

Abstract
A major contributor to cancer mortality is recurrence and subsequent metastatic transformation following therapeutic intervention. Therefore, in order to develop new treatment modalities and improve the efficacy of current ones, it is important to understand the molecular mechanisms that promote resistance to therapy in cancer cells. One pathway contributing to therapy resistance is autophagy, a self-digestive process that can eliminate unnecessary or damaged organelles to protect cancer cells from death. We have found that the VEGF-C/NRP-2 axis is involved in the activation of autophagy, which helps cancer cell survival following treatment. Inhibition of mTOR complex 1 activity by this axis is the underlying mechanism for the activation of autophagy. Furthermore, we identified two VEGF-C/NRP-2-regulated genes, LAMP-2 and WDFY-1 that have previously been suggested to participate in autophagy and vesicular trafficking. Up-regulation of WDFY-1 following VEGF-C or NRP-2 depletion contributes to cytotoxic drug-mediated cell death. Together, these data suggest a link between the VEGF-C axis and cancer cell survival despite the presence of chemotherapy-induced stress. Effective targeting of this pathway may lead to the development of new cancer therapies.
Keywords: VEGF-C, NRP-2, autophagy, therapy resistance
Introduction
A delicate balance between cell survival and cell death is crucial for maintaining normal physiological homeostasis. This balance is dysregulated in cancer cells via alterations in pathways regulating apoptosis, necrosis, and autophagy, which constitutes an important mechanism of therapeutic resistance. The recurrence of cancer after therapy arises from a subset of cells that acquire the ability to survive during therapeutic stress. These cells also show enhanced metastatic properties and lead to cancer mortality (1). A key mechanism that confers stress tolerance and enables cancer cells to survive under stress is macroautophagy, most commonly known as autophagy (2, 3).
Autophagy is a regulated catabolic pathway that promotes lysosomal degradation of damaged proteins, cellular organelles, and other macromolecules (4-9). This self-digestion process, which facilitates the recycling of bioenergetic components, is activated by a number of stimuli, including the presence of reactive oxygen species, deprivation of growth factors, DNA damage, and cytotoxic drugs (10-12). Autophagy dysregulation is associated with a number of disease states, including cancer (6, 12, 13). Autophagy plays different roles during the initiation and progression of cancer (2, 14, 15). While autophagy acts as a tumor suppressor during the initiation phase of cancer, it promotes tumor progression and metastasis in established cancers (2, 16). Metastatic cancer cells that usually grow in a nutrient-poor microenvironment utilize autophagy to fulfill their high metabolic demand. Autophagy can facilitate survival during anchorage-independent growth or anoikis, and promotes therapeutic resistance (17, 18). Furthermore, a recent study indicated that genetic or pharmacologic inhibition of autophagy sensitized tumor cells to anti-cancer treatment (19). During therapy resistance, autophagy protects cancer cells from necrotic death by removing organelles damaged by treatment with chemotherapeutic drugs (2). Autophagy has been demonstrated to be a survival mechanism in castration-resistant prostate cancer cells (20), (21, 22). Additionally, pancreatic ductal adenocarcinoma cells display high basal levels of autophagy, which contributes to their intrinsic treatment resistance (23).
Vascular endothelial growth factor–C (VEGF-C), a member of the VEGF family of proteins, induces the formation of new lymphatic vessels, a process known as lymphangiogenesis (24). VEGF-C binds to a heterodimer, consisting of one of two tyrosine kinase receptors (VEGFR3 or VEGFR2) and a non-tyrosine kinase receptor, neuropilin-2 (NRP-2), on lymphatic endothelial cells (25-27). Notably, VEGF-C has lymphangiogenesis-independent functions. For example, VEGF-C is often overexpressed in glioblastoma patients, even though brain tissue is void of lymphatics. VEGF-C is also a trophic factor for neural progenitors in vertebrate embryonic brain (28); and can stimulate the proliferation and survival of leukemic cells (29, 30), proliferation and migration of Kaposi’s sarcoma cells (31), and the invasion and metastasis of gastric, breast and lung cancer cells (31-33).
Previously, we observed the expression of NRP-2 in cancer cells suggesting an autocrine function of the VEGF-C/NRP-2 axis (34). Interestingly we found that this axis can protect prostate and pancreatic cancer cells during chemotherapeutic stress by activating autophagy. Additionally, we have found evidence that inhibition of mTOR complex 1 (mTORC1) activity by the VEGF-C axis is a potential mechanism through which autophagy is induced in cancer cells for therapy resistance. These findings therefore provide a novel mechanism through which the VEGF-C axis protects cancer cells from chemotherapy-induced stress.
Materials and Methods
Cell culture
Human prostate cancer cell lines PC3 (American Type Culture Collection, Manassas, VA) and Du145 as well as the pancreatic cancer cell line, CaPan-1, (American Type Culture Collection) were cultured at 37°C either in RPMI 1640 with L-glutamine (Invitrogen, Carlsbad, California) or in DMEM (CellGro, Manassas, VA) media supplemented with 10% fetal bovine serum (Invitrogen) and penicillin/streptomycin (Invitrogen). Stably-transfected PC3 cell lines were grown in the presence of 1μg/ml puromycin selection. PC3 and DU145 were purchased from ATCC within last two years. CaPan-1 cell line was received from Dr. Tony Hollingsworth’s laboratory at the University of Nebraska Medical Center (UNMC). STR profiling of CaPan-1 was performed at UNMC with PowerPlex 16 system (Promega, Madison, Wisconsin). An ABI 3130 Genetic Analyzer (Applied Biosystems, Inc., Carlsbad, California) was used for the profiling. The DNA profile data was cross-checked with the ATCC data bank.
Cell transfections and treatment with function-blocking antibodies
Cells were transfected with siRNAs against VEGF-C (Dharmacon RNA Technologies, Chicago, IL and Qiagen, Valencia, CA), NRP-2, VEGF-A, LAMP-2, and WDFY-1 (Dharmacon RNA Technologies, Chicago, IL) using DharmaFECT 1-4 (Dharmacon RNA Technologies). siRNA transfection was allowed to proceed 48-72 h before collection of whole-cell extract or total RNA. Alternatively, cells were treated for 48 hours with 4μg of anti-human VEGF-C (AF752, R&D Systems) or soluble rhNRP-2-Fc fused protein (2215-N2, R&D Systems). The mature form of GFP-mCherry-LC3-expressing plasmid was transfected into PC3 cells by Effectene transfection reagent per manufacturer protocol (Qiagen). Colonies were selected under puromycin selection pressure. Stable clones of PC3 expressing varying levels of GFP-mCherry-LC3 were isolated for subsequent experiments. Rapamycin (100 nM, Sigma, St. Louis, MO) was added to the cells 48hrs after transfection. Cells were harvested 24hrs after rapamycin addition.
mRNA isolation and qRT-PCR
Total RNA was isolated using an RNeasy mini kit (Qiagen, Valencia, CA) per manufacturer protocol. cDNA was prepared from ∼1μg mRNA by Transcriptor First Strand Synthesis Kit (Roche, Mannheim, Germany) with random hexamer primer. Quantiative real-time PCR (qRT-PCR) analyses were performed in an Applied Biosystems machine using Power SYBR®Green master mix following our published protocol (35) (for detail please see the supplement document).
Detail description of the microarray analysis has been provided in the supplement document.
Western blot and ELISA
Western blots were conducted using antibodies against LAMP-2 (The Developmental Hybridoma Bank at the University of Iowa), LC3, , pS6K, S6K (Cell Signaling, Billerica, MA), (Ser308), NRP-2 and rho-GDI (Santa Cruz Biotechnology, Santa Cruz, California), ATG13, WDFY-1 (GeneTex, Irvine, CA) and beta-Actin (Sigma, St. Louis, MO). pATG13 (Ser138) (Rockland Immunochemicals, Gibertsville, Pennsylavnia).
Secreted VEGF-C in conditioned media was analyzed using an ELISA kit (R&D Systems, Inc., Minneapolis, MN) according to manufacturer protocol.
Apoptosis assay and confocal microscopy
Vybrant Apoptosis Assay Kit #7, purchased from Molecular Probes (Invitrogen), was used according to the manufacturer’s protocol. Briefly, PC3, Du145, and CaPan-1 cells were seeded at a density of 1.5 × 105 cells per well of a two-well chamber slide (LabTek, Rochester, NY). VEGF-C, NRP-2, WDFY-1, and LAMP-2 were depleted and 24 hrs later the cells were treated with docetaxel for prostate cancer cell lines and gemcitabine for pancreatic cancer cell lines and 48 hrs later Bafilomycin A1 (BAFM, 10 nM; Sigma, St. Louis, MO) was added. After 24 hrs of BAFM treatment, adherent cells were incubated with 1 μl of Hoechst 33342, YO-PRO-1, and propidium iodide (PI) at room temperature for 15 min. The cells were viewed under a confocal microscope using appropriate filters. The apoptotic cells were stained with green fluorescent YO-PRO-1 dye and the dead cells were stained with red fluorescent PI dye. The blue fluorescent dye, Hoechst 33342, stains the chromatin of the cells. Experiments were repeated at least three times. A t-Test was performed with the data sets obtained to determine whether the differences between experimental groups were significant (p≤0.05).
To determine the autophagic activity, stably-transfected PC3 cells were grown on poly-L-lysine coated coverslips (BD Biosciences, Sparks, MD). VEGF-C and NRP-2 were depleted 72 hr prior to cell fixation. BAFM and docetaxel at the concentrations listed above were added. At the end of the experiment, the cells were fixed with 4% paraformaldehyde and counter stained with DAPI. Respective filters were used to image red and green LC-3II puncta. All confocal images were captured in Zeiss 710 Confocal Laser Scanning Microscope equipped with 4 lasers and images were captured and analyzed using Zeiss Zen 2010 software. For quantification the arithmetic mean intensities of red and green fluorescence images in each field were measured. The ratio of intensities of red fluorescence over green were then calculated and represented graphically. An increased ratio of red to green corresponds to increased autolysosome formation.
Results
Disruption of the VEGF-C/NRP-2 axis alters the expression of autophagy-related genes in prostate cancer cells
We previously observed that the expression of VEGF-C and NRP-2 promoted the survival of prostate cancer cells following treatment with H2O2(34). To identify potential pathways involved in this VEGF-C-mediated cell survival, we performed a microarray study of PC3 prostate cancer cells depleted of either VEGF-C or NRP-2 using SmartPool siRNAs and with scrambled siRNA as a control [Please find the detail microarray data at GEOarchive, Geo accession number GSE36085; NCBI tracking system number 16273855 (36)]. We found 34 gene-tags that were commonly up- or down-regulated in both NRP-2- and VEGF-C-depleted cells. A subset of 10 genes was validated via qPCR (Figure 1A). Two genes, LAMP-2 and WDFY-1, were studied in-depth, based upon previous reports of function and cellular localization of the proteins. LAMP-2 plays an important role in autophagy by facilitating the fusion of autophagosomes to lysosomes (37). WDFY-1 participates in vesicular trafficking (38) and shares homology with WDFY-3, which participates in aggrephagy, a selective process that degrades protein aggregates utilizing the autophagy machinery (39). We found that both the LAMP-2 and WDFY-1 proteins were up-regulated in PC3 cells following VEGF-C/NRP-2 depletion (Figure 1, B and C). Since both these proteins are involved in autophagy, we hypothesized that VEGF-C/NRP-2 axis regulates autophagy. Depletion of VEGF-C was confirmed by both ELISA and qPCR (Supplemental Figures 1A and B), while NRP-2 depletion was confirmed by both qPCR and western blot analysis (Supplemental Figures 1C and D).
Figure 1.
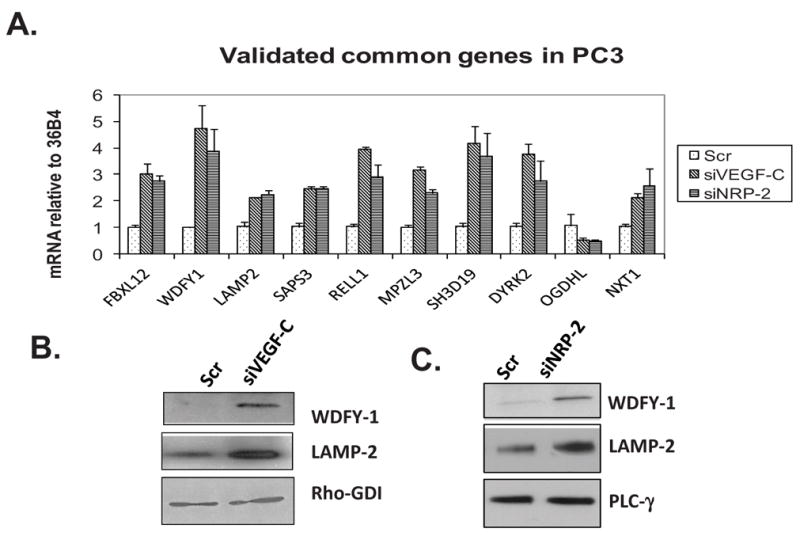
Differential gene expression analysis following depletion of the VEGF-C/NRP-2 Axis: VEGF-C or NRP-2 was depleted from PC3 cells via cocktail siRNAs for 72 hours. A, Total mRNA was isolated from cells, reverse transcribed into cDNA and used in qPCR analysis using primers specific to genes discovered via microarray analysis. B, Immunoblot analysis of WDFY-1 and LAMP-2 following the depletion of VEGF-C. C, Immunoblot analysis of WDFY-1 and LAMP-2 following the NRP-2 depletion.
VEGF-C and NRP-2 promote the activation of autophagy following stress
Starvation of cells initiates autophagy (12); therefore, we examined the effects of starvation on autophagy in VEGF-C-depleted PC3 cells. During autophagy induction microtubule-associated protein 1A/1B-light chain (LC3) becomes post-translationally modified by phosphatidyl ethanolamine and is incorporated into the membrane of the forming autophagosomes. Detection of this lipidated version of LC-3 (LC3-II) by western blot is frequently used in the laboratory to monitor autophagy (12). We monitored the level of LC3-II protein via western blotting in PC3 cells following starvation in phosphate buffer saline (PBS) at various time points. As illustrated in Figure 2A, following VEGF-C depletion, the basal LC3-II level increased compared to controls (scrambled siRNA-treated cells). LC3-II accumulation was maximal at the 1-hr time interval in VEGF-C-depleted samples. Increases in WDFY-1 and LAMP-2 levels were detected following VEGF-C depletion at every time point (data not shown). This increase in LC3-II level does not necessarily represent an enhanced autophagic activation, as LC3-II is eventually degraded by the lysosome during autophagy. Therefore LC3-II accumulates in a time-dependent manner when autophagy is inhibited as a result of decreased lysosomal degradation. In order to determine whether VEGF-C depletion induces or inhibits autophagy we performed autophagic flux experiments. In this assay the degradation of LC3-II is inhibited by treatment with Bafilomycin A1 (BAFM), which prevents the fusion of the autophagosome with the lysosome. The fold-change in LC3-II levels in each experimental condition in the presence or absence of BAFM was calculated. A higher fold-change in LC3-II level in a particular condition compared to control suggests an induction of autophagy and a lower fold-change suggests an inhibition of autophagy (13, 40, 41). We found that VEGF-C depletion blocked the autophagic flux as the fold-change in LC3-II in VEGF-C depleted prostate cancer cells after BAFM treatment were reduced compared to the fold-change in control cells (Figure 2B). We also found a similar decrease in autophagic flux following NRP-2 depletion and nutrient deprivation (Figure 2C). Overall, these results indicate that the depletion of the VEGF-C axis dysregulates autophagy.
Figure 2.
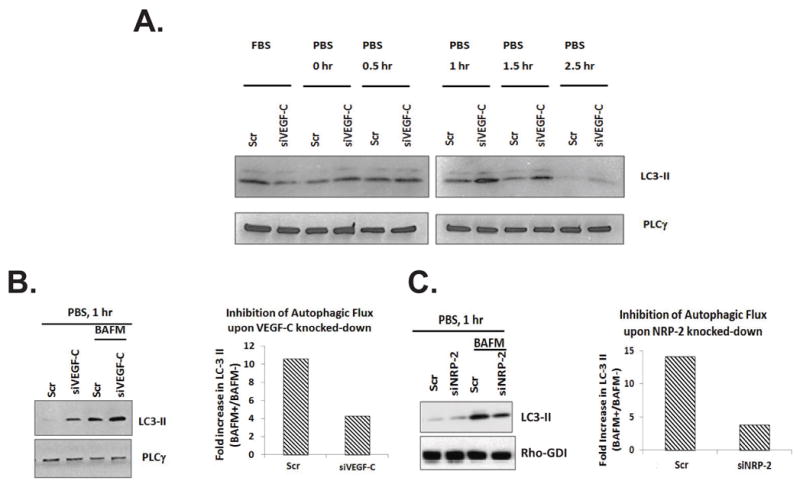
The effects of VEGF-C/NRP-2 depletion and starvation on autophagy: Following the depletion of VEGF-C or NRP-2, cells were maintained in PBS for 30, 60, 90, and 150 minutes. Following incubation in PBS, cells were lysed and A, Immunoblots were performed to examine the level of LC3-II. B, In order to gauge the amount of autophagic flux occurring in the cells, the level of LC3-II in cells treated with Bafilomycin A1 (BAFM) was compared to the level of LC3-II in untreated cells. Immunoblot analysis is shown in the right panel. The density of these bands was calculated and the ratio of LC3-II in BAFM-treated cells was divided by LC3-II in untreated cells. This value is illustrated graphically in the left panel. C, Autophagic flux analysis in starved, NRP-2-depleted cells.
VEGF-C depletion in the presence of therapeutic stress led to a dysregulation of autophagy similar to that observed during starvation. We used docetaxel, which is commonly used to treat patients with metastatic prostate cancer, at different doses (5, 10 and 20 nM) to induce autophagy. Reduced autophagic flux was observed in docetaxel-treated PC3 cells following VEGF-C depletion with smart pool siRNA cocktail (Figure 3A) or with two individual VEGF-C specific siRNAs (Figure 3B). Similar to cocktail siRNA, the individual siRNAs also significantly inhibited VEGF-C expression (Supplemental Figures 2A and B) and were capable of increasing the expression of LAMP-2 and WDFY-1 (Supplemental Figure 2C). NRP-2 depletion in docetaxel-treated PC3 cells also led to an inhibition of autophagy (Figure 3C). We repeated the autophagic-flux experiment in PC-3 cells using function-blocking antibody (anti-human VEGF-C (AF752, R&D Systems) against VEGF-C. Similar to VEGF-C siRNA, the blocking antibodies inhibited the autophagic flux in the presence of docetaxel (Figure 3D). NRP-2 function was specifically inhibited by using soluble NRP-2-Fc fused protein. This soluble chimeric protein competes with the membrane bound receptor for ligand binding and therefore inhibits its function. We once again observed a decrease in autophagic flux in docetaxel-treated PC-3 cells following addition of soluble NRP-2-Fc protein (Figure 3E). Combined, these results suggest the VEGF-C/NRP-2 axis is involved in autophagy maintenance in cancer cells during therapeutic stress.
Figure 3.
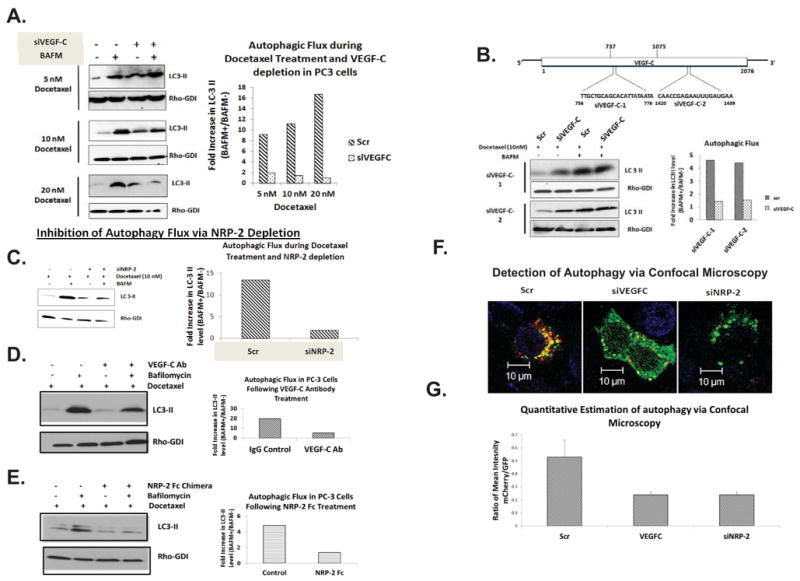
The effect of docetaxel treatment and VEGF-C/NRP-2-depletion on autophagy. A, Immmunoblot of autophagic flux analysis in VEGF-C-depleted PC3 cells treated with 5-, 10-, or 20-nM doses of docetaxel (left) and corresponding quantification (right). B, VEGF-C was also depleted from PC3 cells using two, single siRNAs that bind in different regions of the mRNA. The cells were then treated with docetaxel and the amount of autophagic flux was measured via immunoblot (left) and quantified (right). C, Autophagic flux analysis following the depletion of NRP-2 via both immunoblot analysis (left) with quantification (right). D, PC3 cells were treated with VEGF-C function-blocking antibody along with docetaxel and the amount of autophagic flux was measured via immunoblot (left) and quantified (right). E, PC3 cells were treated with soluble NRP-2 Fc chimera along with docetaxel and the amount of autophagic flux was measured via immunoblot (left) and quantified (right). F, Immunofluorescence analysis of autophagy in PC3 cells. PC3 cells transfected with mCherry-GFP-LC3 were treated with scrambled, VEGF-C, and NRP-2 siRNAs and the morphology and location of LC3 was assessed by the size and color of LC3-positive structures. Representative images are shown. G, Using the data from F, the ratio of mean intensity of mCherry to GFP was calculated and used to determine the amount of LC3 found in later autophagic structures and lysosomes (mCherry-positive) compared to early autophagosomes (GFP-positive).
To confirm that the depletion of VEGF-C or NRP-2 prevents autophagic trafficking, PC3 cells stably expressing a mCherry-GFP-LC3 construct were transfected with VEGF-C or NRP-2 siRNAs, incubated with docetaxel for 48 hrs, and imaged via confocal microscopy. Following autophagic initiation red, yellow or green LC3 punctae are visible (13). While yellow or green puncta corresponding to autophagosomes, as the autophagosomes mature and fuse with the lysosomes (autolysosome), only red puncta should be visible as GFP is acid-labile, and the lysosomal lumen is highly acidified. As shown in Figure 3F, we saw both small green/yellow puncta as well as large, red, perinuclear structures indicative of autophagic activity in control cells. In VEGF-C- and NRP-2-depleted cells, however, we observed only diffuse green staining or green puncta, indicating a decrease in autolysosomal turn-over and therefore an inhibition of autophagy. When we calculated the ratio between the red and green fluorescence intensities (a measure of autophagy activity) in twenty randomly selected fields for control and VEGF-C-/NRP-2 –depleted cells (Figure 3G), we found a decrease in the ratio of red to green which validated our previous results. Representative images for Figure 3G are shown in Supplemental Figure 3. Interestingly VEGF-A, which is functionally redundant with VEGF-C in the regulation of lymphangiogenesis and angiogenesis (42), does not regulate autophagy in PC3 during therapeutic stress. We did not observe any increase in WDFY-1 and LAMP-2 in PC3 cells (Supplemental Figure 4A) and no change in autophagic flux in docetaxel-treated PC3 cells following VEGF-A-depletion (Supplemental Figures 4B-D). These results underscore the specificity of the VEGF-C axis in regulating autophagy during stress.
To confirm that the results we obtained in PC3 cells are neither cell-line nor tumor-type specific, we repeated the autophagic flux assays in Du145 prostate cancer cells treated with 5- and 10-nM doses of docetaxel (Supplemental Figure 5A), as well as CaPan-1 pancreatic cancer cells treated with a 20-nM dose of gemcitabine (Supplemental Figure 5B). Gemcitabine is frequently used to treat pancreatic cancer patients. We found a decrease in autophagic flux following VEGF-C depletion compared to control cells, suggesting that VEGF-C depletion dysregulates autophagy initiated by cytotoxic drugs. NRP-2 depletion in gemcitabine-treated CaPan-1 cells dysregulated autophagy similarly (Supplemental Figure 5B). Similar to PC-3 cells, VEGF-C function-blocking antibody (Supplemental Figure 5C, left panel) and soluble NRP-2-Fc chimera (Supplemental Figure 5C, right panel) also inhibited autophagic flux in CaPan-1 cells and therefore confirmed the siRNA results. Overall, these data indicate that autophagic activation by the VEGF-C axis acts as a generalized stress response in cancer cells and may be responsible for their survival following chemotherapy treatment.
VEGF-C and NRP-2 depletion in cancer cells promotes enhanced cell-death following chemotherapy
To assess whether VEGF-C depletion promotes cancer cell death, VEGF-C-depleted PC3 cells and control cells were treated with docetaxel for 48 hrs prior to staining with Yo-Pro, Hoechst 33342, and Propidium Iodide (PI) and subsequent confocal imaging. Representative images of control and VEGF-C-depleted PC3 cells are shown in Figure 4A, left panel. To quantify the amount of dead and dying cells, the numbers of green, red, and hybrid red-green cells were counted in ten different fields and these numbers were divided by the numbers of blue cells per field (Figure 4A, right panel). We observed enhanced cell death in untreated VEGF-C-depleted cells compared to controls and the addition of docetaxel significantly increased the amount of cell death in VEGF-C-depleted cells versus control cells (p = 0.00584). Similar results were observed in NRP-2-depleted PC3 cells (Figures 4B and C and Supplemental Figures 6 and 7). Interestingly we observed more cell death in untreated PC3 cells following NRP-2 depletion than those in which VEGF-C was depleted (Figures 4A and B). Since NRP-2 is a receptor, we believe its depletion exerts greater downstream effect than VEGF-C and thus a greater consequence to the survival of the PC-3 cells. Therefore, to observe a further increase in cell death in NRP-2-depleted PC3 cells following docetaxel treatment, doses of docetaxel greater than 10 nM (Figure 4B) or the depletion of NRP-2 for shorter time period (48 hours instead of 72 hours; Figure 4C) was required. Similar increases in cell death were observed when the function of VEGF-C or NRP-2 was inhibited in PC-3 cells by VEGF-C function-blocking antibody (Figure 4D) or soluble NRP-2-Fc chimera (Figure 4E). As such these data indicate that signaling from the VEGF-C/NRP-2 axis protects cancer cells from therapy-induced stress. To determine whether autophagy is involved in the response of cancer cells to docetaxel treatment, PC3 cells were incubated with docetaxel, BAFM, or both agents. Autophagic inhibition by BAFM was confirmed by analyzing mCherry-GFP-LC3-expressing cells using confocal microscopy (Supplemental Figure 8A). We observed a significant (p=0.0345) increase in cell death following treatment with both docetaxel and BAFM (Figure 4F and Supplemental Figure 8B), whereas, we did not observe a significant increase in cell death following docetaxel treatment alone. Overall, the extent of cell death was similar to that observed in VEGF-C- or NRP-2- depleted cells. In order to further show that VEGF-C axis regulated autophagy is important for docetaxel resistance, we performed the following experiments. We overexpressed VEGF-C in LNCaP C4-2B and LNCaP C4-2 cells. LNCaP C4-2B is a variant of LNCaP C4-2 cells, which is capable of metastasizing to bone. These cells express low levels of endogenous VEGF-C. Interestingly LNCaP C4-2B expresses similar endogenous level of NRP-2 to PC3. C4-2 expresses detectable level of NRP-2 although lower than both C4-2B and PC3. By overexpressing VEGF-C, we activated the VEGF-C/NRP-2 axis in both LNCaP C4-2B and LNCaP C4-2 cell lines. As expected, the overexpression of VEGF-C induced autophagy and protected the cells from docetaxel-induced death (Figures 4G and H and Supplement Figures 9A, 9B and 10). NRP2 depletion in either cell line prevented VEGF-C-dependent rescue of cell survival. The survival of C4-2B and C4-2 cells due to VEGF-C overexpression was also completely abolished when autophagy was inhibited via treatment with the autophagy inhibitor, Bafilomycin. These results therefore suggest that VEGF-C/NRP-2 axis-dependent autophagy is required for its survival promoting function during chemotherapeutic stress. This conclusion was once again validated in PC3 cells (Supplemental Figure 11). Increased cell survival was observed in VEGF-C siRNA treated PC3 cells, when VEGF-C was exogenously overexpressed. This rescue of cell survival was not detected following autophagy inhibition by Bafilomycin treatment. Thus, we conclude that the cell death we observed in VEGF-C and NRP-2-depleted cells treated with docetaxel is due to an inhibition of autophagy.
Figure 4.
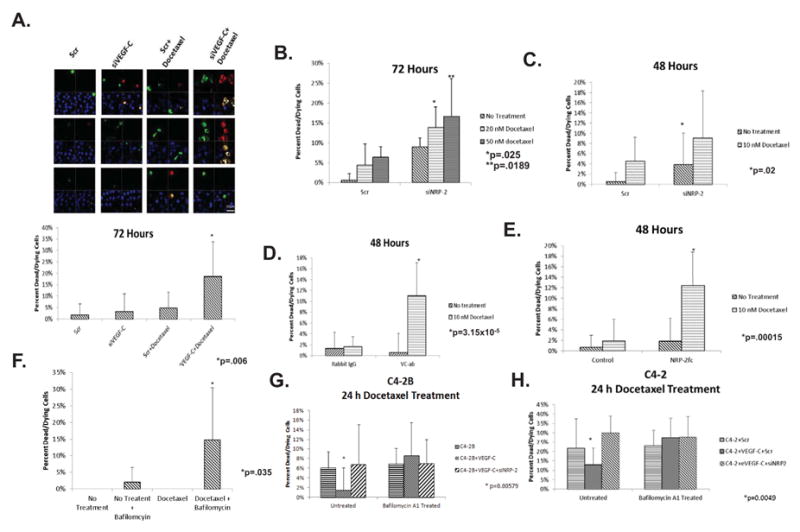
The consequence of VEGF-C/NRP-2 depletion or autophagic blockade, and subsequent drug treatment on cell viability: VEGF-C or NRP-2 were depleted from PC3 for either 72 or 48 hours before treatment with a 10-nM dose of docetaxel. The cells were then stained with YoPro, propidium iodide (PI), and Hoechst 33342. A, Cells were visualized at 63× using an LSM 710 microscope with appropriate filters. Three representative images for each treatment are shown in the left panel. In order to quantify the amount of cell death caused by each treatment, the total number of green, red, and hybrid cells were counted in 10 randomly selected fields and divided by the number of Hoechst-positive cells. The results of this analysis for PC3 cells treated with scrambled- or VEGF-C-sIRNA in the presence and absence of docetaxel are shown in the right panel. B, Cell death analysis in NRP-2-depleted cells treated with 20- and 50-nM doses of docetaxel for 72 hours. C, The resulting cell death in PC3 cells treated with scrambled- or NRP-2 siRNA following treatment with a 10-nM dose of docetaxel for 48 hours. D & E, Cell death analysis in PC3 cells treated with VEGF-C function-blocking antibody or soluble NRP-2-Fc chimera and a 10-nM dose of docetaxel for 48 hours. F. Cells were treated with a 10-nM dose of docetaxel. 24-hours before staining and confocal microscopy, a 10-nM dose of BAFM was added to the cells. G, The effect of BAFM treatment on the survival of LNCapC4-2B cells expressing exogenous VEGF-C and siNRP-2. 18 hours prior to staining, cells were treated with 10-nM doses of docetaxel and BAFM. H, Cell death analysis in LNCapC4-2 cells expressing exogenous VEGF-C. siNRP-2 was added to the cells 48h prior to analysis. 18 h before staining, the cells were treated with 10-nM doses of docetaxel and BAFM. The amount of cell death was quantified in 10 randomly selected fields. Cell death experiments were repeated no fewer than three times. In all samples, standard deviation within the data sets is shown. Furthermore, a t-Test was used to determine whether variation between experimental groups reached statistical significance (p≤0.05).
To demonstrate that autophagic activation by the VEGF-C axis was a general mechanism of tumor cell survival, we repeated the cell-survival experiments in Du145 cells as well as in CaPan-1 cells. In both cell lines, we observed a statistically-significant decrease in survival following VEGF-C depletion and subsequent treatment with chemotherapeutic agents compared to treated control cells (Figures 5A and 5B and Supplemental Figures 12 and 13). Similarly NRP-2 depletion in CaPan-1 cells also leads to increased cell death following gemcitabine treatment (Figure 5C and Supplemental Figure 14). Once again increase in cell death was observed when CaPan-1 cells were treated with VEGF-C function-blocking antibody or NRP-2 Fc chimera along with gemcitabine (Supplemental Figure 15 A and B). These data indicate that the VEGF-C axis promotes autophagy, which contributes to survival in cancer cells exposed to cytotoxic stress.
Figure 5.
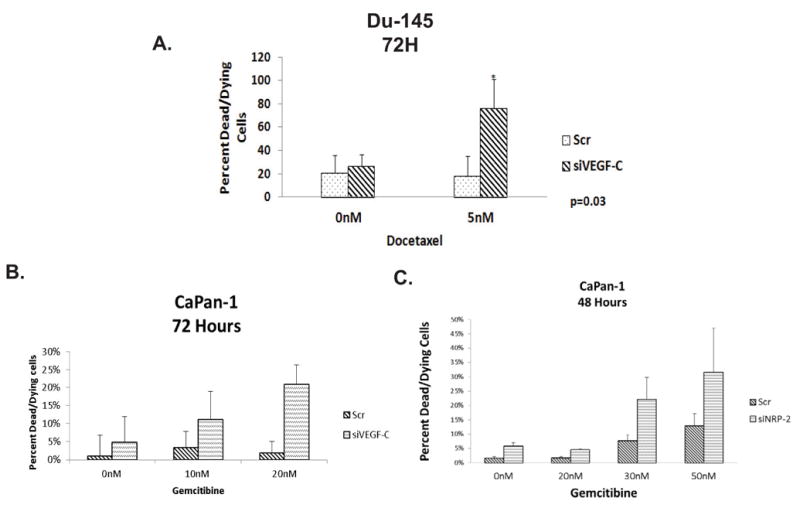
The effect of VEGF-C/NRP-2 depletion on the survival of other cell types. A, Du145 cells were treated with either scrambled or VEGF-C siRNA. 24 hours later, the cells were treated with a 5-nM dose of docetaxel. Cell death was then analyzed using the aforementioned stains and confocal microscopy. The quantification of ten fields is illustrated. B, CaPan-1 cells were seeded onto coverslips and transfected with either scrambled or VEGF-C siRNAs. 24 hours after siRNA treatment, 10- and 20-nM doses of docetaxel were added to the cells. Cell death was then visualized and the quantification of ten fields is shown. C, NRP-2 was depleted from CaPan-1 cells and the cells were then treated with 0-, 20-, 30-, and 50-nM doses of gemcitibine for 48 hours. Cell death was then visualized via confocal microscopy, the quantification of which is shown here.
Up-regulation of WDFY-1 following VEGF-C and NRP-2 depletion contributes to decreased cell viability in chemotherapy-treated cells
We previously demonstrated that LAMP-2 and WDFY-1 levels increase following VEGF-C and NRP-2 depletion (Figure 1). We observed a similar increase in LAMP-2 and WDFY-1 following inhibition of autophagy by BAFM treatment in all cell lines (Figure 6A). We therefore concluded that the upregulation of WDFY-1 and LAMP-2 occur when autophagy is inhibited through any inhibitory mechanism. Next, we tested whether increases in the levels of WDFY-1 and LAMP-2 influence the survival of cancer cells following therapeutic intervention. We found that VEGF-C depletion led to a significant (p = 0.015) increase in cell death following docetaxel treatment. Intriguingly, the co-depletion of VEGF-C and WDFY-1, which we verified by western blot (Figure 6B), maintains cell viability following docetaxel treatment (Figure 6C and Supplemental Figure 16). Enhanced cell viability was also observed in PC3 cells co-depleted of NRP-2 and WDFY-1 following docetaxel treatment than in PC3 cells depleted of NRP-2 alone (Figure 6D and Supplemental Figure 16). Similarly, the depletion of both LAMP-2 and VEGF-C also led to a decrease in cell death compared to the depletion of VEGF-C alone (data not shown), although we did not find that this difference was statistically significant. Together, these results suggest that increased levels of WDFY-1 and LAMP-2 (to a lesser extent) promote cell death following the inhibition of autophagy.
Figure 6.
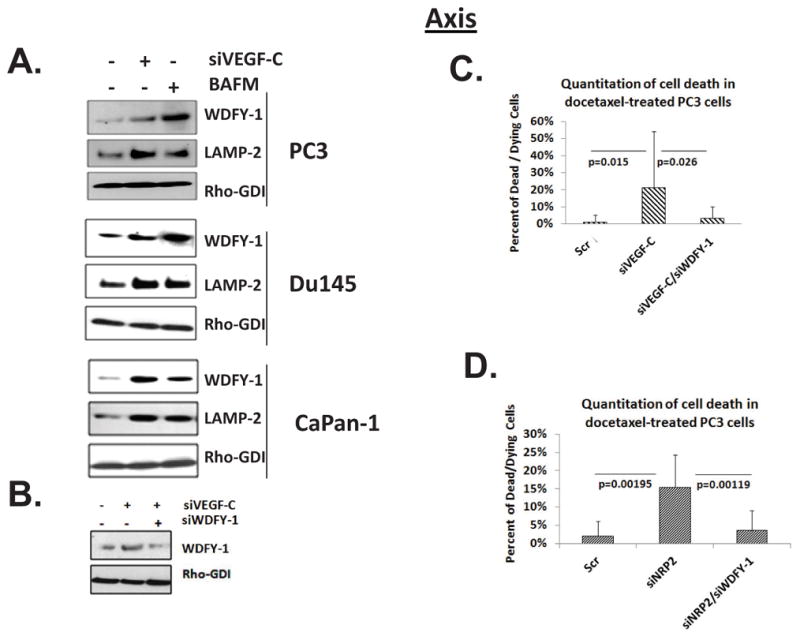
WDFY-1 promotes cell death upon inhibition of the VEGF-C/NRP-2 axis. A, Immunoblot analysis of WDFY-1 and LAMP-2 in PC3, Du145, and CaPan-1 cells treated with BAFM. B, Immunoblot analysis of WDFY-1 expression in PC3 cells depleted of VEGF-C alone or both VEGF-C and WDFY-1 siRNAs. C, PC3 cells were plated and treated with scrambled, VEGF-C, or both VEGF-C and WDFY-1 siRNAs, and the amount of cell death was calculated as described previously. D. Quantification of cell death in PC3 cells depleted of NRP-2 and both NRP-2 and WDFY-1. Again, in all graphs, the standard deviation is depicted. Furthermore, the statistical difference (p≤0.05) between samples was calculated using a t-Test.
Inactivation of mTORC1 activity by the VEGF-C/NRP-2 axis is a potential mechanism for autophagy induction
We have demonstrated that the accumulation of WDFY-1 and LAMP-2 driven by the depletion of VEGF-C and NRP-2 in cancer cells (Figures 6A) might be a consequence of autophagic inhibition. These results therefore indicated that these proteins might not regulate autophagy in cancer cells downstream of the VEGF-C axis. In search of the potential mechanisms through which the VEGF-C axis regulates stress-induced autophagy we turned our attention to our previously reported findings. Earlier, we demonstrated that during oxidative stress the VEGF-C axis maintains mTOR Complex 2 (mTORC2) activity, which is upstream of AKT (34). Interestingly, the downstream mediators of the AKT-mTOR complex 1 (mTORC1) signaling axis, the S6 protein or 4EBP (43), were not activated under these conditions in prostate cancer cells. The reason for this unexpected result remained unanswered in our previous publication (34). As active mTORC1 blocks the induction of autophagy (9), we hypothesized that the VEGF-C/NRP-2 axis actually inhibits mTORC1 activity to promote autophagy during stress (44). Indeed we observed a significant increase in phospho-S6K following the depletion of either VEGF-C or NRP-2 in PC-3 (Figure 7A) or a decrease in phospho-S6K level in docetaxel-treated LNCaP C4-2 cells (34) following VEGF-C addition (Supplemental Figure 17A and B), suggesting an inhibitory role of VEGF-C axis on mTORC1 activity. This increase in S6K phosphorylation upon VEGF-C depletion seemed to be independent of AKT as knocking down AKT level did not significantly influence phospho S6K level (Supplemental Figure 17C). A decrease in ATG13 phosphorylation following the depletion of VEGF-C or NRP-2 in PC-3 treated with docetaxel was observed (Figure 7A). Downregulation of ATG13 was regulated by mTORC1 for the inhibition of autophagy. To further confirm that this increased mTORC1 activity is responsible for the observed dysregulation of autophagy; we used rapamycin to block mTORC1 activity in VEGF-C- and NRP-2-depleted PC3 cells. Rapamycin treatment rescued the autophagic flux in PC3 cells despite decreased levels of VEGF-C and NRP-2 (Figure 7B). Similar results were obtained when we monitored autophagy by confocal microscopy in the docetaxel-treated stablely-expressing mCherry-GFP-LC3 PC3 cell line (Figure 7C). Enhanced mCherry-positive red punctae of autolysosomes, indicative of increased autophagy, were observed in VEGF-C or NRP-2-depleted PC3 cells following treatment with rapamycin. A similar decrease in ATG13 activity and an upregulation of pS6K activity following the inhibition of VEGF-C/NRP-2 axis was also observed in CaPan-1 cells (Figure 7D). Rapamycin also restored the autophagic flux in CaPan-1 cells, despite the depletion of VEGF-C or NRP-2 (Figures 7E and F). We therefore conclude that the VEGF-C/NRP-2 axis promotes autophagy via downregulation of mTORC1 function especially during therapeutic stress.
Figure 7.
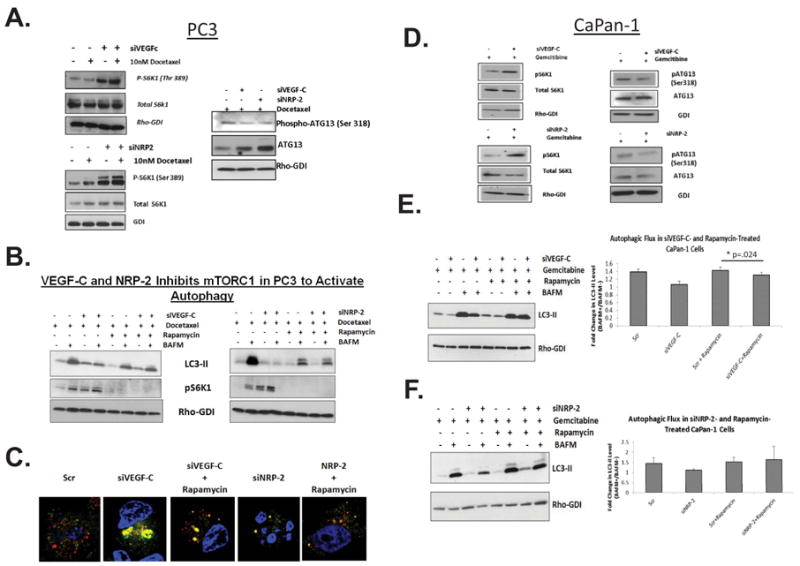
Mechanism through which VEGF-C/NRP-2 regulates autophagy. A, Immunoblot analysis of phospho-S6 kinase (Ser 389), S6K following the depletion of VEGF-C and NRP-2 (left panel) and phospho-ATG13 and ATG13 following VEGF-C and NRP-2 depletion (right panel) in PC-3 cells. B, Immunoblot analysis of LC3-II following the depletion of either VEGF-C or NRP-2 and subsequent treatment with docetaxel and rapamycin. C, Immunofluorescence analysis of autophagy in mCherry-GFP-LC3-expressing PC3 cells following the depletion of either VEGF-C or NRP-2 and subsequent rapamycin treatment. D, Immunoblot analysis of phospho-S6 kinase (Ser 389), S6K following the depletion of VEGF-C and NRP-2 (left panel) and phospho-ATG13 and ATG13 following VEGF-C and NRP-2 depletion (right panel) in CaPan-1 cells. E, Autophagic flux analysis in VEGF-C-depleted CaPan-1 cells treated with both gemcitibine and rapamycin via immunoblot (left panel) with corresponding quantification (right panel). F, Analysis of autophagic flux in gemcitibine- and rapamycicn-treated, NRP-2-depleted CaPan-1 cells via immunoblot (left panel) with corresponding quantification (right panel).
Discussion
The ability of tumor cells to survive under stress is a major obstacle to effective cancer therapy (1, 19). Autophagy has been shown to promote cancer cell survival in the presence of metabolic, hypoxic, or cytotoxic stress (6, 12) that can result in tumor dormancy and subsequent progression and metastasis (2, 17). Previously, we reported a survival-promoting function of the VEGF-C axis during oxidative stress (34). Our subsequent studies suggested that the VEGF-C axis can regulate distinct cellular mechanisms to facilitate the survival of cancer cells under different stress conditions. A clue that the VEGF-C/NRP-2 axis regulates autophagy in cancer cells came from a microarray study to identify genes that are regulated by this axis. We knocked down endogenous VEGF-C and NRP-2 separately in prostate cancer cells and identified genes whose expression was influenced by reduced levels of endogenous VEGF-C and NRP-2. We identified ten genes that were equally up-regulated or down-regulated following the depletion either VEGF-C or NRP-2 and considered them as the genes regulated by VEGF-C axis in prostate cancer. Interestingly, most of these genes are similarly regulated by VEGF-C or NRP-2 in other cancer cells, which suggests the importance of this axis in different cancers. Among these genes, we took particular interest in WDFY-1 and LAMP-2 (Figure 1). LAMP-2 is a lysosomal membrane protein, which plays a role in autophagy (36). WDFY-1 (also known as FENS-1), which contains a single FYVE domain and multiple WD40 repeats, plays a role in vesicular trafficking (37). Interestingly, a related protein WDFY-3 (ALFY) is involved in selective autophagy (aggrephagy) (38), suggesting a potential connection between WDFY-1 and autophagy. We therefore tested the hypothesis that the VEGF-C/NRP-2 axis regulates autophagy and that WDFY-1 and LAMP-2 function as downstream effectors in this pathway.
The experimental proof of this hypothesis came from studies where we tested autophagic activity in VEGF-C- or NRP-2–depleted PC3 cells during nutrient deprivation (Figure 2) and observed an autophagy-inducing function by both VEGF-C and NRP-2. To our knowledge, this is a novel observation and underscores the importance of this axis in cancer stress-resistance. It has been postulated by researchers in this field that autophagy and apoptosis are mutually exclusive (10). Since autophagy is viewed as primarily cytoprotective, it has been hypothesized that its induction should be coupled with the activation of anti-apoptotic pathways (10). Our previous study on the function of the VEGF-C axis in cancer validates this hypothesis (34). We previously demonstrated that the VEGF-C axis activates mTORC2 and promotes cancer cell survival during stress through the activation of anti-apoptotic pathways. Our current study suggests that this axis also controls autophagy. We therefore propose that due to its ability to couple autophagy and anti-apoptotic pathways, the VEGF-C/NRP-2 axis is a significant pathway in promoting cancer cell survival during stress.
We then examined the interaction between the VEGF-C axis and autophagy in the presence of docetaxel-induced cytotoxic stress. Docetaxel has been used to treat patients with metastatic, castration-resistant prostate cancer (45) and independent clinical trials have shown that docetaxel-based treatment can increase median overall survival compared to mitoxantrone and prednisone (46, 47). Docetaxel functions by stabilizing microtubules, thereby inhibiting mitotic spindle depolymerization and cell proliferation. Docetaxel also induces cell death by inhibiting various cell-survival mechanisms. The cell death-inducing function of docetaxel may be therapeutically more significant in prostate cancer as this cancer usually proliferates at a slower rate, and lethality of metastatic castration-resistant prostate cancer is due to its ability to resist death (45). Resistance to docetaxel in men with metastatic castration-resistant disease has been observed in almost 50% of patients. Accordingly, we determined whether the autophagy-inducing activity of the VEGF-C axis in prostate cancer cells is responsible for its ability to resist docetaxel-induced cell death. In VEGF-C- and NRP-2-depleted cells treated with docetaxel, we observed decrease in autophagic flux compared to control cells (Figure 3A-E). We have confirmed this observation morphologically using mCherry-GFP-LC3-expressing cells (Figure 3F and G). As expected the depletion of VEGF-C and NRP-2 in PC3 cells also made them more vulnerable to docetaxel-induced death (Figure 4A-E and Supplemental Figures 6 and 7). A similar increase in cell death was observed when PC3 cells were treated with docetaxel and at the same time autophagy was inhibited chemically (Figure 4F, Supplemental Figures 8A, B and 11). Furthermore the addition of VEGF-C to LNCaP C4-2B and C4-2 cells failed to rescue cell survival following autophagy inhibition (Figure 4G and H, Supplemental Figures 9 and 10). These results indicate a VEGF-C/NRP-2-dependent mechanism of docetaxel resistance in prostate cancer. Since both VEGF-C and NRP-2 are expressed in high levels in metastatic prostate cancer (48, 49), we assume that this axis could contribute to prostate cancer recurrence following docetaxel treatment.
To prove that our observation was not a cell-type specific phenomenon, we performed similar experiments in another prostate cancer cell line, Du145, as well as in a pancreatic cancer cell line, CaPan-1. We selected these lines because of their aggressive nature, metastatic potential, and high endogenous expression of both VEGF-C and NRP-2 (data not shown). We treated CaPan-1 cells with gemcitabine as it is the standard chemotherapeutic agent used to treat metastatic pancreatic cancer (50). Like docetaxel, resistance to gemcitabine is the major reason for its limited therapeutic efficacy (50). We observed a similar decrease in autophagic flux and increase in cell death in both Du145 treated with docetaxel as well as CaPan1 cells treated with gemcitabine (Figure 5 and Supplemental Figures 4, 9-12), which suggests that up-regulation of the VEGF-C axis is a general adaptation that promotes tumor cell survival under adverse conditions. Overall, these data strongly suggest the importance of this axis in autophagic activation.
Although the increased levels of WDFY-1 and LAMP-2 (to a lesser extent) following VEGF-C and NRP-2 depletion can function in the induction of cell death (Figure 6 and Supplemental Figure 13), they are not likely to be involved in regulating autophagy. In future studies we will unravel their roles in promoting cell death following autophagy inhibition. Our finding that the VEGF-C axis inhibits mTORC1 to activate autophagy (Figure 7) is novel and provides a mechanism through which this axis promotes resistance against treatment.
In conclusion, we have established that the VEGF-C/NRP-2 axis is important in the activation of autophagy following treatment with chemotherapy. Our data suggest that VEGF-C-induced autophagy protects cancer cells from the cytotoxic stress-induced death as well as the potential of the VEGF-C axis as a therapeutic target. Our future studies will focus on determining the mechanism(s) through which WDFY-1 activates cell death downstream of autophagy inhibition.
Unraveling these signaling pathways may assist in the development of novel treatments to overcome chemotherapy resistance.
Supplementary Material
Acknowledgments
We thank Janice A. Taylor and James R. Talaska at the Confocal Laser Scanning Microscope Core Facility at the UNMC for providing assistance and the Nebraska Research Initiative and the Eppley Cancer Center for their support of the core facility.
Grant support: NIH grant CA140432 (K Datta), Research Scholar Grant from American Cancer Society (RSG-070944-01-CSM; K Datta); Wilhelm-Sander-Stifung-fur-Krebsforscchung (MHM); NIH grants CA 121277 (D.J.Tindall), CA 91956 (D.J.Tindall), CA 125747 (D.J.Tindall), and the TJ Martell Foundation (FNDT Martell/#1-8; D.J.Tindall).
Footnotes
M.J. Stanton, S. Dutta and H. Zhang contributed equally. K. Datta and M.H. Muders contributed equally as senior authors.
The Authors declare that no conflict of interest exists.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer biology & therapy. 2011;11:127–37. doi: 10.4161/cbt.11.2.14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes & development. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–62. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annual review of biochemistry. 2011;80:125–56. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Current opinion in cell biology. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Archives of biochemistry and biophysics. 2007;462:210–9. doi: 10.1016/j.abb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–39. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 15.Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, et al. The autophagic paradox in cancer therapy. Oncogene. 2011 doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- 16.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. The Journal of clinical investigation. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochemical pharmacology. 2011;82:427–34. doi: 10.1016/j.bcp.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cellular signaling. 2011;23:1466–72. doi: 10.1016/j.cellsig.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes & cancer. 2010;1:40–9. doi: 10.1177/1947601909358324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer research. 2010;70:3667–76. doi: 10.1158/0008-5472.CAN-09-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, Kimmelman AC. A critical role for autophagy in pancreatic cancer. Autophagy. 2011;7:912–3. doi: 10.4161/auto.7.8.15762. [DOI] [PubMed] [Google Scholar]
- 24.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–76. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 25.Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. Embo J. 1997;16:3898–911. doi: 10.1093/emboj/16.13.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. Embo J. 1996;15:1751. [PMC free article] [PubMed] [Google Scholar]
- 27.Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, et al. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. Faseb J. 2006;20:1462–72. doi: 10.1096/fj.05-5646com. [DOI] [PubMed] [Google Scholar]
- 28.Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, et al. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nature neuroscience. 2006;9:340–8. doi: 10.1038/nn1646. [DOI] [PubMed] [Google Scholar]
- 29.Marchio S, Primo L, Pagano M, Palestro G, Albini A, Veikkola T, et al. Vascular endothelial growth factor-C stimulates the migration and proliferation of Kaposi's sarcoma cells. The Journal of biological chemistry. 1999;274:27617–22. doi: 10.1074/jbc.274.39.27617. [DOI] [PubMed] [Google Scholar]
- 30.Dias S, Choy M, Alitalo K, Rafii S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99:2179–84. doi: 10.1182/blood.v99.6.2179. [DOI] [PubMed] [Google Scholar]
- 31.Kodama M, Kitadai Y, Tanaka M, Kuwai T, Tanaka S, Oue N, et al. Vascular endothelial growth factor C stimulates progression of human gastric cancer via both autocrine and paracrine mechanisms. Clin Cancer Res. 2008;14:7205–14. doi: 10.1158/1078-0432.CCR-08-0818. [DOI] [PubMed] [Google Scholar]
- 32.Saintigny P, Kambouchner M, Ly M, Gomes N, Sainte-Catherine O, Vassy R, et al. Vascular endothelial growth factor-C and its receptor VEGFR-3 in non-small-cell lung cancer: concurrent expression in cancer cells from primary tumour and metastatic lymph node. Lung cancer (Amsterdam, Netherlands) 2007;58:205–13. doi: 10.1016/j.lungcan.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 33.Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer cell. 2006;9(3):209–23. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 34.Muders MH, Zhang H, Wang E, Tindall DJ, Datta K. Vascular endothelial growth factor-C protects prostate cancer cells from oxidative stress by the activation of mammalian target of rapamycin complex-2 and AKT-1. Cancer research. 2009;69:6042–8. doi: 10.1158/0008-5472.CAN-09-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Muders MH, Li J, Rinaldo F, Tindall DJ, Datta K. Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer. Cancer research. 2008;68:8770–8. doi: 10.1158/0008-5472.CAN-08-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36085
- 37.Saftig P, Beertsen W, Eskelinen EL. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–2. doi: 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- 38.Ridley SH, Ktistakis N, Davidson K, Anderson KE, Manifava M, Ellson CD, et al. FENS-1 and DFCP1 are FYVE domain-containing proteins with distinct functions in the endosomal and Golgi compartments. Journal of cell science. 2001;114:3991–4000. doi: 10.1242/jcs.114.22.3991. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto A, Simonsen A. Alfy-dependent elimination of aggregated proteins by macroautophagy: can there be too much of a good thing? Autophagy. 7:346–50. doi: 10.4161/auto.7.3.14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang AL, Boulton ME, Dunn WA, Jr, Rao HV, Cai J, Lukas TJ, et al. Using LC3 to monitor autophagy flux in the retinal pigment epithelium. Autophagy. 2009;5:1190–3. doi: 10.4161/auto.5.8.10087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Manseau EJ, et al. VEGF-A induces angiogenesis, arteriogenesis, lymphangiogenesis, and vascular malformations. Cold Spring Harb Symp Quant Biol. 2002;67:227–37. doi: 10.1101/sqb.2002.67.227. [DOI] [PubMed] [Google Scholar]
- 43.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nature reviews. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 44.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature genetics. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 45.Higano CS. New treatment options for patients with metastatic castration-resistant prostate cancer. Cancer treatment reviews. 2011 doi: 10.1016/j.ctrv.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 47.Petrylak DP, Ankerst DP, Jiang CS, Tangen CM, Hussain MH, Lara PN, Jr, et al. Evaluation of prostate-specific antigen declines for surrogacy in patients treated on SWOG 99-16. Journal of the National Cancer Institute. 2006;98:516–21. doi: 10.1093/jnci/djj129. [DOI] [PubMed] [Google Scholar]
- 48.Jennbacken K, Vallbo C, Wang W, Damber JE. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. The Prostate. 2005;65:110–6. doi: 10.1002/pros.20276. [DOI] [PubMed] [Google Scholar]
- 49.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 50.Vaccaro V, Melisi D, Bria E, Cuppone F, Ciuffreda L, Pino MS, et al. Emerging pathways and future targets for the molecular therapy of pancreatic cancer. Expert opinion on therapeutic targets. 2011;15:1183–96. doi: 10.1517/14728222.2011.607438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.