

Significance
Density functional theory studies on the OH state in the catalytic mechanism of cytochrome c oxidase are performed. The proposed structure of OH state and its calculated characteristics are found to comply with the experimental data. The presented hypothesis solves, at least in part, the dilemma of the OH and O states in the catalytic mechanism of cytochrome oxidase.
Keywords: oxygen reduction, electron transfer
Abstract
Complex IV in the respiratory chain of mitochondria and bacteria catalyzes reduction of molecular oxygen to water, and conserves much of the liberated free energy as an electrochemical proton gradient, which is used for the synthesis of ATP. Photochemical electron injection experiments have shown that reduction of the ferric/cupric state of the enzyme’s binuclear heme a3/CuB center is coupled to proton pumping across the membrane, but only if oxidation of the reduced enzyme by O2 immediately precedes electron injection. In contrast, reduction of the binuclear center in the “as-isolated” ferric/cupric enzyme is sluggish and without linkage to proton translocation. During turnover, the binuclear center apparently shuttles via a metastable but activated ferric/cupric state (OH), which may decay into a more stable catalytically incompetent form (O) in the absence of electron donors. The structural basis for the difference between these two states has remained elusive, and is addressed here using computational methodology. The results support the notion that CuB[II] is either three-coordinated in the OH state or shares an OH− ligand with heme a3 in a strained μ-hydroxo structure. Relaxation to state O is initiated by hydration of the binuclear site. The redox potential of CuB is expected, and found by density functional theory calculations, to be substantially higher in the OH state than in state O. Our calculations also suggest that the neutral radical form of the cross-linked tyrosine in the binuclear site may be more significant in the catalytic cycle than suspected so far.
The respiratory chain in the mitochondria of all eukaryotes and many prokaryotes terminates in cytochrome c oxidase (CcO), which conserves free energy from the reduction of O2 to water by linking it to proton pumping across the mitochondrial or bacterial membrane (1, 2). Oxygen reduction takes place at a binuclear center (BNC) consisting of a heme iron (heme a3) and a copper ion (CuB) (Fig. 1 A and B) (1–4). Electrons from cytochrome c are supplied to this site in succession via two other metal sites, CuA and heme a (1, 2). Protons for consumption at the BNC, or to be pumped across the membrane, are taken from the negatively charged N-side of the membrane via the proton transfer pathways D and K, named after conserved aspartate and lysine residues, respectively (1–4). The D pathway has been proposed to be responsible for transferring all of the pumped protons, plus 2–3 “chemical” protons for consumption in O2 reduction, whereas the K pathway is suggested to carry the remaining 1–2 chemical protons to the BNC (5). The D pathway terminates at a highly conserved glutamic acid residue in subunit I, E242 (numbering based on the bovine heart enzyme). From here the protons to be pumped across the membrane are suggested first to be carried to a pump site (also called a proton-loading site, PLS) in a redox-dependent manner (6–10). The proton at the PLS is generally agreed to be ejected to the positively charged P side of the membrane by electrostatic repulsion from uptake of a chemical proton into the BNC, as suggested independently by Morgan et al. (11) and Rich (12).
Fig. 1.

(A and B) The BNC of CcO from Bos taurus (PDB ID code 1V54). The T-shaped three-coordinate structure (A) and near planarity (B) of the CuB center are shown. The copper, its histidine ligands, heme, and cross-linked tyrosine are displayed in orange, blue, gray, and green, respectively. (C) The proposed catalytic cycle is drawn clockwise. The oxidation states of Fe, CuB, and the cross-linked tyrosine in the BNC are shown, along with protonation states of the cross-linked tyrosine and metal ligands. The name of each state is shown in the upper right corner of each yellow rectangle. Blue arrows show proton pumping, whereas electrons and protons consumed at the BNC are shown in red and black, respectively. O2 binds to the BNC in the R→A transition.
The main features of the catalytic cycle of CcO are fairly well established (Fig. 1C). Low-temperature work by Chance et al. (13) identified the O2 adduct of heme a3 (compound A), and partial reversal of the catalytic reaction in mitochondria (14) helped to establish the subsequent intermediates P and F, both of which turned out to have heme a3 in the ferryl state (1, 2, 15, 16). A conserved tyrosine in the BNC is covalently bonded to one of the three histidine ligands of CuB, and forms a neutral tyrosine radical in state PM (Fig. 1 A and C) (2). The PR state differs from PM in having one more electron in the BNC (Fig. 1C and Fig. 2). PR is formed directly after state A when the fully reduced (thus the R subscript) enzyme reacts with O2, and its structure may formally be written as shown in Fig. 1C (2, 17, 18). The next state of the BNC, called F (or FH here, Fig. 1C), differs from PR only by an additional proton, by which the unusual electron paramagnetic resonance (EPR) characteristics of the PR state are lost (2, 19, 20). Recent infrared experiments have suggested that the cross-linked tyrosine in the BNC is anionic in both states PR and F (21), on which basis we concluded that the extra proton in the F state is used to form water from the OH− ligand of CuB (2, 17). Consequently, this water molecule is often formally drawn as a ligand of CuB in state F (17), but it may well dissociate from the copper (in which case we call it state FH here; Fig. 2), which is one of the issues analyzed in the current work.
Fig. 2.

Sequence of events leading to the formation of states OH and O. The protonation of the –OH ligand of CuB in PR leads to the formation of an activated state FH, in which Cu is three-coordinated. Subsequent reduction and protonation of the BNC forms state OH, which has a strained μ-hydroxo–bridged structure (dashed line indicates a weak bonding between CuB and the –OH ligand of Fe; Table S2). Diffusion of a water molecule toward CuB, structural relaxation (expansion) of the active site, and hydrogen bonding rearrangements (all depicted as a dashed vertical arrow) would lead to the formation of O from OH.
The Metastable OH State
Further transfer of one electron and one proton to the BNC in the ferryl/cupric state F leads, in addition to translocation of one proton across the membrane (2), to formation of the ferric/cupric state. The ferric/cupric form of the BNC of “as-isolated” CcO (often called O, for oxidized) cannot be a true catalytic intermediate for several independent reasons (see below in this section), and must therefore be preceded by a metastable activated form of the ferric/cupric site (22, 23), which is denoted OH here. Further reduction of state O is sluggish and not coupled to proton translocation (24, 25), whereas the catalytically relevant one-electron reduction of the activated OH state is much faster, and at least 2–3 orders of magnitude faster than the decay of OH into O (24, 25). Two more observations show that state O cannot be the catalytically active form. First, electron transfer from cytochrome c (Em,7 ∼ 270 mV) to the BNC during reduction of the O state could hardly be coupled to effective translocation of 2 charges/e− against a proton electrochemical gradient of at least 0.15 V across the membrane due to the low redox potential (Em,7 ≤ 0.4 V) of state O (2, 26). Second, the sum of the individual free energy changes of the partial reactions of the catalytic cycle, determined at equilibrium, falls considerably short of the overall free energy change of the net reaction, viz. that of O2 reduction to water (2, 26). Again, this shortfall is due to the low Em,7 values of the O→E and E→R transitions found in equilibrium titrations. All these observations suggest involvement of a metastable activated form of the BNC (OH) during turnover that has a high Em,7, and that decays into a lower energy state O when an electron donor is lacking (e.g., during enzyme isolation).
It has turned out to be difficult to trap the metastable OH state to study its structure, lifetime, and how it differs from state O (27, 28). However, electron photo-injection experiments indeed showed that CuB has a raised redox potential in the OH state: the Em,7 is at least 100 mV higher than the corresponding potentials of hemes a and a3 (25, 29). This observation is interesting, because according to anaerobic redox titrations the Em,7 values of heme a, CuB, and ferric/ferrous heme a3 for adding the first electron to enzyme in state O are all similar, and do not exceed 0.4 V (30)(see also ref. 2). It matches the expectation that the OH state has a raised redox potential and suggests that this is due to CuB. The lifetime of the OH state (in the absence of an electron donor) can be estimated from such experiments, but not very accurately due to technical difficulties, giving a range from 0.2 s to 5 min.
Resonance Raman spectroscopy studies by Rousseau and coworkers (31) have established an intermediate beyond the F state, but before state O. The observed stretching frequency at 450 cm−1 is indicative of a distal heme iron-bound hydroxide that is strongly hydrogen-bonded. The data also showed that the ferric heme iron is high spin in this state, which is consistent with the strongly hydrogen-bonded distal hydroxide. A state with hydroxide-liganded high-spin ferric heme a3 is therefore a possible candidate for state OH (17), as concluded originally (31), and this is the rationale for the subscript H (but see footnote in Table 1). The half-life of the 450 cm−1 species was ∼1 ms (31), which is too short for the OH state. However, this short lifetime may only be apparent, because of exchange of the isotopically labeled hydroxide ligand with water (31). Preliminary data by Han et al. (31) indicated that the ferric hydroxide species was no longer present on a seconds time scale, which would considerably reduce the upper lifetime limit for OH if identical to the ferric hydroxide species, and also tends to exclude a hydroxide ligand of heme iron in the O state. Early spectroscopic data indicated that heme a3 iron is six-coordinated high-spin ferric in state O (32), which suggests that the distal ligand is water.
Table 1.
Energies (E) of various model systems in different spin states (in kcal/mol)
| Models | E | Models | E |
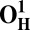 *
*
|
16.1 | O1 | 0.2 |
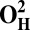 |
28.2 | O2 | 0 |
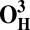 |
19.4 | O3 | 3.8 |
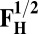 |
5.0 | F1/2 | 0 |
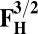 |
5.0 | F3/2 | 0 |
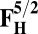 |
17.8 | F5/2 | 14.71 |
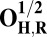 |
6.9 | 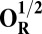 |
0 |
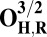 |
29.2 | 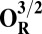 |
25.1 |
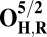 |
13.8 | 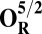 |
9.2 |
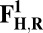 |
2.5 | 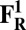 |
1.7 |
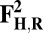 |
14.6 | 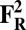 |
24.3 |
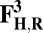 |
47.8 | 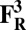 |
0 |
The energies (E) reported in the table are obtained at the B3LYP (Becke, three-parameter, Lee-Yang-Parr)/def2–TZVP (triple zeta valence plus polarization)/dielectric constant ε = 4 level of theory. The energies of all OH and O, FH ,and F, OH,R and OR, and FH,R and FR spin states are relative to the corresponding lowest energy O, F, OR, and FR states (i.e., O2, F1/2, 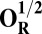 , and
, and 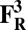 ).
).
The H subscript in OH state denotes the hydroxyl ligand of heme a3 Fe (see The Metastable OH State). For all other states, subscript H is retained for consistency and to distinguish them from “inactivated” four-coordinated states (O, F, OR, and FR).
Rationale
Taken together, the experimental evidence suggests that the BNC in the catalytically relevant OH state may have high-spin ferric heme a3 with a strongly hydrogen-bonded distal hydroxide ligand, and cupric CuB with a raised redox potential. The cross-linked tyrosine is unprotonated (21) so that the cupric CuB will have either a water molecule as the fourth ligand or no fourth ligand at all. The reason for this is that the oxidation of the fully reduced enzyme is coupled to net uptake of no more than two protons, which occurs in the steps P→F and F→O (Fig. 1C). The current work was spurred by the observation of a near-planar T-shaped geometry for CuB (Fig. 1 A and B) with its three nitrogenous histidine ligands (3, 4), by the known rather T-shaped tris-histidyl copper (CuH) coordination in the enzyme peptidylglycine α-hydroxylating monooxygenase (33), and by the recognition of the fact that inorganic cuprous complexes prefer a three-coordinated planar geometry, whereas cupric complexes prefer a higher coordination number (34–36). Due to this preference, a near-planar three-coordinated form of CuB[II] would be expected to have a much higher Em,7 than the four-coordinated variant, a prediction that is reminiscent of an earlier “rack” hypothesis (36). If so, the requirements of a raised Em,7 of the OH state and the experimental observation of an elevated Em,7 of CuB in that state would all be satisfied. We therefore decided to test this hypothesis using computational methodologies.
Results
Based on the above rationale, the OH/O difference might simply relate to the absence/presence of a fourth oxygenous ligand of CuB. To test this possibility we constructed model systems corresponding to the two states (OH and O), and their precursors FH and F, as described in Methods (SI Methods). We then systematically compared the properties of the binuclear site structures in these states. In addition, we calculated redox potentials—that is, we gathered information on the relative electron affinities of these states by analyzing the corresponding structures with an additional electron (i.e., FR, OR vs. FH,R, OH,R), but before subsequent protonation. These latter states may be considered putative intermediates between states F(FH) and O(OH), and between O(OH) and E(EH), in the catalytic cycle (Fig. 1C). The total charge and spin of the model systems tested are listed in Table S1, and Table 1 lists the calculated energies.
The constructed OH states are consistently found to be 12–19 kcal/mol higher in energy than the corresponding O states (Table 1). Neglecting zero-point vibrational contributions, and assuming an entropy loss of 3 kcal/mol upon binding of a water molecule to CuB[II] (37) in state O (Fig. 2 and Fig. S1), the OH state is still 9–17 kcal/mol higher in free energy than the O state.
Geometric analysis of the optimized structures of the different redox states (Table S2) shows a root mean square deviation of ≤0.7 Å after superposition, which suggests high overall structural similarity. In most of the states where CuB lacks a fourth oxygenous ligand (OH, FH, OH,R, FH,R), the copper attains near-planar geometry with the dihedral angle NδHis244–NεHis291–NεHis290–CuB approaching 10°, which is about 10° smaller than what is observed in the four-coordinate O, F, OR, and FR states (Table S2). The Fe–O bond lengths in the O and OH states, and in the F and FH states (Table S2), are also in agreement with previously reported bond lengths in similar systems (38–40).
Redox potentials calculated for the lowest energy states are listed in Table 2. Even though such density functional theory (DFT) calculations are challenging (41, 42), our data systematically suggests a higher redox potential of the BNC in the activated OH and FH states relative to the corresponding O and F states. It is noteworthy that this effect is much larger for the O/OH→OR/OH,R transitions (∼0.43 V) than for the corresponding transitions of states F/FH into FR/FH,R (∼0.18 V). Analysis of the optimized geometries of the O states provides additional reasons for their lower redox potential. In the low energy O2 and O3 states, the hydrogen on the water molecule ligating the CuB ion (H2) shifts toward the distal hydroxyl ligand of Fea3 (Fig. S1, see also Table S2) in such a way that the formal structure should be described as Fe[III]–OH2, CuB[II] –OH−. The OH− ligand of CuB is the likely cause for the lowered redox potentials (Table 2). Such a scenario is not possible in the OH states, where no water molecule ligates the CuB ion, which thereby maintains planarity and a high redox potential (but see below in this section). In support of our computational results, elevated Cu(II)/Cu(I) redox potentials, ≥0.7 V vs. normal hydrogen electrode (NHE), are in fact known for rare examples where synthetic coordination complexes with copper ion are found to be ligated by three neutral nitrogenous ligand donors in a three-coordinate near-planar coordination geometry (35, 43, 44).
Table 2.
Redox potentials (Eo) of different redox couples (in volts)
| Redox couple* | Eo† |
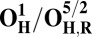 |
0.037 |
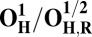 |
0.338 |
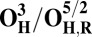 |
0.180 |
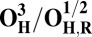 |
0.481 |
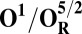 |
−0.452 |
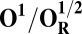 |
−0.053 |
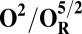 |
−0.460 |
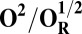 |
−0.060 |
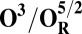 |
−0.293 |
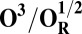 |
0.105 |
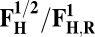 |
0.144 |
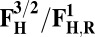 |
0.146 |
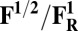 |
−0.037 |
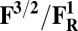 |
−0.036 |
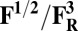 |
0.034 |
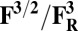 |
0.036 |
Redox potentials (Eo) are evaluated for the low energy spin states of reduced/oxidized forms in Table 1.
Energies used in evaluating the redox potentials are calculated at the B3LYP/def2–TZVP/ε = 4 level of theory. It is especially noteworthy that these are the midpoint redox potentials without charge-compensating protonation.
Analysis of the electronic structures of the OH and O states also reveals critical differences. The extensive delocalized spin density on the cross-linked tyrosine in the OH states is markedly different from the near absence of such spin density on the same moiety in the O states (Fig. S2 and Table S3). Consequently (although not shown in Fig. 1C), the CuB system in the OH state may have significant characteristics of a cuprous/neutral tyrosyl radical state, which may contribute to its higher free energy and higher redox potential. Overall, analyses of energies (Table 1), redox potentials (Table 2), and geometrical data (Table S2) together suggest that the likely reasons for the higher free energy and higher redox potential of the OH state relative to state O are geometrical as well as electronic in nature.
To understand why the metastable OH state would preferentially form in the catalytic cycle, instead of the lower energy O state, we also analyzed the preceding F state of the catalytic cycle, as well as its one-electron reduced derivative (FR). In contrast to the large energy differences obtained between the OH and O states, the difference between the F and FH states is much smaller, ca. 5 kcal/mol (Table 1), and only around 2–3 kcal/mol when entropic effects are included (37). Due to near degeneracy of the FH and F states, they would be equally favored from a thermodynamic viewpoint. However, the higher redox potential of the FH state (Table 2), and hence the higher driving force, may render it kinetically more competent than F in accepting the electron. Geometric analysis (Table S2) suggests that the water molecule is indeed weakly bonded to the CuB ion in different spin states of F, with d(Cu–O) ∼ 2.2 Å (45, 46). In contrast, in forming the O state, the copper-bound water is effectively stabilized as a strongly bonded hydroxide ligand due to proton transfer to the distal OH− ligand of iron, as depicted in Fig. 2, Fig. S1, and Table S2.
The spin density analysis of the O/F and OH/FH states and their one-electron–reduced counterparts, OR/FR and OH,R/FH,R, revealed some further interesting details. Upon one-electron reduction of OH to OH,R, the electron transfer primarily occurs to the neutral tyrosine radical, whereas it takes place to CuB[II] in the case of reduction of state O (Table S3). This is likely one of the reasons for the large redox potential difference observed between the redox couples OH/OH,R and O/OR (Table 2). Similarly, there is more spin density on the active site tyrosine in the two low-energy FH states, relative to the corresponding spin states of F (Table S3). Hence, FH may at least in part be characterized as Fe[IV] = O Cu[I], Tyr-O*, where the copper is reduced and the tyrosine is in the form of the neutral radical. When this state receives an electron to yield FH,R, the lowest energy states all have contributions from Cu[I] and Tyr-O−, indicating that a significant part of this process has been the transfer of the electron to the tyrosine (Table S3). In contrast, the lowest energy FR state is one where part of the reduction has occurred on the heme iron (Fe[III]–OH− Cu[II] –OH−, TyrO−), allowing local protonation of the oxo ligand by the water ligand of the copper.
The above results are in good agreement with the expected structure of state OH, but a detailed structural analysis yields an unexpected result. In the OH3 state the heme iron is slightly pulled out of the heme plane in the distal direction, and the copper is also pulled toward the iron, so that a weak bond may be formed between the ferric OH ligand and the copper (Fig. 2 and Table S2). Such a μ-hydroxo bridge between iron and copper has been anticipated in the past for the BNC, and model compounds of this kind have been produced (47, 48). Note that this state no longer conforms to the idea of three-coordinate cupric copper, nor to a planar copper geometry, and that the distance between the proximal histidine nitrogen and heme iron has lengthened considerably (Table S2 and Discussion).
The above analysis does not address the issue pertaining to the lifetime of the OH state. To be coupled to proton translocation, the OH state must receive an electron to form EH before it relaxes to the O state. The OH→O relaxation would hence have to have an activation energy barrier of at least ∼16 kcal/mol at 310 K. According to our model of OH, the relaxation barrier would comprise at the least hydrogen-bonding rearrangements as well as diffusion of a water molecule (49) toward CuB. Structural rearrangements (50) may further be required, and the calculated optimum structure of the OH3 state indeed exhibits a much tighter BNC than those found in the crystal structures (3, 4, 51), the Fe–Cu distance being only 4.01 Å (Table S2). The thermodynamic costs associated with the H-bonding and structural rearrangements are likely to contribute to the lifetime of OH (SI Methods), which would be significantly longer if a water molecule is recruited from outside the catalytic cavity (49). Finally, when considering the lifetime of the OH state and its relaxation to state O, it should be noted that the latter could be a catalytically relevant state in which the proton-translocating efficiency is reduced, for example under conditions of a high protonmotive force (8).
Discussion
The structural basis of the heterogeneity of the oxidized CcO, especially the difference between the form encountered immediately after oxidation of the reduced enzyme by O2 (OH) and the as-isolated form (O), has so far escaped recognition. A previous spectroscopic study on the bovine heart enzyme revealed no difference between the two states (27). However, in that study the observed rate of reduction of the BNC in the presumed OH state was much slower than reported in ref. 52 for the bovine and in ref. 29 for the Paracoccus enzyme.
Based on the known chemistry of inorganic copper complexes and the available experimental observations, we have here considered the simple hypothesis that state OH may be a dehydrated form of state O such that CuB[II] is three-coordinated planar in the former case, but has a fourth oxygenous ligand in the latter. This hypothesis was tested by DFT calculations.
With the exception of unlikely S = 0 states, and after correction for an entropy change due to water dissociation, the calculated free energy of the OH states lies ca. 9–17 kcal/mol higher than for states O (Table 1), in good agreement with OH being an “activated” metastable form of the ferric/cupric center. Moreover, due to trigonal near-planar geometry of CuB, the cuprous form is favored over the cupric, and the redox potential of the OH/OH,R couple is much higher than for the four-coordinated O/OR couple (Table 2). It is important to note that these calculated redox potentials differ substantially from the experimentally determined values (26) due to the fact that the latter are equilibrium potentials where charge compensation by protonation has occurred. The calculated OH/OH,R redox potentials are consistently ∼0.4 V higher than for the O/OR redox couples, which is in accord with the experimental observations of an elevated redox potential of CuB in the OH state (29, 52). The 0.4 V higher redox potential of the activated form solves the dilemma, at least in part, of an apparently far too low redox potential of the O→E transition to be consistent with proton pumping, and with the overall energetics of the catalytic cycle (2) (see The Metastable OH State).
The notion of a trigonal near-planar CuB in the FH and OH states has additional interesting corollaries. Electron transfer to the BNC in the PM state is primarily expected to yield PR (Fig. 1C), even though this has not been explicitly shown in experiments (2, 17). Instead, PR is known to form directly from intermediate A in experiments where the fully reduced enzyme reacts with O2 (opening paragraphs); at the same time, the PLS is loaded by an internal proton transfer from E242 (2) (not shown in Fig. 1C). The notion of a neutral radical form of the cross-linked tyrosine is strong for state PM (17), and the electron transfer to this state reduces the radical to the tyrosinate anion in PR: Resonance Raman, EPR, and infrared spectroscopic data prove that PR has ferryl heme iron, cupric copper, and tyrosinate, respectively (see references in refs. 2, 17). PR is well known to be followed by proton uptake from the solution to form state F. Here, our DFT calculations suggest that if the water molecule formed in this reaction dissociates from CuB (forming FH; Figs. 1C and 2), the tyrosine tends once again to have some characteristics of a neutral radical (Table S3). The next electron input would in part occur to this radical (to yield FH,R), followed by net proton uptake to yield the OH state. Even in state OH at least part of the population seems to have reduced CuB and a neutral tyrosine radical (Table S3). Hence, the OH state may differ from O, not only with regard to the coordination of CuB but also with respect to the redox nature of the tyrosine and copper. Altogether, our results suggest a more central role in catalysis of the neutral radical state of the cross-linked tyrosine than previously expected.
FTIR data (21) suggested that the cross-linked tyrosine is a deprotonated tyrosinate in both the F and O states. The proton taken up into the BNC upon reduction of state F is thus likely to form a ferric heme a3 hydroxide (Fig. 2), which agrees very well with the resonance Raman data of Han et al. (31), according to which the hydroxide ligand is strongly hydrogen-bonded to yield the Raman resonance at 450 cm−1 and a high-spin ferric heme (S = 5/2). We assume that the hydrogen bonding is to one or two water molecules between the hydroxide and the tyrosinate (51) (Fig. S1). Interestingly, the most likely states corresponding to our view of OH have high-spin ferric heme iron (S = 5/2) and CuB[II] (S = 1/2), for which we have calculated the ferromagnetically coupled form (S = 3; Table S3), but the antiferromagnetically coupled form (S = 2) is likely to have very similar energetics and structural properties (Table S4). Unexpectedly, this form of OH appears to be a μ-hydroxo–bridged compound with a very compact BNC. Thus, CuB actually has a weak fourth ligand that it shares with the high-spin heme iron, the copper geometry is no longer planar, the iron is pulled out of the heme plane on the distal side, and the distance between Fe and the proximal histidine nitrogen is very long (Table S2). In this case our original hypothesis of a three-coordinated CuB in the OH state fails, and this state is instead “strained” with a very short distance between Fe and Cu. The heme iron appears to be five-coordinated as anticipated from μ-hydroxo–bridged models of the BNC (47, 48). However, the Raman bands typical of five-coordinated high-spin hemes (53) are not observed in the spectra of the ferric heme a3 hydroxide intermediate described by Han et al. (31), which suggest six-coordination in that state. The true structure of state OH may thus be a compromise where the bond still persists between the iron and the proximal histidine nitrogen.
The calculations presented here do not address the existence of a similar activated E state. The experimental evidence for EH is not as strong as for OH (24, 25), and part of the driving force for the proton pumping E/EH→R reaction is likely to emerge from the exergonic R→PM transition (∼–10 kcal/mol based on ref. 54).
In summary, the primary finding here is that a subtle change in the hydration of the BNC could account for the difference between the metastable catalytically active OH state and its low-energy counterpart, state O. The high energy of OH and high redox potential of CuB in that state may stem from a three-coordinated near-planar geometry of CuB[II]. Alternatively, the BNC may be compact in state OH with a very short Fe–Cu distance, a μ-hydroxo ligand bridging the two metals, and Cu pulled out from the plane of its nitrogen ligands. Finally, our DFT data suggest that a contributing reason for the high redox potential of the FH and OH states may be that the tyrosine has some characteristics of the neutral free radical, and that this state of the cross-linked tyrosine is more important than considered previously. Validation or falsification of our hypothesis requires further spectroscopic studies of the OH state. It may be of particular interest to search for Raman evidence for a lengthened Fe–N bond to the proximal histidine, to test whether the lifetime of the ferric heme a3-hydroxide species (31) extends beyond the 100 ms domain, and whether a partial tyrosine radical/cuprous copper feature can be distinguished by EPR. An extended X-ray absorption fine structure (EXAFS) study of the ligands of CuB would also be of interest, but would require a heme-copper oxidase variant in which the CuA center is lacking.
Methods
The details of DFT calculations are presented in SI Methods. Figures in Supporting Information show DFT-optimized structures and spin-density distribution in relevant redox states. Tables in Supporting Information comprise description of model systems, their energies, geometric data and spin-density analysis.
Supplementary Material
Acknowledgments
M.W. acknowledges helpful discussions with Drs. Gerhard Hummer [National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH)] and Denis Rousseau (Albert Einstein College of Medicine). Center for Scientific Computing, the Finnish IT Center for Science, is acknowledged for computational resources. This work was supported by grants from the Academy of Finland, Biocentrum Helsinki, and the Sigrid Jusélius Foundation. V.S. acknowledges funding from the European Research Council project CROWDED-PRO-LIPIDS. K.D.K. acknowledges financial support from NIH (R01GM060353).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220379110/-/DCSupplemental.
References
- 1.Ferguson-Miller S, Babcock GT. Heme-copper terminal oxidases. Chem Rev. 1996;96(7):2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Kaila VRI, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110(12):7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 3.Yoshikawa S, et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280(5370):1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 4.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376(6542):660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 5.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94(17):9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wikström M, Krab K. Proton-pumping cytochrome c oxidase. Biochim Biophys Acta. 1979;549(2):177–22. doi: 10.1016/0304-4173(79)90014-4. [DOI] [PubMed] [Google Scholar]
- 7.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2003;1604(2):61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 8.Wikström M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta. 2004;1655(1-3):241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Popović DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome C oxidase. J Am Chem Soc. 2004;126(6):1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 10.Popović DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Lett. 2004;566(1-3):126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Morgan JE, Verkhovsky MI, Wikström M. The histidine cycle: A new model for proton translocation in the respiratory heme-copper oxidases. J Bioenerg Biomembr. 1994;26(6):599–608. doi: 10.1007/BF00831534. [DOI] [PubMed] [Google Scholar]
- 12.Rich PR. Towards an understanding of the chemistry of oxygen reduction. Aust J Plant Physiol. 1995;22(3):479–486. [Google Scholar]
- 13.Chance B, Saronio C, Leigh JS., Jr Functional intermediates in reaction of cytochrome oxidase with oxygen. Proc Natl Acad Sci USA. 1975;72(4):1635–1640. doi: 10.1073/pnas.72.4.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wikström M. Energy-dependent reversal of the cytochrome oxidase reaction. Proc Natl Acad Sci USA. 1981;78(7):4051–4054. doi: 10.1073/pnas.78.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weng LC, Baker GM. Reaction of hydrogen peroxide with the rapid form of resting cytochrome oxidase. Biochemistry. 1991;30(23):5727–5733. doi: 10.1021/bi00237a014. [DOI] [PubMed] [Google Scholar]
- 16.Proshlyakov DA, et al. Selective resonance Raman observation of the “607 nm” form generated in the reaction of oxidized cytochrome c oxidase with hydrogen peroxide. J Biol Chem. 1994;269(47):29385–29388. [PubMed] [Google Scholar]
- 17.Wikström M. Active site intermediates in the reduction of O(2) by cytochrome oxidase, and their derivatives. Biochim Biophys Acta. 2012;1817(4):468–475. doi: 10.1016/j.bbabio.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Brzezinski P, Gennis RB. Cytochrome c oxidase: Exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40(5):521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan JE, Verkhovsky MI, Palmer G, Wikström M. Role of the PR intermediate in the reaction of cytochrome c oxidase with O2. Biochemistry. 2001;40(23):6882–6892. doi: 10.1021/bi010246w. [DOI] [PubMed] [Google Scholar]
- 20.Blair DF, Witt SN, Chan SI. Mechanism of cytochrome c oxidase-catalyzed dioxygen reduction at low temperatures. Evidence for two intermediates at the three-electron level and entropic promotion of the bond-breaking step. J Am Chem Soc. 1985;107(25):7389–7399. [Google Scholar]
- 21.Gorbikova EA, Wikström M, Verkhovsky MI. The protonation state of the cross-linked tyrosine during the catalytic cycle of cytochrome c oxidase. J Biol Chem. 2008;283(50):34907–34912. doi: 10.1074/jbc.M803511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antonini E, Brunori M, Colosimo A, Greenwood C, Wilson MT. Oxygen “pulsed” cytochrome c oxidase: Functional properties and catalytic relevance. Proc Natl Acad Sci USA. 1977;74(8):3128–3132. doi: 10.1073/pnas.74.8.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belevich I, Verkhovsky MI. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxid Redox Signal. 2008;10(1):1–29. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- 24.Verkhovsky MI, Jasaitis A, Verkhovskaya ML, Morgan JE, Wikström M. Proton translocation by cytochrome c oxidase. Nature. 1999;400(6743):480–483. doi: 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- 25.Bloch D, et al. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci USA. 2004;101(2):529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wikström M, Verkhovsky MI. Towards the mechanism of proton pumping by the haem-copper oxidases. Biochim Biophys Acta. 2006;1757(8):1047–1051. doi: 10.1016/j.bbabio.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Jancura D, et al. Spectral and kinetic equivalence of oxidized cytochrome C oxidase as isolated and “activated” by reoxidation. J Biol Chem. 2006;281(41):30319–30325. doi: 10.1074/jbc.M605955200. [DOI] [PubMed] [Google Scholar]
- 28.Brudvig GW, Stevens TH, Morse RH, Chan SI. Conformations of oxidized cytochrome c oxidase. Biochemistry. 1981;20(13):3912–3921. doi: 10.1021/bi00516a039. [DOI] [PubMed] [Google Scholar]
- 29.Belevich I, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci USA. 2007;104(8):2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorbikova EA, Vuorilehto K, Wikström M, Verkhovsky MI. Redox titration of all electron carriers of cytochrome c oxidase by Fourier transform infrared spectroscopy. Biochemistry. 2006;45(17):5641–5649. doi: 10.1021/bi060257v. [DOI] [PubMed] [Google Scholar]
- 31.Han S, Takahashi S, Rousseau DL. Time dependence of the catalytic intermediates in cytochrome c oxidase. J Biol Chem. 2000;275(3):1910–1919. doi: 10.1074/jbc.275.3.1910. [DOI] [PubMed] [Google Scholar]
- 32.Wikström M, Krab K, Saraste M. Cytochrome Oxidase: A Synthesis. London: Academic; 1981. [Google Scholar]
- 33.Chufán EE, et al. Differential reactivity between two copper sites in peptidylglycine α-hydroxylating monooxygenase. J Am Chem Soc. 2010;132(44):15565–15572. doi: 10.1021/ja103117r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Himes RA, Park GY, Siluvai GS, Blackburn NJ, Karlin KD. Structural studies of copper(I) complexes of amyloid-beta peptide fragments: Formation of two-coordinate bis(histidine) complexes. Angew Chem Int Ed Engl. 2008;47(47):9084–9087. doi: 10.1002/anie.200803908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Himes RA, Park GY, Barry AN, Blackburn NJ, Karlin KD. Synthesis and X-ray absorption spectroscopy structural studies of Cu(I) complexes of histidylhistidine peptides: The predominance of linear 2-coordinate geometry. J Am Chem Soc. 2007;129(17):5352–5353. doi: 10.1021/ja0708013. [DOI] [PubMed] [Google Scholar]
- 36.Gray HB, Malmström BG, Williams RJP. Copper coordination in blue proteins. J Biol Inorg Chem. 2000;5(5):551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 37.Dunitz JD. The entropic cost of bound water in crystals and biomolecules. Science. 1994;264(5159):670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 38.Wirstam M, Blomberg MRA, Siegbahn PER. Reaction mechanism of compound I formation in heme peroxidases: A density functional theory study. J Am Chem Soc. 1999;121(43):10178–10185. [Google Scholar]
- 39.Heimdal J, Rydberg P, Ryde U. Protonation of the proximal histidine ligand in heme peroxidases. J Phys Chem B. 2008;112(8):2501–2510. doi: 10.1021/jp710038s. [DOI] [PubMed] [Google Scholar]
- 40.Kaukonen M. Calculated reaction cycle of cytochrome c oxidase. J Phys Chem B. 2007;111(43):12543–12550. doi: 10.1021/jp070578w. [DOI] [PubMed] [Google Scholar]
- 41.Baik M, Freisner RA. Computing redox potentials in solution: Density functional theory as a tool for rational design of redox agents. J Phys Chem A. 2002;106(32):7407–7412. [Google Scholar]
- 42.Roy LE, Jakubikova E, Guthrie MG, Batista ER. Calculation of one-electron redox potentials revisited. Is it possible to calculate accurate potentials with density functional methods? J Phys Chem A. 2009;113(24):6745–6750. doi: 10.1021/jp811388w. [DOI] [PubMed] [Google Scholar]
- 43.Martens CF, et al. Redox behaviour of novel copper(II) crown ether-pyrazole complexes. J Chem Soc Chem Commun. 1993;1:88–90. [Google Scholar]
- 44.Martens CF, et al. X-ray structures and redox properties of copper(II) bis(pyrazole) complexes. Inorg Chem. 1995;34(19):4735–4744. [Google Scholar]
- 45.Chen P, Bell J, Eipper BA, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine alpha-hydroxylating monooxygenase. Spectroscopic definition of the resting sites and the putative CuIIM-OOH intermediate. Biochemistry. 2004;43(19):5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 46.Pettingill TM, Strange RW, Blackburn NJ. Carbonmonoxy dopamine beta-hydroxylase. Structural characterization by Fourier transform infrared, fluorescence, and x-ray absorption spectroscopy. J Biol Chem. 1991;266(26):16996–17003. [PubMed] [Google Scholar]
- 47.Fox S, Nanthakumar A, Wikström M, Karlin KD, Blackburn NJ. XAS structural comparisons of reversible inter-convertible oxo- and hydroxo-bridged heme-copper oxidase model compounds. J Am Chem Soc. 1996;118(1):24–34. [Google Scholar]
- 48.Obias HV, et al. Heterobinucleating ligand-induced structural and chemical variations in [(L)FeIII-O-CuII]+ μ-oxo complexes. J Am Chem Soc. 1998;120(37):9696–9697. [Google Scholar]
- 49.Baron R, McCammon JA. Dynamics, hydration, and motional averaging of a loop-gated artificial protein cavity: The W191G mutant of cytochrome c peroxidase in water as revealed by molecular dynamics simulations. Biochemistry. 2007;46(37):10629–10642. doi: 10.1021/bi700866x. [DOI] [PubMed] [Google Scholar]
- 50.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Designing a functional type 2 copper center that has nitrite reductase activity within α-helical coiled coils. Proc Natl Acad Sci USA. 2012;109(52):21234–21239. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin L, et al. Redox-dependent conformational changes in cytochrome C oxidase suggest a gating mechanism for proton uptake. Biochemistry. 2009;48(23):5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brand SE, et al. A new ruthenium complex to study single-electron reduction of the pulsed O(H) state of detergent-solubilized cytochrome oxidase. Biochemistry. 2007;46(50):14610–14618. doi: 10.1021/bi701424d. [DOI] [PubMed] [Google Scholar]
- 53.Rousseau DL, Ching YC, Brunori M, Giacometti GM. Axial coordination of ferric Aplysia myoglobin. J Biol Chem. 1989;264(14):7878–7881. [PubMed] [Google Scholar]
- 54.Sharma V, Wikström M, Kaila VR. Stabilization of the peroxy intermediate in the oxygen splitting reaction of cytochrome cbb(3) Biochim Biophys Acta. 2011;1807(7):813–818. doi: 10.1016/j.bbabio.2011.02.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.