new DNA sequencing technologies are paving the way for studies of human diversity based on whole genomes33. Hence in the context of a pathogen like MTBC, future studies should be based on paired human- and bacterial whole-genome information collected prospectively. Such an integrated approach will allow investigating the molecular determinants of host-pathogen co-evolution in human tuberculosis and other diseases.
The accumulation of more than 30 thousand SNPs by human MTBC strains over the proposed timeframe of 70 thousand years corresponds to a long-term genome-wide substitution rate of 2.58 × 10−9 substitutions per site per year (95% HPD 1.66 × 10−9 to 2.89 × 10−9, Table 1). This is much lower than recent estimates of short-term substitution rates in experimental models and human outbreaks24,25. A decrease in substitution rates measured over increasing time intervals is a common feature of phylogenetic analyses27, and an exponential decrease is observed in the substitution rate with time when we pool our data with other similar genome-based studies published recently (correlation coefficient = −0.9614, P < 0.0001, Figure 4). Fixation or removal of single nucleotide changes by natural selection can contribute to this phenomenon, though retention of a high proportion of nonsynonymous mutations suggests that this has had a low impact on MTBC8. Alternative mechanisms to account for the reduction of genetic diversity over long timescales include serial founder effects linked to sequential expansions of human subpopulations and their associated pathogenic and commensal microbial flora50.
Figure 4. Time-dependent decay of substitution rates in bacteria based on whole-genome datasets.

Scatter plot graph representing the relationship between substitution rate and time span between the most recent ancestor and the last sampling date for each studied pathogen. Values were extracted from relevant publications that use whole-genome representative datasets and coalescent analysis of substitution rates (for a complete list of references see Supplementary Table 9).
In conclusion, we propose that MTBC has been a constant companion of anatomically modern humans during our evolution and global dissemination over the last 70 thousand years. Furthermore, MTBC has been able to adapt to changing human populations. Exploration of changes that have occurred in this interaction over time may help predict future patterns of disease and to design rational strategies to bring an end to this historic partnership.
Online methods
Datasets
1. MTBC datasets
We have analyzed a total of 259 MTBC strains (including one Mycobacterium canettii strain used as outgroup). We used two different strains sets for different aspects of the analyses:
-
-
Global MTBC dataset (n = 220): This dataset represents a global collection of MTBC clinical strains covering all the known phylogenetic lineages of MTBC and including representatives from 46 countries. In addition, three strains from the animal-adapted lineage (including one strain of the Mycobacterium bovis BCG vaccine) were included as reference, and one strain of Mycobacterium canettii, which was used as the outgroup. More detailed information can be found in Supplementary Table 1.
-
-
MTBC Lineage 2 enriched dataset (n = 75): To explore the evolution of MTBC in a regional setting, we extended our collection of 36 MTBC strains from Lineage 2 with an additional 39 strains which represent the population diversity of Lineage 2 in China based on standard genotyping (Supplementary Table 1).
Illumina reads of the strains described in this study have been deposited under the project number ERP001731.
2. Human mitochondrial dataset
For the comparison with human genetic diversity, we analyzed large datasets of complete mitochondrial genomes (described below). There are limitations inherent to mtDNA. First, estimating the most frequent mtDNA haplogroup in a particular country is always difficult and sampling-dependent. Second, mtDNA harbours limited phylogenetic information. However, the reasons to focus on a mitochondrial marker rather than on a chromosomal marker are 1) availability of information for most of the regions/countries in terms of mtDNA haplogroup frequencies and 2) the possibility to compare with previous published studies dealing with human mtDNA haplogroups, human migrations and population dynamics. We used three different sets of human mitochondrial genomes that were available in public repositories. These are listed in Supplementary Tables 3, 6 and 7.
-
-
Global reference dataset of human mtDNAs (n = 4,955): This dataset is a compilation of most of the publicly available human mitochondrial (mtDNA) genomes for which the haplogroup has been determined18. This dataset includes representatives of most known human mitochondrial macro-haplogroups and derived haplogroups.
-
-
Neolithic population expansion dataset of human mtDNAs (n = 423): This second dataset is derived from the dataset reported by Gignoux 201138, and includes selected representative haplogroups known to have their origin either before, during or shortly after the Neolithic period, and therefore maximized to detect signatures of population expansions around that period that could be obscure by earlier expansion events.
-
-
East Asia enriched Neolithic dataset of human mtDNAs (n = 72): As for MTBC Lineage 2, we complemented the dataset for East Asia by adding any newly published human mitochondrial genome from the mtDNA haplogroups of interest (B4a1, F1a1, E1a, E1b).
Sequencing of MTBC strains
The majority of MTBC strains were sequenced during the present project at different sequencing centres (GATC, Germany; Wellcome Trust Sanger Institute, United Kingdom; and Southern Genome Center, China); a few additional sequences were retrieved from publicly available databases. MTBC DNA was extracted using standard procedures. Single- or Paired-end multiplexed Illumina sequencing was performed as described previously51. Briefly, sequencing was performed on a HiScanSQ instrument and TruSeq SBS Kit – HS chemistry (Illumina, USA) to generate between 51 and 100 bases long sequencing reads depending on the strain. Average genome coverage was 146.5 of the reference genome (the strain-specific genome coverage is shown in Supplementary Table 1).
Mapping Illumina sequencing reads and SNP calling
Sequencing reads of each MTBC strain were mapped to the inferred most recent common ancestor (MRCA) of MTBC as previously determined52 (the sequence of the MTBC MRCA can be found as fasta formatted as part of the Supplementary information). We used two mapping approaches; the un-gapped MAQ53 algorithm and the Burrows-Wheeler algorithm described in BWA54 and MAQ SNP caller and Samtools55 to generate two different lists of SNPs. We kept those polymorphic positions called by both approaches. For a complete description of the SNP calling procedure and annotation of the positions see Supplementary Text as well as Supplementary Figure 10 for a workflow of the SNP calling procedure.
Phylogenetic and principal component analyses
Human mtDNA datasets were obtained from the database of variant positions used by Behar et al.18. For the population expansion dataset of human mtDNA during Neolithic, the relevant accession numbers described in Gignoux et al.38 were downloaded and the genomes aligned using the ClustalW56 implementation in BioEdit package57 followed by manual curation. We removed the poorly aligned region known as D-loop and kept the polymorphic sites for subsequent phylogenetic and coalescent analyses. For the MTBC datasets, we used the variable positions for all downstream analyses. In both cases we applied phylogenetic distance as well as maximum-likelihood methods. For a complete description of the phylogenetic analyses, the identification of homoplastic sites and the principal component analysis of the SNPs used see Supplementary Text.
Phylogeographic analyses
For the phylogeographic analyses we used the BSSVS model implemented in BEAST 1.658. We also used RASP59 that implements both Bayesian and parsimony approaches to analyze the ancestral geographic ranges of MTBC lineages. We sub-divided the world map into seven broad geographic areas and used them as a proxy for the most likely origin of each strain (see Supplementary Fig. 1 for the world sub-division and Supplementary Table 1 for patient origin). We used broad geographic areas instead of the exact location because the high number of locations to consider, and hence the exchange rates to estimate, would be unmanageable if using all individual countries. Predefined geographic areas were introduced for each MTBC strain according to the patient’s country of origin. See Supplementary Text for a complete description of the settings of the different phylogeographic analyses.
MTBC-mtDNA association test
We tested the hypothesis that modern Lineages 2, 3 and 4, Lineage 1 and the African Lineages 5 and 6 are associated with the N, M and L human mtDNA lineages. To this end, we assigned for each MTBC strain from a given country an mtDNA haplogroup according to the frequency of the haplogroup in that country based on a review of the published literature (Supplementary Table 5). Only the two most frequent MTBC lineages of a country and the two most frequent mtDNA haplogroups were considered, unless only one MTBC lineage occurred in the country, in which case it was assigned to the most frequent mtDNA of the country (Supplementary table 4). We used BaTs (Bayesian Tip-association significance testing) 60 to test whether the main lineages of MTBC for each country tend to be associated with a particular human mtDNA macro-haplogroup (L, M, N) or haplogroup (A, B, D, E, F, G, H, K, L, M, R, U) (Supplementary Fig. 4 and Supplementary Tables 4 and 5). For the tests we assumed that there was no MTBC lineage that corresponded to the L0, L1, L2 and L4 human mitochondrial lineages based on the fact that no Lineage 5 or 6 strains are found outside of West Africa where human mitochondrial L3 have the highest frequency31. However even when we introduce L0, L1, L2 and L4 the results of the tests do not change. BaTs implements two association indexes, the Parsimony Score (PS) that quantifies the number of state changes in the phylogeny (low number indicates high clustering of states) and the Association Index (AI) that looks at internal nodes and records the most frequent state in the taxa downstream of the node. A statistical test was carried out by reshuffling the various states across the phylogeny. Given the constrained phylogeographic distribution of Lineage 5 and 6 (i.e. Mycobacterium africanum) to West Africa and their basal but close position to all the "Out-of-Africa" lineages, these M. africanum lineages correlate best with the human mitochondrial L3 haplogroup which shows remarkable similarities.
BEAST analyses
We used BEAST v. 1.619 to date the evolutionary events and population dynamics of MTBC and the human mtDNA haplogroups. BEAST implements the joint sampling of the posterior distribution of different evolutionary parameters like the substitution rate or the population size under a coalescent framework. In all cases, we used a skyline prior to look for changes of population size over time. For MTBC, we used two datasets: to explore different dating hypotheses, we used the complete MTBC dataset, a total of 216 strains excluding the outgroup (M. canettii) and the animal strains. We used an uncorrelated log-normal distribution for the substitution rate in all cases. We imposed different prior values to the coalescent times of the Lineages 5 and 6 according to plausible time estimates. Because no fossil records or good substitution rate estimates are available for MTBC, we used this approach as a way to narrow down the origin and age of the extant strains of MTBC. We imposed normal distributions in the coalescent time of Lineages 5 and 6, as time estimates for mitochondrial haplogroups are usually given in coalescent times and not times of splitting events between groups: 185 (+/− 20kya), 70 (+/− 10kya) and 10 (+/−2kya). We also added as a second anchor point the split of MTBC Lineage 1 with a normal prior of 65 +/− 10 kya, based on the co-incident geographic distribution of Lineage 1 with human mitochondrial macro-haplogroup M. Similar approaches were followed to analyzed the mtDNA datasets where we used both a molecular clock approach (by specifying a published substitution rate31) and a dating approach (by assuming the height of the phylogeny distributed normally around 185 kya as a mean +/− 20 kya). Both approaches yielded similar results, and we report the results for the dating analyses. Similarly for the East Asia clade, we specified priors for the age of the whole dataset (60 +/− 10 kya) and for the individual haplogroups as described in the literature (B4a1: 11 +/− 3 kya; E1a: 9 +/− 3 kya; E1b: 6 +/− 3 kya)18. For a detailed description of the models and the statistical comparison of skyline plots see Supplementary Notes.
Supplementary Material
Acknowledgements
We thank Doron Behar and Sharon Rosset for providing the mitochondrial genome sequences and Christopher Gignoux for advise on the mitochondrial neolithic dataset, Nerges Mistry from The Foundation for Medical Research for providing bacterial strains, and Chris Dye, Francois Balloux and Lucy Weinert for comments on the manuscript. This work was supported by the Medical Research Council UK (grant U.1175.02.002.00015.01 to S.G. and U117581288 to D.Y.), the Swiss National Science Foundation (PP0033-119205 to S.G.), the National Institutes of Health (AI090928 and HHSN266200700022C to S.G.), the Leverhulme-Royal Society Africa Award (AA080019 to S.G.), and the Natural Science Foundation of China Grants (#91231115 to Q.G). DNA sequencing was partially supported by core funding of the Wellcome Trust (grant number 098051) and by a FP7 project of the European Community (SysteMTb HEALTH-F4-2010-241587 to D.G.). IC is supported by European Union funding from the Mari Curie FP7 actions (project number 272086) and Project BFU2011-24112 from the Ministerio de Economía y Competitividad (Spain).
Footnotes
Accession codes
The sequencing reads of those strains that are not already publicly available can be found under study number ERP001731. For a complete list of strains sequenced in this study refer to Supplementary Table 1.
The authors declare no competing financial interests.
Author contribution
IC, QG, DY and SG designed and supervised the study. MC, SBO, KEH, MK-M, JP, BM, SBO, GT, DY-M, GB, JM, LW, SBE, SH, SN, RD, AA, QG and SG provided MTBC strains/reagents. JP, SBE, and SH contributed to the genome sequencing. IC, MC and TL analysed the data. IC, MC, TL, SBO, KEH, JP, SB, GT, DY-M, SBE, SH, SN, AA, QG, DY and SG contributed to the manuscript writing. All authors read and approved the manuscript.
References
- 1.Wilson LG. Commentary: Medicine, population, and tuberculosis. Int. J Epidemiol. 2005;34:521–524. doi: 10.1093/ije/dyh196. [DOI] [PubMed] [Google Scholar]
- 2.WHO G. The Global Plan to STOP TB 2011–2015. WHO. 2011
- 3.Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diamond J. Guns, Germs, and Steel: The Fates of Human Societies. Vol. 496. W. W. Norton & Company; 1999. Apr 1, [Google Scholar]
- 5.Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- 6.Berg G. The prognosis of open pulmonary tuberculosis: a clinical-statistical analysis. JAMA. 1940;114:1954–1955. [Google Scholar]
- 7.Barry CE, 3rd, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Mic. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershberg R, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PloS Biol. 2008;6:e311. doi: 10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mostowy S, Cousins D, Brinkman J, Aranaz A, Behr MA. Genomic deletions suggest a phylogeny for the Mycobacterium tuberculosis Complex. J. Infect. Dis. 2002;186:74–80. doi: 10.1086/341068. [DOI] [PubMed] [Google Scholar]
- 10.Wirth T, et al. Origin, spread and demography of the Mycobacterium tuberculosis Complex. PLoS Pathogens. 2008;4:e1000160. doi: 10.1371/journal.ppat.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brosch R, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci U.S.A. 2002;99:3684–3689. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez MC, et al. Ancient origin and gene mosaicism of the progenitor ofMycobacterium tuberculosis . PLoS Pathogens. 2005;1:e5. doi: 10.1371/journal.ppat.0010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gagneux S, et al. Variable host–pathogen compatibility inMycobacterium tuberculosis . Proc. Natl. Acad. Sci U.S.A. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Firdessa R, et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis in Ethiopia. Emerg. Infect. Dis. 2013;19:460–463. doi: 10.3201/eid1903.120256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Namouchi A, Didelot X, Schöck U, Gicquel B, Rocha EPC. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome res. 2012;22:721–734. doi: 10.1101/gr.129544.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci U.S.A. 2004;101:4871–4876. doi: 10.1073/pnas.0305627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comas I, Gagneux S. The past and future of tuberculosis research. PLoS Pathogens. 2009;5:e1000600. doi: 10.1371/journal.ppat.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behar DM, et al. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 2012;90:675–684. doi: 10.1016/j.ajhg.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PloS Biol. 2006;4(5):e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bos KI, et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mutreja A, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morelli G, et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 2010;42:1140–1143. doi: 10.1038/ng.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djelouadji Z, Raoult D, Drancourt M. Palaeogenomics of Mycobacterium tuberculosis: epidemic bursts with a degrading genome. Lancet Infect. Dis. 2011;11:641–650. doi: 10.1016/S1473-3099(11)70093-7. [DOI] [PubMed] [Google Scholar]
- 24.Ford CB, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 2011;43:482–186. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker TM, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 2012 doi: 10.1016/S1473-3099(12)70277-3. in press, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morelli G, et al. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6:e1001036. doi: 10.1371/journal.pgen.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho SYW, et al. Time-dependent rates of molecular evolution. Molecular ecology. 2011;20:3087–3101. doi: 10.1111/j.1365-294X.2011.05178.x. [DOI] [PubMed] [Google Scholar]
- 28.Holt KE, et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 2012;44:1056–1059. doi: 10.1038/ng.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croucher NJ, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soares P, et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 2009;84:740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soares P, et al. The expansion of mtDNA haplogroup L3 within and out of Africa. Mol. Biol. Evol. 2012;29:915–927. doi: 10.1093/molbev/msr245. [DOI] [PubMed] [Google Scholar]
- 33.Rasmussen M, et al. An Aboriginal australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henn BM, Cavalli-Sforza LL, Feldman MW. The great human expansion. Proc. Natl. Acad. Sci U.S.A. 2012;109:17758–17764. doi: 10.1073/pnas.1212380109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart JR, Stringer CB. Human evolution Out of Africa: the role of refugia and climate change. Science. 2012;335:1317–1321. doi: 10.1126/science.1215627. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, Su B. Natives or immigrant: modern human origins in East Asia. Nat. Rev. Genet. 2000;1:126–133. doi: 10.1038/35038565. [DOI] [PubMed] [Google Scholar]
- 37.Bellwood P, Oxenham M. The expansions of farming societies and the role of the Neolithic Demographic Transition. The Neolithic Demographic Transition and its Consequences. 2008:13–34. [Google Scholar]
- 38.Gignoux CR, Henn BM, Mountain JL. Rapid, global demographic expansions after the origins of agriculture. Proc. Natl. Acad. Sci U.S.A. 2011;108:6044–6049. doi: 10.1073/pnas.0914274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parwati I, van Crevel R, van Soolingen D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancer Infect. Dis. 2010;10:103–111. doi: 10.1016/S1473-3099(09)70330-5. [DOI] [PubMed] [Google Scholar]
- 40.Barton L, et al. Agricultural origins and the isotopic identity of domestication in northern China. Proc. Natl. Acad. Sci U.S.A. 2009;106:5523–5528. doi: 10.1073/pnas.0809960106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atkinson QD, Gray RD, Drummond AJ. mtDNA variation predicts population size in humans and reveals a major Southern Asian chapter in human prehistory. Mol. Biol. Evol. 2008;25:468–474. doi: 10.1093/molbev/msm277. [DOI] [PubMed] [Google Scholar]
- 42.Wei W, et al. A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res. 2012 doi: 10.1101/gr.143198.112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton MJ, Milne BT, Walker RS, Burger O, Brown JH. The complex structure of hunter–gatherer social networks. Proc. R. Soc. London Ser. B. 2007;274:2195–2203. doi: 10.1098/rspb.2007.0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linz B, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell host & microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perry S, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PloS one. 2010;5:e8804. doi: 10.1371/journal.pone.0008804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jong BC, et al. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J. Infect. Dis. 2008;198:1037–1043. doi: 10.1086/591504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martineau AR, et al. Reciprocal seasonal variation in vitamin D status and tuberculosis notifications in Cape Town, South Africa. Proc. Natl. Acad. Sci U.S.A. 2011;108:19013–19017. doi: 10.1073/pnas.1111825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnes I, Duda A, Pybus OG, Thomas MG. Ancient urbanization predicts genetic resistance to tuberculosis. Evolution. 2011;65:842–848. doi: 10.1111/j.1558-5646.2010.01132.x. [DOI] [PubMed] [Google Scholar]
- 50.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci U.S.A. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quail MA, et al. A large genome center ’s improvements to the Illumina sequencing system. Nature Met. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Comas I, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010;42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Durbin R. Fast and accurate long-read alignment with Burrows – Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 57.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- 58.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu Y, Harris, a J, He X. S-DIVA (Statistical Dispersal-Vicariance Analysis): a tool for inferring biogeographic histories. Mol. Phylogenet. Evol. 2010;56:848–850. doi: 10.1016/j.ympev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 60.Parker J, Rambaut A, Pybus OG. Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008;8:239–246. doi: 10.1016/j.meegid.2007.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.