

Abstract
The presence of tissue specific precursor cells is an emerging concept in organ formation and tissue homeostasis. Several progenitors are described in the kidneys. However, their identity as a true stem cell remains elusive. Here, we identify a neonatal kidney-derived c-kit+ cell population that fulfills all of the criteria as a stem cell. These cells were found in the thick ascending limb of Henle's loop and exhibited clonogenicity, self-renewal, and multipotentiality with differentiation capacity into mesoderm and ectoderm progeny. Additionally, c-kit+ cells formed spheres in nonadherent conditions when plated at clonal density and expressed markers of stem cells, progenitors, and differentiated cells. Ex-vivo expanded c-kit+ cells integrated into several compartments of the kidney, including tubules, vessels, and glomeruli, and contributed to functional and morphological improvement of the kidney following acute ischemia-reperfusion injury in rats. Together these findings document a novel neonatal rat kidney c-kit+ stem cell population that can be isolated, expanded, cloned, differentiated, and employed for kidney repair following acute kidney injury. These cells have important biological and therapeutic implications.
Keywords: C-kit, kidney stem cells, clonogenicity, self-renewal, multipotentiality, regenerative potential
Introduction
The search for putative stem cells or precursors within the kidney has been the focus of extensive research. The identification of a kidney stem cell would provide important biological insights and could therapeutically be utilized to generate new tubular, glomerular, and vascular cells in the treatment of both acute and chronic kidney injuries.
To date several possible kidney stem cell candidates have been described. These include label retaining cells (LRCs) or slow-cycling cells identified by using bromodeoxyuridine (BrdU) pulse-chase analyses. LRCs are detected in proximal tubules, thick ascending limb of Henle, distal tubules, and collecting ducts after a short (2-week) chase period [1] or in the interstitium and in the papillary tubules after a long (2-month) chase period [2,3]. Other stem cell candidates include kidney cells expressing surface markers and found in different locations, such as the interstitium (Sca1) [4,5], Bowman's capsule (CD24, CD133) [6-9], papilla (Nestin,CD133) [10], and proximal tubular compartment (CD24, CD133) [11,12] (or only CD133) [13]. These studies demonstrate multipotentiality in vitro and the capacity of these cells to integrate into the kidney during development or in response to injury.
However, kidney epithelial tubular regeneration has been the subject of intense debate generating multiple hypotheses. Cell-tracking studies using transgenic mice provide strong evidence in favor of an intra-tubular regeneration source, suggesting that differentiated epithelial cells that survive acute injury undergo proliferative expansion [14,15]. More recently, a study involving two-step sequences of nucleotide analogue pulses following murine ischemia-reperfusion injury further suggests an absence of kidney stem cells in the adult kidney [16]. Moreover, telomerase activity-expressing cells were reported in 5% of the LRCs, but are not involved in kidney repair [17].
These studies generated controversy in the field, because they challenged the significance of work from many groups investigating the existence and the role of putative post-natal kidney stem cells. Notably, Lin et al. (2005) and Humphreys et al. (2008 and 2011) do not provide conclusive evidence for the absence of post natal kidney stem cells and they do not eliminate the possibility of a tubular stem cell population, possibly of more limited potency. Those cells derived from the Six-2+ cap mesenchyma or expressing kidney specific-cadherin would be identically labeled in the regenerating tubules. Furthermore, there is evidence that in the renal papilla, LRCs or their immediate progeny are able to proliferate and migrate, as demonstrated in transgenic mice conditionally expressing GFP fused to histone 2B [18]. Additionally, the SDF-1/CXCR4 axis is involved in the papillary LRC activation after acute kidney injury [19].
Studies of other organs have engendered similar controversy. In the pancreas, studies using a transgenic reporter mouse strain showed that the major source of new β-cells during adult life and after pancreatectomy arose from the proliferation of terminally differentiated β-cells rather than from pluripotent stem cells [20]. However, more recently, rare pancreas-derived multipotent precursor cells that form spheres, express insulin and generate multiple pancreatic and neural cell types in vivo were observed in adult human tissue [21]. The presence of differentiation markers was also described in human neuronal stem cells that display morphologic and molecular characteristics of differentiated astrocytes [22].
Expression of c-kit receptor, a tyrosine kinase receptor, is detected in differentiated cells that do not exhibit stem cell properties, such as mast cells, germ cells, melanocytes, gastrointestinal Cajal cells, fetal endothelial cells, and epithelial cells, including breast ductal, sweat gland, some cells of skin adnexa, and cerebellum neurons [23]. However, c-kit+ cells have been described as a marker of stem cells in many organs and tissues, such as bone marrow [24], liver [25], heart [26], amniotic fluid [27], and lungs [28].
C-kit+ cells have also been identified during metanephric mesenchyme (MM) development and the ligand for c-kit, stem cell factor (SCF), is abundantly expressed in the ureteric bud. [29]. Therefore, we hypothesized that c-kit+ cells isolated from neonatal rat kidney could represent a population of stem cells. Here, we show that c-kit+ cells possess the stem cell properties, including self-renewal capacity, clonogenicity, and multipotentiality. Furthermore, they exhibit the potential to treat renal failure by multi-compartment engraftment, e.g., tubular, vascular, and glomerular following acute ischemia-reperfusion injury.
Material and Methods
Explant culture of neonatal rat kidney
Neonatal rat kidneys from Sprague-Dawley (SD; n=6-8) were harvested, chopped, digested with collagenase II, and incubated in red blood cell lysing buffer (Sigma-Aldrich). Expansion medium included Dulbecco's Modified Eagle Medium (DMEM/F12), 20% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/mL streptomycin (Sigma-Aldrich).
Immunopanning and FACS (fluorescence-activated cell sorting)
C-kit+ cells were isolated by immunopanning using rabbit polyclonal c-kit antibody (H300, Santa Cruz) and further selected using FACS (BD FACSAria™, University of Miami). Depletion of hematopoietic stem cells lineage was also performed to ensure that c-kit+ cells came from kidney and that bone marrow-derived cells were removed. Therefore, the APC lineage antibody cocktail was used, which depletes CD3e, CD11b, CD45R/B220, erythroid cells, and Ly-6G and Ly-6C (BD Pharmigem), and the anti-mouse CD117-PE conjugated (eBioscience).
Expansion of c-kit+ cells
Sorted c-kit+/Lin− cells were plated and cultured in DMEM/F12 supplemented with 10% FBS, 10 ng/mL bFGF, 20 ng/mL EGF, 40 ng/mL SCF (stem cell factor) (PeproTech), 10 ng/mL LIF (leukemia inhibitory factor; Millipore), ITS (insulin-transferrin-selenium A liquid media supplement; Invitrogen), and antibiotics.
Quantitative real-time PCR (qPCR) and immunofluorescence
All experiments for c-kit characterization were performed 5-10 passages after sorting. For qPCR, total RNA was extracted from cells using Pure-Link Micro-to-Midi Total RNA Purification System (Qiagen) and reverse-transcribed using High capacity cDNA reverse transcription kit (Applied Biosystems). All samples were treated with Turbo™ DNase (Ambion). qPCR was performed in triplicate using a 20 μl reaction mixture containing 10 ng cDNA, TaqMan Universal PCR Master Mix (Roche) and primers/probes sets for specific genes (TaqMan Gene Expression Assays, Applied Bio systems). As an internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or 18s was determined in each reaction. Reactions conditions were performed according to the manufacturer: 1 cycle of 50°C for 2 minutes, 1 cycle of 90°C for 10 minutes and 40 cycles of 95°C for 15 seconds and 60° for 1 minute. Software from iQ5 multicolor real-time PCR detection system (Bio-Rad) was used for PCR analyses. Relative fold change was calculated by 2ΔΔCt method and compared to baseline values (set at 1). For immunofluorescence, cells were fixed in paraformaldehyde 4%, blocked with BSA 1% and Tween-20 0.5%, incubated with primary antibodies, and then incubated with 488 or 568-conjugated secondary antibodies (Invitrogen).
Fragments of kidney were fixed overnight in neutral buffered formalin 10%, dehydrated in alcohol, and embedded in paraffin. Sections of 4-5μm thickness were stained with hematoxylin and eosin (H&E) and Periodic Acid Schiff (PAS) reagent. For immunofluorescence, renal sections were deparaffinized with xylene and rehydrated in alcohol series and water. The sections were subsequently microwaved twice for 10 min in citrate buffer, and then blocked for 1h in donkey serum. Primary antibodies were applied for 1 hour at room temperature or overnight at 4°C. Incubations for 1 hour using 488 or 568-conjugated secondary antibodies (Invitrogen) were then performed. For GFP staining, an antibody goat or rabbit polyclonal anti-GFP FITC-conjugated (Abcam) was applied for 1 hour at 37°C. Nuclei labeling was obtained with DAPI. After that, slides were incubated with Sudan Black 0.1% (Sigma-Aldrich) at room temperature. In control experiments, the first antibody was omitted.
FACS
A total of 1-2×106 c-kit+ cells were used for FACS characterization. For intra-nuclear markers, cells were fixed in cold 80% ethanol, washed twice with 1 ml PBS during centrifugation for 10 min at 200x g, incubated 1 hour with FACS buffer (1% bovine serum albumin and 5% FBS diluted in distilled water) on ice, and subsequently 1 hour with the primary and secondary antibodies. For intra-cellular markers, we used the BD Cytofix/Cytoperm™ Fixation/Permeabilization kit (BD Pharmigem); for surface markers, periods of incubation with FACS buffer for 1h, and primary and secondary antibodies incubation were also performed. Each analysis included at least 10,000 events and was performed on at least three separate cell preparations.
In vitro differentiation
For endothelial differentiation, c-kit+/Lin− cells were cultured in Endothelial Cell Growth Medium-2 (EGM-2; Lonza) supplemented with 2% FBS, EGF, VEGF, IGF, bFGF, hydrocortisone, ascorbic acid, and heparin for 1-4 weeks. For in vitro Matrigel tube formation assay, cells were trypsinized and plated on 300 μl Matrigel, reduced growth factor (BD Pharmigem). Seeding density was 10×104 cells/well and endothelial cell basal medium without supplements was added in 0.1% bovine serum albumin. Tube formation was analyzed at 2 time points; 6h and 24h. Number of tubes and tube length (μm) were calculated by using image J software. For epithelial differentiation, cells were incubated in DMEM containing 10% FBS, 50 ng/ml bFGF, 20 ng/ml LIF, and 5 ng/ml TGF-β for 3 weeks. For adipogenic differentiation, cells were incubated in DMEM high glucose containing 10% FBS, 1μM dexamethasone, 0.5 μM 1-methyl-3-isobuthylxanthine, 10 μg/mL insulin, and 100 μM indomethacin (Sigma-Aldrich) for 2 weeks. For osteogenic differentiation, cells were cultured in α-MEM and 10% FBS that contained 10-7 M dexamethasone, 0.2 mM ascorbic acid and 10 mM β-glycero-phosphate (Sigma-Aldrich) for 4 weeks. Oil Red O and Alizarin S staining quantification protocols are described in the Supplemental Data. We used green fluorescent protein (GFP) transgenic rat to isolate mesenchymal stem cells (MSCs) from bone marrow and perform mesodermic differentiation as a positive control. For neuronal differentiation, c-kit+ cells were plated on fibronectin-coated dishes at a seeding density of 5×103 cells/cm2 in DMEM/F12 supplemented with 5% FBS and 100 ng/mL bFGF (Pepro Tech) for 2 weeks.
GFP labeling
For cell tracking, c-kit+ cells were transfected with a lentiviral vector carrying the gene encoding GFP. The protocol is described in the Supplemental Data.
In vivo endothelial differentiation
All procedures involving animals were approved by the respective Institutional Animal Care and Use Committees of the University of Miami. C-kit+/Lin− cells were labeled with GFP and cultured in endothelial medium for 1 week. They were then subcutaneously injected into NOD-SCID mice (n=3; 2×106 cells) in a Matrigel plug. Negative control included the Matrigel plug containing only EGM-2 (n=3). Positive control included HUVEC (Human Umbilical Vein Endothelial Cells).
Calcium (Ca2+) transient
Intracellular Ca2+ was measured using the Ca2+-sensitive dye Fura-2 and a dual-excitation spectrofluorometer (IonOptix LLC) and is described in the Supplemental Data.
Sphere-forming assay
Dissociated single c-kit+ cells obtained from c-kit sheets and from clones obtained in 96-well plates were plated on ultra-low attachment 6-well plates (Corning, Costar) at clonal density, 1×103 cells/well, in DMEM-F12 medium containing 20% knockout serum replacement, 10 mM MEM nonessential amino acids, 0.2 mM β-mercaptoethanol (GIBCO), L-glutamine (Sigma-Aldrich), and 20 ng/ml bFGF. Medium was changed every 3 days. After 12 days, cultures were assessed for nephrosphere number. Nephrospheres were defined as free-floating spheres of > 40 μm diameter and results were expressed as a percentage of the plated cells. To assess size and number, spheres were visualized with a Nikon Eclipse TS100 inverted microscopic fitted with a Nikon digital camera image capture system and analyzed with image J software.
Acute kidney ischemia-reperfusion injury (IRI)
Briefly, we applied vascular clamps across both renal pedicles for 35 minutes in female 2 months-SD rats weighing 200-250g (Charles River). After removing the clamps, reperfusion was visually observed. Subsequently, 2×106 cells (GFP- labeled c-kit+ cells or MSCs from GFP-SD) or saline were injected directly into the abdominal aorta above the renal arteries, after application of a vascular clamp to the abdominal aorta below the renal arteries. The needle used for cell injection was 31G, length 8mm (5/16″) insulin syringe needle (BD Biosciences). Cells were then resuspended in 300 μl of saline. The same volume of saline (300 μl) was injected into aorta in the control group. Blood collection was performed at baseline, days 1, 2, 4, and 8 post ischemia-reperfusion injury for creatinine and blood urea nitrogen (BUN) measurement (Products Vitros Chemistry). Kidneys were harvested after 8 days for histological analyses.
Morphologic studies, immunofluorescence, and PCNA (proliferating cell nuclear antigen) index on kidney tissue
Acute tubular necrosis (ATN) was assigned by semi-quantitative analysis of each individual variable (casts, brush border loss, tubular dilation, necrosis, and calcification) to augment the ATN score (maximum 7). Scoring for PCNA-positive cells was carried out by counting the number of positive nuclei in four randomly chosen kidney cortex and outer medulla sections using X20 magnification and after applying a rabbit polyclonal PCNA antibody (Santa Cruz). Data from all fields and all kidneys were pooled to obtain PCNA score.
Statistics
Error bars represent mean ± SEM or mean ± SD. The means of two populations were compared by Student's t-test or Mann-Whitney test. For multiple comparisons, analysis of variance was employed. Differences were considered significant at P<0.05.
Results
Neonatal rat kidneys contain c-kit+ stem cells
Cells expressing the c-kit epitope on their cell surface were widely distributed in the neonatal rat kidney, localized not only to renal papilla (Fig. 1A), but also to the medulla and the nephrogenic zone. These cells expressed E-cadherin (Fig. 1B) and N-cadherin (Fig. 1C). C-kit+ cells were located primarily within a laminin-positive membrane, indicating that they are epithelial cells (Fig. 1D). In contrast, c-kit did not co-localize with Dolichos biflorus agglutinin (DBA) (Fig. 1E), a marker of ureteric bud and its derivates, or with the Na-Cl co-transporter (NCCT/SLC12A3), a distal tubule marker (Fig. 1F). However, c-kit co-localized at the apical membrane of epithelial cells of the thick ascending limb (TAL) of Henle's loop with the Na-K-2Cl co-transporter (NKCC2/SCL12A1) in both nephrogenic cortex (Fig. 1G) and medulla (Fig. 1H). Aquaporin 1 (AQP1) did not co-localize with c-kit (Fig. 1I). C-kit+ cells were not detected in vessels or glomeruli. Importantly, in the adult rat kidney, c-kit+ cells exhibited identical distribution as found in neonatal rat kidney, e.g., co-localization with NKCC2 in the TAL (Fig. S1).
Figure 1. C-kit+ cells are expressed in neonatal rat kidney and can be isolated by FACS.
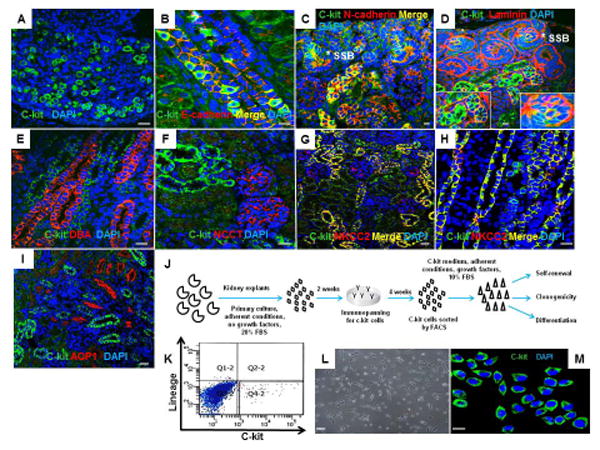
(A) C-kit+ cells in the renal papilla.
(B,C) C-kit+ cells in the medullary rays and nephrogenic cortex exhibiting co-localization with E-cadherin and N-cadherin, respectively.(*) represents S-shaped bodies (SSB).
(D) Section of the nephrogenic cortex costained for c-kit and laminin in SSB (*) and tubules. Insets show regions at higher magnification.
(E,F) C-kit+ cells did not stain for Dolichos biflorus agglutinin (DBA) or Na-Cl co-transporter (NCCT), respectively.
(G,H) Section of the nephrogenic cortex and medulla costained for c-kit and Na-K-2Cl co-transporter (NKCC2) in the thick ascending limb of Henle's loop.
(I) C-kit+ cells did not stain for aquaporin-1 (AQP1).
(J) Schematic of the experimental procedure for c-kit isolation from neonatal rat kidneys.
(K) Isolation of c-kit+/Lin− cells by FACS in the quadrant Q4-2 with depletion of lineage cells.
(L) C-kit+/Lin- cells in monolayer culture on plastic after sorting.
(M) Sorted cells were positive for the c-kit receptor.
Cell nuclei are stained blue with DAPI. Scale bars represent 20μm (A-I, M) and 50 μm (L).
Next, we isolated c-kit+ cells and evaluated their stemness properties in vitro. C-kit+ cells were isolated from neonatal rat kidney explants (n=6-8) by immunopanning and fluorescence activating cell sorting (FACS). These cells were found to be Lin− (lineage cells depleted) (Fig. 1J) and represented 1.1% of the cells (∼0.15%/kidney) (Fig. 1K). These cells exhibited the ability to self-replicate and grow in a monolayer on plastic (Fig. 1L). After sorting, the c-kit epitope remained detectable by immunofluorescence microscopy (Fig. 1M). By FACS, 88.6±5.5% of the cells were positive for c-kit, and this high level of c-kit positivity continued up to 50 passages (76.2± 8.6%) (Fig. S2).
Characterization of c-kit+/Lin− cells
We observed that c-kit+/Lin− cells expressed proteins associated with early stem cells and reprogramming genes, such as Oct4, sex-determining-region Y-box 2 (Sox2), c-myc, and Kruppel-like factor 4 (Klf4) (Fig. 2A and Fig. S3A). Negative control was obtained by omitting the primary antibodies. All those markers were confirmed by immunofluorescence staining (Fig. 2B) and by qPCR (Fig. 2C and Table S1). Vascular (von Willebrand factor-vWF, isolectin, α-actin 2-Acta2), epithelial (ZO-1, NKCC2, NCCT, AQP1), neuronal (nestin and neurofilament-heavy chain, NF-H), and mesenchymal (CD73, CD90, vimentin) markers were also detected (Fig. 2B, C and Fig. S3B). A low percentage of kidney-derived c-kit+/Lin− cells expressed CD24 (<10%), CD133 (∼30%), and Pax2 (∼30%) (Fig. S3B).
Figure 2. Characterization of c-kit+/Lin− cells.
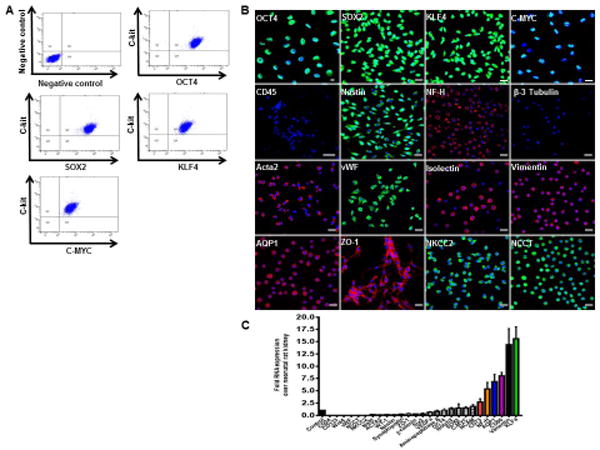
(A) Representative FACS analyses of c-kit, OCT4, SOX2, KLF4, and C-MYC. Negative control was obtained by omitting the primary antibodies.
(B) C-kit+ cells in culture express OCT4, SOX2, KLF4, C-MYC, nestin, NF-H, Acta2, vWF, isolectin, vimentin, ZO-1, NKCC2, AQP1, and NCCT. CD45 and β-3 tubulin are negative in C-kit+ cells.
(C) Gene expression by real-time PCR analyzed by the fold change (2ΔΔCt) in undifferentiated c-kit+ cells over neonatal rat kidney. Error bars represent mean ± SEM.
Cell nuclei are stained blue with DAPI. Scale bars represent 20 μm. Abbreviations: octamer-binding transcription factor (Oct3/4); sex-determining-region Y-box 2 (Sox2); Kruppel-like factor 4 (Klf4), neurofilament heavy chain (NF-H), alpha-actin 2 (Acta2), von Willebrand factor (vWF), zonula occludens-1 (ZO-1), Na-K-2Cl co-transporter (NKCC2), aquaporin-1 (AQP1), and Na-Cl co-transporter (NCCT).
By qPCR, CD73, NF-H, AQP1, CD90, vimentin, and Klf4 expression was at least 2.5 fold higher than neonatal rat kidney (Fig. 2C). C-kit+/Lin− cells were negative for CD45, a marker of hematopoietic cells (Fig. 2B).
C-kit+/Lin− cells were subcultured for more than a year (> 100 passages) without any evidence of senescence or growth arrest (Fig. S4A). Cells frozen at different passages and thawed 6 and 12 months later retained their original characteristics. Similar telomerase activity was detected at different passages of c-kit+/Lin− cells (Fig. S4B). Moreover, c-kit+/Lin− cells exhibited a normal karyotype (Fig. S4C).
Non-clonal c-kit+-derived cells differentiate into mesoderm and neuroectoderm lineages but not into endoderm
To assess their plasticity, c-kit+ cell monolayers were treated for 1-4 weeks with differentiation media to promote adipogenic, osteogenic, neuronal, epithelial, or endothelial differentiation (Fig. 3A). The cells successfully differentiated and expressed markers for these cell types, as assessed by immuno- and -histochemical stainings, and qPCR (Figs. 3B-E).
Figure 3. Non-clonal c-kit+/Lin− cells show multilineage differentiation.
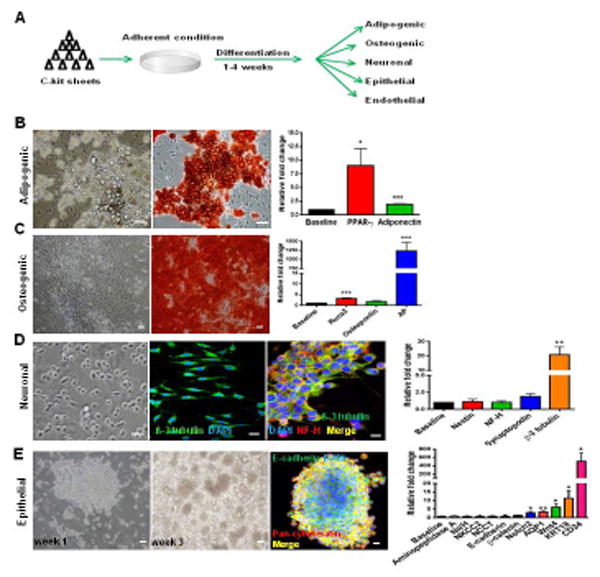
(A) Schematic of the experimental procedure.
(B) Adipogenic differentiation exhibited lipid droplet accumulation, Oil Red O positivity, and PPAR-γ (*P=0.026) and adiponectin (***P<0.0001) up-regulation.
(C) Osteogenic differentiation exhibited Alizarin Red S positivity and Runx2 (***P<0.0001), osteopontin (P=0.077), and alkaline phosphatase (AP;***P<0.0001) up-regulation.
(D) Monolayer of c-kit+ /Lin- cells exhibiting prolongations in neuronal medium. β-3 tubulin was positive and co-localized with NF-H. β-3 tubulin was up-regulated (**P=0.0071).
(E) C-kit+ cells after 3 weeks in epithelial medium formed embryoid body-like structures that stained for E-cadherin and pan-cytokeratin. Notch2 (*P=0.034), AQP1 (**P=0.0056), Wnt4 (*P=0.039), KRT18 (*P=0.028), and CD24 (*P=0.035) were up-regulated.
Cell nuclei are stained blue with DAPI. Scale bars represent 50 μm (B-E) and 20 μm for confocal images (D,E). Error bars represent mean ± SEM.
C-kit+/Lin− cells grown in adipogenic medium for 2 weeks accumulated lipid droplets, that stained positive for Oil-Red O, and up-regulated PPAR-γ and adiponectin (Fig. 3B). Later passage cells continued to show commitment to adipogenic differentiation, although it was less pronounced (Fig. S5A). Mesenchymal stem cells (MSCs), the positive control for mesoderm differentiation, significantly exhibited higher lipid accumulation than c-kit+ early and late passage cells (Fig. S5B). PPAR-γ up-regulation was comparable between c-kit+ cells and MSCs, whereas adiponectin was higher in c-kit+ differentiated cells. (Fig. S5C).
Growing c-kit+/Lin− cells in osteogenic medium for 4 weeks resulted in Alizarin Red S positivity, indicative of mineralization, which correlated with a significant up-regulation of Runx2 and alkaline phosphatase (AP) expression (Fig. 3C). Osteopontin expression was not significantly up-regulated. Later passages also exhibited Alizarin Red S positivity (Fig. S5D). Similar to adipogenic differentiation, MSCs had more Alizarin Red S staining compared to c-kit+ cells, and a greater up-regulation of Runx2 and osteopontin (Fig. S5E). At baseline, AP was expressed at low levels in c-kit+ cells, as opposed to MSCs; after differentiation, however, AP up-regulation was more pronounced in c-kit+ cells (Fig. S5F).
After 2 weeks in the neuronal medium, c-kit+/Lin− cells at low density decreased their proliferation and exhibited prolongations (Fig. 3D). Cells were positive for β-3 tubulin which co-localized with NF-H (Fig. 3D). β-3 tubulin was significantly up-regulated (Fig. 3D).
Epithelial differentiation was induced by growing c-kit+/Lin− cells in medium containing bFGF, TGF-β3 [30], and LIF [31] for 3 weeks. After 1 week in epithelial medium, the morphology of c-kit+/Lin− cells changed and they started to form packed clusters. These clusters detached after 3 weeks and acquired an embryoid body-like morphology (Fig. 3E). Even late passage (P50-52) cells acquired this morphology (Fig. S5G). CD24, cytokeratin 18 (KRT18), Wnt4, Notch2, and AQP1 were all up-regulated (Fig. 3E), suggesting mesenchymal-epithelial transition [32,33]. In these epithelial spheres, E-cadherin co-localized with pan-cytokeratin (Fig. 3E).
C-kit vascular differentiation is associated with functional activity in vitro
Based on the presence of vascular markers, we performed in vitro endothelial differentiation by culturing c-kit+/Lin− cells in endothelial basal medium supplemented with growth factors (VEGF, bFGF, IGF-1, and EGF) for 1-4 weeks. Between the third and fourth week, myotube-like structures appeared (Fig. 4A), which stained for Acta2 and co-stained for vWF (Fig. 4B).
Figure 4. Non-clonal c-kit+/Lin− cells undergo vascular differentiation and start to respond to angiotensin II (Ang II) via AT1a receptor.
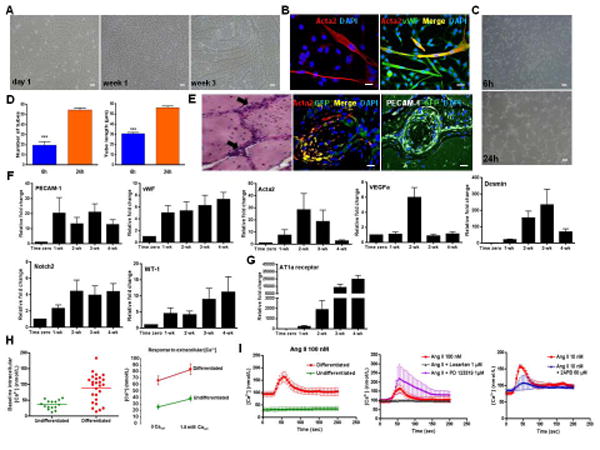
(A) Monolayer of c-kit+ cells in endothelium medium (EGM-2) grown for the indicated period of time. Myotube-like structures were observed after 3 weeks.
(B) Myotube-like structures stained for Acta2 and co-localized with vWF.
(C) In vitro endothelial differentiation assessed by the Matrigel tube formation assay.
(D) Matrigel assay produced more (***P<0.0001) and longer (***P<0.0001) tubes at 24h compared to 6h. Pictures represent three different experiments each done in triplicate.
(E) In vivo vascular differentiation. GFP-labeled c-kit+ cells embedded in Matrigel and injected into NOD-SCID mice. H&E staining of the implants containing c-kit+/Lin− cells showed network connections (arrows). Matrigel plug stained for GFP, Acta2 and PECAM-1 (platelet-endothelial cell adhesion molecule-1).
(F) Relative fold change with time in EGM-2 of PECAM-1 (P=0.055), vWF (P=0.008), Acta2 (P=0.48), VEGFa (P=0.0022), Desmin (P<0.0001), Notch2 (P=0.034), and WT-1 (P=0.0001). qPCR data reflects five different experiments each done in triplicate.
(G) Relative fold change of AT1a receptor with time in EGM-2 (P<0.0001).
(H) Differentiated cells exhibited higher intracellular calcium (Ca2+) concentration at baseline and their response to extracellular Ca2+ was more pronounced compared to undifferentiated cells (P<0.0001).
(I) Differentiated (red line) and undifferentiated cells (green line) responses to Ang II (100 nM) stimulus (P<0.0001). The response to Ang II (red line) was selectively blocked by Losartan (1 μM; blue line) suggesting that Ang II is mediated by AT1a receptor (P=0.0008); AT2 receptor antagonist, PD123319 (1 μM; purple line), had no effect on Ang II response. 2-APB (blue line; 60 μM), a specific blocker of inositol-1,4,5-triphosphate (IP3) receptor, reduced the increase in intracellular Ca2+ caused by Ang II (P<0.0001).
Cell nuclei are stained blue with DAPI. Scale bars represent 50 μm A,C and E (H&E staining) and 20 μm for confocal images (B,E). Error bars represent mean ± SD for (D) and mean ± SEM for (F,G).
Endothelial tubes were observed at two time-points (6h and 24h) by the in vitro Matrigel assay performed on c-kit+/Lin− cells (Fig. 4C). They were more abundant and longer after 24h compared to 6h (Fig. 4D). Both early (P15-20) and late (P50-71) passage cells formed endothelial tubes on Matrigel (Fig. S6A). C-kit+/Lin− cells produced significantly more but shorter tubes compared to MSCs at 24h (Fig. S6B). In vivo endothelial differentiation demonstrated that GFP-labeled c-kit+/Lin− cells embedded in Matrigel formed network-like connections when injected into NOD-SCID (non-obese diabetic severe combined immunodeficiency) mice (Fig. 4E). These connections were positive for Acta2 and PECAM-1 (platelet endothelial cell adhesion molecule-1) (Fig. 4E). HUVEC (human umbilical vein endothelial cells), a positive control, exhibited pronounced connections in the Matrigel plug, while no connections were seen when Matrigel plug containing only EGM-2 was injected (Figs. S6C-F).
qPCR data of in vitro endothelial differentiation showed a time-dependent up-regulation of vWF, VEGFa (vascular endothelial growth factor a), and desmin (P<0.05), a marginal up-regulation of PECAM-1 (P=0.055), and no significant change in the expression of Acta2 (Fig. 4F). Notch2 and WT-1 genes were also time-dependently regulated. Podocytes markers were not expressed.
After growing c-kit+/Lin− cells in EGM-2 for 1 week, they began to express angiotensin II (Ang II) type 1a (AT1a) receptor and its expression increased significantly with time (Fig. 4G). In contrast, Ang II type 2 receptor was not detected after differentiation (Table S1). Calcium (Ca2+) gradient analysis demonstrated higher intracellular Ca2+ concentration in differentiated cells at baseline and their response to extracellular Ca2+ was more pronounced compared to undifferentiated cells (Fig. 4H). Therefore, responsiveness to Ang II was assessed. Differentiated cells exhibited higher depolarization following Ang II administration (100 nM), a response that was selectively blocked by Losartan, but not by PD123319 (Fig. 4I), confirming the involvement of the Ang II type 1a and not Ang II type 2 receptor. Antagonism of inositol-1,4,5-triphosphate (IP3) receptor by 2-aminoethoxydiphenylborane (2-APB; 60 μM) decreased Ca2+ dependent influx from the sarcoplasmic reticulum after Ang II administration [34] (Fig. 4I). The dose-response to Ang II is described in Fig. S7A. The response to endothelin (ET) via ETA and ETB receptors and to prostaglandin F-2α (PGF2α) was more intense in differentiated compared to undifferentiated cells, a response that was attenuated by 2-APB, as well as by the specific antagonists (BQ -123, BQ-788, and SQ 29,548, respectively) (Figs. S7B-J). Notably, extracellular Ca2+ absence attenuated endothelin response in both differentiated and undifferentiated cells (Figs. S7D-E). However, that effect was more pronounced in the undifferentiated cells. The response to bradykinin was more intense in undifferentiated cells and was attenuated by 2-APB (Figs. S7K-L). That response was specifically mediated by B2 receptor, since HOE 140 decreased the Ca2+ influx (Figs. S7M-O). ETA, PGF2α, B2 bradykinin receptors were up-regulated with time in endothelial medium (Fig. S7P). Together these results support the differences in the intracellular Ca2+ and in the responses to extracellular Ca2+ and to vasoactive agents between undifferentiated and differentiated cells.
C-kit+/Lin− cells are clonogenic and c-kit-derived clones exhibit the capacity for multipotent differentiation
To further substantiate the stemness of these cells, we documented clonogenicity. We obtained c-kit+/Lin− single cells by carrying out two serial dilutions in 96-well plates (Fig. 5A). After obtaining one cell per well, we picked three faster proliferating clones and expanded them under non-adherent conditions. All three clones exhibited c-kit epitope by immunofluorescence (Fig. S8).
Figure 5. Clonal–derived c-kit+ cells present multipotent differentiation capacity.
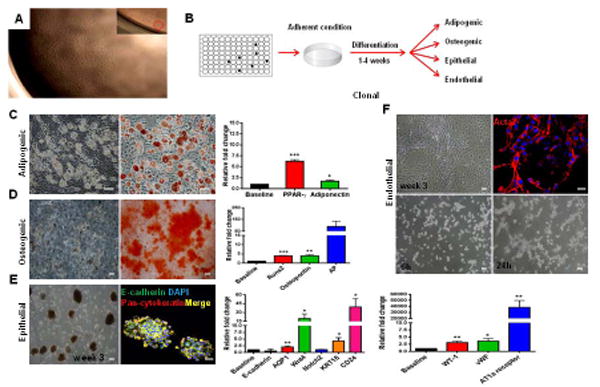
(A) C-kit+ derived clones were obtained by carrying out two serial dilutions in a 96-well plate. Magnification 4x. Insert shows 1 cell/well.
(B) Schematic representation of the experimental procedure.
(C) Adipogenic differentiation exhibited lipid droplet accumulation, Oil Red O positivity, and PPAR-γ (***P<0.0001) and adiponectin (*P=0.036) up-regulation.
(D) Osteogenic differentiation demonstrated Alizarin Red S positivity, and Runx2 (***P<0.0001), osteopontin (**P=0.0012), and AP (P=0.059) up-regulation.
(E) C-kit+ clonal cells formed packed clusters that detached and acquired an embryoid body-like morphology after 3 weeks. Epithelial spheres stained for E-cadherin and pan-cytokeratin. AQP1 (**P=0.009), Wnt4 (*P=0.023), Notch2 (P=0.8), KRT18 (**P=0.0044), and CD24 (*P=0.011) were up-regulated.
(F) In endothelial medium, c-kit+ cells formed myotube-like structures that stained for Acta2. Matrigel assay revealed tube formation at 24h. WT-1 (**P=0.0036), vWF (*P=0.017), and AT1a receptor (**P=0.0065) were up-regulated.
Cell nuclei are stained blue with DAPI. Scale bars represent 50 μm for C-F and 20 μm for confocal images (E,F). Error bars represent mean ± SEM.
After growing the c-kit+-derived clones in differentiation media (Fig. 5B), they all exhibited plasticity. In adipogenic medium, they accumulated lipid droplets that stained for Oil Red O, and exhibited up-regulation of PPAR-γ and adiponectin (Fig. 5C). In osteogenic medium, Runx2, osteopontin, and AP were up-regulated, and cultures stained for Alizarin Red S (Fig. 5D). In epithelial medium, embryoid body-like morphology was observed, and the genes involved in epithelial commitment were up-regulated (Fig. 5E). Co-localization of pan-cytokeratin and E-cadherin was also observed in the epithelial spheres (Fig. 5E).
C-kit+ cells form nephrospheres when grown in non-adherent conditions
Sphere-forming assays have been used, both retrospectively and prospectively, to investigate stem cells and progenitors in many tissues during development and in the adult [35]. A central tenet of sphere-forming assays is that each sphere is derived from a single cell and is therefore clonal.
Accordingly, non-clonal (non-single cell derived) and clonal (single cell-derived) c-kit+ cells, previously grown in adherent conditions, were dissociated into single cells and grown at clonal density (1×103 cells/well), in 6-well plates, in nonadherent conditions (Fig. 6A). Primary spheres were formed by proliferation instead of aggregation and were visible after 4 days (Fig. 6B) at a frequency of ∼2.5% of the initially plated cells (Fig. 6C). The majority of the spheres were small, measuring 40-100 μm (Fig. 6C). The spheres were passaged a minimum of three times, demonstrating self-renewal capacity. Hence, c-kit+ cells clearly are clonogenic supporting their identity as a true stem cell. However, the higher proliferation rate was observed when plating cells in adherent conditions, likely reflecting the importance of cell-cell interaction and cell adhesion for c-kit+/Lin− cell growth.
Figure 6. Non-clonal and clonal-derived c-kit+ cells form nephrospheres.
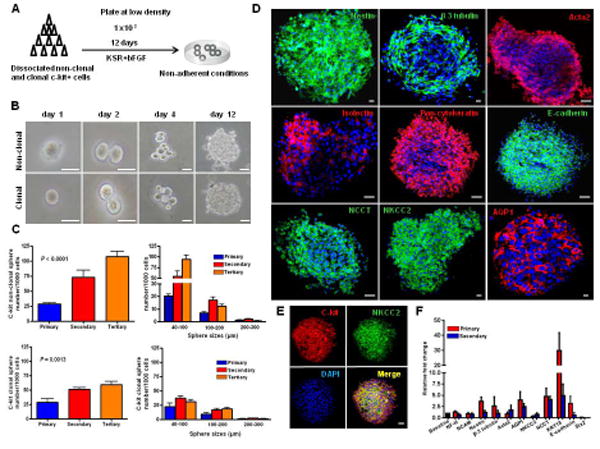
(A) Experimental procedure used to generate nephrospheres from c-kit+ cells.
(B) An example of clonal sphere growth obtained from non-clonal and clonal c-kit+ cells.
(C) Primary spheres were formed at a frequency of ∼2.5% of the initially plated cells. When dissociated to single cells, secondary and tertiary spheres arose from both non-clonal and clonal c-kit+ cells, and measured on average 40-100 μm.
(D,E) Nephrospheres stained for nestin, β-3 tubulin, Acta2, isolectin, pan-cytokeratin, E-cadherin, NCCT, NKCC2, and AQP1. C-kit costained for NKCC2.
(F) Gene expression by real-time PCR analyzed by the fold change (2ΔΔCt) in primary and secondary spheres. Nestin (P=0.008), NCCT (P=0.043), KRT18 (P=0.008) were significantly up-regulated, while SIX2 was down-regulated (P<0.0001).
Scale bars represent 50 μm (B) and 20 μm for confocal images (D,E). Error bars represent mean ± SEM.
C-kit-derived spheres exhibited markers from both neuroectoderm and mesoderm progeny (Fig. 6D), including nestin, β-3 tubulin, Acta2, isolectin, pan-cytokeratin, E-cadherin, and markers found in the kidney (NKCC2, NCCT, and AQP-1). Co-localization of c-kit receptor and NKCC2 was observed in the spheres (Fig. 6E). Up-regulation of those genes was observed in primary and secondary spheres (Fig. 6F).
Regenerative capacity of c-kit+/Lin− cells after acute ischemia-reperfusion injury
As a final test of the regeneration capacity of kidney neonatal c-kit+ stem cells, we assessed their ability for in vivo tissue repair. To evaluate the potential of c-kit+/Lin− cells to improve renal function in vivo, we utilized the model of acute ischemia-reperfusion injury (IRI) [36]. This model which mainly affects proximal tubular function, also affects the glomeruli involving podocyte foot process effacement [37].
We injected c-kit+/Lin− cells (n=8), MSCs (n=6) or saline (n=12) following IRI into the aorta immediately upstream of the renal arteries, while gently clamping the aorta below the kidneys. Animals were followed for 8 days. C-kit+/Lin− cells promoted renal functional recovery as demonstrated by improvement of creatinine and BUN at day 4. (Fig. 7A). MSCs improved renal function at day 2. C-kit+/Lin− cells and MSCs-treated animals exhibited not only a higher proliferation of surviving epithelial tubular cells in comparison to the control (Fig. 7B) but also a less severe kidney injury score (Fig. 7C), as indicated by less tubular damage compared to the control group (Fig. 7D).
Figure 7. C-kit+/Lin − cells present multi-compartment engraftment after acute ischemia-reperfusion injury (IRI).
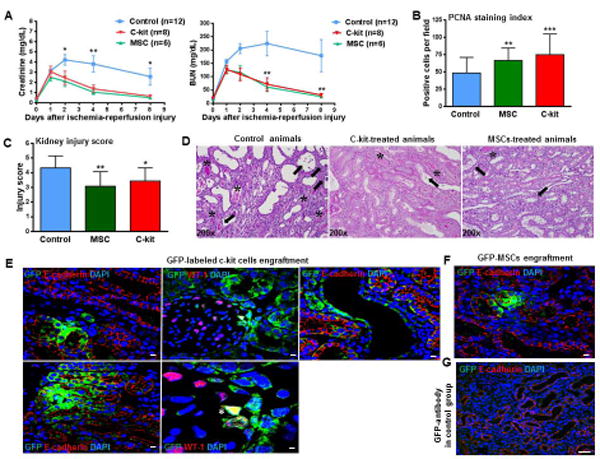
(A) C-kit+ cells and MSCs (mesenchymal stem cells) administration immediately after reflow to animals with IRI improved renal function as assessed by creatinine (*P<0.05, MSC-treated animals versus saline at day 2; **P<0.01, MSC- and c-kit-treated animals versus control at day 4, *P<0.05, MSC- and c-kit-treated animals versus control at day 8) and blood urea nitrogen measurements (BUN) (**P<0.01 MSC- and c-kit-treated animals versus control at days 4 and 8).
(B) MSC- and c-kit+-treated animals showed significantly higher number of proliferating cells compared to the control group (**P<0.01 and ***P<0.001, respectively).
(C) Injury score in the cortex and outer medulla was lower in MSC- and c-kit+- treated animals compared to the control group (**P<0.01 and *P<0.05, respectively).
(D) Representative PAS (Periodic Acid Schiff) staining in all three groups (asterisk, casts; arrows, brush border loss, necrosis and/or tubular dilation), magnification 200x.
(E) GFP-labeled c-kit+ cells exhibited tubular (left panels), glomerular (middle panels), and vascular (right panel) engraftment. These cells were integrated into the tubular wall and exhibited co-localization with E-cadherin. In glomeruli, GFP-labeled c-kit+ cells were found in Bowman's capsule and a few of them stained for WT-1 (*).
(F) GFP-MSCs exhibited tubular engraftment and stained for E-cadherin.
(G) GFP-antibody in saline group.
Scale bars represent 20 μm for confocal images (E-G). Error bars represent mean ± SEM in (A) and mean ± SD in (B,C).
Immunofluorescence staining with an anti-GFP antibody and staining for E-cadherin indicated that c-kit+/Lin− cells were integrated into tubules in all 8 animals studied (Fig. 7E). C-kit+/Lin− cells also engrafted into glomeruli and vessels in 3 of 8 animals (Fig. 7E). Most of GFP-labeled c-kit+ cells engrafted into glomeruli were found in Bowman's capsule, while a few of them were also seen in podocytes, as demonstrated by the co-localization with WT-1 (Fig. 7E). On day 8 after ischemia-reperfusion injury, the number of GFP-positive c-kit cells expressing E-cadherin was 11.5± 1.1% of all tubular cells counted in 20x fields with 2x zoom (∼1000 epithelial tubular cells counted). For MSCs, that number was significantly lower: 7.7 ± 1.5% (∼800 epithelial tubular cells counted; P=0.044) (Fig. 7F). No engraftment into vessels and glomeruli in MSCs-treated animals was observed. There were also GFP+-cells observed within the lumen of the tubules, indicating that some cells may have been eliminated in the urine Anti-GFP antibody in saline sections was used as a control (Fig. 7G).
Discussion
Here, we demonstrate that c-kit+ cells found in neonatal rat kidneys exhibit the fundamental properties of stem cells, including clonogenicity, self-renewal, multipotential capacity for commitment to mesoderm and neuroectoderm progeny, and contribute to kidney repair.
We detected c-kit+ cells in the thick ascending limb (TAL) of Henle's loop, a MM-derived structure. MM-derived cells also express epithelial, mesenchymal, endothelial, neuronal, and renal differentiated cell markers [32,38-40], as did our neonatal c-kit+ cells. Notably, immature tubules can also express vimentin and epithelial markers, such as ZO-1 [39]. More recently, human inducible pluripotent stem cells-derived OSR1+ cells differentiated into intermediate mesoderm, the precursor structure of the MM and ureteric bud, and exhibited not only tubular markers but also glomerular and vascular markers [41].
During neonatal rat kidney development, intense proliferation is seen in S-shaped bodies, immature tubules, and undifferentiated cells [42]. Several genes are up-regulated during that period, including the c-kit receptor in the MM and ureteric bud [29]. Exogenous SCF expands c-kit+ population from both renal interstitium and hemangioblasts, and can accelerate kidney development [43]. Additionally, studies on transgenic mice confirmed c-kit expression in hemangioblasts, MM, and also in the epithelial cells of the distal tubules, collecting ducts, ureter and bladder [44]. However, further studies on in vivo lineage tracing of endogenous c-kit+ population would bring additional information whether c-kit+ cells invade the condensing MM from outside, are a mesenchymal population of cells distinct from the MM cells that will became renal epithelia, or derive from some distinct MM progenitor cell [43,45-47].
The TAL segments of the nephron reside in areas of low oxygen tension, suggesting that these segments may represent an hypoxic niche of stem cells, as described in other organs and tissues [48]. Recently, the adult progenitor/stem cell marker Lgr5 was also identified in S-shaped body segments dedicated to generating the TAL [49]. However, Lgr5 positive cells exhibited a different gene profile from the c-kit+ population and their expression was restricted to cell clusters within developing nephrons in the cortex until post natal day 7, as opposed to c-kit+ cells that were detected in adult TAL segments.
Moreover, early stem cells and reprogramming genes Oct4, Sox2, Klf4, and c-myc are detected in developing kidneys according to the GUDMAP database [50]. This is interesting in light of studies showing that inducible pluripotent stem cells were obtained from proximal tubular cells with only two transcription factors (Oct4 and Sox2) [51] and four transcription factors (Oct4, Sox2, Klf4, c-myc) [52] from mesangial cells, suggesting that epigenetic memory might also exist in the kidney. Yet, Oct4 is dispensable for both self-renewal and maintenance of somatic stem cells in the adult mammal [53]. Furthermore, Klf4 regulates kidney epithelial tubular differentiation [54], while c-myc promotes proliferation of renal progenitors [55].
Sphere-forming assays were tested in different adult murine and human tissues, including anterior pituitary, prostate, dermis, pancreas, cornea, retina, breast and heart, and are a useful tool to test the potential of cells to exhibit stem cells traits, although not considered a read-out of in vivo stem cell activity [35]. Here, we showed that c-kit-derived nephrospheres exhibited markers of neuroectoderm and mesoderm progeny. It is noteworthy that common sphere features include the presence of stem cells, progenitors, and differentiated cells, the expression of nestin, routinely used for detection of neural stem cells but also characteristic for progenitor epithelial cells, and the ability of the sphere-derived cells to differentiate into other cell types in addition to their own tissue-specific cell type. More recently, E-cadherin and KRT18 were described as early differentiation markers in embryonic stem cells [56], although contrasting data showed E-cadherin involvement in somatic cell reprogramming [57].
In the present study, c-kit-mediated kidney regeneration involved multi-compartment engraftment. Importantly, our study did not rule out a paracrine effect [58] or the intrinsic mechanism of repair due to the proliferating capacity of surviving tubular epithelial cells [15,59]. Further lineage tracing studies could evaluate the involvement of c-kit+ cells in that mechanism. Engraftment of c-kit+ cells into Bowman's capsule and podocytes suggest that these cells may also play a role in repopulating kidney stem cell niches, as the one described in Bowman's capsule [7,8]. Furthermore, c-kit+ cells from different organs, including the biliary [25], bronchiolar [28], and renal epithelia [60] exhibit stem cell characteristics and epithelial differentiation. Cardiac c-kit+ cells have also been therapeutically employed after myocardium infarction [61,62]. Moreover, c-kit+ cells can promote kidney regeneration by an autocrine mechanism, as demonstrated by the shift of these cells from the papilla and medullary rays to the corticomedullary area following acute ischemia-reperfusion injury [63]. However, c-kit+ cells described here are distinct from the kidney c-kit+ side population, because the latter exhibited variable differentiation potential and failed to integrate into tubules [64,65].
Conclusion
Here, we identified a neonatal kidney-derived c-kit+ cell population that fulfills all of the criteria as a stem cell. These cells were found in the thick ascending limb of Henle's loop and exhibited clonogenicity, self-renewal, and multipotentiality with differentiation capacity into mesoderm and ectoderm progeny. Ex-vivo expanded c-kit+ cells not only exhibited a paracrine effect but also integrated into several compartments of the kidney, including tubules, vessels, and glomeruli, and contributed to functional and morphological improvement of the kidney following acute ischemia-reperfusion injury in rats. Taken together, kidney-derived c-kit+ cells have important biological and therapeutic implications in regenerative medicine.
Supplementary Material
Supplementary Fig. 1 – C-kit expression in adult rat kidney in the thick ascending limb of Henle's loop. (A,B) C-kit+ cells (green) were detected in the cortex and in the medulla, and exhibited co-localization with E-cadherin (red, upper panels) and Na-K-2Cl co-transporter (red, lower panels).
Supplementary Fig. 2- FACS analyses for c-kit positivity at different passages of c-kit+ cells in comparison to rat bone cells and using anti-mouse CD117-PE conjugated antibody. The vertical or horizontal axes on each histogram indicate the proportion of positive cells. (A) In early passage cells, the left panel reflects the isotype control and the right panel shows the proportion of positive cells (88.6 ± 5.5%). (B) In late passage cells, the left panel reflects the isotype control and the right panel shows the proportion of positive cells (76.2 ±8.6%). (C) Rat bone cells were isolated and comprised two different populations according to the size (first panel). The middle panels correspond to the isotype controls and the lower panels show the proportion of c-kit positive cells on the horizontal axis (average 7.6 ± 6.8%).
Supplementary Fig. 3 – Characterization of c-kit+/Lin− cells by flow cytometric analyses. (A) Flow cytometric analysis of c-kit+/Lin− cells: The horizontal axis of each histogram indicates the proportion of positive cells for each intra-nuclear marker OCT4, SOX2, KLF4, and C-MYC (orange line) in comparison to unstained cells (red line) and to secondary antibody alone (blue line). All secondary antibodies were Alexa-Fluor 568. Values are expressed as mean ± SD percentage of positive cells found. (B) Flow cytometric analysis of c-kit+/Lin− cells for Nestin, NF-H, Pax2, CD24, CD133, PECAM-1, Acta2, and CD90. The horizontal axis of each histogram indicates the proportion of positive cells for each intracellular or surface protein (orange line) in comparison to unstained cells (red line) and to secondary antibody alone (blue line). All secondary antibodies were Alexa-Fluor 568, except CD90-conjugtaed FITC. Values are expressed as mean ± SD percentage of positive cells found. Abbreviations: Neurofilament-heavy chain (NF-H), paired-box gene 2 (Pax2), platelet-endothelial cell adhesion molecule (PECAM-1), and alpha-actin 2 (Acta2).
Supplementary Fig. 4 – Characterization of c-kit+/Lin− cells in culture. (A) c-kit+/Lin− late passage cells grow in monolayers. C-kit+/Lin− cells were subcultured for more than a year (> 100 passages) without any evidence of senescence or growth arrest. The image reflects an example of a late passage cell (passage 50). Scale bar represents 100 μm. (B) Telomerase activity. Positive telomerase activity expressed by Log10 (delta Fluorescein/delta Rhodamine) in relative fluorescence units analyzed in passages (P) 11-24, 43-52, 65-66 of c-kit+/Lin− cells in comparison to the positive control (cell extract) and to the neonatal rat kidney (P = NS). (C) Karyotyping by G-banding method: C-kit+/Lin− cells exhibited normal karyotyping ∼5 passages after sorting.
Supplementary Fig. 5 - C-kit+ early and late passage cells undergo adipogenic, osteogenic, and epithelial differentiation. (A) C-kit+ early (P17-P22) and late (P48-P50) passage cells exhibited lipid droplet accumulation that stained positive for Oil Red O after adipogenic differentiation. Mesenchymal stem cells (MSCs) were used as a positive control. (B) Oil Red O quantification by absorbance (at 510 nm) showing a higher degree of staining in MSCs group in comparison to early passage and late passage cells (**P<0.01 and ***P<0.001, respectively). (C) PPAR-γ and adiponectin calculated as relative fold change in differentiated c-kit+ cells in comparison to MSCs (P=0.11 and **P=0.0024, respectively). (D) C-kit+ early (P17-P22) and late (P46-P50) passage cells exhibited calcium deposition that was positive for Alizarin Red S. MSCs were used as a positive control. (E) Alizarin Red S quantification by absorbance (at 550 nm) exhibiting a higher degree of staining in MSCs in comparison to early and late passage cells (***P<0.0001). (F) Runx2, osteopontin, and alkaline phosphatase (AP) calculated as relative fold change in differentiated c-kit+ cells in comparison to MSCs (*P=0.029, *P=0.012, ***P=0.0007, respectively). (G) C-kit+ late passage cells (P50-P52) formed packed clusters that detached and acquired an embryoid body-like morphology after 3 weeks growing in epithelium medium.
Scale bars represent 50 μm. Error bars represent mean ± SEM.
Supplementary Fig. 6 –In vitro and in vivo endothelial differentiation of control cells. (A) In vitro Matrigel tube formation assay performed with c-kit+ late passage (P50-71) cells (scale bar represents 50 μm). (B) Matrigel assay at 6 hours showing that the number of tubes was not different between c-kit+ early (P15-P20) and late passage (P50-P55) cells, but was lower than MSCs (***P<0.001 and ***P<0.001, respectively). At 24h, both c-kit+ early and late passage cells formed more tubes than MSCs (***P<0.001 and ***P<0.001, respectively), although the number of tubes in c-kit+ late passage cells were higher than c-kit+ early passage cells (**P<0.01). MSCs formed longer tubes in comparison to c-kit+ early and late passage cells at both 6h (***P<0.001 and ***P<0.001, respectively) and 24h (***P<0.001 and ***P<0.001, respectively). Pictures represent three different experiments in triplicate. (C) Human umbilical vein endothelial cells (HUVEC) were used as a positive control of in vivo study of endothelial differentiation. They grew for 1 week in endothelial medium (EGM-2), were embedded in a Matrigel plug (n=2; 2×106 cells), and then injected into NOD-SCID mice. After 2 weeks, the Matrigel plug was removed, and stained with H&E. HUVEC formed network connections inside the Matrigel plug (scale bars represent 50 μm). (D) GFP-labeled HUVEC stained for vWF inside the Matrigel plug (confocal images, scale bars represent 10 μm). (E) GFP-labeled HUVEC stained for Acta2 inside the Matrigel plug (scale bar represents 10 μm). (F) Negative control of endothelial differentiation based on the injection of a Matrigel plug containing only EGM-2 into NOD-SCID mice. There were a few cells infiltrating the Matrigel plug and they did not form network connections (H&E staining scale bar represents 50 μm).
Supplementary Fig. 7 - Calcium (Ca2+) transient of undifferentiated and differentiated c-kit+ cells, response of these cells to different vasoactive agents, and qPCR data after differentiation according to time in endothelial medium (EGM-2). (A) There was no difference on Ca2+ peak after Ang II administration at doses of 10 nM and 100 nM (P=0.19) in differentiated cells. (B) The response to endothelin-1 (ET-1; 100 nM) was more intense after differentiation (P<0.0001). (C) ET-1 response was dose-dependent (P<0.0001). (D) ET-1 response was related to extracellular Ca2+ (1.8 nM) and sodium (137 mnol/L), since their absence in the solution attenuated the response in differentiated cells (P<0.0001). (E) Extracellular Ca2+ and sodium absence attenuated ET-1 response more intensely in undifferentiated cells in comparison to differentiated cells (P<0.0001). (F) Endothelin Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (G) There was a biphasic attenuation of ET-1 response after antagonists administration; ETB antagonist (BQ-788 1 μM; P<0.0001) attenuated the response earlier whereas ETA antagonist (BQ -123 1 μM; P<0.0001) had a later effect. (H) The response to prostaglandin F2α (10 μM) was higher in differentiate cells (P<0.0001). (I) Prostaglandin F2α (PGF-2α, 0.1 μM) Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (J) Prostaglandin F2α response was attenuated after SQ 29,548 administration (P<0.0001). (K) The response to bradykinin (100 nM) was higher in undifferentiated cells (green line) in comparison to differentiated cells (red line; P<0.0001). (L) Bradykinin Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (M) The response to bradykinin was specifically mediated by B2 receptor, since HOE 140 (10 μM) attenuated Ca2+ influx (P<0.0001). (N) That response was dose-dependent, since HOE 140 1 μM had no effect. (O). B1 antagonist Lys-(des-Arg9, Leu8)-bradykinin trifluoracetate salt had no effect on bradykinin administration. (P) Gene expression by real-time PCR analyzed by the fold change (2ΔΔCt) according to time in the endothelium medium for endothelin type Ia receptor (P=0.0001), PGF-2α receptor (P=0.0001), and bradykinin B2 receptor (P=0.066). Data represent mean ± SEM.
Supplementary Fig. 8 - C-kit expression in different clones (1 cell/well) obtained in 96-well plates. C-kit receptor (red) was detected by immunofluorescence in three different clones (single-cell derived) obtained by serial dilutions in 96-well plates. Cell nuclei are stained blue with DAPI.
Table S1. Gene expression of c-kit+ /Lin- cells by quantitative PCR (qPCR). Results are expressed according to the mean Ct (threshold cycle).
Acknowledgments
This work was supported by a postdoctoral research fellowship grant (1KD07-33958) from the James and Esther King Florida Biomedical Research Program to E.B.R., and National Institutes of Health RO1 grants HL107110 and AG025017 to J.M.H.
Special thanks to Carmen Perez for preparing the histologic sections, Irene Margitich for technical support, Wayne Balkan for carefully reading the paper, Shannon Opiela and Jay Enten for the assistance at FACS facility, and Qinghua Hu for providing the GFP-MSCs cells.
Footnotes
Author contributions: E.B.R.: conception and design, data collection and analysis, manuscript writing; S.A.G.: conception and design, data collection and analysis; R. D.: conception and design, data collection and analysis; C.P.: data collection; C.R.: conception and design; R.M.K.T.: data collection; B.O.: data collection; D.C.: data collection; P.R.: data analysis. J.R.: manuscript writing; J.H.: conception and design, manuscript writing
Competing interests: The authors declare that they have no competing interests.
Conflict of interest: The authors declare that they have no conflict of interest.
References
- 1.Maeshima A, Yamashita S, Nojima Y. Identification of renal progenitor-like tubular cells that participate in the regeneration processes of the kidney. J Am Soc Nephrol. 2003;14:3138–3146. doi: 10.1097/01.asn.0000098685.43700.28. [DOI] [PubMed] [Google Scholar]
- 2.Oliver JA, Maarouf O, Cheema FH, et al. The renal papilla is a niche for adult kidney stem cells. J Clin Invest. 2004;114:795–804. doi: 10.1172/JCI20921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maeshima A, Sakurai H, Nigam SK. Adult kidney tubular cell population showing phenotypic plasticity, tubulogenic capacity, and integration capability into developing kidney. J Am Soc Nephrol. 2006;17:188–198. doi: 10.1681/ASN.2005040370. [DOI] [PubMed] [Google Scholar]
- 4.Hishikawa K, Marumo T, Miura S, et al. Musculin/MyoR is expressed in kidney side population cells and can regulate their function. J Cell Biol. 2005;169:921–928. doi: 10.1083/jcb.200412167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dekel B, Zangi L, Shezen E, et al. Isolation and characterization of nontubular sca-1+lin- multipotent stem/progenitor cells from adult mouse kidney. J Am Soc Nephrol. 2006;17:3300–3314. doi: 10.1681/ASN.2005020195. [DOI] [PubMed] [Google Scholar]
- 6.Sagrinati C, Netti GS, Mazzinghi B, et al. Isolation and characterization of multipotent progenitor cells from the Bowman's capsule of adult human kidneys. J Am Soc Nephrol. 2006;17:2443–2456. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- 7.Lazzeri E, Crescioli C, Ronconi E, et al. Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J Am Soc Nephrol. 2007;18:3128–3138. doi: 10.1681/ASN.2007020210. [DOI] [PubMed] [Google Scholar]
- 8.Ronconi E, Sagrinati C, Angelotti ML, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20:322–332. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Appel D, Kershaw DB, Smeets B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20:333–343. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward HH, Romero E, Welford A, et al. Adult human CD133/1(+) kidney cells isolated from papilla integrate into developing kidney tubules. Biochim Biophys Acta. 2011;1812:1344–1357. doi: 10.1016/j.bbadis.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sallustio F, De BL, Castellano G, et al. TLR2 plays a role in the activation of human resident renal stem/progenitor cells. FASEB J. 2010;24:514–525. doi: 10.1096/fj.09-136481. [DOI] [PubMed] [Google Scholar]
- 12.Lindgren D, Bostrom AK, Nilsson K, et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol. 2011;178:828–837. doi: 10.1016/j.ajpath.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bussolati B, Bruno S, Grange C, et al. Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005;166:545–555. doi: 10.1016/S0002-9440(10)62276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest. 2005;115:1756–1764. doi: 10.1172/JCI23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphreys BD, Valerius MT, Kobayashi A, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 16.Humphreys BD, Czerniak S, DiRocco DP, et al. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A. 2011;108:9226–9231. doi: 10.1073/pnas.1100629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song J, Czerniak S, Wang T, et al. Characterization and fate of telomerase-expressing epithelia during kidney repair. J Am Soc Nephrol. 2011;22:2256–2265. doi: 10.1681/ASN.2011050447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliver JA, Klinakis A, Cheema FH, et al. Proliferation and migration of label-retaining cells of the kidney papilla. J Am Soc Nephrol. 2009;20:2315–2327. doi: 10.1681/ASN.2008111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliver JA, Maarouf O, Cheema FH, et al. SDF-1 activates papillary label-retaining cells during kidney repair from injury. Am J Physiol Renal Physiol. 2012;302:F1362–F1373. doi: 10.1152/ajprenal.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dor Y, Brown J, Martinez OI, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 21.Smukler SR, Arntfield ME, Razavi R, et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell. 2011;8:281–293. doi: 10.1016/j.stem.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez-Buylla A, Seri B, Doetsch F. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57:751–758. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 23.Miettinen M, Lasota J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol. 2005;13:205–220. doi: 10.1097/01.pai.0000173054.83414.22. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa M, Nishikawa S, Yoshinaga K, et al. Expression and function of c-Kit in fetal hemopoietic progenitor cells: transition from the early c-Kit-independent to the late c-Kit-dependent wave of hemopoiesis in the murine embryo. Development. 1993;117:1089–1098. doi: 10.1242/dev.117.3.1089. [DOI] [PubMed] [Google Scholar]
- 25.Crosby HA, Kelly DA, Strain AJ. Human hepatic stem-like cells isolated using c-kit or CD34 can differentiate into biliary epithelium. Gastroenterology. 2001;120:534–544. doi: 10.1053/gast.2001.21175. [DOI] [PubMed] [Google Scholar]
- 26.Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 27.De CP, Bartsch G, Jr, Siddiqui MM, et al. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol. 2007;25:100–106. doi: 10.1038/nbt1274. [DOI] [PubMed] [Google Scholar]
- 28.Kajstura J, Rota M, Hall SR, et al. Evidence for human lung stem cells. N Engl J Med. 2011;364:1795–1806. doi: 10.1056/NEJMoa1101324. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Schmidt-Ott KM, Yang J, Chen X, et al. Novel regulators of kidney development from the tips of the ureteric bud. J Am Soc Nephrol. 2005;16:1993–2002. doi: 10.1681/ASN.2004121127. [DOI] [PubMed] [Google Scholar]
- 30.Sakurai H, Barros EJ, Tsukamoto T, et al. An in vitro tubulogenesis system using cell lines derived from the embryonic kidney shows dependence on multiple soluble growth factors. Proc Natl Acad Sci U S A. 1997;94:6279–6284. doi: 10.1073/pnas.94.12.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barasch J, Yang J, Ware CB, et al. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell. 1999;99:377–386. doi: 10.1016/s0092-8674(00)81524-x. [DOI] [PubMed] [Google Scholar]
- 32.Nishinakamura R. Stem cells in the embryonic kidney. Kidney Int. 2008;73:913–917. doi: 10.1038/sj.ki.5002784. [DOI] [PubMed] [Google Scholar]
- 33.Ivanova L, Hiatt MJ, Yoder MC, et al. Ontogeny of CD24 in the human kidney. Kidney Int. 2010;77:1123–1131. doi: 10.1038/ki.2010.39. [DOI] [PubMed] [Google Scholar]
- 34.Stockand JD, Sansom SC. Glomerular mesangial cells: electrophysiology and regulation of contraction. Physiol Rev. 1998;78:723–744. doi: 10.1152/physrev.1998.78.3.723. [DOI] [PubMed] [Google Scholar]
- 35.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Togel F, Hu Z, Weiss K, et al. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–F42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]
- 37.Wagner MC, Rhodes G, Wang E, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 2008;283:35579–35589. doi: 10.1074/jbc.M805507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metsuyanim S, Harari-Steinberg O, Buzhor E, et al. Expression of stem cell markers in the human fetal kidney. PLoS One. 2009;4:e6709. doi: 10.1371/journal.pone.0006709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliver JA, Barasch J, Yang J, et al. Metanephric mesenchyme contains embryonic renal stem cells. Am J Physiol Renal Physiol. 2002;283:F799–F809. doi: 10.1152/ajprenal.00375.2001. [DOI] [PubMed] [Google Scholar]
- 40.Batchelder CA, Lee CC, Martinez ML, et al. Ontogeny of the kidney and renal developmental markers in the rhesus monkey (Macaca mulatta) Anat Rec (Hoboken ) 2010;293:1971–1983. doi: 10.1002/ar.21242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mae S, Shono A, Shiota F, et al. Monitoring and robust induction of nephrogenic intermediate mesoderm from human pluripotent stem cells. Nat Commun. 2013;4:1367. doi: 10.1038/ncomms2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marquez MG, Cabrera I, Serrano DJ, et al. Cell proliferation and morphometric changes in the rat kidney during postnatal development. Anat Embryol (Berl) 2002;205:431–440. doi: 10.1007/s00429-002-0262-9. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt-Ott KM, Chen X, Paragas N, et al. c-kit delineates a distinct domain of progenitors in the developing kidney. Dev Biol. 2006;299:238–249. doi: 10.1016/j.ydbio.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 44.Bernex F, De SP, Kress C, et al. Spatial and temporal patterns of c-kit-expressing cells in WlacZ/+ and WlacZ/WlacZ mouse embryos. Development. 1996;122:3023–3033. doi: 10.1242/dev.122.10.3023. [DOI] [PubMed] [Google Scholar]
- 45.Velagapudi C, Nilsson RP, Lee MJ, et al. Reciprocal induction of simple organogenesis by mouse kidney progenitor cells in three-dimensional co-culture. Am J Pathol. 2012;180:819–830. doi: 10.1016/j.ajpath.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Little M, Georgas K, Pennisi D, et al. Kidney development: two tales of tubulogenesis. Curr Top Dev Biol. 2010;90:193–229. doi: 10.1016/S0070-2153(10)90005-7. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi A, Valerius MT, Mugford JW, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Barker N, Rookmaaker MB, Kujala P, et al. Lgr5(+ve) Stem/Progenitor Cells Contribute to Nephron Formation during Kidney Development. Cell Rep. 2012;2:540–552. doi: 10.1016/j.celrep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 50.Harding SD, Armit C, Armstrong J, et al. The GUDMAP database--an online resource for genitourinary research. Development. 2011;138:2845–2853. doi: 10.1242/dev.063594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montserrat N, Ramirez-Bajo MJ, Xia Y, et al. Generation of induced pluripotent stem cells from human renal proximal tubular cells with only two transcription factors, oct4 and sox2. J Biol Chem. 2012;287:24131–24138. doi: 10.1074/jbc.M112.350413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song B, Niclis JC, Alikhan MA, et al. Generation of induced pluripotent stem cells from human kidney mesangial cells. J Am Soc Nephrol. 2011;22:1213–1220. doi: 10.1681/ASN.2010101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lengner CJ, Camargo FD, Hochedlinger K, et al. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell. 2007;1:403–415. doi: 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saifudeen Z, Dipp S, Fan H, et al. Combinatorial control of the bradykinin B2 receptor promoter by p53, CREB, KLF-4, and CBP: implications for terminal nephron differentiation. Am J Physiol Renal Physiol. 2005;288:F899–F909. doi: 10.1152/ajprenal.00370.2004. [DOI] [PubMed] [Google Scholar]
- 55.Couillard M, Trudel M. C-myc as a modulator of renal stem/progenitor cell population. Dev Dyn. 2009;238:405–414. doi: 10.1002/dvdy.21841. [DOI] [PubMed] [Google Scholar]
- 56.Galat V, Malchenko S, Galat Y, et al. A model of early human embryonic stem cell differentiation reveals inter- and intracellular changes on transition to squamous epithelium. Stem Cells Dev. 2012;21:1250–1263. doi: 10.1089/scd.2010.0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Redmer T, Diecke S, Grigoryan T, et al. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 2011;12:720–726. doi: 10.1038/embor.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perin L, Sedrakyan S, Giuliani S, et al. Protective effect of human amniotic fluid stem cells in an immunodeficient mouse model of acute tubular necrosis. PLoS One. 2010;5:e9357. doi: 10.1371/journal.pone.0009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vogetseder A, Picard N, Gaspert A, et al. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am J Physiol Cell Physiol. 2008;294:C22–C28. doi: 10.1152/ajpcell.00227.2007. [DOI] [PubMed] [Google Scholar]
- 60.Perin L, Giuliani S, Jin D, et al. Renal differentiation of amniotic fluid stem cells. Cell Prolif. 2007;40:936–948. doi: 10.1111/j.1365-2184.2007.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oskouei BN, Lamirault G, Joseph C, et al. Increased potency of cardiac stem cells compared with bone marrow mesenchymal stem cells in cardiac repair. Stem Cells Transl Med. 2012;1:116–124. doi: 10.5966/sctm.2011-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams AR, Hatzistergos KE, Addicott B, et al. Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation. 2013;127:213–223. doi: 10.1161/CIRCULATIONAHA.112.131110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stokman G, Stroo I, Claessen N, et al. Stem cell factor expression after renal ischemia promotes tubular epithelial survival. PLoS One. 2010;5:e14386. doi: 10.1371/journal.pone.0014386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwatani H, Ito T, Imai E, et al. Hematopoietic and nonhematopoietic potentials of Hoechst(low)/side population cells isolated from adult rat kidney. Kidney Int. 2004;65:1604–1614. doi: 10.1111/j.1523-1755.2004.00561.x. [DOI] [PubMed] [Google Scholar]
- 65.Challen GA, Bertoncello I, Deane JA, et al. Kidney side population reveals multilineage potential and renal functional capacity but also cellular heterogeneity. J Am Soc Nephrol. 2006;17:1896–1912. doi: 10.1681/ASN.2005111228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 – C-kit expression in adult rat kidney in the thick ascending limb of Henle's loop. (A,B) C-kit+ cells (green) were detected in the cortex and in the medulla, and exhibited co-localization with E-cadherin (red, upper panels) and Na-K-2Cl co-transporter (red, lower panels).
Supplementary Fig. 2- FACS analyses for c-kit positivity at different passages of c-kit+ cells in comparison to rat bone cells and using anti-mouse CD117-PE conjugated antibody. The vertical or horizontal axes on each histogram indicate the proportion of positive cells. (A) In early passage cells, the left panel reflects the isotype control and the right panel shows the proportion of positive cells (88.6 ± 5.5%). (B) In late passage cells, the left panel reflects the isotype control and the right panel shows the proportion of positive cells (76.2 ±8.6%). (C) Rat bone cells were isolated and comprised two different populations according to the size (first panel). The middle panels correspond to the isotype controls and the lower panels show the proportion of c-kit positive cells on the horizontal axis (average 7.6 ± 6.8%).
Supplementary Fig. 3 – Characterization of c-kit+/Lin− cells by flow cytometric analyses. (A) Flow cytometric analysis of c-kit+/Lin− cells: The horizontal axis of each histogram indicates the proportion of positive cells for each intra-nuclear marker OCT4, SOX2, KLF4, and C-MYC (orange line) in comparison to unstained cells (red line) and to secondary antibody alone (blue line). All secondary antibodies were Alexa-Fluor 568. Values are expressed as mean ± SD percentage of positive cells found. (B) Flow cytometric analysis of c-kit+/Lin− cells for Nestin, NF-H, Pax2, CD24, CD133, PECAM-1, Acta2, and CD90. The horizontal axis of each histogram indicates the proportion of positive cells for each intracellular or surface protein (orange line) in comparison to unstained cells (red line) and to secondary antibody alone (blue line). All secondary antibodies were Alexa-Fluor 568, except CD90-conjugtaed FITC. Values are expressed as mean ± SD percentage of positive cells found. Abbreviations: Neurofilament-heavy chain (NF-H), paired-box gene 2 (Pax2), platelet-endothelial cell adhesion molecule (PECAM-1), and alpha-actin 2 (Acta2).
Supplementary Fig. 4 – Characterization of c-kit+/Lin− cells in culture. (A) c-kit+/Lin− late passage cells grow in monolayers. C-kit+/Lin− cells were subcultured for more than a year (> 100 passages) without any evidence of senescence or growth arrest. The image reflects an example of a late passage cell (passage 50). Scale bar represents 100 μm. (B) Telomerase activity. Positive telomerase activity expressed by Log10 (delta Fluorescein/delta Rhodamine) in relative fluorescence units analyzed in passages (P) 11-24, 43-52, 65-66 of c-kit+/Lin− cells in comparison to the positive control (cell extract) and to the neonatal rat kidney (P = NS). (C) Karyotyping by G-banding method: C-kit+/Lin− cells exhibited normal karyotyping ∼5 passages after sorting.
Supplementary Fig. 5 - C-kit+ early and late passage cells undergo adipogenic, osteogenic, and epithelial differentiation. (A) C-kit+ early (P17-P22) and late (P48-P50) passage cells exhibited lipid droplet accumulation that stained positive for Oil Red O after adipogenic differentiation. Mesenchymal stem cells (MSCs) were used as a positive control. (B) Oil Red O quantification by absorbance (at 510 nm) showing a higher degree of staining in MSCs group in comparison to early passage and late passage cells (**P<0.01 and ***P<0.001, respectively). (C) PPAR-γ and adiponectin calculated as relative fold change in differentiated c-kit+ cells in comparison to MSCs (P=0.11 and **P=0.0024, respectively). (D) C-kit+ early (P17-P22) and late (P46-P50) passage cells exhibited calcium deposition that was positive for Alizarin Red S. MSCs were used as a positive control. (E) Alizarin Red S quantification by absorbance (at 550 nm) exhibiting a higher degree of staining in MSCs in comparison to early and late passage cells (***P<0.0001). (F) Runx2, osteopontin, and alkaline phosphatase (AP) calculated as relative fold change in differentiated c-kit+ cells in comparison to MSCs (*P=0.029, *P=0.012, ***P=0.0007, respectively). (G) C-kit+ late passage cells (P50-P52) formed packed clusters that detached and acquired an embryoid body-like morphology after 3 weeks growing in epithelium medium.
Scale bars represent 50 μm. Error bars represent mean ± SEM.
Supplementary Fig. 6 –In vitro and in vivo endothelial differentiation of control cells. (A) In vitro Matrigel tube formation assay performed with c-kit+ late passage (P50-71) cells (scale bar represents 50 μm). (B) Matrigel assay at 6 hours showing that the number of tubes was not different between c-kit+ early (P15-P20) and late passage (P50-P55) cells, but was lower than MSCs (***P<0.001 and ***P<0.001, respectively). At 24h, both c-kit+ early and late passage cells formed more tubes than MSCs (***P<0.001 and ***P<0.001, respectively), although the number of tubes in c-kit+ late passage cells were higher than c-kit+ early passage cells (**P<0.01). MSCs formed longer tubes in comparison to c-kit+ early and late passage cells at both 6h (***P<0.001 and ***P<0.001, respectively) and 24h (***P<0.001 and ***P<0.001, respectively). Pictures represent three different experiments in triplicate. (C) Human umbilical vein endothelial cells (HUVEC) were used as a positive control of in vivo study of endothelial differentiation. They grew for 1 week in endothelial medium (EGM-2), were embedded in a Matrigel plug (n=2; 2×106 cells), and then injected into NOD-SCID mice. After 2 weeks, the Matrigel plug was removed, and stained with H&E. HUVEC formed network connections inside the Matrigel plug (scale bars represent 50 μm). (D) GFP-labeled HUVEC stained for vWF inside the Matrigel plug (confocal images, scale bars represent 10 μm). (E) GFP-labeled HUVEC stained for Acta2 inside the Matrigel plug (scale bar represents 10 μm). (F) Negative control of endothelial differentiation based on the injection of a Matrigel plug containing only EGM-2 into NOD-SCID mice. There were a few cells infiltrating the Matrigel plug and they did not form network connections (H&E staining scale bar represents 50 μm).
Supplementary Fig. 7 - Calcium (Ca2+) transient of undifferentiated and differentiated c-kit+ cells, response of these cells to different vasoactive agents, and qPCR data after differentiation according to time in endothelial medium (EGM-2). (A) There was no difference on Ca2+ peak after Ang II administration at doses of 10 nM and 100 nM (P=0.19) in differentiated cells. (B) The response to endothelin-1 (ET-1; 100 nM) was more intense after differentiation (P<0.0001). (C) ET-1 response was dose-dependent (P<0.0001). (D) ET-1 response was related to extracellular Ca2+ (1.8 nM) and sodium (137 mnol/L), since their absence in the solution attenuated the response in differentiated cells (P<0.0001). (E) Extracellular Ca2+ and sodium absence attenuated ET-1 response more intensely in undifferentiated cells in comparison to differentiated cells (P<0.0001). (F) Endothelin Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (G) There was a biphasic attenuation of ET-1 response after antagonists administration; ETB antagonist (BQ-788 1 μM; P<0.0001) attenuated the response earlier whereas ETA antagonist (BQ -123 1 μM; P<0.0001) had a later effect. (H) The response to prostaglandin F2α (10 μM) was higher in differentiate cells (P<0.0001). (I) Prostaglandin F2α (PGF-2α, 0.1 μM) Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (J) Prostaglandin F2α response was attenuated after SQ 29,548 administration (P<0.0001). (K) The response to bradykinin (100 nM) was higher in undifferentiated cells (green line) in comparison to differentiated cells (red line; P<0.0001). (L) Bradykinin Ca2+ influx-induced was attenuated by 2-APB (60 μM) (P<0.0001). (M) The response to bradykinin was specifically mediated by B2 receptor, since HOE 140 (10 μM) attenuated Ca2+ influx (P<0.0001). (N) That response was dose-dependent, since HOE 140 1 μM had no effect. (O). B1 antagonist Lys-(des-Arg9, Leu8)-bradykinin trifluoracetate salt had no effect on bradykinin administration. (P) Gene expression by real-time PCR analyzed by the fold change (2ΔΔCt) according to time in the endothelium medium for endothelin type Ia receptor (P=0.0001), PGF-2α receptor (P=0.0001), and bradykinin B2 receptor (P=0.066). Data represent mean ± SEM.
Supplementary Fig. 8 - C-kit expression in different clones (1 cell/well) obtained in 96-well plates. C-kit receptor (red) was detected by immunofluorescence in three different clones (single-cell derived) obtained by serial dilutions in 96-well plates. Cell nuclei are stained blue with DAPI.
Table S1. Gene expression of c-kit+ /Lin- cells by quantitative PCR (qPCR). Results are expressed according to the mean Ct (threshold cycle).